

Abstract
Objective:
To describe immunologic, virologic, and clinical HIV disease progression by HIV-1 subtype among Africans with well documented estimated dates of HIV infection (EDIs).
Design:
Prospective cohort.
Methods:
Adults and youth with documented HIV-1 infection in the past 12 months were recruited from seroincidence cohorts in East and Southern Africa and followed at 3–6 month intervals. Blood for lymphocyte subset and viral load determination was collected at each visit. Pol was sequenced from the first positive specimen to ascertain subtype. Preantiretroviral therapy disease progression was measured by three time-to-event endpoints: CD4+ cell count 350 cells/μl or less, viral load measurement at least 1 × 105 copies/ml, and clinical AIDS.
Results:
From 2006 to 2011, 615 participants were enrolled at nine research centers in Kenya, Rwanda, South Africa, Uganda, and Zambia; 579 (94.1%) had viral subtyping completed. Predominant subtypes were C (256, 44.2%), A (209, 36.1%), and D (84, 14.5%). After adjustment for age, sex, and human leukocyte antigen alleles in Cox regression analyses, subtype C-infected participants progressed faster than subtype A to all three endpoints [CD4+ hazard ratio 1.60, 95% (confidence interval) CI 1.16, 2.20; viral load hazard ratio 1.59, 95% CI 1.12, 2.25; and AIDS hazard ratio 1.60, 95% CI 1.11, 2.31). Subtype D-infected participants reached high viral load more rapidly (hazard ratio 1.61, 95% CI 1.01, 2.57) and progressed nearly twice as fast to AIDS compared to subtype A (hazard ratio 1.93, 95% CI 1.21, 3.09).
Conclusion:
Subtype-specific differences in HIV disease progression suggest that the local subtype distribution be considered when planning HIV programs and designing and defining clinical endpoints for HIV prevention trials.
Keywords: Africa, AIDS, CD4+ cell count, HIV disease progression, HIV-1 subtype, HIV-serodiscordant couples, men who have sex with men, viral load
Introduction
Although several efficacious HIV prevention interventions have been identified [1], their impact has been insufficient to end the pandemic. Improved methods, including a well tolerated and efficacious preventive vaccine, are needed. HIV type 1 (HIV-1) uses myriad strategies to evade host immune defenses. HIV-1 mutates rapidly, generating multiple subtypes (A–D, F–H, and J), six sub-subtypes (A1–4 and F1–2), circulating recombinant forms, and unique recombinant forms of HIV-1 Group M worldwide [2]. Sub-Saharan Africa (SSA) is home to only 12% of the world's population [3], yet carries the burden of 72% of HIV-infected individuals [4]. With its diversity in HIV virology, human genetics, and a heterogeneous epidemic, understanding HIV epidemiology in Africa is vital to characterize the interplay between viral subtype and host immunology.
HIV disease progression has been well described for HIV-1 subtype B in Europe and North America [5–7], subtype B′ and CR01_AE (formerly subtype E) in Thailand [8,9], subtype C in India and Brazil [10–12], HIV-1 subtypes G and F in West Africa [13], and HIV-2 in west Africa [14]. The epidemiology and disease course in East and Southern Africa, where HIV-1 subtypes A, D, and C predominate [2], have also been characterized [15–19]. Subtype C predominates in Southern Africa, where HIV prevalence is higher than in East Africa, for reasons that may include differential transmissibility or pathogenesis. Direct comparison between subtype C and other subtypes is less available. Published data describe HIV-1 to be more pathogenic than HIV-2, HIV-1 subtype D to be more pathogenic than subtype A [15–17,19–23], and subtype C more pathogenic than A and D [24].
In preparation for HIV vaccine efficacy trials in SSA, the International AIDS Vaccine Initiative (IAVI) partnered with research centers in Kenya, Rwanda, South Africa, Uganda, and Zambia to develop prospective HIV incidence cohorts of adult participants at risk for HIV-1. We describe the immunologic, virologic, and clinical disease progression for HIV in this cohort.
Methods
Ethical considerations
All participants provided informed consent. This study was approved by: the Kenya Medical Research Institute Ethical Review Committee, the Kenyatta National Hospital Ethical Review Committee of the University of Nairobi, the Rwanda National Ethics Committee, the Uganda Virus Research Institute Science and Ethics Committee, the Uganda National Council of Science and Technology, the University of Cape Town Health Science Research and Ethics Committee, the University of Zambia Research Ethics Committee, the Bio-Medical Research Ethics Committee at the University of KwaZulu Natal, and the Emory University Institutional Review Board.
Study population and clinical procedures
This is a prospective, multicenter cohort study of HIV-infected men and women aged 16–60 years with recent HIV infection of known duration. The network comprised nine clinical research centers (CRCs) in five countries (Kenya: the Kenya Medical Research Institute/Centre for Geographic Medicine Research – Coast, Kilifi, and Mtwapa, and University of Nairobi Kenya AIDS Vaccine Initiative, Nairobi; Rwanda: Project San Francisco, Kigali; South Africa: University of Cape Town Desmond Tutu HIV Centre, Cape Town, and The Aurum Institute, Rustenburg Research Centre; Uganda: Medical Research Council of Uganda, Masaka, and the Uganda Virus Research Institute/IAVI program, Entebbe; Zambia: Zambia-Emory HIV Research Project, Lusaka, Ndola, and Kitwe).
Study eligibility required a documented HIV-negative test within the previous year and a subsequent HIV-positive test. Study participants were primarily recruited from prospective incidence cohorts of HIV-negative individuals who received HIV prevention counseling and testing and condoms quarterly or monthly. Key populations included HIV-negative members of HIV-serodiscordant couples, men who have sex with men (MSM), youth (16–24 years), men or women who reported a recent sexually transmitted infection (STI) or unprotected sex with multiple partners in exchange for money or gifts, and women from high prevalence communities. A few participants (6%) were recruited from HIV voluntary counseling and testing centers.
Participants with a positive p24-antigen ELISA or HIV-antibody test were invited to enroll. The estimated date of HIV infection (EDI) was defined as the midpoint between the last negative and first positive HIV-antibody test, 14 days before the first positive p24 antigen test, 10 days before the first positive viral load test in the absence of p24 antigen or rapid HIV antibodies, or the date of a high-risk exposure event. All HIV-1 infection was confirmed by viral load testing.
Participants were seen monthly until 3 months after EDI, then quarterly until 24 months, and semiannually thereafter. Participants underwent a complete (entry visit) or symptom-directed physical examination and a detailed complete or interim medical history including onset of HIV-related illness, a blood draw, and a urine pregnancy test (women). Participants were provided acute clinical care at the study clinic or referred for treatment as needed. Non-HIV STIs were managed syndromically or treated per national guidelines. If HIV care, including antiretroviral therapy (ART), was available near the CRC, study participants were referred. Otherwise, HIV care, including ART, was provided by the CRC.
Laboratory procedures
Laboratory equipment and procedures were standardized across CRCs. Study clinic staff were trained in good clinical practice (GCP) and laboratory staff in good clinical laboratory practices (GCLP) [25]. Laboratories that performed study procedures participated in internal and external quality control and assurance processes and were GCLP accredited.
Laboratory testing including CD4+ cell counts and routine laboratory tests were conducted at the CRCs and viral load testing [Roche Amplicor Monitor v1.5; Roche Diagnostics, Indianapolis, USA (February 2006–January 2011) or Abbott Real Time HIV-1 v1.0 Abbott Molecular Diagnostics, Mississauga, Canada, thereafter] at Clinical Laboratory Services, Johannesburg [26]. A positive annual syphilis test [rapid plasma reagin, Biotec Laboratories, Inc., UK] was confirmed by Treponema pallidum hemagglutination assay. Abnormal urinalysis was investigated by microscopy.
All p24-positive, antibody negative specimens were repeat tested, and, if positive, early HIV infection was suspected [27]. If participants did not report ART but showed an abrupt drop in viral load to less than 2000 RNA copies/ml, antiretroviral drug level testing was conducted on the first available sample following the drop [26].
HIV-1 viral subtype analysis
The REGA HIV-1 subtyping tool (http://hivdb.stanford.edu/) was used to analyze sequence from the pol gene from the first specimen after HIV-infection [26]. Additional phylogenetic analysis was done if REGA results were indeterminate.
Human leukocyte antigen class I testing
Human leukocyte antigen (HLA) genotyping included HLA-A, HLA-B, and HLA-C, as described by Tang et al.[28]. Allelic variants were resolved to four-digit specificities using PCR and defined according to current guidelines [28].
Data and statistical analyses
Data were collected on case report forms, verified, and faxed (Clinical DataFax Systems, Inc., Hamilton, Canada) to a central database at Perinatal HIV Research Unit, University of Witswatersrand, Johannesburg RSA. Data analyses were conducted using STATA version 12 (Stata Corporation, College Station, Texas, USA) and R version 2.15.0 (http://CRAN.R-project.org).
Demographic variables, method to determine EDI, and baseline characteristics were summarized by HIV-1 subtype. For continuous variables, the median and interquartile range was determined. The F-test of no difference in the mean values across infecting subtypes was performed. For categorical variables, the frequencies and percentages were calculated. Pearson's chi square test of no association between response and subtype was conducted. Variables associated with infecting subtype were considered potential confounders in subsequent analyses. MSM were examined as a separate category to evaluate potentials subgroup differences.
Disease progression was measured by three endpoints: immunologic, virologic, and clinical. Immunologic progression was defined as time from EDI to the first of two consecutive CD4+ cell counts 350 cells/μl or less. Virologic progression was defined as time from EDI to the first of two consecutive viral load measurements at least 1 × 105 copies/ml. Clinical progression was defined by the 1993 CDC definition for AIDS [29], or the time from EDI to the first occurrence of a Category C event, a CD4+ cell count lower than 200 cells/μl, or a CD4% lower than 14. To avoid confounding by the transient events of acute infection, CD4+ cell counts and viral load measurements from the first 69 days post-EDI were excluded from analysis. Day 70 begins the window for the month 3 visit, by which time viral load should be past its initial peak and CD4+ cell count past its acute phase nadir [30]. Except for those on short-course antiretroviral drug for prevention of mother-to-child HIV transmission, participants were censored at time of self-reported ART initiation or detection of blood antiretroviral drug levels. To evaluate the potential bias of early or late ART initiation or any country or regional differences in ART provision, the endpoint of time to CD4+ cell count 350 cells/μl or less or ART initiation was also examined.
The log-rank test was used to assess the association between infecting HIV-1 subtype and disease progression without stratification for baseline covariates. Using subtype A-infected participants as the referent group, Cox proportional hazards models were applied for all three endpoints, if HLA results and at least one CD4+ cell count and viral load measurement after day 70 post EDI were available. Unadjusted and adjusted analyses were performed to assess baseline covariates of age at EDI, sex, risk group, BMI, male circumcision, delayed enrolment (more than 6 months post-EDI), education, hemoglobin, marital status, method of HIV detection, report of acute retroviral syndrome, source of recruitment, and HLA type on risk of disease progression. Baseline variables significant in univariate analysis were included in the final adjusted model.
Linear mixed effects models were used to assess the change in log10 transformed and untransformed CD4+ cell count after day 70. Visual inspection of CD4+ cell count trajectories suggested a steeper decline between months 3 and 12 versus after month 12. A piecewise linear mixed effects model of CD4+ cell count over time with a single knot at month 12 was fit to the data. HIV-1 subtype, age group, sex, and risk group were examined as fixed effects. All models included a random intercept and slope for each participant to account for individual set points and correlated outcomes. An individual's intercept and slope were assumed uncorrelated. Other knots for change in CD4+ cell count were explored.
Several types of sensitivity analyses were performed. Analyses restricted to East African cohorts were performed to assess potential confounding by geographic location. Because preinfection CD4+ cell counts were not measured, CD4+ cell count at month 3 was examined as a predictor of disease progression. Although CD4+ cell count and viral load at month 3 were highly predictive of disease progression, they were excluded from the final Cox models because both reflect disease progression. The Cox models were restricted to participants from discordant couples only and to volunteers with and without delayed enrollment, to assess the robustness of the study's conclusions.
Results
Cohort description and follow-up
From February 2006 to December 2011, 615 participants with incident HIV infection enrolled: 256 (41.6%) women, 92 (15.0%) MSM, and 264 (42.9%) from Southern Africa. The majority (74.8%) was enrolled from HIV-serodiscordant couple cohorts. The median age at EDI was 29 years (range 16–58); men were older than women (median 31 versus 27 years, respectively, P < 0.001). Three participants were adolescents at the time of enrolment; all were women, from Cape Town, and aged 16 (1) or 17 (2) years. (See Table, Supplemental Digital Content, which shows the population characteristics for all 615 enrolled volunteers.)
The median time from EDI to enrolment was 54 days (range: 10–388). From 2005 to 2006, 78 (12.7%) participants diagnosed with HIV infection before study initiation delayed enrolment by 6 months to 1 year. The study included 1790 person-years of ART-free follow-up after EDI (median per-volunteer person-years duration 2.8, range 0.1–6.9 years).
Overall, 204 participants discontinued follow-up. Forty-seven (23.0%) initiated ART, 41 (20.1%) were lost to follow-up for unknown reasons, 33 (9.3%) were dropped due to CRC closure, 27 (13.2%) were discontinued per investigator discretion, 22 (10.8%) moved out of the study area, 16 (7.8%) died, 14 (6.97.3%) withdrew voluntarily, and four (2.0%) were incarcerated. Of the 16 deaths, seven were AIDS-related.
Infecting HIV-1 subtype
Viral subtyping was available for 579 (94.1%) participants. The predominant HIV-1 infecting subtypes were C (256, 44.2%), A (209, 36.1%), and D (84, 14.5%). Subtype was strongly associated with geographic location and therefore also key population status (Table 1). The majority of subtype C infections occurred in Southern Africa whereas subtypes A and D predominated in East Africa. Compared to subtype A-infected participants, subtype C individuals were older (32 versus 28 years, from median age at EDI) and had a lower month 3 CD4+ cell count (503 versus 595 cells/μl). Despite small numbers, a lower CD4+ cell count was also seen among subtype C-infected participants when the analysis was restricted to East Africa. (See Table, Supplemental Digital Content 1, which shows the Cox proportional hazards models for participants in East Africa only.) Bivariate analysis of other baseline characteristics showed that age at infection, sex, marital status, education, male circumcision status, method used to determine EDI, and 3-month viral load values among non-MSM males were all significantly associated with subtype (Table 1).
Table 1.
Baseline characteristics of the African Early HIV Infection Cohort by infecting HIV-1 subtype, N = 579.
| Infecting HIV-1 subtype | |||||||||||
| Total | A | C | D | Othera | |||||||
| Characteristic | N | Percentage of total | N | % | N | % | N | % | N | % | P |
| Total | 579 | 100.0 | 209 | 36.1 | 256 | 44.2 | 84 | 14.5 | 30 | 5.2 | |
| Sex | |||||||||||
| Women | 235 | 40.6 | 72 | 30.6 | 115 | 48.9 | 40 | 17.0 | 8 | 3.4 | <0.001b |
| Men | 346 | 59.8 | 137 | 39.6 | 142 | 41.0 | 45 | 13.0 | 22 | 6.4 | |
| Non-MSM | 255 | 44.0 | 73 | 28.6 | 133 | 52.2 | 36 | 14.1 | 13 | 5.1 | |
| MSM | 90 | 15.5 | 64 | 71.1 | 9 | 10.0 | 8 | 8.9 | 9 | 10.0 | |
| Clinical research center | |||||||||||
| Kilifi, Kenya | 87 | 15.0 | 63 | 72.4 | 8 | 9.2 | 8 | 9.2 | 8 | 9.2 | <0.001 |
| Nairobi, Kenya | 24 | 4.1 | 18 | 75.0 | 1 | 4.2 | 4 | 16.7 | 1 | 4.2 | |
| Kigali, Rwanda | 92 | 15.9 | 74 | 80.4 | 10 | 10.9 | 2 | 2.2 | 6 | 6.5 | |
| Rustenburg, South Africa | 12 | 2.1 | 0 | 0 | 12 | 100 | 0 | 0 | 0 | 0 | |
| Cape Town, South Africa | 6 | 1.0 | 0 | 0 | 6 | 100 | 0 | 0 | 0 | 0 | |
| Masaka, Uganda | 95 | 16.4 | 29 | 30.5 | 4 | 4.2 | 54 | 56.8 | 8 | 8.4 | |
| Entebbe, Uganda | 45 | 7.8 | 25 | 55.6 | 1 | 2.2 | 16 | 35.6 | 3 | 6.7 | |
| Lusaka, Zambia | 141 | 24.4 | 0 | 0 | 138 | 97.9 | 0 | 0.0 | 3 | 2.1 | |
| Copperbelt, Zambia | 78 | 13.5 | 0 | 0 | 77 | 98.7 | 0 | 0.0 | 1 | 1.3 | |
| Age at infection (years) | |||||||||||
| 16 to <40 | 493 | 85.1 | 185 | 37.5 | 214 | 43.4 | 70 | 14.2 | 24 | 4.9 | NSc |
| ≥40 | 86 | 14.9 | 24 | 27.9 | 42 | 48.8 | 14 | 16.3 | 6 | 7.0 | |
| Median | 29 | – | 28 | – | 32 | – | 29 | – | 27 | – | <0.001 |
| Range (min-max) | (16–58) | – | (17–58) | – | (16–58) | – | (17–58) | – | (18–48) | – | |
| Marital status at enrollment | |||||||||||
| Never married | 114 | 19.7 | 71 | 62.3 | 27 | 23.7 | 8 | 7.0 | 8 | 7.0 | <0.001 |
| Currently married | 433 | 74.8 | 126 | 29.1 | 220 | 50.8 | 67 | 15.5 | 20 | 4.6 | |
| Monogamous | 380 | 87.8 | 105 | 27.6 | 212 | 55.8 | 47 | 12.4 | 16 | 4.2 | |
| Polygamous | 53 | 12.2 | 21 | 39.6 | 8 | 15.1 | 20 | 37.7 | 4 | 7.5 | |
| Previously marriedd | 32 | 5.5 | 12 | 37.5 | 9 | 28.1 | 9 | 28.1 | 2 | 6.3 | |
| Highest education level achieved at enrollment | |||||||||||
| None | 44 | 7.6 | 20 | 45.5 | 7 | 15.9 | 13 | 29.5 | 4 | 9.1 | 0.001 |
| Some primary | 312 | 53.9 | 114 | 36.5 | 138 | 44.2 | 48 | 15.4 | 12 | 3.8 | |
| Some secondary | 189 | 32.6 | 58 | 30.7 | 98 | 51.9 | 22 | 11.6 | 11 | 5.8 | |
| More than secondarye | 35 | 6.0 | 17 | 48.6 | 14 | 40.0 | 1 | 2.9 | 3 | 8.6 | |
| Circumcision (men only) | |||||||||||
| No | 195 | 76.5 | 54 | 27.7 | 108 | 55.4 | 22 | 11.3 | 11 | 5.6 | <0.001 |
| Yes | 112 | 43.9 | 69 | 61.6 | 15 | 13.4 | 20 | 17.9 | 8 | 7.1 | |
| Missing | 38 | 14.9 | 14 | 36.8 | 19 | 50.0 | 2 | 5.3 | 3 | 7.9 | |
| Reported risk category | |||||||||||
| HIV-serodiscordant couple | 428 | 73.9 | 117 | 27.3 | 238 | 55.6 | 54 | 12.6 | 19 | 4.4 | <0.001 |
| MSM | 90 | 15.5 | 64 | 71.1 | 9 | 10.0 | 8 | 8.9 | 9 | 10.0 | |
| Other heterosexualf | 48 | 8.3 | 22 | 45.8 | 9 | 18.8 | 16 | 33.3 | 1 | 2.1 | |
| Unknown/Not reported | 13 | 2.2 | 6 | 46.2 | 0 | 0.0 | 6 | 46.2 | 1 | 7.7 | |
| Days from EDI at enrollment | |||||||||||
| <70 | 360 | 62.2 | 134 | 37.2 | 149 | 41.4 | 57 | 15.8 | 20 | 5.6 | NS |
| 70–168 | 152 | 26.3 | 56 | 36.8 | 68 | 44.7 | 20 | 13.2 | 8 | 5.3 | |
| >168 | 66 | 11.4 | 19 | 28.8 | 38 | 57.6 | 7 | 10.6 | 2 | 3.0 | |
| Method used to estimate EDI | |||||||||||
| Antibody test results | 380 | 65.6 | 103 | 27.1 | 198 | 52.1 | 61 | 16.1 | 18 | 4.7 | <0.001 |
| p24 test results | 81 | 14.0 | 38 | 46.9 | 25 | 30.9 | 13 | 16.0 | 5 | 6.2 | |
| PCR test results | 48 | 8.3 | 38 | 79.2 | 5 | 10.4 | 1 | 2.1 | 4 | 8.3 | |
| Other | 69 | 11.9 | 30 | 43.5 | 27 | 39.1 | 9 | 13.0 | 3 | 4.3 | |
| CD4+ cell count at month 3g post-EDIh, median (IQR)i in cells/μl | |||||||||||
| All | 532 (431–698) | – | 595 (453–730) | – | 503 (406–646) | – | 557 (457–726) | – | 555 (337–704) | – | 0.02 |
| Women | 569 (476–758) | – | 629 (487–740) | – | 553 (460–748) | – | 564 (476–804) | – | 555 (477–594) | – | NS |
| Males | 512 (412–673) | – | 579 (440–716) | – | 481 (377–580) | – | 522 (405–674) | – | 522 (405–674) | – | 0.003 |
| N on-MSM | 503 (408–648) | – | 549 (445–705) | – | 479 (369–578) | – | 516 (405–688) | – | 631 (337–868) | – | 0.01 |
| MSM | 574 (428–697) | – | 586 (436–736) | – | 486 (429–600) | – | 574 (487–598) | – | 383 (295–684) | – | NS |
| Viral load at month 3 post-EDI, median (IQR) in 1 × 105 copies/ml | |||||||||||
| All | 4.6 (3.9–5.2) | – | 4.5 (3.8–5.1) | – | 4.7 (4.1–5.2) | – | 4.6 (3.8–5.3) | – | 4.5 (4.0–4.9) | – | NS |
| Women | 4.4 (3.7–5.1) | – | 4.4 (3.8–5.0) | – | 4.5 (3.6–5.1) | – | 4.4 (3.5–5.3) | – | 4.4 (4.0–5.3) | – | NS |
| Non-MSM | 4.8 (4.2–5.3) | – | 4.5 (4.0–4.9) | – | 4.8 (4.3–5.3) | – | 4.8 (4.1–5.3) | – | 4.8 (3.9–4.9) | – | 0.05 |
| MSM | 4.5 (3.8–5.0) | – | 4.3 (3.8–5.2) | – | 4.8 (4.5–4.8) | – | 3.4 (2.4–4.3) | – | 4.5 (4.5–4.6) | – | NS |
aOther subtypes include B, G, CRF02_AG, CRF11_CPX, recombinant forms A1A2D, A1C, A1CD, A1D, CK, and CD. Volunteers with missing subtype data are not shown in this table.
bComparison of women, non-MSM males, and MSM. Comparison of all men to all women is also significant (P = 0.03).
cNot statistically significant at P < 0.05 level.
dIncludes divorced, separated, and widowed.
eIncludes technical training, apprenticeships and other schooling beyond secondary school.
fIncludes multiple sexual partners, recent sexually transmitted infection, sex in exchange for money or gifts, or sexually active youth.
gDefined as day 84 ± 14 days post-EDI.
hEDI, estimated date of infection.
iIQR, interquartile range.
Disease progression
After adjustment for age, sex, and specific HLA types, subtype C-infected participants progressed 59–60% faster to immunologic, virologic, and clinical endpoints compared to subtype A (Table 2). Despite similar rates of immunologic progression to subtype A-infected participants, subtype D-infected participants progressed toward faster virologic progression and had nearly twice the rate of clinical progression to AIDS. Sensitivity analyses gave similar results. (See Table, Supplemental Digital Content 2, which shows the Cox proportional hazards models among study participants from discordant couples.)
Table 2.
Cox proportional hazard ratios for time to event from estimated date of HIV-1 infection among cohort participants with human leukocyte antigena data, n = 491.
| 2a: Time to CD4+ cell count ≤350 cells/μl | 2b: Time to viral load ≥1 × 105 copies/ml | 2c: Time to AIDSb | ||||||||||||||||||||
| Unadjusted analysis | Adjusted analysis | Unadjusted analysis | Adjusted analysis | Unadjusted analysis | Adjusted analysis | |||||||||||||||||
| Characteristic | n | events | HRc | 95% CId | P | HR | 95% CI | P | Events | HR | 95% CI | P | HR | 95% CI | P | Events | HR | 95% CI | P | HR | 95% CI | P |
| Age at EDIe (years) | ||||||||||||||||||||||
| <40 | 416 | 167 | 1 | – | – | 1 | – | – | 151 | 1 | – | – | 1 | – | – | 134 | 1 | – | – | 1 | – | – |
| ≥40 | 75 | 41 | 1.46 | (1.04, 2.05) | 0.03 | 1.35 | (0.94, 1.93) | 0.11 | 36 | 1.40 | (0.97, 2.01) | 0.07 | 1.12 | (0.77, 1.63) | 0.56 | 34 | 1.40 | (0.96, 2.04) | 0.08 | 1.43 | (0.96, 2.12) | 0.08 |
| Sex | ||||||||||||||||||||||
| Women | 200 | 75 | 1 | – | – | 1 | – | – | 53 | 1 | – | – | 1 | – | – | 56 | 1 | – | – | 1 | – | – |
| Non-MSMf | 232 | 111 | 1.43 | (1.06, 1.91) | 0.02 | 1.32 | (0.97, 1.79) | 0.08 | 115 | 2.15 | (1.55, 2.98) | <0.001 | 2.10 | (1.51, 2.93) | <0.001 | 87 | 1.43 | (1.02, 2.00) | 0.04 | 1.36 | (0.96, 1.91) | 0.09 |
| MSM | 59 | 22 | 1.10 | (0.68, 1.77) | 0.70 | 1.23 | (0.75, 2.03) | 0.41 | 19 | 1.31 | (0.78, 2.22) | 0.31 | 1.49 | (0.86, 2.60) | 0.14 | 25 | 1.94 | (1.21, 3.12) | 0.006 | 2.42 | (1.47, 3.99) | <0.001 |
| HLA B*57 allele | ||||||||||||||||||||||
| No | 450 | 195 | 1 | – | – | 1 | – | – | 179 | 1 | – | – | 1 | – | – | 162 | 1 | – | – | 1 | – | – |
| Yes | 41 | 13 | 0.60 | (0.34, 1.05) | 0.07 | 0.60 | (0.34, 1.06) | 0.08 | 8 | 0.38 | (0.19, 0.78) | 0.008 | 0.39 | (0.19, 0.79) | 0.009 | 6 | 0.30 | (0.13, 0.68) | 0.004 | 0.30 | (0.13, 0.69) | 0.004 |
| HLA B*45 allele | ||||||||||||||||||||||
| No | 415 | 164 | 1 | – | – | 1 | – | – | 150 | 1 | – | – | 1 | – | – | 138 | 1 | – | – | 1 | – | – |
| Yes | 76 | 44 | 1.71 | (1.22, 2.39) | 0.002 | 1.60 | (1.14, 2.25) | 0.007 | 37 | 1.58 | (1.10, 2.27) | 0.01 | 1.47 | (1.01, 2.14) | 0.04 | 30 | 1.48 | (0.99, 2.20) | 0.05 | 1.33 | (0.89, 1.99) | 0.16 |
| HIV-1 subtype | ||||||||||||||||||||||
| A | 177 | 65 | 1 | – | – | 1 | – | – | 53 | 1 | – | – | 1 | – | – | 50 | 1 | – | – | 1 | – | – |
| C | 243 | 117 | 1.58 | (1.16, 2.14) | 0.003 | 1.60 | (1.16, 2.20) | 0.004 | 105 | 1.57 | (1.13, 2.19) | 0.008 | 1.59 | (1.12,2.25) | 0.009 | 88 | 1.41 | (0.99, 2.00) | 0.05 | 1.60 | (1.11, 2.31) | 0.01 |
| D | 71 | 26 | 1.05 | (0.67, 1.65) | 0.84 | 1.04 | (0.65, 1.66) | 0.87 | 29 | 1.46 | (1.00, 2.46) | 0.05 | 1.61 | (1.01, 2.57) | 0.05 | 30 | 1.81 | (1.15, 2.85) | 0.01 | 1.93 | (1.21, 3.09) | 0.006 |
aHLA, human leukocyte antigen.
b1993 CDC Case Definition; majority of endpoints were CD4+ cell count ≤200 cells/μl.
cHR, hazard ratio.
dCI, confidence interval.
eEDI, estimated date of infection.
fNon-MSM: a male who does not report sex with men.
In unadjusted analyses, non-MSM showed faster immunologic progression than women or MSM. This increase was most likely due to non-MSM being older, consequently having lower month 3 CD4+ cell counts. In adjusted analysis, women, non-MSM, and MSM did not differ in immunologic progression; however, non-MSM had twice the rate of virologic progression and MSM had over twice the rate of clinical progression to AIDS versus women. Adjusted analyses combining non-MSM and MSM (see Table, Supplemental Digital Content 3, which shows Cox proportional hazards models comparing all men to women) showed no difference in immunologic progression between males and women but indicated faster virologic and clinical progression for males.
Having a B∗57 allele was protective against virologic and clinical progression; allele-positive participants progressed at a rate 61 and 70% slower than negative participants, respectively. The effect of B∗57 on immunologic progression was more modest and did not achieve statistical significance most likely due to the small number of participants with this allele. Conversely, presence of the B∗45 allele was associated with faster immunologic and virologic progression. In adjusted analyses, B∗45-positive participants progressed 60 and 47% faster than negative participants, respectively, but was not associated with a significant trend toward faster clinical progression (Table 2). (See Table, Supplemental Digital Content 4, which shows the distribution of HLA types identified in the study by infecting HIV-1 subtypes.)
Figure 1 shows the Kaplan–Meier curves for the proportion of participants reaching each disease progression endpoint by infecting HIV-1 subtype and the P-value from the corresponding log-rank test. The significant P-values for time to CD4+ cell count 350 cells/μl or less and time to viral load at least 1 × 105 copies/ml reflect the different rates of progression between subtype A-infected and C-infected participants. However, both subtype C-infected and D-infected participants progressed faster to AIDS than those with subtype A.
Fig. 1.
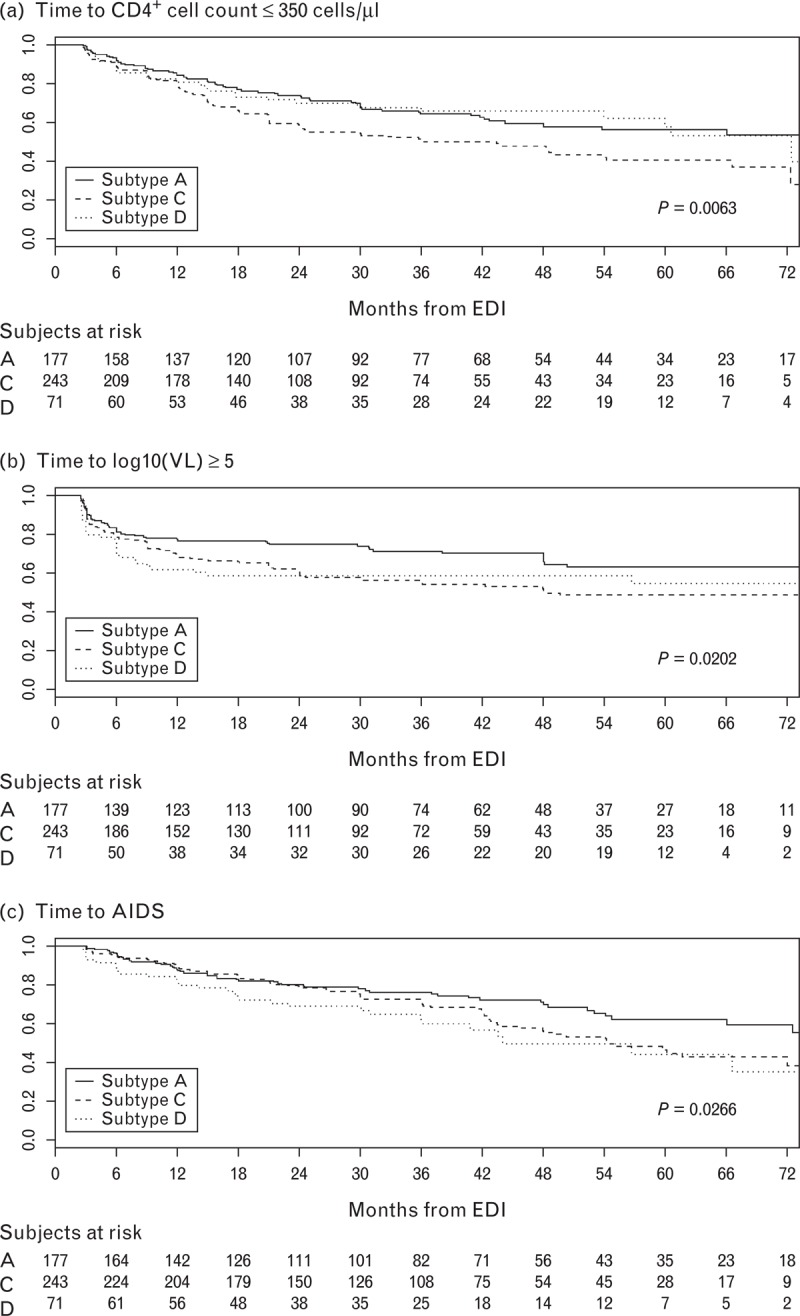
Kaplan–Meier survival curves. (a) Time to CD4+ cell count ≤ 350 cells/μl, (b) time to viral load (VL) ≥105 copies/ml, and (c) time to AIDS by HIV-infecting subtype among participants with human leukocyte antigen (HLA) data (n = 491).
In the analysis of clinical progression, 168 (34.2%) participants developed AIDS. Among the AIDS cases, 28 (16.7%) experienced a Category C event before reaching a CD4+ cell count lower than 200 cells/μl or a CD4% less than 14% including tuberculosis (14), herpes simplex (seven), recurrent pneumonia (four), esophageal candidiasis (two), and recurrent Salmonella septicemia (one). Another 12 participants experienced Category C events after CD4+ cell count decline. Thus, clinical progression primarily reflects immunologic progression.
Linear mixed effects models that controlled for sex and infecting subtype found subtype C-infected participants had 106 ± 18 fewer CD4+ cells at month 3 post-EDI versus subtype A. There was no difference in month 3 CD4+ cell count between subtype D and A participants. From months 3–12, CD4+ cell count declined by 124 ± 10 cells/year with no difference in the rate of decline by sex or subtype. After month 12, CD4+ cell count declined by 13 ± 15 cells/year. Although subtype C-infected participants reached a CD4+ cell count of 350 cells/μl sooner than subtype A-infected participants, this is largely due to subtype C-infected participants having lower CD4+ cell counts at month 3 post-EDI. A Cox regression analysis that included month 3 CD4+ cell count showed no difference in the rate of CD4+ cell count decline between subtype A and C individuals [adjusted hazard ratio 1.11, 95% confidence interval (CI) 0.73–1.69].
Among the 140 (28.5%) participants initiating ART, country-specific differences were observed in time of ART initiation, most likely reflecting differences in national treatment guidelines and programs. Ugandans and Rwandans were two and three times more likely than Kenyans to initiate ART; Zambians and South Africans had similar rates of ART uptake to Kenyans. Censoring of participants who initiated ART at CD4+ cell count higher than 350 cells/μl did not significantly change the time to the combined endpoint of CD4+ cell count 350 cells/μl or less or ART initiation. (See table, Supplementary Digital Content 5, which shows the Cox regression analysis of disease progression with the combined endpoints of CD4 reaching ≤350 cells/μl or ART initiation.)
Just over 10% of the cohort was enrolled more than 6 months post-EDI (Table 1); however, delayed enrollment was not associated with infecting HIV subtype nor was it associated with any of the three outcomes. The method used to determine volunteer EDI (antibody versus p24 versus PCR test results, or volunteer report) was associated with infecting subtype, with a significantly higher proportion of subtype-A infected volunteers having their EDI determined by p24 antigen or PCR test result (Table 1). In the multivariable analysis, this variable was not associated with any of the three outcomes and therefore did not appear to confound any of the observed relationships.
Discussion
This early HIV infection cohort in SSA is notable for its size, extensive follow-up, well documented time of HIV infection, diversity of geographic region and infecting HIV-1 subtypes, and characterization of HLA class I alleles. These factors allowed robust characterization of predictors of HIV disease progression pre-ART initiation in Africa. We observed significantly faster progression to CD4+ cell count 350 cells/μl or less, viral load at least 1 × 105 copies/ml, and AIDS endpoints among subtype C-infected versus A-infected participants, supporting the hypothesis that HIV-1 subtype C may be more pathogenic [24]. Subtype D-infected participants progressed significantly faster versus subtype A-infected participants to viral load at least 1 × 105 copies/ml and AIDS; there was no difference in immunologic progression.
Although they reached a CD4+ cell count of 350 cells/μl sooner after infection than subtype A-infected participants, subtype C-infected participants also had lower CD4+ cell counts at 3 months post-EDI. (See Table, Supplemental Digital Content 6, which shows median CD4+ cell counts at month 3 among participants in East Africa by sex and infecting subtype.) Because preseroconversion CD4+ cell counts were not available, it is difficult to determine whether this lower CD4+ cell count predated HIV acquisition. However, a study of laboratory references ranges in healthy, HIV-uninfected African adults of comparable age groups also sponsored by IAVI and conducted at six of the CRCs participating in this study showed no significant differences in median CD4+ cell counts by geographic region, suggesting that preinfection CD4+ cell counts may not significantly differ in this study [31].
Kaleebu and Kiwanuka have reported faster disease progression with subtype D versus A infections partially explained by co-receptor tropism [15,21]. Baeten et al.[32] found no difference between subtype C and A infections, but the study sample size was modest. The dominance of HIV-1 subtype C in much of Africa and Asia has caused speculation that the overall pathogenicity or transmissibility of subtype C is greater than other subtypes [33]. We showed that infection with subtype C is associated with a more rapid virologic disease progression, and high viral load is known to be associated with increased transmission [34]. A study of Zambian participants in this cohort showed that viral load of both the transmitter and the newly infected partner is predicted, at least in part, by the intrinsic replication capacity of the transmitted subtype C virus [35]. A similar evaluation of subtype A virus will be of interest.
Study bias may have arisen in a number of ways. Differences in clinical evaluation across CRCs may have introduced misclassification bias in clinical endpoints. Most Kenyan participants were MSM, who tend to delay ART initiation and may not have optimal health seeking behaviors due at least in part to stigma [36]. Pol sequencing for subtype determination does not detect recombinants in env or other regions. However, missed recombinations in other genes should have been randomly distributed. Specimens from participants with delayed enrolment were sequenced at a longer time from EDI. Interclade recombinants from superinfection or dual infection, while uncommon, are probably more frequent in East Africa than Southern Africa with more than 99% subtype C.
Selection bias is also possible. CRCs differed in duration of follow-up, time from EDI to study enrolment, age, sex, and risk groups. Healthier participants may have out-migrated for work and rapid progressors may have initiated ART or been too ill to enroll; however, sensitivity analyses between participants with delayed versus immediate enrolment produced no significant changes to our conclusions. Because of the strong association between HIV-1 infecting subtype and geographic region, the faster disease progression observed among subtype C-infected participants could be due to population differences for which we could not adequately control. Additionally, the modest number of subtype D-infected participants somewhat limited our ability to evaluate comparative disease progression in this subgroup.
In the context of a diverse HIV-1 genome, understanding the role of infecting subtype on disease progression is essential to guide public health policy, plan regionally appropriate HIV care and treatment programs, and define appropriate populations and endpoints for HIV prevention efficacy trials. Our findings also suggest that monitoring a relative loss of CD4 +cell count per volunteer may also be informative, as time to a CD4+ cell count threshold may be confounded by infecting subtype, age, and sex.
Identifying individuals with early HIV infection, while challenging, creates great opportunity for immunologic and genetic investigation, along with the potential for health benefits and reduction of HIV transmission. Substantial investment in infrastructure, community and national level engagement, and access to services are prerequisites to establish longstanding, prospective cohorts in resource-poor settings. Additional benefits of this work include both the establishment of HIV incidence cohorts suitable for efficacy trials of HIV prevention interventions in diverse at-risk populations and the collection of specimens that will contribute to improved understanding of the HIV immunology, virology, and pathology, including acute infection, development of broadly neutralizing HIV antibodies, and development and evaluation of new assays [37–39]. Observational epidemiology provides ideal research platforms and facilitates HIV care [40].
Conclusion
Our study demonstrates subtype-specific differences in HIV disease progression. To guide public health policy and clinical trial design, it is essential to understand the virologic, immunologic and clinical events that occur by infecting HIV-1 subtype. Our work suggests that HIV vaccine efficacy and relevant prevention trials should include diverse HIV-1 subtypes, have adequate sample sizes to evaluate this viral diversity, and consider potential subtype-specific differences when defining clinical endpoints.
Acknowledgements
The authors are grateful to numerous people, whose contributions have made this study possible: All of the Study Participants, Drs James Tang and Richard Kaslow (University of Alabama at Birmingham for HLA typing), Dr Ed Acosta (University of Alabama at Birmingham School of Medicine for antiretroviral drug testing), staff at the Central Laboratory Services and the Perinatal HIV Research Unit, South Africa; staff at the IAVI Human Immunology Laboratory, Imperial College, London; the Africa-based IAVI staff; Helen Thomson, Marietta Krebs, Leslie Nielsen, Melissa Simek, Lisa Stoll, Paul Sayer, Jan de Bont, Andrea von Lieven, Sarah Yates, Elise van der Elst, Dr Nzeera Ketter, and Dr Chrispin Kambili.
We also thank the study funders: Becton, Dickinson and Company; Bill and Melinda Gates Foundation; Bristol-Meyers Squibb; Canadian International Development Agency; Dutch Product Development Partnership Fund; European Commission South Africa; Foundation for the National Institute of Health; John Evans Foundation; Medical Research Council; Norwegian Agency for Development Corporation; Organization of the Petroleum Exporting Countries (OPEC) Fund for International Development; Pfizer, Inc.; Spain Ministry of Foreign Affairs; Swedish International Development Agency; United Kingdom Department for International Development; United States Agency for International Development (USAID); and World Bank.
Funding source: Primary: United States Agency for International Development (USAID).
This work was made possible in part by the generous support of the American people through the United States Agency for International Development (USAID). The contents are the responsibility of the study authors and do not necessarily reflect the views of USAID or the United States Government. This report was published with permission from KEMRI.
The IAVI Africa HIV Prevention Partnership includes: Mary Mwangome (KEMRI), Elizabeth Wahome (KEMRI), Gwynn Stevens (IAVI), Pontiano Kaleebu (MRC/UVRI), Heeran Makkan (AI), Gaudensia Mutua (KAVI), Eugene Ruzagira (MRC), Ubaldo Bahemuka (MRC), Rogers Twesigye (MRC), Agnes Bwanika (MRC), Freddie Kibengo (MRC), Roger Bayingana (PSF), Eric Hunter (PSF, ZEHRP, Emory University), Mubiana Inambao (ZEHRP), Kayitesi Kayitenkore (PSF).
Conflicts of interest
There are no conflicts of interest
Supplementary Material
Footnotes
Correspondence to Matt A. Price, International AIDS Vaccine Initiative 125 Broad Street, 9th Floor New York, NY 10004, USA. Tel: +1 646 752 0255; e-mail: mprice@iavi.org
Contributor Information
Collaborators: for the IAVI Africa HIV Prevention Partnership
References
- 1.Laga M, Piot P. Prevention of sexual transmission of HIV: real results, science progressing, societies remaining behind. AIDS 2012; 26:1223–1229 [DOI] [PubMed] [Google Scholar]
- 2.Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006; 20:W13–23 [DOI] [PubMed] [Google Scholar]
- 3.Population Reference Bureau World Population Data Sheet, 2012: Washington, DC: Population Reference Bureau; 2012 [Google Scholar]
- 4.UNAIDS Global AIDS epidemic facts and figures, 2012: Geneva, Switzerland; UNAIDS; 2012 [Google Scholar]
- 5.Hessol NA, Koblin BA, van Griensven GJ, Bacchetti P, Liu JY, Stevens CE, et al. Progression of human immunodeficiency virus type 1 (HIV-1) infection among homosexual men in hepatitis B vaccine trial cohorts in Amsterdam, New York City, and San Francisco, 1978–1991. Am J Epidemiol 1994; 139:1077–1087 [DOI] [PubMed] [Google Scholar]
- 6.Veugelers PJ, Strathdee SA, Kaldor JM, Shafer KA, Moss AR, Schechter MT, et al. Associations of age, immunosuppression, and AIDS among homosexual men in the Tricontinental Seroconverter Study. J Acquir Immune Defic Syndr Hum Retrovirol 1997; 14:435–441 [DOI] [PubMed] [Google Scholar]
- 7.Prins M, Veugelers PJ. Comparison of progression and nonprogression in injecting drug users and homosexual men with documented dates of HIV-1 seroconversion. European Seroconverter Study and the Tricontinental Seroconverter Study. AIDS 1997; 11:621–631 [DOI] [PubMed] [Google Scholar]
- 8.Gao F, Robertson DL, Morrison SG, Hui H, Craig S, Decker J, et al. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J Virol 1996; 70:7013–7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amornkul PN, Tansuphasawadikul S, Limpakarnjanarat K, Likanonsakul S, Young N, Eampokalap B, et al. Clinical disease associated with HIV-1 subtype B′ and E infection among 2104 patients in Thailand. AIDS 1999; 13:1963–1969 [DOI] [PubMed] [Google Scholar]
- 10.Csillag C. HIV-1 subtype C in Brazil. Lancet 1994; 344:1354. [PubMed] [Google Scholar]
- 11.Djomand G, Duerr A, Faulhaber JC, Struchiner CJ, Pacheco AG, Barroso PF, et al. Viral load and CD4 count dynamics after HIV-1 seroconversion in homosexual and bisexual men in Rio de Janeiro, Brazil. J Acquir Immune Defic Syndr 2006; 43:401–404 [DOI] [PubMed] [Google Scholar]
- 12.Mehendale SM, Bollinger RC, Kulkarni SS, Stallings RY, Brookmeyer RS, Kulkarni SV, et al. Rapid disease progression in human immunodeficiency virus type 1-infected seroconverters in India. AIDS Res Hum Retroviruses 2002; 18:1175–1179 [DOI] [PubMed] [Google Scholar]
- 13.Laurent C, Bourgeois A, Faye MA, Mougnutou R, Seydi M, Gueye M, et al. No difference in clinical progression between patients infected with the predominant human immunodeficiency virus type 1 circulating recombinant form (CRF) 02_AG strain and patients not infected with CRF02_AG, in Western and West-Central Africa: a four-year prospective multicenter study. J Infect Dis 2002; 186:486–492 [DOI] [PubMed] [Google Scholar]
- 14.Marlink R, Kanki P, Thior I, Travers K, Eisen G, Siby T, et al. Reduced rate of disease development after HIV-2 infection as compared to HIV-1. Science 1994; 265:1587–1590 [DOI] [PubMed] [Google Scholar]
- 15.Kaleebu P, Nankya IL, Yirrell DL, Shafer LA, Kyosiimire-Lugemwa J, Lule DB, et al. Relation between chemokine receptor use, disease stage, and HIV-1 subtypes A and D: results from a rural Ugandan cohort. J Acquir Immune Defic Syndr 2007; 45:28–33 [DOI] [PubMed] [Google Scholar]
- 16.Kaleebu P, Ross A, Morgan D, Yirrell D, Oram J, Rutebemberwa A, et al. Relationship between HIV-1 Env subtypes A and D and disease progression in a rural Ugandan cohort. AIDS 2001; 15:293–299 [DOI] [PubMed] [Google Scholar]
- 17.Kiwanuka N, Laeyendecker O, Robb M, Kigozi G, Arroyo M, McCutchan F, et al. Effect of human immunodeficiency virus Type 1 (HIV-1) subtype on disease progression in persons from Rakai, Uganda, with incident HIV-1 infection. J Infect Dis 2008; 197:707–713 [DOI] [PubMed] [Google Scholar]
- 18.Neilson JR, John GC, Carr JK, Lewis P, Kreiss JK, Jackson S, et al. Subtypes of human immunodeficiency virus type 1 and disease stage among women in Nairobi, Kenya. J Virol 1999; 73:4393–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasan A, Renjifo B, Hertzmark E, Chaplin B, Msamanga G, Essex M, et al. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin Infect Dis 2006; 42:843–852 [DOI] [PubMed] [Google Scholar]
- 20.Kaleebu P, French N, Mahe C, Yirrell D, Watera C, Lyagoba F, et al. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J Infect Dis 2002; 185:1244–1250 [DOI] [PubMed] [Google Scholar]
- 21.Kiwanuka N, Robb M, Laeyendecker O, Kigozi G, Wabwire-Mangen F, Makumbi FE, et al. HIV-1 viral subtype differences in the rate of CD4+ T-cell decline among HIV seroincident antiretroviral naive persons in Rakai district, Uganda. J Acquir Immune Defic Syndr 2010; 54:180–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuritzkes DR. HIV-1 subtype as a determinant of disease progression. J Infect Dis 2008; 197:638–639 [DOI] [PubMed] [Google Scholar]
- 23.Kanki PJ, Travers KU, MBoup S, Hsieh CC, Marlink RG, Gueye-NDiaye NA, et al. Slower heterosexual spread of HIV-2 than HIV-1. Lancet 1994; 343:943–946 [DOI] [PubMed] [Google Scholar]
- 24.Gray CM, Williamson C, Bredell H, Puren A, Xia X, Filter R, et al. Viral dynamics and CD4+ T cell counts in subtype C human immunodeficiency virus type 1-infected individuals from southern Africa. AIDS Res Hum Retroviruses 2005; 21:285–291 [DOI] [PubMed] [Google Scholar]
- 25.Stiles T, Grant V, Mawbey N. Good clinical laboratory practice (GCLP): A quality system for laboratories that undertake the analysis of samples from clinical trials. Suffolk, United Kingdom:British Association of Research Quality Assurance (BARQA); 2003 [Google Scholar]
- 26.Fabiani M, Ble C, Grivel P, Lukwiya M, Declich S. 1989–1996 HIV-1 prevalence trends among different risk groups in Gulu District, North Uganda. J Acquir Immune Defic Syndr Hum Retrovirol 1998; 18:514. [DOI] [PubMed] [Google Scholar]
- 27.Mulenga J, Hunter E, Manigart O, Stevens G, Allen S, RZ HRG. P24 antigen screening for early detection of HIV infection in discordant heterosexual couples in Zambia. AIDS Vaccine Conference 2006; Amsterdam, The Netherlands [Google Scholar]
- 28.Tang J, Cormier E, Gilmour J, Price MA, Prentice HA, Song W, et al. Human leukocyte antigen variants B∗44 and B∗57 are consistently favorable during two distinct phases of primary HIV-1 infection in sub-Saharan Africans with several viral subtypes. J Virol 2011; 85:8894–8902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.CDC 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep 1992; 41 (RR-17):1–19 [PubMed] [Google Scholar]
- 30.Cohen MS, Shaw GM, McMichael AJ, Haynes BF. Acute HIV-1 Infection. N Engl J Med 2011; 364:1943–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karita E, Ketter N, Price MA, Kayitenkore K, Kaleebu P, Nanvubya A, et al. CLSI-derived hematology and biochemistry reference intervals for healthy adults in eastern and southern Africa. PLoS ONE 2009; 4:e4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baeten JM, Chohan B, Lavreys L, Chohan V, McClelland RS, Certain L, et al. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J Infect Dis 2007; 195:1177–1180 [DOI] [PubMed] [Google Scholar]
- 33.Essex M. Human immunodeficiency viruses in the developing world. Adv Virus Res 1999; 53:71–88 [DOI] [PubMed] [Google Scholar]
- 34.Quinn TC, Wawer MJ, Sewankambo N, Serwadda D, Li C, Wabwire-Mangen F, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N Engl J Med 2000; 342:921–929 [DOI] [PubMed] [Google Scholar]
- 35.Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, et al. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog 2012; 8:e1003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham SM, Mugo P, Gichuru E, Thiong’o A, Macharia M, Okuku HS, et al. Adherence to antiretroviral therapy and clinical outcomes among young adults reporting high-risk sexual behavior, including men who have sex with men, in coastal Kenya. AIDS Behav 2013; 17:1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilcher C, Busch M, Welte A, Facente S, Kassanjee R, Keating S, et al. A novel specimen and data repository for evaluation of HIV incidence assays. 19th International AIDS Conference 2012; Washington, DC, USA [Google Scholar]
- 38.Manak M, Sina S, Anekella B, Hewlett I, Sanders-Buell E, Ragupathy V, et al. Pilot studies for development of an HIV subtype panel for surveillance of global diversity. AIDS Res Hum Retroviruses 2012; 28:594–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spentzou A, Bergin P, Gill D, Cheeseman H, Ashraf A, Kaltsidis H, et al. Viral inhibition assay: a CD8 T cell neutralization assay for use in clinical trials of HIV-1 vaccine candidates. J Infect Dis 2010; 201:720–729 [DOI] [PubMed] [Google Scholar]
- 40.Ngongo BP, Priddy F, Park H, Becker J, Bender B, Fast P, et al. Developing standards of care for HIV prevention research in developing countries – a case study of 10 research centers in Eastern and Southern Africa. AIDS Care 2012; 24:1277–1289 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.