

Abstract
FOXP3 is an X-linked tumor suppressor gene and a master regulator in T regulatory cell function. This gene has been found to be mutated frequently in breast and prostate cancers and to inhibit tumor cell growth, but its functional significance in DNA repair has not been studied. We found that FOXP3 silencing stimulates homologous recombination-mediated DNA repair and also repair of γ-irradiation-induced DNA damage. Expression profiling and chromatin-immunoprecipitation analyses revealed that FOXP3 regulated the BRCA1-mediated DNA repair program. Among 48 FOXP3-regulated DNA repair genes, BRCA1 and 12 others were direct targets of FOXP3 transcriptional control. Site-specific interaction of FOXP3 with the BRCA1 promoter repressed its transcription. Somatic FOXP3 mutants identified in breast cancer samples had reduced BRCA1 repressor activity, while FOXP3 silencing and knock-in of a prostate cancer-derived somatic FOXP3 mutant increased the radioresistance of cancer cells. Together our findings provide a missing link between FOXP3 function and DNA repair programs.
Introduction
Radiation has been part of therapies for about 50% of cancer patients (1, 2). Despite well demonstrated clinical efficacy, many cancers are resistant (2, 3). The mechanisms for cancer resistance to irradiation remain poorly understood. Several mechanisms have been proposed. First, irradiation may not be effective against cancer stem cells that are in a dormant state (4). This interpretation is supported by an enrichment of CD133+ cancer stem cells in glioma lesion after radiotherapy (5). Second, genetic alteration in cancer cells may make them more resistant to irradiation. Tumor suppressors that are involved in radiation-induced apoptosis and/or DNA repairs, such as TP53, ATM, BRAC1, BRAC2, are often inactivated during cancer development. Among them, targeted mutation of TP53 increase cellular resistance to irradiation (6). On the other hand, BRCA1/2, whose germline mutations are responsible for a major portion of hereditary breast and ovarian cancer (7, 8), are directly involved in DNA repair (9–11). BRCA1 has been shown to regulate tumor cell sensitivity to irradiation (11). Therefore, the mutations that induce BRCA1 expression may confer resistance to irradiation and chemically induced DNA damage. However, because the regulation of BRCA1 expression is poorly understood, the relationship between BRCA1 gene transcriptional regulation and DNA repair is largely unexplored.
FOXP3 is the X-linked tumor suppressor for both breast and prostate cancers (12–14). About 13% of prostate and breast cancers deleted the FOXP3 locus, while 25–35% of cancer samples have somatic FOXP3 mutations (12, 13). In addition, high proportion of cancer samples, including breast, prostate, melanoma and pancreatic cancer cells express FOXP3 in the cytoplasm (12, 15–18). The aberrant cytoplasmic FOXP3 likely reflects functional inactivation of the protein as FOXP3 is a transcription factor that normally resides in the nuclei (12). FOXP3 inhibits tumor cell growth by induction of tumor suppressors and repression of oncogenes (13, 19–21). Apart from tumor cell growth inhibition, FOXP3 may regulate response to therapy as it is induced by both stress-inducing and DNA damaging drugs (22, 23). A search of FOXP3 targets based on our recently reported databases (20, 24) reveal that FOXP3 regulates a large number of genes associated with DNA damage response, in particular those that are involved in BRCA1-mediated DNA repair program and thus prompt us to investigate the role for FOXP3 in BRCA1-mediated DNA repair program.
Materials and Methods
Mice
Rag2−/− Foxp3+/+, Rag2−/− Foxp3+/y, Rag2−/− Foxp3sf/sf and Rag2−/− Foxp3sf/y BALB/c mice have been previously described (25). Four-month-old virgin mice were used to analyze the effect of FoxP3 mutation on Brca1 expression. All animal experimental procedures have been approved by the Institutional Animal Care and Use Committee of University of Michigan.
Cell culture
Breast cancer cell line MCF-7 and immortalized mammary epithelial cell line MCF-10A, human sarcoma cell line U2OS, human colon cancer cell line HCT116 and mouse mammary tumor cell line 4T1 were purchased from the American Type Culture Collection, none of the cells have been in tissue culture for more than six month prior to use. Mouse mammary cell line TSA has been described (26). A tet-off FOXP3 expression system in the MCF-7 cells has been previously established (13). Transfections were performed using Lipofectamine 2000 (6 μl/106 cells). Cell banks were created after cells were received. Early passages of cells were used for the study. No reauthentification of cells has been done since receipt.
FOXP3 silencing
The human FOXP3 silencing vectors were previously described (20). The mouse Foxp3 shRNA, Brca1 shRNA and control lentiviral vectors pLKO.1 were purchased from Open Biosystems. The targeting sequences are listed in supplemental Table S4.
Western blot
The anti-FOXP3/Foxp3 (hFOXY, eBioscience, 1:100), anti-Brca1/BRCA1 (Cell Signaling, 1–1000) and anti-actin (Sigma, 1:3,000) were used as primary antibodies. Anti-rabbit or mouse IgG horseradish peroxidase–linked secondary antibody at 1:3,000 to 1:5,000 dilutions (Cell Signaling) was used.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was carried out according to published procedure (20). Briefly, the FOXP3-transfected tet-off cells were sonicated and fixed with 1% paraformaldehyde. The anti-FOXP3, and anti-IgG (Santa Cruz Biotechnology) antibodies were used to pull down chromatin associated with FOXP3. The amounts of the specific DNA fragment were quantitated by real-time PCR and normalized against the genomic DNA preparation from the same cells. The same PCR primers were used to measure ChIP and input genomic DNA. The ChIP real-time PCR primers are listed in Supplemental Table S4.
Quantitative real-time PCR
Relative quantities of mRNA expression were analyzed using real-time PCR (ABI Prism 7500 Sequence Detection System, Applied Biosystems). The SYBR (Applied Biosystems) green fluorescence dye was used in this study. The primer sequences are listed in Supplementary Table S4.
Tumorigenicity assay
TSA cells (106/mice) were injected into mammary fatpads of syngeneic BALb/c mice. The tumor volumes defined as 0.75πr3, where r is radius. The tumor diameters were derived from the average of largest diameters in two dimensions.
Site-directed mutagenesis
All mutants were generated using mutgenesis kit from Stratagene (Catalog #210518). The mutating sequence Brca1 promoter mutants were from GTCAACA to GTCGGCA. The primers for site-directed mutagenesis of FOXP3 are listed in Table S4.
Immunohistochemistry
Immunohistochemistry was performed by the avidin-biotin complex method. Expression of FOXP3 in human breast cancer or normal tissue samples was determined using immunohistochemistry, as described (12). The rabbit anti-BRCA1 monoclonal antibody (Cell Signaling, 1:200), and as secondary antibodies, biotin goat anti-mouse IgG were obtained from Santa Cruz and used at 1:200. FOXP3 and BRCA1 staining of TMA samples (US Biomax, Inc., Rockville, MD) were scored double blind.
HR DNA repair Assay
U2OS with integrated DR-GFP was used as the reporter cells. The cells were infected shRNAi scramble and FOXP3 ShRNA. Then the stable clones were treated with BRCA1 siRNA. 24 hour after RNAi treatment, the cells were infected with an adenovirus encoding I-SceI endonuclease (1000 MOI). At 48 hours after infection, the GFP+ cells were quantified by flow cytometry.
Comet assay
The experiment was conducted according to a published procedure (27). Briefly, U2OS cells were irradiated for 60Gy and cultured 0, 1 or 3 hours. The cells were harvested and mixed in agarose gel and plated on a glass slide, and digested by proteinase K overnight. The agarose gel slides were applied for electrophoresis. The DNA was stained by PI. The distance of nuclear DNA movement, which is called DNA momentum, was used as indicator for DNA damage and repair.
Knockin of a somatic FOXP3 mutation into cancer cell line
The somatic mutation in genomic DNA was generated by site directed mutagenesis. The vector construction and knockin by homologous recombination have been described in detail (28). Briefly, the targeting vector was constructed by polymerase chain reaction (PCR) with 203G>R mutated genomic DNA as the template for the homologous left arm. Homologous recombination was screened by genomic PCR with primers derived from the neomycin resistance gene and the upstream region of the left homologous arm. Two homologous recombination clones were identified from two 96-well-plates of G418 resistant single clones. The neomycin resistance gene was excised by Cre recombinase.
Results
FOXP3 inhibits DNA repair in tumor cells
In order to determine if FOXP3 regulate DNA repair following irradiation, we use the U2OS-DR-GFP reporter system in which expression of GFP depends repair of double strand DNA breaks (DSB) caused by ectopic expression of restriction enzyme I-Sce I. The U2OS-DR-GFP cell line has an out-of-frame GFP reporter due to an engineered I-SceI recognition sites that can be corrected by homologous recombination DNA repair (29). The efficiency of repairing the DNA break was assessed by counting GFP positive cells by flow cytometry (29). We use shRNA to knockdown FOXP3 in the U2OS cells before infecting them with adenovirus vector encoding I-SceI. Thus, the % of GFP+ cells was used to measure the efficacy of DNA repair. A representative FACS profile is presented in Fig. 1A and summary data are presented Fig. 1B. In comparison to cells expressed scrambled ShRNA, FOXP3 silencing doubled the efficiency of DNA repair. Since cell cycle affects homologous recombination, we compared cell cycle of WT and FOXP3 silenced U2OS cells. As shown in supplemental Fig. S1, FOXP3 silencing had no effect on cell cycle. These data indicated an important role for FOXP3 in DSB repair by homologous recombination.
Figure 1.

FOXP3 represses DNA repairs. A. Representative histograms depicting distribution of GFP signals. U2OS cells with integrated DR-GFP reporter were stably transduced with scrambled or FOXP3 shRNA lentiviral vectors. Then the cells were infected with adenovirus carried restriction enzyme I-Sce I. Since the HR DNA repair corrects the GFP open-reading frame, the repair efficiency was measured by GFP+ cells recorded with flow cytometry.. B. Upper panel shows summary data on DNA repairing efficiency as measured by GFP+ cells. Data shown are means +/− S.D. from 3 independent experiments. *** P < 0.001. Lower panel shows FOXP3 protein expressions detected by Western blot, as shown in the lower panel. C. Comet assay of DNA damage response. U2OS cells stably transduced with scramble or FOXP3 shRNA were irradiated for 60GY. The cells were cultured for the given periods and then subject to agarose gel electrophoresis. The photographs show the momentum of the nuclear DNA movement. D. Summary data of momentum of cellular DNA. Data shown are means +/− S.D. from three independent experiments. *** P< 0.001.
To extend the spectrum of FOXP3 impact, we used a comet assay to evaluate the repair of irradiation-induced DSB. Since damaged DNA forms a comet-like trail in agarose gel electrophoresis, the lengths of DNA tail (called momentum) could be used to assess the DSB repair. In comparison to scrambled ShRNA-transfected cells, the FOXP3 shRNA transfectants showed significantly reduced momentum (Fig. 1C, D). Therefore, FOXP3 represses DNA repair following irradiation damage.
FOXP3 is a transcriptional repressor of BRCA1
To understand how FOXP3 regulates DNA repair, we searched our previously reported expression profiles (20) for the effect of FOXP3 on 248 genes associated with DNA damage responses listed in the UniProt database. Surprisingly, using P<0.001 as statistical endpoint, 48 of the 252 genes are significantly affected following Tet-off induction of FOXP3 (Fig. 2A and supplemental Table S1). Pathway analyses suggest that FOXP3 regulates multiple DNA repair pathways, including mismatch repair, DNA double-strand break repair and nucleotide excision repair pathways (Supplemental Figure S2). The most prominent signature, however, is found in the BRCA1-mediated DNA repair pathway (Supplemental Figure S3).
Figure 2.

Regulation of DNA damage response by FOXP3. A. Gene expression profile of DNA repair-related genes. DNA repair related genes were selected by UniProt database and their gene expression levels are shown as a heat map. Gene expression levels without FOXP3 induction were normalized to 1.0. Color scale is indicated at the bottom of the figure. Using P<0.001 as statistical endpoint, 48/247 DNA repair genes are affected by FOXP3. See Table S1 and Figures S1 for details. B. Gene expression profile of DNA repair-related genes among direct targets of FOXP3 revealed by ChIP-seq. Gene names are indicated at the right side of the heat map. See Table S2 and Figure S2 for details. C. FOXP3 binding site at the BRCA1 promoter identified by a FOXP3-ChIP-seq analysis. A single peak is identified with 15 reads in ChIP-seq. DNA sequence around the FOXP3-binding site is shown as a bottom panel. “GTCAACA” marked as a red box is a known Forkhead binding motif (FKH).
By searching our recently reported FOXP3 chromatin immunoprecipitation deep sequencing data (24), we found that 13 significantly regulated genes are bound to FOXP3 (Fig. 2B). These targets are involved in multiple DNA repair pathways (Supplemental Figure S2 and Table S3). These genes are considered direct target of FOXP3. Among them, 10 were significantly repressed, while 3 were induced by FOXP3. One of the directly repressed genes is BRCA1. As shown Fig. 2C, a single peak of 15 reads was found in the BRCA1 promoter. An authentic forkhead domain-binding motif was found in the BRCA1 promoter.
To determine whether endogenous Foxp3 regulates Brca1 in murine mammary epithelial cells, we compared the Foxp3-expressing mammary epithelial cells (25) from the Scurfy (sf) mice, which have a spontaneous di-nucleotide insertion of Foxp3 (30), with the age- and sex-matched WT mice for expression of Brca1. Since the null mutation caused lethal autoimmune diseases in the immune competent mice (30), we crossed the mutation into Rag2-deficient mice lacking T and B cells. We isolated mammary epithelial cells (MEC) and prostate epithelial cells (PEC) from Rag2−/− Foxp3+/+ and Rag2−/− Foxp3sf/sf mice and compared the levels of Brca1 transcripts by real-time PCR. As shown in Fig. 3A, Foxp3sf/sf MEC showed a 4-fold increase of Brca1 mRNA. Foxp3 mRNA was reduced in the Foxp3sf/sf MEC, presumably due to nonsense-mediated mRNA decay (30). Likewise, Brca1 mRNA was also elevated in PEC (Fig. 3B). Consistent with changes in mRNA levels, the Foxp3 mutation also increased the Brca1 protein in both MEC and PEC (Fig. 3C). Based on the immunohistochemistry data, Brca1 protein elevation in Foxp3sf/sf mammary gland was limited to epithelial cells (Fig. 3D), which selectively expressed Foxp3 (25).
Figure 3.

Foxp3 repressed Brca1 expression in mammary and prostate epithelial cells in the mice. A. Mouse mammary epithelial cells (MEC) were isolated from Rag2−/− Foxp3+/+ and Rag2−/− FoxP3sf/sf mammary glands. Brca1(left panel) and Foxp3 (right panel) mRNA levels were determined by real-time RT-PCR. Data shown were means+/−S.D. of mRNA, presented as %GAPDH and have been repeated three times. ***, P< 0.001. B. Mouse prostate epithelial cells (PEC) were isolated from Rag2−/− Foxp3+/y and Rag2−/− Foxp3sf/y prostates. Brca1(left panel) and Foxp3 (right panel) mRNA levels were determined by real-time RT-PCR. Data shown were means+/−S.D. of mRNA, presented as % Gapdh. The results have been repeated three times. C. The left panel shows Brca1 and Foxp3 expression in MEC by Western blot, while the right panel shows those in PEC. D. Immunohistochemical analysis for Brca1protein in benign mammary tissue from WT and Foxp3sf/sf mice. E–H. FOXP3 represses the BRCA1 gene in normal and malignant human mammary epithelial cells. E. Silencing FOXP3 in MCF10A cells up-regulates BRCA1 mRNA level. The levels of BRCA1 mRNA (left panel) and FOXP3 (right panel) in two scrambled or two FOXP3 siRNA transfected cells as determined by real-time RT-PCR. Data shown are means+/−S.D. of three independent experiments. F. Under the same conditions as in E, MCF10A cells were lysed for Western blot to detect BRCA1 and FOXP3 protein expression as indicated. G. Reduction of BRCA1 mRNA by FOXP3 in MCF-7 Tet-off expression system. MCF-7 cells with Tet-off expression of either GFP control or GFP in conjunction with FOXP3 were cultured for 48 hours in the presence or absence of doxycyclin. The BRCA1 mRNA was quantified by real-time PCR(left panel). The induction of FOXP3 mRNA was shown in the right panel. Data shown are means+/−S.D. of three independent experiments. ***: p value< 0.001. H. BRCA1 and FOXP3 protein were detected by Western blot as shown.
In order to study the regulation of Brca1 expression by FOXP3 in human cells, we tested the effect of FOXP3 silencing on immortalized human epithelial cell line MCF10A. As shown in Fig. 3E, FOXP3 shRNA silencing caused an approximately 10-fold increase of BRCA1 transcript. A corresponding increase in BRCA1 protein was observed in Western blot (Fig. 3F). As a complementary approach, we used a Tet-off system to test the effect of inducible expression of FOXP3 on BRCA1 transcripts in human breast cancer cell line MCF-7. As shown in Fig. 3G, the induction of FOXP3 in the MCF-7 breast cancer cell line significantly inhibited BRCA1 expression. The decrease of BRCA1 protein was observed by western blot (Fig. 3H). Using a prostate cancer tissue microarray, we observed a strong inverse correlation between nuclear FOXP3 and BRCA1 levels (Table S3).
To validate the ChIP-seq data, we searched the 5′ region of the BRCA1 locus based on the FKH motifs 5′ RYMAAYA or 3′ YRKTTRT, R=A, G; Y=C, T; M=A, C; K=G, T.) and identified 5 potential FOXP3-binding sites (Figure 4A, top panel). We then used real-time PCR to determine the amounts of specific DNA sequence in chromatin immunoprecipitates of anti-FOXP3 mAb. The MCF-7 tumor cells with induced FOXP3 expression were used as source of chromatin. Of the 5 regions analyzed, the motifs located at about 0.5 Kb 5′ upstream of BRCA1 gene showed a specific binding to FOXP3. This overlaps with the sequence identified by ChIP-Seq (Fig. 2C) and thus confirmed specificity of FOXP3 binding site. In order to identify a functional element for FOXP3-mediated regulation of BRCA1, we generated luciferase reporters that covers all identified forkhead binding motifs and tested the function of FOXP3. As shown in Fig. 4B, FOXP3 significantly inhibited BRCA1 promoter activity. To determine if the FKH (GTCAACA) at 0.5Kb upstream of Brca1 gene is responsible for functional FOXP3 binding, we mutated GTCAACA to GTCGGCA in BRCA1 promoter and tested its response to FOXP3 gene expression. As shown in Fig. 4C, the mutation abrogated FOXP3 repression of BRCA1 promoter. Therefore, the FKH at 0.5kb upstream of BRCA1 TSS is the essential FOXP3 responsive element in the BRCA1 promoter.
Figure 4.
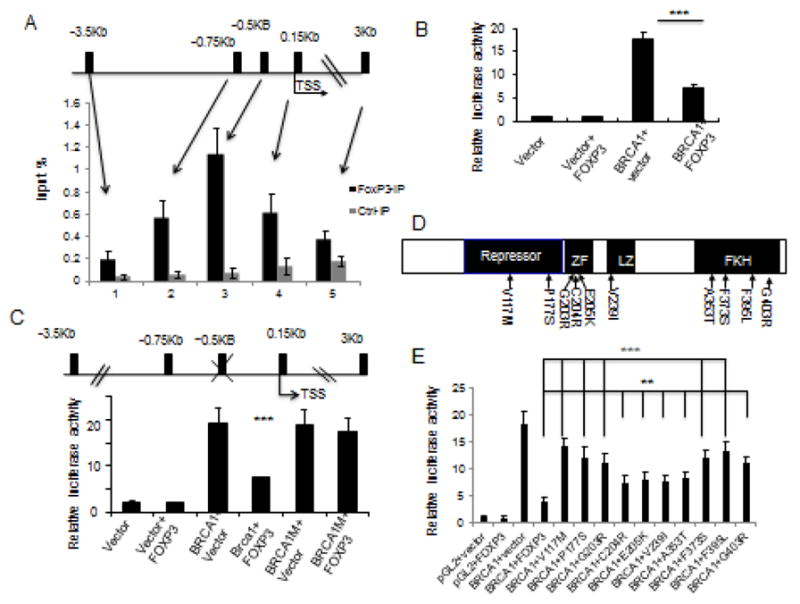
Identification of a functional FOXP3 binding site in the BRCA1 gene. A. The top panel shows a diagram of the potential forkhead binding motif in BRCA1 promoter, as marked black bars, while the lower panel shows that the % of input DNA precipitated by anti-FOXP3 or control IgG. Data shown are means+/−S.D. of % input DNA, from three independent experiments. TSS: transcriptional start site. B. Relative luciferase activity assay of BRCA1 promoter. 3.5 kB DNA upstream of BRCA1 TSS was cloned into luciferase reporter vector pGL2 transfected into 293 cells. Luciferase activity was assayed at 48 hours after transfection. Data shown are means+/−S.D. of three independent experiments. C. Identification of FOXP3 responsive element by mutational analysis. The FKH at −0.5 kB of BRCA1 TSS (GTCAACA) was mutated to GTCGGCA. The constructs used are diagrammed on the upper panel, while the transcriptional activity in the presence or absence of FOXP3 is presented in the lower panel. Data shown are means+/−S.D. of three independent analyses. D. Diagram depicting the position of somatic FOXP3 mutants from breast cancer samples used in this study. E. FOXP3 mutations abrogated its repression of BRCA1 promoter. Somatic mutations of FOXP3 from human breast and prostate cancer samples were tested for their repression of BRCA1 promoter using a luciferase reporter assay. Data shown are means+/−S.D. of triplicate sample analyses. **P<0.01, ***P<0.001.
We have reported a large number of missense mutations in breast and prostate cancer samples (12, 13). To assess the effects of these mutations on BRCA1 transcription, we tested a panel of10 somatic mutants (Fig. 4D) for their repressor activity. As shown in Fig. 4E, all 10 FOXP3 mutants have significantly reduced repressor activity on the BRCA1 promoter. Thus, FOXP3 binding to a specific FKH site in the BRCA1 promoter mediates its repressor activity. The significance of the repression is bolstered by the fact that most of the somatic mutations inactivate the repressor activity.
FOXP3 regulates BRCA1-dependent DNA repair
Using the DR-GFP U2OS cells, we tested if BRCA1 repression is the underlying cause for FOXP3-mediated repression of HR DNA repair. As shown in Fig. 5A, B, BRCA1 knockdown reduced the DNA repair efficiency while FOXP3 knockdown increased it. Importantly, the impact of FOXP3 silencing is significantly abrogated by BRCA1 silencing. Therefore, the FOXP3 repression of HR repair depends on its regulation of BRCA1.
Figure 5.

FOXP3-BRCA1 interaction regulates DNA repair in U2OS cells. A, B. U2OS cells integrated with DR-GFP reporter were infected either with scrambled or FOXP3 shRNA. Transfectants were selected by puromycin for two weeks. The U2OS cells with scramble and FOXP3 shRNA were treated with scrambled RNAi (S) or BRCA1 RNAi. A. FOXP3 and BRCA1 expression. B. Repair of DSB as measured by GFP positive cells with flow cytometry. Data shown in B are means +/− S.D. from 3 independent experiments. C. Comet assay to measure repair of γ-ray-induced DNA damages. U2OS cells with scramble and FOXP3 shRNA, FOXP3 and/or BRCA1 ShRNAs were irradiated for 60 Gy. The cells were cultured for given periods and used for comet assays. Data showed were +/−S.D. means from three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Our data in Fig. 1 show that FOXP3 silencing increased repair of DNA damage induced by γ-irradiation in U2OS cell line. To determine if FOXP3-BRCA1 interaction explains the role for FOXP3 in DNA repair following irradiation, we evaluate the impact of FOXP3 silencing on DNA repair when BRCA1 expression is silenced by siRNA. FOXP3 and BRCA1 were efficiently silenced using ShRNA and siRNA respectively (Fig. 5A). As shown in Fig. 1B, the increase of HR in U2OS cells is significantly blunted by BRCA1 silencing. Remarkably, despite lack of significant effect of BRCA1 silencing alone in DNA momentum over a 3-hour period, BRAC1 silencing significantly blunted the impact of FOXP3 silencing at 1 hour, and essentially ablated the impact by 3 hours (Fig. 5C). Therefore, to a large extent, FOXP3 regulates repair of irradiation-induced DNA damage via a BRCA1-dependent mechanism.
FOXP3 regulates cancer cell resistance to γ-irradiation through BRCA1
We transduced the TSA cells with scramble, Foxp3 shRNA, Foxp3 shRNA/Brca1 shRNA and Brca1 shRNA. After confirming the efficacy of the silencing vectors (Fig. 6A), the 4 cell lines were exposed to given doses of γ–irradiation. The tumor cell survival was determined using colony formation assay. As shown in Fig. 6B, while Foxp3 silencing had no effect in the number of colonies from non-irradiated TSA cells, it increased the number of TSA colonies at all doses of irradiations tested. To determine whether Foxp3-Brca1 interaction affects tumorigenity of the irradiated TSA cells, we injected the 4 tumor lines with or without 15Gy irradiation into mammary fat-pad of syngeneic mice and monitored tumor growth. Foxp3 silencing somewhat increased the growth rate of tumors originated from non-irradiated tumor cells. This impact is abrogated by Brca1 silencing (Fig. 6C). A very striking impact of Foxp3 silencing was observed when TSA cells were subjected to 15G irradiation. As shown in Fig. 6D, while the irradiation eliminated tumorigenicity of the scrambled shRNA transduced TSA, mice that received irradiated Foxp3-silenced TSA developed tumors, albeit at a slower pace than those that received non-irradiated tumor cells. Again, the impact of Foxp3 silencing was abrogated by concurrent Brca1 silencing. The dramatic effect demonstrates that Foxp3-Brca1 interaction is a major determinant of cancer cell resistancy to irradiation.
Figure 6.

Foxp3-Brca1 interaction regulates survival and tumorigenicity of mouse mammary tumor cell line TSA. A. Efficacy of ShRNA silencing of Foxp3 and siRNA silencing of Brca1, as detected by Western blot. B. Foxp3-Brca1 interaction determines TSA resistance to irradiation, as measured by colony formation assay. TSA with ShRNA silencing of Foxp3 and/or Brca1 received 0, 5, 10, 15, or 20Gy of γ–irradiation. The irradiated cell lines were cultured for a week before the colonies were counted. The data shown are from those from triplicate cultures. ***: P <0.001. C and D. Foxp3-Brca1 interaction regulates tumorigencity of γ-irradiated TSA. TSA cell lines that were stably transfected with scrambled, Foxp3 shRNA and/or Brca1shRNA were either unirradiated (C) or irradiated with 15Gy of γ–ray (D) were transplanted into mammary fatpads of syngeneic BALB/c mice. The tumor sizes were monitored over 5 weeks. Data shown are means+/−S.D. of tumor volumes and have been repeated three times.
Given the high rates of the somatic FOXP3 mutations in both breast and prostate cancer samples, it is of interest whether these mutations would increase cancer cell resistance to irradiation. Since somatic mutation is best mimicked by homologous recombination, we test the impact of somatic mutation in HCT116 as the method of gene knockin was established for this line (28). As diagramed in Fig. 7A,B we took an adenovirus-associated virus-based approach to knock in a mutant allele uncovered from prostate cancer cell sample into the HCT116 cells. Since HCT116 has a single X-chromosome, only one allele of FOXP3 is present. As shown in Fig. 7C, mutation of FOXP3 resulted in significant increase of BRCA1 mRNA. The WT and two independent mutant HCT116 cell clones were given 5Gy, 10Gy, or 15Gy or irradiation and the survival of HCT116 cells were determined by colony formation assay. As shown in Fig. 7D, knockin of mutant alleles increased the % survival by 2–4 folds across the irradiation doses. Thus, a somatic mutation uncovered from cancer samples increased cancer cell resistance to irradiation.
Figure 7.

Cancer-related somatic mutation of FOXP3 increases cancer cell resistance to irradiation. Somatic missense mutation uncovered from prostate cancer tissue (203G>R) were knocked into colon cancer cell line, as diagramed in A. The position of primers used for both screening of homologous recombinant and for Cre-mediated excision of Neo cassette is marked with bold arrows. Red color is used for vector sequence, while the black color is used for sequence of endogenous locus. B. Confirmation of homologous recombination and excision of Neo cassette by PCR. Two homologous recombination clones were identified from two 96-well-plates of G418 resistant single clones. The neomycin resistance gene was excised by Cre recombinase carried by adenovirus. Similar data were obtained for the homologous recombination in the right arm. C. Knockin of mutant FOXP3 increase BRCA1 transcripts as determined by real-time PCR. D. Survival of cancer cells after given doses of γ-irradiation. Data shown are means and S.D. of triplicated culture. *P<0.05, ***P<0.001.
Discussion
Taken together, the data presented in this manuscript revealed that FOXP3 is a major regulator for cellular response to double-stranded DNA break. This function is mediated through transcriptional repression of BRCA1. Using the DR-GFP transfected U2OS cells, we were able to show that FOXP3 inhibits the HR DNA repair. However, since BRCA1 has also been implicated in non-homologous end-joint DNA repair (31–33), the FOXP3-regulated repair of γ-irradiation-induced DNA damage may involve both non-homologous end joint and HR repairs.
Given the relevance of γ-irradiation in cancer therapy, we have focused on both DNA repair and cancer cell survival following γ-irradiation. Our data show that FOXP3 is upregulated following γ-irradiation, which is consistent with FOXP3’s role in cellular response to irradiation. Although we have not investigated the mechanism of the induction, our recent studies implicated that FOXP3 transcription is upregulated by an ATF-2/JNK dependent mechanism (23). Given the recent report that γ- and UV-irradiation induces ATF2 transcription (34, 35), it is worth exploring whether induction of ATF2 may contribute to induction of FOXP3. In addition, FOXP3 is also regulated by NFκB (36), which in turn have been shown to be upregulated by irradiation (37).
Our data also show that γ-irradiation upregulates BRCA1 transcripts. Given the role for FOXP3 in repressing BRCA1 expression, the paralleled upregulation of both genes is somewhat unexpected. Nevertheless, FOXP3 must have restrained irradiation-induced BRCA1 expression as shRNA silencing of FOXP3 result in even more BRCA1 induction.
Using both tumorigenicity and colony-forming assays, we demonstrate that defective FOXP3 expression leads to increased radioresistance of cancer cells. Given the wide-spread abnormal FOXP3 expression in cancers, including from breast, prostate and pancreatic cancers and melanoma (12, 15–18), it is of great interest to determine whether FOXP3 defects contribute to cancer radioresistance. We used homologous recombination to knockin a missense FOXP3 mutant from prostate cancer and demonstrated the impact of FOXP3 mutation in cancer resistance to irradiation. The clear increase in cancer cell resistance to irradiation raised the possibility that cancer lesion associated with FOXP3 mutations may require higher doses of irradiation. The fact that 8 of 9 missense mutant tested have significantly reduced repressor activity for BRCA1 promoter is consistent with this view.
It is worth pointing out that although we have focused on FOXP3-BRCA1 interaction in radioresistance, our systemic approach based on gene expressing profiling and ChIP-seq demonstrated that FOXP3 can regulate DNA repair through a multitude of mechanisms. As shown in Table S2, among 12 other direct FOXP3 targets that are implicated in DNA damage response, FOXP3 repressed 9 genes that are involved either in common steps of DNA repair (RBM14) (38), or pathway specific for homologous recombination repair (RAD54B, NFKBIL2) (39, 40), nucleotide excision repair (CHAF1A, POLD1, POLD3, and DDB) (41–43) and base excision repair (LIG1, FEN1) (44, 45). In contrast, the three genes that are upregulated by FOXP3 (JMY, PAXIP/TPIP, and RRM2B) (46–48) are involved in P53 checkpoint of DNA damage responses. Therefore, in addition to previous described function of FOXP3 in growth inhibition, FOXP3 may suppress tumor cells both by preventing repair of DNA damage and promote apoptosis associated with DNA damage.
Apart from the direct targets, FOXP3 also indirectly regulates expression of a large number of genes involved in DNA repair. It is of particularly interest that by repressing key regulators of BRCA1 DNA repair pathway, such as BRCA2 and BARD, FOXP3 has exerted the most profound effect on BRCA1-mediaed DNA repair (P<10−34). Much like the direct targets, the indirect FOXP3 targets broadly affect mismatch DNA repair, nucleotide excision repair, HR and NHEJ repair. Since many chemotherapeutics exert their effects through tumor cell DNA damage, it is likely that FOXP3, which is also induced by chemotherapeutic drugs and stress (22, 23), may emerge as a key regulator for cancer response to chemotherapeutics. In support of this concept, a significant correlation between FOXP3 expression (judged by both nuclear and cytoplasmic staining) and better survival of anthracyclines-treated primary breast cancer patients was recently reported (49).
Supplementary Material
Acknowledgments
We thank Dr. Fei Tang for assistance in cell cycle analysis, Dr. Runhua Liu for the FOXP3 G203R mutant, Darla Kroft for editorial assistance. This work is supported by grants from National Institute of Health (CA120901) and US Department of Defense (W81XWH-08-1-0537 and W81XWH-09-0153).
Footnotes
The authors have no conflict of interest in this study.
References
- 1.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–53. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 2.Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104:1129–37. doi: 10.1002/cncr.21324. [DOI] [PubMed] [Google Scholar]
- 3.Bhide SA, Nutting CM. Recent advances in radiotherapy. BMC Med. 2010;8:25. doi: 10.1186/1741-7015-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008;8:545–54. doi: 10.1038/nrc2419. [DOI] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–9. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 7.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 8.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–92. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 9.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–75. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 10.Scully R, Ganesan S, Brown M, De Caprio JA, Cannistra SA, Feunteun J, et al. Location of BRCA1 in human breast and ovarian cancer cells. Science. 1996;272:123–6. doi: 10.1126/science.272.5258.123. [DOI] [PubMed] [Google Scholar]
- 11.Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–9. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Liu R, Li W, Chen C, Katoh H, Chen GY, et al. Somatic Single Hits Inactivate the X-Linked Tumor Suppressor FOXP3 in the Prostate. Cancer Cell. 2009;16:336–46. doi: 10.1016/j.ccr.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell. 2007;129:1275–86. doi: 10.1016/j.cell.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Wang L, Zheng P. X-linked tumor suppressors: perplexing inheritance, a unique therapeutic opportunity. Trends Genet. 2010;26:260–5. doi: 10.1016/j.tig.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebert LM, Tan BS, Browning J, Svobodova S, Russell SE, Kirkpatrick N, et al. The regulatory T cell-associated transcription factor FoxP3 is expressed by tumor cells. Cancer Res. 2008;68:3001–9. doi: 10.1158/0008-5472.CAN-07-5664. [DOI] [PubMed] [Google Scholar]
- 16.Ladoire S, Arnould L, Mignot G, Coudert B, Rebe C, Chalmin F, et al. Presence of Foxp3 expression in tumor cells predicts better survival in HER2-overexpressing breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 2011;125:65–72. doi: 10.1007/s10549-010-0831-1. [DOI] [PubMed] [Google Scholar]
- 17.Merlo A, Casalini P, Carcangiu ML, Malventano C, Triulzi T, Menard S, et al. FOXP3 expression and overall survival in breast cancer. J Clin Oncol. 2009;27:1746–52. doi: 10.1200/JCO.2008.17.9036. [DOI] [PubMed] [Google Scholar]
- 18.Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344–50. doi: 10.1158/0008-5472.CAN-06-3304. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Wang L, Katoh H, Liu R, Zheng P, Liu Y. Identification of a tumor suppressor relay between the FOXP3 and the Hippo pathways in breast and prostate cancers. Cancer Res. 2011;71:2162–71. doi: 10.1158/0008-5472.CAN-10-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu R, Wang L, Chen G, Katoh H, Chen C, Liu Y, et al. FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res. 2009;69:2252–9. doi: 10.1158/0008-5472.CAN-08-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuo T, Liu R, Zhang H, Chang X, Liu Y, Wang L, et al. FOXP3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. J Clin Invest. 2007;117:3765–73. doi: 10.1172/JCI32538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung DJ, Jin DH, Hong SW, Kim JE, Shin JS, Kim D, et al. Foxp3 expression in p53-dependent DNA damage responses. J Biol Chem. 2010;285:7995–8002. doi: 10.1074/jbc.M109.047985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Wang Y, Li W, Zheng P, Liu Y. Activating transcription factor 2 and c-Jun-mediated induction of FoxP3 for experimental therapy of mammary tumor in the mouse. Cancer Res. 2009;69:5954–60. doi: 10.1158/0008-5472.CAN-09-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katoh H, Qin ZS, Liu R, Wang L, Li W, Li X, et al. FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol Cell. 2011;44:770–84. doi: 10.1016/j.molcel.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen GY, Chen C, Wang L, Chang X, Zheng P, Liu Y. Cutting Edge: Broad Expression of the FoxP3 Locus in Epithelial Cells: A Caution against Early Interpretation of Fatal Inflammatory Diseases following In Vivo Depletion of FoxP3-Expressing Cells. J Immunol. 2008;180:5163–6. doi: 10.4049/jimmunol.180.8.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giovarelli M, Musiani P, Modesti A, Dellabona P, Casorati G, Allione A, et al. Local release of IL-10 by transfected mouse mammary adenocarcinoma cells does not suppress but enhances antitumor reaction and elicits a strong cytotoxic lymphocyte and antibody-dependent immune memory. J Immunol. 1995;155:3112–23. [PubMed] [Google Scholar]
- 27.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–9. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Guo C, Chen Y, Shulha HP, Schnetz MP, LaFramboise T, et al. Epitope tagging of endogenous proteins for genome-wide ChIP-chip studies. Nat Methods. 2008;5:163–5. doi: 10.1038/nmeth1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–8. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 31.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. 2009;7:1110–8. doi: 10.1158/1541-7786.MCR-09-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19:524–9. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arora H, Qureshi R, Park AK, Park WY. Coordinated regulation of ATF2 by miR-26b in gamma-irradiated lung cancer cells. PLoS One. 2011;6:e23802. doi: 10.1371/journal.pone.0023802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herdegen T, Blume A, Buschmann T, Georgakopoulos E, Winter C, Schmid W, et al. Expression of activating transcription factor-2, serum response factor and cAMP/Ca response element binding protein in the adult rat brain following generalized seizures, nerve fibre lesion and ultraviolet irradiation. Neuroscience. 1997;81:199–212. doi: 10.1016/s0306-4522(97)00170-x. [DOI] [PubMed] [Google Scholar]
- 36.Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–31. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 37.Veeraraghavan J, Natarajan M, Aravindan S, Herman TS, Aravindan N. Radiation-triggered tumor necrosis factor (TNF) alpha-NFkappaB cross-signaling favors survival advantage in human neuroblastoma cells. J Biol Chem. 2011;286:21588–600. doi: 10.1074/jbc.M110.193755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwasaki T, Chin WW, Ko L. Identification and characterization of RRM-containing coactivator activator (CoAA) as TRBP-interacting protein, and its splice variant as a coactivator modulator (CoAM) J Biol Chem. 2001;276:33375–83. doi: 10.1074/jbc.M101517200. [DOI] [PubMed] [Google Scholar]
- 39.Miyagawa K, Tsuruga T, Kinomura A, Usui K, Katsura M, Tashiro S, et al. A role for RAD54B in homologous recombination in human cells. EMBO J. 2002;21:175–80. doi: 10.1093/emboj/21.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Donnell L, Panier S, Wildenhain J, Tkach JM, Al-Hakim A, Landry MC, et al. The MMS22L-TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell. 2010;40:619–31. doi: 10.1016/j.molcel.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kadyrova LY, Blanko ER, Kadyrov FA. CAF-I-dependent control of degradation of the discontinuous strands during mismatch repair. Proc Natl Acad Sci U S A. 2011;108:2753–8. doi: 10.1073/pnas.1015914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell. 2010;37:714–27. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 43.Stoyanova T, Yoon T, Kopanja D, Mokyr MB, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol Cell Biol. 2008;28:177–87. doi: 10.1128/MCB.00880-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma W, Panduri V, Sterling JF, Van Houten B, Gordenin DA, Resnick MA. The transition of closely opposed lesions to double-strand breaks during long-patch base excision repair is prevented by the coordinated action of DNA polymerase delta and Rad27/Fen1. Mol Cell Biol. 2009;29:1212–21. doi: 10.1128/MCB.01499-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomkinson AE, Levin DS. Mammalian DNA ligases. Bioessays. 1997;19:893–901. doi: 10.1002/bies.950191009. [DOI] [PubMed] [Google Scholar]
- 46.Coutts AS, Boulahbel H, Graham A, La Thangue NB. Mdm2 targets the p53 transcription cofactor JMY for degradation. EMBO Rep. 2007;8:84–90. doi: 10.1038/sj.embor.7400855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–9. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 48.Wu J, Prindle MJ, Dressler GR, Yu X. PTIP regulates 53BP1 and SMC1 at the DNA damage sites. J Biol Chem. 2009;284:18078–84. doi: 10.1074/jbc.M109.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ladoire S, Mignot G, Dalban C, Chevriaux A, Arnould L, Rebe C, et al. FOXP3 expression in cancer cells and anthracyclines efficacy in patients with primary breast cancer treated with adjuvant chemotherapy in the phase III UNICANCER-PACS 01 trial. Ann Oncol. 2012 doi: 10.1093/annonc/mds028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.