

Abstract
Derivation of induced pluripotent stem (iPS) cells is mainly an epigenetic reprogramming process. It is still quite controversial how genomic imprinting is reprogrammed in iPS cells. Thus, we derived multiple iPS clones from genetically identical mouse somatic cells. We found that parentally inherited imprint was variably lost among these iPS clones. Concurrent with the loss of DNA methylation imprint at the corresponding Snrpn and Peg3 imprinted regions, parental origin-specific expression of the Snrpn and Zim1 imprinted genes was also lost in these iPS clones. This loss of parental genomic imprinting in iPS cells was likely caused by the reprogramming process during iPS cell derivation because extended culture of iPS cells did not lead to significant increase in the loss of genomic imprinting. Intriguingly, one to several paternal chromosomes appeared to have acquired de novo methylation at the Snrpn and Zac1 imprinted regions in a high percentage of iPS clones. These results might have some implications for future therapeutic applications of iPS cells. Since DNA methylation imprint can be completely erased in some iPS clones at multiple imprinted regions, iPS cell reprogramming may also be employed to dissect the underlying mechanisms of erasure, reacquisition and maintenance of genomic imprinting in mammals.
Introduction
Induced pluripotent stem (iPS) cells were derived from somatic cells directly with four transcription factors (Oct4, Sox2, C-Myc and Klf4) (Okita et al., 2007; Takahashi et al., 2007; Takahashi and Yamanaka, 2006; Wernig et al., 2007). This epigenetic reprogramming process is rapid and stochastic (Yamanaka, 2009). Genomic imprinting is an epigenetic phenomenon that is characterized by parental origin-dependent expression of the imprinted genes (Barlow, 2011; Bartolomei, 2009; Bartolomei and Ferguson-Smith, 2011; Ferguson-Smith, 2011; Li, 2013). Since many imprinted genes play an important role in development and diseases, it is important to know whether genomic imprinting is properly reprogrammed in iPS cells (Tomizawa and Sasaki, 2012).
About 150 imprinted genes have been discovered in mammals so far (see http://www.mousebook.org/catalog.php?catalog=imprinting). Some are singleton imprinted genes (Bartolomei, 2009). Most are clustered and co-regulated by a cis-acting imprinting control region (ICR) that is methylated on the maternal or paternal chromosomes (Barlow, 2011; Bartolomei and Ferguson-Smith, 2011; Ben-Porath and Cedar, 2000; Lewis and Reik, 2006; Li, 2013). DNA methylation imprint at the ICRs is reset during gametogenesis (Li, 2013). Differentially methylated region (DMR) is essential for maintaining genomic imprinting in somatic cells. The loss of DNA methylation imprint at the DMR results in the loss of mono-allelic expression of the corresponding imprinted genes in these imprinted domains (Li et al., 1993, 2008).
It is quite controversial how iPS reprogramming may affect expression of the imprinted genes. To further examine how genomic imprinting may be perturbed in iPS cells, we derived multiple iPS clones from genetically identical hybrid MEF cells carrying single nucleotide polymorphisms (SNPs) at some imprinted regions. We analyzed DNA methylation imprint by Combined Bisulfite Restriction Analysis (COBRA). Taking advantage of the SNPs at two DMRs, we examined the inheritance of parental DNA methylation imprint at the Snrpn and Zac1 imprinted regions in these iPS clones. In addition, we performed allele-specific RT-PCR analysis to determine if mono-allelic expression of the Snrpn and Zim1 imprinted genes was retained in iPS cells and their progeny.
Materials and methods
Timed mouse mating for MEF cells
The transgenic mice carrying the TetO-OSKM transgene and the Rosa-rtTA transgene as well as the DBA/2 female mice were obtained from the Jackson Laboratories. These two transgenic mice were originally generated in the Jaenisch lab (Carey et al., 2010). Timed mating was set up between the wild-type DBA/2 female mice and the male mice that were homozygous for the Rosa-rtTA transgene and the TetO-OSKM transgene at the Col1A1 locus. The male mice with the Rosa-rtTA transgene and the TetO-OSKM transgene were primarily on a 129 genetic background (129*) based on the information provided by the Jackson laboratories. Noon of the day when vaginal plug was found in the female mice was counted as half day of pregnancy. Pregnant female mice from this cross were sacrificed at E13.5 for live embryos that were used for deriving hybrid (DBA/129*) MEF cells carrying a Rosa-rtTA transgene and a TetO-OSKM transgene.
Derivation of iPS clones
Hybrid (DBA/129*) MEF cells carrying a Rosa-rtTA transgene and a TetO-OSKM transgene were used for the derivation of iPS clones. The MEF cells were plated on irradiated SNL feeder cells at 100,000 MEF cells/10-cm dish plate with the addition of doxycycline at a final concentration of 2 μg/ml. ES cell medium was used for MEF cells cultured on irradiated SNL feeder cells that constitutively express leukemia inhibitory factor (LIF) (McMahon and Bradley, 1990). The medium was changed every 2–3 days and 2 μg/ml of doxycycline was included in the ES cell medium for 3–4 weeks until ES cell-like iPS colonies were picked individually. After trypsin digestion, individual iPS cell colonies were resuspended by pipetting and plated on irradiated SNL feeder cells in one well of a 96-well plate. When iPS cell colonies became confluent, they were digested with trypsin. Resuspended iPS cells were transferred to one well of a 24-well plate, and subsequently to one well of a 6-well plate for expansion.
Three iPS clones (D15D, D36 and DL3) originated from the hybrid OSKM/+ MEF cells plated on the irradiated feeder cells that were derived from primary MEF cells. These three iPS clones were expanded on the irradiated SNL feeder cells after being picked. All other iPS clones were derived from the hybrid OSKM/+ MEF cells plated on the irradiated SNL feeder cells and subsequently expanded on SNL feeder cells following the procedure described above.
Culture of iPS clones on feeder cells or on gelatin-coated plates
The standard ES cell medium was used for the iPS cell culture. It was made of DMEM plus 15% of FCS, l-glutamine, non-essential amino acids, β-mercaptoethanol and leukemia inhibitory factor (LIF). The medium was changed daily. The iPS clones were passaged once every 3–5 days when they were grown on irradiated SNL feeder cells. They were diluted and plated on gelatin-coated 6-well plates until the iPS cell culture became confluent for DNA, protein or total RNA sample preparation.
Embryoid body (EB) culture
About 1–2 million of early-passage (P < 5) iPS cells grown on irradiated SNL feeder cells were plated on non-adherent 10-cm bacterial dish plates coated with poly-hema (Sigma). ES cell medium without LIF was used for the EB culture. The medium was changed once every 2–3 days without removing the aggregated EBs. Mature EBs were harvested at different intervals from day 8 until day 16 in suspension culture for total RNA sample preparation. They were dissolved in Trizol reagent (Invitrogen).
Bisulfite mutagenesis
Genomic DNA samples from the parental cells as well as the iPS clones were subjected to bisulfite mutagenesis with the EZ DNA Methylation-Gold™ Kit (Zymo Research). The bisulfite mutagenized DNA was subjected to COBRA analysis and bisulfite colony sequencing.
Combined Bisulfite Restriction Analysis (COBRA) of the DMR regions
PCR product was derived from the bisulfite mutagenized DNA samples with the primers covering a portion of the DMR regions (Zuo et al., 2012). The amplified bisulfite PCR product was used for restriction digestion with the restriction enzymes targeting the CpG sites within the DMR regions. Since the restriction enzyme sites were lost (or gained) when the unmethylated C was mutagenized to become U, the PCR product from the unmethylated DNA displayed different restriction enzyme digestion patterns on the agarose gel than the PCR product from the methylated DNA.
Bisulfite sequencing
The PCR product from the bisulfite mutagenized DNA samples covering a portion of the DMR regions was cloned into the pGEM-T vector system (Promega). Bacterial colonies were sent for direct sequencing (Zuo et al., 2012). The sequence result was analyzed with the web-based bisulfite DNA sequence analysis program called QUMA (see the website: http://quma.cdb.riken.jp/).
Allele-specific RT-PCR analysis of the imprinted genes
RT-PCR for the Snrpn and Zim1 imprinted genes was performed with the primers spanning at least one intron for the total RNA samples isolated from the EBs generated with iPS cells or from iPS cells grown on gelatin-coated plates. The purified RT-PCR product was directly sent for sequencing to detect the SNPs present in the original transcript for these two imprinted genes.
Results
Established iPS clones are pluripotent
To assess how genomic imprinting is reprogrammed in iPS cells, we employed two different strains of mice to generate hybrid MEF cells that carry the SNPs at the imprinted regions. A total of 12 iPS clones were derived from the hybrid (DBA/129*) MEF cells carrying two transgenes for doxycycline-inducible expression of the reprogramming factors (Fig. 1A). When they were plated on the irradiated SNL feeder cells (McMahon and Bradley, 1990), these iPS clones appeared to have similar morphology to undifferentiated wild-type ES cells (Figs. 1B and 1SA). All iPS clones readily formed embryoid bodies (EBs) on non-adherent bacterial plates (Fig. 1SB). Similar to the EBs of wild-type ES cells (Lane 3), mature EBs at day 13 of the iPS clone L7 (Lane 7) were shown to up-regulate the expression of two cardiac marker genes, cardiac troponin T (cTnT) and myosin light chain 2 atrial transcripts (Mlc2a), based on semi-quantitative RT-PCR analysis (Fig. 1SC). These results suggest that the isolated iPS clones possess differentiation potential.
Figure 1.

A diagram is shown for the experimental procedure in the derivation of iPS clones. A, schematic experimental procedure. Timed mating was set up between the wild-type DBA/2 female mice (DBA female) and the male mice primarily on a 129 genetic background (129*) carrying the OSKM transgene at the Col1A1 locus on both chromosomes (Carey et al., 2010). E13.5 embryos were used to derive hybrid MEF (DBA/129*) cells carrying one copy of the OSKM transgene from the pregnant female mice. B, an iPS clone (D15D, left panel) with similar morphology to the wild-type TC1 ES cells (ES, right panel). The microscope images are shown for D15D and TC1 on the irradiated SNL feeder cells.
The cells for these iPS clones expressed two pluripotency marker proteins (Oct4 and Nanog) when stained with the antibodies against these two proteins (Fig. 2SA). Expression of Oct4 was also confirmed by western blotting (Fig. 2SB). ZFP57, a master regulator in genomic imprinting (Li et al., 2008; Mackay et al., 2008), was not expressed in parental MEF cells but was turned on in the iPS cells (Fig. 2SB). Indeed, ZFP57 is highly enriched in undifferentiated ES cells and it maintains DNA methylation imprint in undifferentiated ES cells as well as in mouse (Li et al., 2008; Li and Leder, 2007; Zuo et al., 2012). These results suggest that these iPS clones are truly reprogrammed iPS cells that express pluripotency marker genes.
Examining paternal methylation imprint by COBRA
DNA methylation imprint for four imprinted domains is known to be established during spermatogenesis (Li, 2013; Watanabe et al., 2011). We employed COBRA to examine the IG-DMR of Dlk1-Dio3, the H19 DMR of Igf2-H19 and the Rasgrf1 DMR regions in these iPS clones (Kobayashi et al., 2006).
Methylation at the IG-DMR was largely maintained
The IG-DMR, located between the Dlk1 and Gtl2 imprinted genes, is the ICR of the Dlk1-Dio3 imprinted domain (Hiura et al., 2007; Lin et al., 2003; Schmidt et al., 2000). Substantial methylation was retained in most iPS clones (Lanes 5–6, 8–14 of Fig. 2A). However, little methylation was observed in two iPS clones (Lanes 7,15 of Fig. 2A). All iPS clones were less methylated than wild-type ES cells in Lane 16 (Fig. 2A). By contrast, hypermethylation was observed in the parental mouse tail samples and MEF cells (Lanes 1–4 of Fig. 2A).
Figure 2.
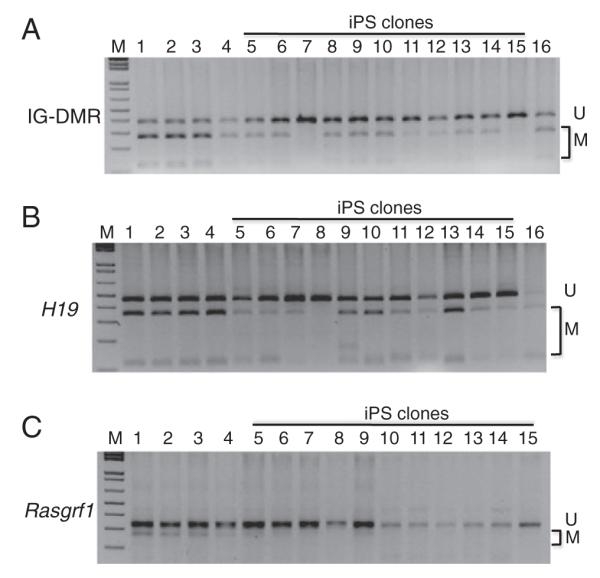
COBRA analysis of paternally inherited DNA methylation imprint. Genomic DNA was isolated from the iPS clones (Lanes 5–15), mouse tail of the mother (Lane 1) and father (Lane 2), MEF cells without doxycycline (Lane 3), MEF cells with doxycycline (Lane 4) as well as from the wild-type TC1 ES cells (Lane 16). COBRA was performed for three DMR regions (IG-DMR of the Dlk1-Dio3 imprinted region, H19 DMR, Rasgrf1 DMR). The iPS clones used for COBRA are: L7 (Lane 5), L9 (Lane 6), B11 (Lane 7), E7 (Lane 8), D15D (Lane 9), D36 (Lane 10), C8D (Lane 11), E7D (Lane 12), S4D (Lane 13), S5D (Lane 14) and n2 (Lane 15). U, unmethylated product. M, methylated product. A, IG-DMR with TaqI digestion. B, H19 DMR with ClaI digestion. C, Rasgrf1 DMR with BstUI digestion.
The H19 DMR was partially methylated in most iPS clones
The H19 DMR is the ICR for the Igf2-H19 imprinted region (Bartolomei, 2009). Based on COBRA, substantial methylation was observed in three iPS clones (Lanes 9–10, 13 of Fig. 2B). Partial methylation was present in six iPS clones (Lanes 5–7, 11–12, 14 of Fig. 2B). Little methylation was observed in two other iPS clones (Lanes 8, 15 of Fig. 2B).
Methylation at the Rasgrf1 DMR was mostly lost
Like the IG-DMR and H19 DMR, methylation at the Rasgrf1 DMR is also inherited on the paternal chromosome (Kobayashi et al., 2006; Watanabe et al., 2011). According to COBRA, we found that it was partially methylated in the parental MEF cells and mouse tail samples (Lanes 1–4 of Fig. 2C). Partial methylation was observed in three iPS clones (Lanes 5–6, 9 of Fig. 2C). Methylation was almost completely lost in other iPS clones (Fig. 2C).
Taken together, these results suggest that paternally inherited DNA methylation imprint was largely maintained at the IG-DMR and H19 DMR regions but mostly lost at the Rasgrf1 DMR during iPS cell derivation.
Examining maternal methylation imprint by COBRA
DNA methylation imprint at most ICRs is inherited on the maternal chromosome (Kobayashi et al., 2006; Li, 2013). To gauge how these ICRs were reprogrammed in iPS cells, we analyzed five imprinted domains (Peg1, Peg3, Peg10, Snrpn and Zac1) by COBRA.
Hypermethylation at the Peg1 DMR was stably maintained
According to COBRA, Peg1 DMR was mostly methylated in the parental mouse tail samples (Lanes 1–2 of Fig. 3A). This was also true in the hybrid MEF cells with or without doxycycline (Lanes 3–4) and TC1 ES cells (Lane 16). This hypermethylation was completely maintained in all iPS clones (Lanes 5–15 of Fig. 3A). These results suggest that Peg1 DMR was not sensitive to demethylation during iPS reprogramming.
Figure 3.

COBRA analysis of maternally inherited DNA methylation imprint. Genomic DNA was isolated from the iPS clones (Lanes 5–15 for Fig. 3A–C and Lanes 5–16 for Fig. 3D–E), mouse tail of the mother (Lane 1) and father (Lane 2), MEF cells without doxycycline (Lane 3), MEF cells with doxycycline (Lane 4) as well as from the wild-type TC1 ES cells (Lane 16 for A, B and C, Lane 17 for D and E). COBRA was performed for five DMR regions (Peg1, Peg3, Snrpn, Zac1 and Peg10). The iPS clones used for COBRA are: L7 (Lane 5), L9 (Lane 6), B11 (Lane 7), E7 (Lane 8), D15D (Lane 9), D36 (Lane 10), C8D (Lane 11), E7D (Lane 12), S4D (Lane 13), S5D (Lane 14), n2 (Lane 15) and DL3 (Lane 16 for D and E). U, unmethylated product. M, methylated product. A, Peg1 DMR with ClaI digestion. B, Peg3 DMR with TaqI digestion. C, Snrpn DMR with HhaI digestion. D, Zac1 DMR with TaqI digestion. E, Peg10 DMR with HhaI digestion.
Methylation at the Peg3 DMR was erased in most iPS clones
As shown in Fig. 3B, only two iPS clones in Lanes 5 and 7 had methylation at the Peg3 DMR that was comparable to that of TC1 ES cells (Lane 16) or the parental cells (Lanes 1–4). Methylation was almost completely missing in other iPS clones (Fig. 3B), suggesting that the Peg3 DMR was very sensitive to demethylation during iPS reprogramming.
Methylation at the Snrpn DMR was variably lost in iPS cells
Like Peg1 and Peg3 DMRs, DNA methylation imprint at the Snrpn DMR is stably maintained on the maternal chromosome in somatic cells. Based on COBRA, significant methylation was observed in seven iPS clones (Lanes 5, 7–10, 12–13 of Fig. 3C). Little methylation was present in three iPS clones (Lanes 6, 11, 15 of Fig. 3C). No methylation was observed in Lane 14. The COBRA result indicates that methylation at the Snrpn DMR was variably lost during iPS reprogramming.
The Zac1 DMR was partially methylated in some but lost in other iPS clones
Similar to what had been observed at the Snrpn DMR region, methylation at the Zac1 DMR was also variably lost in these iPS clones in comparison to that of their parental cells (Fig. 3D). Substantial methylation was present in four iPS clones (Lanes 5–6, 12–13 of Fig. 3D). Four iPS clones (Lanes 9–11, 14) were slightly methylated at the Zac1 DMR. Little or no methylation was observed in four iPS clones (Lanes 7–8, 15–16).
Peg10 DMR was almost completely unmethylated
As shown in Fig. 3E, parental mouse tail samples and MEF cells displayed both methylated and unmethylated COBRA product of the Peg10 DMR (Lanes 1–4). Peg10 DMR was also methylated in TC1 ES cells (Lane 17). However, all tested iPS clones except two had almost no methylated COBRA product although the iPS clone in Lane 8 failed to give rise to any bisulfite PCR product after several tries (Fig. 3E). These results suggest that Peg10 DMR was very sensitive to demethylation during iPS cell derivation, similar to Peg3 DMR (Fig. 3B).
Bisulfite sequencing of four DMR regions
According to COBRA, there was significant but variable loss of methylation at most imprinted regions in these iPS clones. However, COBRA only analyzes one or several CpG sites embedded within the restriction enzyme sites. It is formally possible that methylation levels may be different at other CpG sites. To test this possibility, we subjected the bisulfite PCR product to bacterial colony sequencing. We sequenced two paternal (IG-DMR of Dlk1-Dio3 and Rasgrf1) and two maternal (Snrpn and Zac1) DMR regions. The results are illustrated below in Figs. 3S, 4S, 4 and 5.
Figure 4.

Maternally inherited DNA methylation imprint was variably lost at the Snrpn DMR in iPS clones. Bacterial colony sequencing of the bisulfite PCR product was performed for genomic DNA samples from the parental cells as well as from the iPS clones at early passages (<P5). Filled circle, methylated CpG. Unfilled circle, unmethylated CpG. Cross (X), CpG site without a clear methylation status. Each row stands for a sequenced DNA template molecule from a single bacterial colony. M, maternal chromosome with an “A” at the nucleotide position 267 and a “T” at the nucleotide position 275 of the 471-bp bisulfite PCR product with a DBA/2 origin. P, paternal chromosome with a “G” at the nucleotide position 270 and another “G” at the nucleotide position 278 of the 474-bp bisulfite PCR product with a 129 origin. A, bisulfite sequencing results for the parental cells. Mother, mouse tail of the mother. Father, mouse tail of the father. MEF, MEF cells. MEF + Dox, MEF cells cultured with doxycycline. B, three iPS clones (B11, E7D and L7) with relatively high methylated Snrpn DMR on the maternal chromosome (M). C, four iPS clones (S4D, D15D, D36 and DL3) with partially methylated Snrpn DMR on the maternal chromosome (M). D, five iPS clones (L9, C8D, E7, S5D and n2) with unmethylated Snrpn DMR on the maternal chromosome (M).
Figure 5.

Maternally inherited DNA methylation imprint was variably lost at the Zac1 DMR region in the iPS clones. Bacterial colony sequencing of the bisulfite PCR product was performed for genomic DNA samples from the parental cells as well as from the iPS clones at early passages (<P5). Filled circle, methylated CpG. Unfilled circle, unmethylated CpG. Cross (X), CpG site without a clear methylation status. Each row stands for a sequenced DNA template molecule from a single bacterial colony. M, maternal chromosome with a “G” at the nucleotide position 169 of the 317-bp bisulfite PCR product with a DBA/2 origin. P, paternal chromosome with an “A” at the nucleotide position 169 of the 317-bp bisulfite PCR product with a 129 origin. A, bisulfite sequencing results for the parental cells. Mother, mouse tail of the mother. Father, mouse tail of the father. MEF, MEF cells. MEF + Dox, MEF cells cultured with doxycycline. B, six iPS clones (L7, E7D, D15D, S4D, C8D and L9) with partially methylated Zac1 DMR on the maternal chromosome (M). C, six iPS clones (B11, E7, S5D, D36, n2 and DL3) with unmethylated Zac1 DMR on the maternal chromosome (M).
Bisulfite sequencing of the IG-DMR
We subjected the bisulfite PCR product for the M5 region of the IG-DMR to bacterial colony sequencing that is a part of the ICR for the Dlk1-Dio3 imprinted region (Takada et al., 2002). We found that most iPS clones had significant methylation at the M5 region of the IG-DMR although two iPS clones (n2 and B11) had almost no methylation (Fig. 3S). Our sequencing results were in agreement with the COBRA results. In both assays, methylation at the IG-DMR was largely maintained but partially lost in most iPS clones in comparison to that of the parental MEF cells or mouse tail samples.
Bisulfite sequencing confirmed loss of methylation at the Rasgrf1 DMR
Bisulfite PCR product covering a portion of the Rasgrf1 DMR region was sent for bacterial colony sequencing (Kobayashi et al., 2006; Watanabe et al., 2011). As shown in Fig. 4SA, parental mouse tail samples and MEF cells were similarly methylated at the Rasgrf1 DMR region. Two iPS clones (L9 and DL3) had close to normal methylation and three (E7, L7 and D15D) had partial methylation at this DMR (Fig. 4SB). But it was largely missing in other seven iPS clones (Fig. 4SB). The sequencing results were generally consistent with the COBRA result for this DMR region (Fig. 2C).
Bisulfite sequencing of the Snrpn DMR
In our previous study, we had utilized SNPs within the Snrpn DMR to distinguish its parental origin (Li et al., 2008). Similarly, we confirmed the presence of the SNPs within the Snrpn DMR in these iPS clones by direct sequencing of the genomic DNA samples for these iPS clones as well as their parental cells. Then we determined the parental origin of the methylated or unmethylated Snrpn DMR after bisulfite sequencing. As expected, both methylated and unmethylated Snrpn DMR with the ratio close to 1:1 was present in the parental mouse tail samples (Fig. 4A). In the parental MEF cells, only the maternal chromosome with the DBA/2 origin contained the methylated Snrpn DMR whereas the paternal chromosome with a 129 origin did not have any methylated Snrpn DMR (Fig. 4A). Doxycycline had no effect on methylation at the Snrpn DMR in parental MEF cells (Fig. 4A). However, methylation at the Snrpn DMR on the maternal chromosome was variably lost in these iPS clones (Fig. 4B–D). It was largely intact in three iPS clones (B11, E7D, L7) although there was already some loss of maternal methylation imprint at the Snrpn DMR even in these three iPS clones (Fig. 4B). Maternal methylation imprint was partially maintained in four iPS clones (S4D, D15D, D36 and DL3) (Fig. 4C). Maternally derived DNA methylation imprint at the Snrpn DMR was completely missing in other iPS clones (L9, C8D, E7, S5D and n2) (Fig. 4D).
In general, the Snrpn COBRA results correlated with the bisulfite sequencing data for these iPS clones except for E7 (compare Figs. 3C and 4). Although significant methylation was found for E7 by COBRA (Lane 8 of Fig. 3C), not many methylated DNA molecules were discovered for E7 by bisulfite sequencing (Fig. 4D). This discrepancy was due to an abnormally high percentage of methylation at the CpG within the target site of restriction enzyme HhaI used in COBRA (see the 9th CpG site of the analyzed Snrpn DMR in E7 of Fig. 4D). Interestingly, some iPS clones appeared to have one (B11, D36, L9, C8D, E7 and n2) or two (DL3) methylated Snrpn DMR carrying a SNP that would be expected to be derived from a paternal chromosome of the 129 origin rather than from a maternal chromosome of the DBA/2 origin (Figs. 4B–D).
Some iPS clones were cultured on SNL feeder cells until Passage 20 (P20). Then iPS cells at P20 were grown on gelatin-coated plates before being harvested for bisulfite sequencing. We found that the maternal Snrpn DMR became somewhat more methylated in the iPS clone L7 at P20 than it was at earlier passage (compare Fig. 5SA with Fig. 4B). However, methylation was somewhat reduced in E7D and completely lost in S4D at P20 (Fig. 5SA), whereas E7D was relatively highly methylated and S4D was partially methylated at early passages (<P5) (Figs. 4B–C). Compared to early passages (Fig. 4D), methylation remained absent or largely lost at the Snrpn DMR on the maternal chromosome in other iPS clones (E7, L9 and S5D) at P20 (Fig. 5SA). These results suggest that partially methylated Snrpn DMR may be more dynamic than unmethylated or fully methylated Snrpn DMR. Methylation can be either increased or decreased at a partially methylated Snrpn DMR.
Bisulfite sequencing of the Zac1 DMR
We identified a SNP at the Zac1 DMR that allowed us to determine its parental origin. As shown in Fig. 5, maternally inherited DNA methylation imprint at the Zac1 DMR was also variably lost in these iPS clones. Parental mouse tail samples contained about 40% of methylated and 60% of unmethylated Zac1 DMR (Fig. 5A). As expected, only the maternal chromosome of the DBA/2 origin was methylated at the Zac1 DMR in the hybrid (DBA/129*) MEF cells with or without doxycycline. Six iPS clones (L7, E7D, D15D, S4D, C8D and L9) displayed partial loss of methylation at the maternal Zac1 DMR (Fig. 5B), whereas methylation was completely lost at the maternal Zac1 DMR in six other iPS clones (B11, E7, S5D, D36, n2 and DL3) (Fig. 5C).
Bisulfite sequencing of the Zac1 DMR confirmed the COBRA results for all iPS clones except for D15D (compare Fig. 3D with Fig. 5). Many methylated DNA molecules were discovered for D15D by bisulfite sequencing (Fig. 5B), but little methylated product was identified by COBRA (Lane 9 of Fig. 3D). This discrepancy was caused by almost a complete absence of methylation at the second CpG site of the analyzed Zac1 DMR in D15D (Fig. 5B). This CpG site was the target site of TaqI and that was why there was very little methylated product by COBRA (Lane 9 of Fig. 3D). Intriguingly, a few methylated Zac1 DMR carried a SNP of an apparent 129 origin rather than one of an apparent DBA/2 origin (Figs. 5B and 5C).
We also analyzed the Zac1 DMR for some iPS clones at P20 by bisulfite sequencing. We found that maternal DNA methylation imprint was partially maintained at the Zac1 DMR in four iPS clones (L7, L9, S4D and E7D) (Fig. 5SB). Methylation was completely lost in two iPS clones (E7 and S5D) at P20 (Fig. 5SB). Compared to those at early passages, DNA methylation imprint was somewhat increased for L9 at P20 and reduced for E7D and S4D at P20 (compare Fig. 5 with Fig. 5SB). It remained almost the same for L7, E7 and S5D at P20 (compare Fig. 5 with Fig. 5SB). Again, it seems that partially methylated Zac1 DMR in the iPS clones L9, E7D and S4D was more dynamic during extended culture than highly methylated Zac1 DMR in L7 or unmethylated Zac1 DMR in E7 and S5D.
Allele-specific expression analysis of imprinted genes
After sequencing genomic DNA samples, we found that there is a SNP in an exon of two imprinted genes (Snrpn and Zim1) in these iPS clones that are located in the Snrpn and Peg3 imprinted regions, respectively. However, we did not find any SNP in the exons of other imprinted genes located in the imprinted regions that were examined in this study. These SNPs allowed us to distinguish whether the transcript for Snrpn and Zim1 was transcribed from the maternal allele of a DBA/2 origin or from the paternal allele of a 129 origin.
RT-PCR analysis of the Snrpn imprinted gene
Snrpn is usually transcribed from its paternal allele at the Snrpn imprinted region in somatic cells. Indeed, we found that it was exclusively expressed from the paternal allele in the MEF cells regardless of doxycycline (Fig. 6A). Total RNA isolated from mature EBs of these iPS clones was subjected to RT-PCR analysis. Two iPS clones (B11 and L7) retained this paternal allele-specific expression of the imprinted Snrpn gene and 90% or more of the transcript for Snrpn was expressed from the paternal allele carrying the SNP of a 129 origin (Fig. 6A). Snrpn was preferentially (70% or more of the transcript) expressed from the paternal allele in three iPS clones (E7, C8D and S4D) (Fig. 6B). However, the rest of these iPS clones either displayed only slight preference for the paternal allele (60–65%G for D15D, E7D, D36 and L9) or were almost completely bi-allelic (50–55%G for S5D, n2 and DL3) with respect to the expression of Snrpn (Fig. 6B).
Figure 6.

RT-PCR analysis of the Snrpn imprinted gene in MEF cells as well as in embryoid bodies (EBs) derived from the iPS cells. Total RNA was isolated from MEF cells and EBs derived from 12 iPS clones generated from the hybrid MEF cells. Typically, iPS clones at earlier passages (P < 5) were used for EB derivation and mature EBs were harvested between day 12 and day 16 in suspension culture for total RNA preparation. The RT-PCR product of normally paternally expressed Snrpn imprinted gene was generated from these total RNA samples and subjected to direct sequencing after agarose gel purification. A portion of the sequencing result containing the SNP of the Snrpn imprinted gene is shown for each sample, together with the part of the chromatogram that harbors the SNP. The estimated percentage of G nucleotide at the SNP underneath the chromatogram indicates the percentage of the Snrpn transcript that was transcribed from the paternal 129 allele of Snrpn in EBs of each iPS clone or in the parental hybrid MEF cells. The maternal allele of Snrpn with a DBA/2 origin has a T at the corresponding nucleotide position. A, sequencing results for the parental MEF cells as well as two iPS clones (B11 and L7) that displayed almost exclusive expression from the paternal allele of Snrpn. MEF, MEF cells. MEF + Dox, MEF cells cultured in the presence of doxycycline. B, sequencing results for ten other iPS clones that showed partial or complete bi-allelic expression of Snrpn.
We also isolated total RNA from iPS clones directly plated on gelatin-coated plates from early (<P5) to late passages (P20). We analyzed a few iPS clones at earlier passages (P3–P5) (Fig. 6S). Indeed, Snrpn was exclusively expressed from the paternal allele in B11 (100%G, 129 origin), and preferentially expressed from the paternal allele in L7 (85%G), E7 (80%G) and E7D (80%G) (Fig. 6S). The expression for Snrpn became more or less bi-allelic for four iPS clones (S4D, D15D, L9 and S5D), ranging from 50%G to 60%G of the transcript with a 129 origin (Fig. 6S).
We cultured a few iPS clones until P20. The expression patterns of Snrpn for these iPS clones were similar to those of EBs or iPS clones at earlier passages (see Figs. 6, 6S and 7S). In the iPS clone L7 at P20, Snrpn was exclusively expressed from the paternal allele (100%G of a 129 origin) (Fig. 7S). It was preferentially expressed from the paternal allele in E7D at P20 (80%G) (Fig. 7S). Partial bi-allelic expression was observed in E7 (65%G), S4D (65%G), D36 (60%G), L9 (60%G), S5D (60%G), n2 (60%G) and DL3 (55%G) (Fig. 7S).
RT-PCR analysis of the Zim1 imprinted gene
Zim1 in the Peg3 imprinted region is maternally expressed in somatic cells. Indeed, it was exclusively expressed from the maternal allele in the MEF cells irrespective of doxycycline (Fig. 7A). When differentiated as EBs, complete maternal allele-specific expression of Zim1 was observed in two iPS clones (L7 and S4D, both 100%G) (Fig. 7A). It was preferentially expressed from the maternal allele of Zim1 in three other iPS clones (85%G for D36, 80%G for B11 and 75%G for E7D) (Fig. 7A). Bi-allelic expression of Zim1 was observed in the rest of iPS clones (40–50%G) (Fig. 7B).
Figure 7.

RT-PCR analysis of the Zim1 imprinted gene at the Peg3 imprinted region in MEF cells as well as in EBs. Total RNA was isolated from MEF cells and EBs derived from 12 iPS clones. Typically, iPS clones at earlier passages (P < 5) were used for EB derivation and mature EBs were harvested between day 12 and day 16 in suspension culture for total RNA preparation. The RT-PCR product of normally maternally expressed Zim1 imprinted gene was sent for direct sequencing after agarose gel purification. The estimated percentage of G nucleotide at the SNP underneath the chromatogram indicates the percentage of the Zim1 transcript that was transcribed from the maternal DBA/2 allele in EBs of each iPS clone or in the parental hybrid MEF cells. The paternal allele of Zim1 with a 129 origin has an A at the corresponding nucleotide position. A, sequencing results for the parental hybrid MEF cells as well as five iPS clones that displayed exclusive (L7 and S4D) or preferential (D36, B11 and E7D) mono-allelic expression from the maternal allele of Zim1. MEF, MEF cells. MEF + Dox, MEF cells cultured with doxycycline. B, sequencing results for seven other iPS clones that showed almost complete bi-allelic expression of Zim1.
We examined the expression of Zim1 in some iPS clones that were directly plated on gelatin-coated plates after being grown on feeder cells. Out of eight iPS clones examined at early passages (within P5) (Fig. 8S), only B11 still maintained somewhat maternal allele-specific expression of Zim1 (75%G at P5). Bi-allelic expression of Zim1 was observed in the rest of iPS clones at early passages (40–50%G) (Fig. 8S).
We also analyzed allele-specific expression of Zim1 in iPS clones at P20 plated on gelatin-coated plates. Only L7 retained some maternal allele-specific expression of Zim1 (75%G) (Fig. 9S). All other iPS clones became bi-allelic at P20 (40–50%G) (Fig. 9S).
Discussion
Our comprehensive DNA methylation analysis in many iPS clones has demonstrated that parentally inherited DNA methylation imprint was variably lost at many imprinted regions in iPS cells (Table 1S). Some imprinted regions such as the paternally inherited Rasgrf1 DMR and maternally inherited Peg3 and Peg10 DMRs were more susceptible to this presumably genome-wide demethylation process during iPS cell reprogramming (Figs. 2, 3 and 4S). Except for one or two iPS clones, most iPS clones had no or very little DNA methylation imprint retained in these three imprinted regions based on COBRA and bisulfite sequencing analyses. DNA methylation imprint was relatively more stably maintained in the maternally inherited Peg1 DMR and the paternally inherited H19 DMR and IG-DMR of the Dlk1-Dio3 imprinted region (Figs. 2 and 3). Indeed, Peg1 DMR seemed to be properly methylated in all iPS clones examined in this study. Whereas several iPS clones had lost methylation, most iPS clones had partial or close to normal levels of methylation at the H19 DMR and IG-DMR (Figs. 2 and 3S). By contrast, methylation was variably lost at the Snrpn DMR and Zac1 DMR in almost all iPS clones isolated in this study (Figs. 3–5). These results suggest that different imprinted regions may possess different mechanisms in maintaining DNA methylation imprint.
Several iPS clones retained most of maternally inherited DNA methylation imprint at either the Snrpn or Zac1 or both DMR regions. For example, maternally inherited DNA methylation imprint was largely intact at both the Snrpn and Zac1 DMR regions in the iPS clone L7 (Figs. 4B and 5B). The iPS clone B11 only retained DNA methylation imprint at the Snrpn but not at the Zac1 DMR (Figs. 4B and 5C). Some iPS clones maintained partial methylation at one or both DMR regions. The rest of them had completely lost the maternally inherited DNA methylation imprint at either the Snrpn or Zac1 or both DMRs. These results also indicate that independent epigenetic mechanisms may be present at different imprinted regions in the maintenance of parental DNA methylation imprint.
Most iPS clones (8 out of 12) used in this study were derived from the same population of MEF cells that were originated from a single mouse embryo. Thus, they were genetically identical. However, significant difference was observed among these eight iPS clones in maintaining parental DNA methylation imprint during reprogramming of iPS cells. Four other iPS clones (S4D, D15D, D36 and S5D) were derived from the MEF cells of another embryo. These four genetically identical iPS clones also exhibited variable loss of methylation at both Snrpn and Zac1 DMRs (Figs. 4–5) as well as other imprinted regions (Figs. 2–3). These results suggest that this variable loss of DNA methylation imprint at different imprinted regions was likely caused by the variations in epigenetic reprogramming during the derivation of iPS cells.
It has been reported previously that Dlk1-Dio3 imprinted domain may be hypermethylated in iPS clones (Liu et al., 2010; Stadtfeld et al., 2010, 2012). However, hypermethylation at the IG-DMR of the Dlk1-Dio3 region was found to be mainly caused by different expression levels of transgenes used for iPS cell derivation in another study (Carey et al., 2011). The iPS clones we derived with the transgenes developed by the Jaenisch laboratory did not display any hypermethylation at the IG-DMR either (Figs. 2A and 3S). In fact, the IG-DMR was somewhat hypomethylated in most iPS clones we isolated compared with that of the parental cells as well as the wild-type TC1 ES cells. Several iPS clones may have lost methylation at the IG-DMR. These results suggest that the dosage of the reprogramming factors as well as culture conditions may contribute to the variations of DNA methylation at the Dlk1-Dio3 imprinted region among different studies.
Interestingly, we found that IG-DMR was hypermethylated in the MEFs and mouse tail cells in comparison to ES cells (Figs. 2A and 3SA). This may be a result of IG-DMR imprinting in a tissue-specific manner. Indeed, it had been reported before that IG-DMR is hypermethylated in postnatal neural stem cells (NSCs) and niche-astrocytes but differentially methylated on the paternal chromosome in embryonic NSCs (Ferron et al., 2011). Likewise, IG-DMR may not be imprinted in MEFs or in mouse tail, meaning that both maternal and paternal IG-DMR regions are partially or completely methylated. But it is imprinted in ES cells and only the paternal IG-DMR is methylated. That is why we found IG-DMR exhibited hypermethylation in the MEF and mouse tail samples, compared with about 50% of methylation in the ES cell sample (Fig. 3SA). It has been documented that pluripotent stem cells obtained via iPS reprogramming harbor more residual DNA methylation signatures of the parental somatic cells than nuclear-transfer-derived pluripotent stem cells (Kim et al., 2010). Thus, it is possible that some iPS clones isolated with certain approaches may have retained more parental imprinting memory at this Dlk1-Dio3 imprinted region than other iPS clones derived with other approaches.
We generated multiple iPS clones from the hybrid MEF cells derived from the cross between a DBA/2 female mouse and a 129 male mouse. The SNPs between these two mouse strains allowed us to distinguish the parental origins of several ICRs. Indeed, we found that maternally inherited DNA methylation imprint at both the Snrpn and Zac1 DMRs was partially or completely lost in all iPS clones examined. Even for several iPS clones with relatively high methylation levels at these two DMR regions, partial loss of maternally inherited DNA methylation imprint was still evident. These results indicate that DNA methylation imprint is susceptible to the genome-wide demethylation process during iPS derivation. Thus, an approach using iPS cell derivation may be employed to dissect the underlying mechanism in the maintenance and erasure of genomic DNA methylation imprint in somatic cells.
Curiously, one or two DNA molecules carrying the SNPs of an apparent 129 origin were methylated at the Snrpn or Zac1 DMR based on bisulfite sequencing data (Figs. 4, 5 and 5S). This could mean that DNA methylation imprint was established de novo on the paternal chromosome for these two imprinted regions during iPS cell derivation. Alternatively, the DNA molecules containing the SNPs of a 129 origin could come from the trace amount of contaminated genomic DNA of the SNL feeder cells carried over with the iPS cells when they were diluted onto the gelatin-coated plates for genomic DNA isolation. However, only a small number of feeder cells were typically transferred to the gelatin-coated plates and the irradiated feeder cells did not proliferate because of radiation-induced cell cycle arrest. By contrast, iPS cells proliferate rapidly. We usually did not harvest the iPS cells on gelatin-coated plates for DNA preparation until they reached confluency. Thus, it is unlikely that the carryover feeder cells would comprise a significant portion of the iPS cell culture on gelatin-coated plates before genomic DNA preparation.
Therefore, we would favor the first model that de novo methylation occurred on the paternal chromosome for these two imprinted regions in a small percentage of iPS cells. We hypothesize that this may have occurred immediately after the original parental DNA methylation imprint was erased during the process of iPS cell derivation from the MEF cells. This suggests that some aspects of the iPS reprogramming process may mimic what happens in the germ line during the resetting of DNA methylation imprint. If that is the case, it might be possible to manipulate genomic imprinting in vitro so that human cells with genomic imprinting defect can be fixed in iPS cells before being put back into human patients for cell-based therapies. It also indicates that iPS reprogramming can be harnessed as an approach to dissect the underlying mechanisms in the establishment of DNA methylation imprint.
The results for allele-specific RT-PCR analysis of both Snrpn and Zim1 imprinted genes are consistent with the DNA methylation analysis of these two ICRs in most iPS clones with a few notable exceptions. The iPS clones with more intact DNA methylation imprint had the tendency to maintain the preferential mono-allelic expression patterns of both imprinted genes. This was true for Snrpn in the iPS cells at early-passage (<P5), late-passage (P20) or in EBs (Figs. 6, 6S and 7S). Indeed, two iPS clones (B11 and L7) that had retained most of the maternally derived DNA methylation imprint maintained mono-allelic expression of Snrpn at all tested stages as well as in EBs (Figs. 6A, 6SA and 7S).
Although it had maintained most of the original DNA methylation imprint at early passages (Fig. 4B), the iPS clone E7D did not show much mono-allelic expression of Snrpn in EBs derived from the iPS cells at early passages (Fig. 6B). Interestingly, Snrpn was more preferentially expressed from the paternal allele in the iPS cells of E7D grown on gelatin-coated plates at P5 or P20 (Figs. 6SA and 7S). This suggests that some imprinting marks other than DNA methylation imprint (such as histone modifications) might have been lost in E7D when differentiated as EBs. Consistent with this, maternally derived DNA methylation imprint was not stably maintained at the Snrpn DMR in E7D during its extended culture on feeder cells (compare Fig. 4B with Fig. 5SB). However, the expression of Snrpn in E7D at P20 was almost identical to what it was at P5 (80%G for E7D in both Figs. 6S and 7S). Even more intriguingly, Snrpn was preferentially expressed from the paternal allele in EBs as well as in iPS cells at early passages for the iPS clone E7 even though it had lost almost all maternally inherited DNA methylation imprint at the Snrpn DMR (see Figs. 6B, 6S and 4D). These results imply that other imprinting marks such as histone modifications may preserve the preferential mono-allelic expression patterns of the imprinted genes even when DNA methylation imprint may be compromised at the Snrpn imprinted region. Indeed, there is DNA methylation-independent imprinting memory at the Snrpn DMR in mouse embryos based on our previously published study (Li et al., 2008). It will be interesting to find out the nature of imprinting memory at the Snrpn DMR that maintains the mono-allelic expression patterns of the imprinted genes. In this regard, the iPS clones E7 and E7D may serve as useful model systems for dissecting out this kind of imprinting memory in the future.
Zim1 in the Peg3 imprinted region seemed to be more preferentially expressed from the maternal allele for four iPS clones (L7, S4D, D36 and E7D) in the cells differentiated as EBs than in the iPS cells differentiated on gelatin-coated plates (compare Fig. 7A with Figs. 8S and 9S). Thus, Zim1 appeared to be more inclined to be preferentially expressed from the maternal allele in EBs for these iPS clones. This means that the imprinting marks for mono-allelic expression of Zim1 may be stabilized during EB differentiation. Interestingly, little DNA methylation imprint was detected in three iPS clones (S4D, D36 and E7D) based on COBRA (Fig. 3B), suggesting that there may be DNA methylation-independent imprinting memory at the Peg3 DMR region as well. Indeed, imprinted regulation at the Peg3 DMR appears to be quite unusual. It was recently reported that deleting a fragment of 2.5 kb within the Peg3 DMR surprisingly had no effect on the methylation at this imprinted domain in the progeny carrying this deletion irrespective of its parental origin (Kim et al., 2012). This hints that chromatin marks or other epigenetic factors may be involved in maintaining differential DNA methylation as well as mono-allelic expression of the imprinted genes at the Peg3 imprinted region. Interestingly, the methylated maternal Peg3 DMR was thought to be non-functional at some stages but functional at other stages of development (Kim et al., 2012). Thus, it is possible that maternal Peg3 DMR may be non-functional in undifferentiated iPS or ES cells but it becomes functional when differentiated as EBs. As a result, Zim1 was largely bi-allelically expressed in almost all iPS clones in the undifferentiated state but it was preferentially expressed from the maternal allele in 5 out of 12 iPS clones when differentiated as EBs (compare Fig. 7 with Figs. 8S and 9S).
It was reported before that genomic imprinting was also variably lost in the mouse ES cell lines derived via nuclear transfer (nt) (Humpherys et al., 2001). Similar to what we found with the iPS clones in this study, expression of the Peg1 imprinted gene appeared to be stably maintained in ntES clones whereas methylation at the H19 DMR as well as the expression of Igf2 and H19 imprinted genes were variably affected in ntES clones (Humpherys et al., 2001). Expression of Peg3 and Snrpn was reported to be less affected in ntES clones. In contrast, methylation at the Peg3 DMR was largely missing and methylation at the Snrpn DMR was variably lost in iPS clones. Mono-allelic expression of the Snrpn and Peg3 imprinted genes was also variably lost in mouse iPS clones, concurrent with the loss of DNA methylation imprint at these two imprinted regions. Thus, reprogramming via ntES cells shares some similarities to iPS reprogramming. Both reprogramming processes will result in a variable loss of genomic imprinting at some common imprinted regions. At least one imprinted region (Peg1) seems to be resistant to reprogramming. But other imprinted regions exhibit different susceptibilities with some being more sensitive to one reprogramming process than the other or vice versa. This implies that epigenetic modifiers may be differentially expressed in these two systems so that differential loss of imprinting was observed at some imprinted regions.
Conclusions
Genomic imprinting is absolutely essential for the embryonic development in mammals including humans (Barlow, 2011; Bartolomei and Ferguson-Smith, 2011; Li, 2013). Improper expression of the imprinted genes causes a variety of human diseases (Das et al., 2009; Mackay et al., 2008; Robertson, 2005; Tomizawa and Sasaki, 2012). For instance, the Snrpn imprinted region is associated with Prader-Willi and Angelman Syndromes (Horsthemke and Wagstaff, 2008; Jiang et al., 1998; Kantor et al., 2006; Mabb et al., 2011). About 20% of the cases of transient neonatal diabetes have been linked with hypomethylation at the Zac1/PLAGL1 imprinted region in humans (Mackay et al., 2008). Based on this study, both imprinted regions suffered variable loss of maternally inherited DNA methylation imprint. In addition, the Snrpn imprinted gene lost its normal paternal allele-specific expression pattern and became more or less bi-allelic in most iPS clones. We also found that DNA methylation imprint was almost completely missing in most iPS clones at some imprinted regions such as Rasgrf1, Peg3 and Peg10 DMRs (Figs. 2C, 3B and 3E). Consistent with our findings in mouse iPS cells, genomic imprinting defect has been shown to be present in human iPS cells as well (Nazor et al., 2012; Pick et al., 2009). Therefore, caution must be taken in assessing whether certain iPS clones are properly reprogrammed to have normal developmental potentials if the imprinting status of all imprinted regions has not been thoroughly examined. Future research focused on how genomic imprinting is reprogrammed during iPS cell derivation may lead to eventual production of therapeutically suitable iPS cells.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.scr.2013.05.011.
Supplementary Material
Acknowledgments
The work in the author’s laboratory is currently supported by the grants from NIH (GM093335), New York State (NYSTEM Contract# C026434) and American Heart Association (09SDG2400151). YS is partly supported by NIH T32 Cancer Biology Training Grant. XL and ST conceived the study. XL and CR wrote the manuscript.
References
- Barlow DP. Genomic imprinting: a mammalian epigenetic discovery model. Annu. Rev. Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009;23:2124–2133. doi: 10.1101/gad.1841409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Cedar H. Imprinting: focusing on the center. Curr. Opin. Genet. Dev. 2000;10:550–554. doi: 10.1016/s0959-437x(00)00126-x. [DOI] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Beard C, Hanna J, Jaenisch R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods. 2010;7:56–59. doi: 10.1038/nmeth.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Hanna JH, Faddah DA, Buganim Y, Kim J, Ganz K, Steine EJ, Cassady JP, Creyghton MP, et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell. 2011;9:588–598. doi: 10.1016/j.stem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Das R, Hampton DD, Jirtle RL. Imprinting evolution and human health. Mamm. Genome. 2009;20:563–572. doi: 10.1007/s00335-009-9229-y. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- Ferron SR, Charalambous M, Radford E, McEwen K, Wildner H, Hind E, Morante-Redolat JM, Laborda J, Guillemot F, Bauer SR, et al. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature. 2011;475:381–385. doi: 10.1038/nature10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiura H, Komiyama J, Shirai M, Obata Y, Ogawa H, Kono T. DNA methylation imprints on the IG-DMR of the Dlk1-Gtl2 domain in mouse male germline. FEBS Lett. 2007;581:1255–1260. doi: 10.1016/j.febslet.2007.02.034. [DOI] [PubMed] [Google Scholar]
- Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader–Willi/Angelman region. Am. J. Med. Genet. A. 2008;146A:2041–2052. doi: 10.1002/ajmg.a.32364. [DOI] [PubMed] [Google Scholar]
- Humpherys D, Eggan K, Akutsu H, Hochedlinger K, Rideout WM, 3rd, Biniszkiewicz D, Yanagimachi R, Jaenisch R. Epigenetic instability in ES cells and cloned mice. Science. 2001;293:95–97. doi: 10.1126/science.1061402. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Tsai TF, Bressler J, Beaudet AL. Imprinting in Angelman and Prader–Willi syndromes. Curr. Opin. Genet. Dev. 1998;8:334–342. doi: 10.1016/s0959-437x(98)80091-9. [DOI] [PubMed] [Google Scholar]
- Kantor B, Shemer R, Razin A. The Prader–Willi/Angelman imprinted domain and its control center. Cytogenet. Genome Res. 2006;113:300–305. doi: 10.1159/000090845. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ekram MB, Kim H, Faisal M, Frey WD, Huang JM, Tran K, Kim MM, Yu S. Imprinting control region (ICR) of the Peg3 domain. Hum. Mol. Genet. 2012;21:2677–2687. doi: 10.1093/hmg/dds092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Suda C, Abe T, Kohara Y, Ikemura T, Sasaki H. Bisulfite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet. Genome. Res. 2006;113:130–137. doi: 10.1159/000090824. [DOI] [PubMed] [Google Scholar]
- Lewis A, Reik W. How imprinting centres work. Cytogenet. Genome. Res. 2006;113:81–89. doi: 10.1159/000090818. [DOI] [PubMed] [Google Scholar]
- Li X. Genomic imprinting is a parental effect established in mammalian germ cells. Curr. Top. Dev. Biol. 2013;102:35–59. doi: 10.1016/B978-0-12-416024-8.00002-7. [DOI] [PubMed] [Google Scholar]
- Li X, Leder P. Identifying genes preferentially expressed in undifferentiated embryonic stem cells. BMC Cell Biol. 2007;8:37. doi: 10.1186/1471-2121-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev. Cell. 2008;15:547–557. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, Cavaille J, Ferguson-Smith AC. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat. Genet. 2003;35:97–102. doi: 10.1038/ng1233. [DOI] [PubMed] [Google Scholar]
- Liu L, Luo GZ, Yang W, Zhao X, Zheng Q, Lv Z, Li W, Wu HJ, Wang L, Wang XJ, et al. Activation of the imprinted Dlk1-Dio3 region correlates with pluripotency levels of mouse stem cells. J. Biol. Chem. 2010;285:19483–19490. doi: 10.1074/jbc.M110.131995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34:293–303. doi: 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 2008;40:949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Muller FJ, Wang YC, Boscolo FS, et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Pick M, Stelzer Y, Bar-Nur O, Mayshar Y, Eden A, Benvenisty N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells. 2009;27:2686–2690. doi: 10.1002/stem.205. [DOI] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Matteson PG, Jones BK, Guan XJ, Tilghman SM. The Dlk1 and Gtl2 genes are linked and reciprocally imprinted. Genes Dev. 2000;14:1997–2002. [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Ferrari F, Choi J, Walsh RM, Chen T, Ooi SS, Kim SY, Bestor TH, Shioda T, et al. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nat. Genet. 2012;44(398–405):S391–S392. doi: 10.1038/ng.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada S, Paulsen M, Tevendale M, Tsai CE, Kelsey G, Cattanach BM, Ferguson-Smith AC. Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: implications for imprinting control from comparison with Igf2-H19. Hum. Mol. Genet. 2002;11:77–86. doi: 10.1093/hmg/11.1.77. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tomizawa S, Sasaki H. Genomic imprinting and its relevance to congenital disease, infertility, molar pregnancy and induced pluripotent stem cell. J. Hum. Genet. 2012;57:84–91. doi: 10.1038/jhg.2011.151. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, Iida N, Hoki Y, Murphy PJ, Toyoda A, et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science. 2011;332:848–852. doi: 10.1126/science.1203919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- Yamanaka S. Elite and stochastic models for induced pluripotent stem cell generation. Nature. 2009;460:49–52. doi: 10.1038/nature08180. [DOI] [PubMed] [Google Scholar]
- Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, Bell FT, Iacovino M, Kyba M, Xu G, et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J. Biol. Chem. 2012;287:2107–2118. doi: 10.1074/jbc.M111.322644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.