

Abstract
Purpose
BRAFV600E mutations are associated with poor clinical prognosis in colorectal cancer (CRC). Whereas selective BRAF inhibitors are effective for treatment of melanoma, comparable efforts in CRC have been disappointing. Here, we investigated potential mechanisms underlying this resistance to BRAF inhibitors in BRAFV600E CRC.
Experimental Design
We examined phosphatidyl inositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling in BRAFV600E CRC cell lines after BRAF inhibition and cell viability and apoptosis after combined BRAF and PI3K/mTOR inhibition. We assessed the efficacy of in vivo combination treatment using a novel genetically engineered mouse model (GEMM) for BRAFV600E CRC.
Results
Western blot revealed sustained PI3K/mTOR signaling upon BRAF inhibition. Our BRAFV600E GEMM presented with sessile serrated adenomas/polyps, as seen in humans. Combination treatment in vivo resulted in induction of apoptosis and tumor regression.
Conclusions
We have established a novel GEMM to interrogate BRAFV600E CRC biology and identify more efficacious treatment strategies. Combination BRAF and PI3K/mTOR inhibitor treatment should be explored in clinical trials.
Keywords: colon cancer, mouse models, targeted therapy
Introduction
BRAF is a critical component of the mitogen-activated protein kinase (MAPK) signaling pathway.1 BRAFV600E mutations have been identified in melanoma, papillary thyroid carcinoma, and CRC, and result in constitutive MAPK signaling that promotes carcinogenesis.2 BRAFV600E mutations are seen in 15% of CRC and are associated with decreased survival.3 In particular, microsatellite stable (MSS) BRAFV600E CRC is associated with these poor clinical outcomes.4 With respect to CRC with high microsatellite instability (MSI-H) in the adjuvant setting (mainly stage II and III CRC), patients have better outcomes and the presence of BRAF mutation does not affect outcomes. 5 However, in the setting of metastatic tumor, BRAFV600E/MSI-H tumors have poorer outcomes than BRAF wild-type/MSI-H tumors.6,7
As more than 600,000 people worldwide die from CRC each year, robust therapies targeting MSS BRAFV600E CRC is a critical unmet clinical need.8 The development of selective BRAF inhibitors is a compelling goal for drug discovery.9 For instance, a recent phase III melanoma clinical trial resulted in a 48% response following treatment with the BRAF inhibitor PLX4032 (vemurafenib) versus 5% for standard of care dacarbazine chemotherapy.10 However, acquired resistance to vemurafenib quickly emerges.11,12 Potential resistance mechanisms include: 1) secondary mutations in RAS, HRAS, NRAS, or MEK1; 2) activation of COT-1 kinase; and 3) induction of PI3K/mTOR signaling through recruitment of PDGFRβ or IGF-1R.13-16
CRC treatment with BRAF inhibitors has been disappointing. For example, vemurafenib treatment in 19 BRAFV600E CRC patients yielded only one partial and four minor responses. Furthermore, five patients presented with a mixed response pattern of both regressing and progressing lesions.17 Whereas these results highlight the therapeutic potential for such BRAF inhibitors, they underscore the presence of molecular features in BRAFV600E CRC that restrict their clinical efficacy. Unlike melanoma, initiation of CRC carcinogenesis derives from perturbation of Wnt signaling.18 As such, one hypothesis for the disparate clinical responses in melanoma and CRC could include the recruitment of additional signaling pathways. Indeed, two recent publications have reported a mechanism in BRAFV600E CRC that following BRAF-inhibition, phospho-EGFR activity increases leading to reactivation of the MAPK pathway.19,20
Whereas PI3K/mTOR signaling has been implicated in resistance to BRAF inhibitors in melanoma, it also plays a dominant role in CRC carcinogenesis, being constitutively activated in approximately 30% of all patients.21 However, 60% of BRAFV600E CRCs have been reported to have increased p-AKT expression, suggesting that the PI3K/mTOR axis is activated in a significant proportion of BRAFV600E CRC.22 Taken together, these findings suggest that PI3K/mTOR signaling may represent a significant resistance mechanism to BRAF inhibitor treatment in BRAFV600E CRC.
We have described multiple novel genetically engineered mouse models (GEMMs) for sporadic CRC.23,24 These GEMMs are ideally suited for preclinical trials, using optical colonoscopy to assess dynamic tumor responses to treatment. In our studies, we used in vitro and in vivo experiments incorporating human CRC cell lines and a new GEMM for BRAFV600E CRC that recapitulates the sessile serrated adenoma/polyp pathway seen in humans, respectively. Our findings provide compelling preclinical evidence that combination BRAF and PI3K/mTOR inhibition should be explored in clinical trials. Furthermore, our results establish a novel GEMM for investigation of the sessile serrated adenoma/polyp pathway, interrogation of BRAFV600E CRC biology, and identification of more efficacious treatment strategies.
Materials and Methods
In vitro treatment of human CRC cell lines
The following cell lines were used in this study: VACO432, RKO, VT1, T29, HCT-116 and DLD-1. VACO432, RKO, VT1, and T29 cell lines (a kind gift from B. Vogelstein) were maintained in McCoy’s (Invitrogen) with 10% FBS (Invitrogen) and 1x Penicillin/Streptomycin (Invitrogen). HCT-116 and DLD-1 (ATCC) were maintained in DMEM (Invitrogen), as described above. VACO432/VT1 and RKO/T29 are isogenic pairs.25 VACO432 and RKO are homozygous for BRAFV600E, while both VT1 and T29 have the BRAFV600E allele knocked out. VACO432 and VT1 are heterozygous for a PIK3CAP124T mutation (this mutation is uncharacterized). RKO and T29 are homozygous for the PIK3CAH1047R activating mutation. RKO is also heterozygous for 2 frame shift mutations in NF1. HCT-116 and DLD-1 are heterozygous for the PIK3CAH1047R activating mutation. All mutations were confirmed by DNA sequence analysis using published primers flanking each mutated exon. After treatment with GDC-0879 (a kind gift from Genentech) and/or NVP-BEZ235 (LC Labs) for 48 hours, cell viability was measured by MTS assay, as previously described.24 All experiments were performed a minimum of three times in quadruplicate.
Immunoblotting
Cells were lysed in lysis buffer (20 mM Tris, 150 mM NaCl, 1% Nonidet P-40, 0.1 mM EDTA, and protease and phosphatase inhibitors), incubated on ice for 10 m and centrifuged at 14,000 rpm for 10 m. Tumors were homogenized with a TH tissue homogenizer (Omni International) in lysis buffer, incubated for 10 m on ice, and vigorously shaken for 30 m at 4 °C. Concentrations of whole cell or tumor lysates were determined by Bio-Rad Protein Assay (Bio-Rad). 10 μg and 25 μg protein lysate for whole cell and tumor, respectively, were separated on 10% SDS/PAGE gel, transferred to nitrocellulose membrane, blocked in 1% BSA for one hour, incubated at room temperature for two hours with primary antibody and one hour with secondary antibody. Detection was performed using the Amersham™ ECL™ Western Blot Detection Reagents (GE Healthcare). p-MEK, total MEK, p-ERK, total ERK, p-AKT Thr473, total AKT, p-S6 Ser240/244, total S6 were obtained from Cell Signaling Technologies (Beverly, MA).
Apoptosis assay
106 VACO432 and RKO cells were treated with 2μM GDC-0879, 100nM NVP-BEZ235, and combination in 0.1% FBS for 16 hours. Cells were washed, pelleted, and resuspended in Cell Lysis Buffer (Invitrogen). Caspase-3 activity was measured by the CaspACE™ Assay System, Colorimetric (Promega), as per manufacturer’s instructions. All experiments were performed a minimum of three times in duplicate.
BrafV600E GEMM
Mice with conditional Apc alleles (Apc CKO) were crossed to those with a latent BrafV600E allele (BrafCA) (a kind gift from M. McMahon) to generate ApcCKO/CKO; BrafCA/+ mice (Apc-Braf).26,27 Colonic tumors were induced using adenovirus expressing Cre recombinase (AdCre) and followed by optical colonoscopy, as previously described.23 As a tumor size metric, the Tumor Size Index (TSI) was calculated as (tumor area/colonic lumen area) × 100 (%), as we have validated previously.24 Ten tumors were genotyped for the recombined BrafV600E allele using the following primers and cycling conditions: Braf:AD FwdA1, 5′ -TGAGTATTTTTGTGGCAACTGC and Braf:AD RevB1, 5′ -CTCTGCTGGGAAAGCGGC; 94 °C for 2m, 94 °C for 30s, 60 °C for 90s, 72 °C for 1m, 72 °C for 10m. The product sizes are as follows: wild type Braf is 185bp, conditionally-targeted Braf is 308bp, and BrafV600E is 335bp.26 The ten tumors were sequenced for mutations in exons 9 and 20 of the Pik3ca gene using the following primers: 9F: 5′ GCAGTGTGGTGAAGTTTCCA 3′, 9R: 5′ TGGCCAATCCTTTGATTTGT 3′, 20F: 5′ ACTGCGTGGCAACCTTTATC 3′, and 5′ TGATGGTGTGGAAGATCCAA 3′. Tumor-bearing mice were treated with 100 mg/kg GDC-0879 and/or 25mg/kg NVP-BEZ235 in 0.5% methylcellulose/0.2% Tween 80 by daily gavage for 28 days. All protocols were approved by the Tufts Institutional Animal Care and Use Committee.
Statistical Analysis
Data are expressed as means ± standard deviation. Comparisons of tumor size, proliferation, and apoptosis between control and treated cohorts were calculated using two-tailed Independent-Samples T Tests. Percentage of adenoma vs. carcinoma in Apc and Apc-Braf group was compared using Fisher Exact Test. P < 0.05 was considered significant for all analyses. All analyses were calculated using SPSS 18.0 for Windows (IBM, Inc).
Microsatellite instability (MSI) testing
MSI was assessed using a fluorescently-labeled primer mBat-59 (F –GTAATCCCTTTATTCCATTTAGCA, R -GGCTCACAACCATCCGTAACAAGA) and Platinum PCR SuperMix High Fidelity (Invitrogen), as previously described.28 Tumors were scored as MSI positive if one or more novel alleles were present in tumor but not the matched tail.
Histopathology and immunohistochemistry
Please see Supplemental Information for details regarding histopathology and immunohistochemistry.
Results
BRAF inhibition decreases cell viability in BRAFV600E CRC
The BRAF inhibitors PLX4032, PLX4720, and GDC-0879 have comparable mechanism and efficacy across different BRAFV600E cancer cell lines.29,30 We used the tool compound GDC-0879 to examine BRAF inhibition in BRAF mutant (VACO432 and RKO), BRAF/KRAS wild-type (VT1 and T29), and KRAS mutant (DLD-1 and HCT-116) CRC cell lines. GDC-0879 is a potent BRAF inhibitor in the nanomolar range and has a high degree of selectivity, as demonstrated by screening of a representative panel of 140 full-length protein kinases.31 These studies revealed that BRAF inhibition decreases viability in BRAFV600E (IC50 < 1.5μM), whereas BRAF/KRAS wild-type and KRAS mutant CRC are significantly less sensitive (IC50 > 20μM) (Figure 1A). Western blot analysis of VACO432 and RKO after 0, 2, and 10 μM GDC-0879 treatment for 2 hours revealed decreased p-MEK and p-ERK (Figure 1B), with a rebound in signal seen at 24 hours. GDC-0879 treatment in a BRAF/KRAS wild-type cell line showed an induction of p-MEK and p-ERK signaling likely through activation of CRAF as reported by others19, while a KRAS mutant cell line was not affected by BRAF-inhibitor treatment. These results demonstrate that in vitro BRAF inhibition decreases MAPK signaling and cell viability in a BRAFV600E-specific manner.
Figure 1. GDC-0879 treatment is specific for BRAFV600E CRC.

(A) Cell viability in BRAF mutant (VACO432 and RKO), BRAF/KRAS wild-type (VT1 and T29), and KRAS mutant (DLD-1 and HCT-116) cells was examined after GDC-0879 treatment for 48 hours (As KRAS mutant cell lines were insensitive to BRAF inhibition, formal IC50 values could not be calculated and are presented simply as >10μM). (B) p-MEK and p-ERK were examined by western blot in BRAF mutant (VACO432 and RKO), BRAF/KRAS wild-type (VT1), and KRAS mutant (DLD-1) cells after treatment with 0, 2, and 10 μM GDC-0879 for 2 hours. (C) Caspase-3 activity was assessed after treatment of BRAF mutant (VACO432 and RKO) and BRAF/KRAS wild-type (VT1 and T29) cells with 2μM GDC-0879 for 16 hours. * P < .05.
Isolated BRAF inhibition does not induce apoptosis
As previous studies have shown that BRAF inhibition induces apoptosis in BRAFV600E melanoma cells, we examined caspase-3 activity after treatment of BRAF mutant (VACO432 and RKO) and BRAF/KRAS WT (VT1 and T29) cells with 2μM GDC-0879 for 16 hours, which revealed no significant induction of caspase-3 activity in the BRAF mutants (Figure 1C). Similarly, others have confirmed that BRAF inhibitor treatment in BRAF mutant CRC cell lines is not sufficient to induce apoptosis, as these cells show minimal sensitivity to single-agent treatment.32
PI3K/mTOR pathway sustains signaling after BRAF inhibition
To identify signaling pathways that might promote resistance to BRAF inhibition, we examined the PI3K/mTOR pathway. We performed western blot analyses to assess p-AKT Thr473 and p-S6 Ser240/244 in VACO432 and RKO after treatment with 0, 2, and 10 μM GDC-0879 for 2 hours. While it was expected that RKO would show activation of p-AKT and p-S6 given the presence of an activating PIK3CA mutation, these studies revealed sustained p-AKT and p-S6 after BRAF inhibition in VACO432, as well, suggesting that continued PI3K/mTOR may limit the effectiveness of BRAF inhibition (Figure 2A). BRAF wild-type and KRAS mutant cells, VT1 and DLD-1, respectively, also showed sustained p-AKT and p-S6. Recent publications have indicated that receptor tyrosine kinases (RTKs), such as EGFR and IGFR, can become activated in the BRAFV600E cell lines following BRAF inhibitor treatment,14,19,20 and therefore, the residual activity of p-AKT and p-S6 could be a result of RTK activation. To address the possibility that continued PI3K/mTOR may limit the effectiveness of GDC-0879 treatment, we examined the effects of concomitant PI3K/mTOR and BRAF inhibition by treating VACO432 and RKO with GDC-0879 and the dual PI3K/mTOR inhibitor NVP-BEZ235. NVP-BEZ235 is a potent PI3K/mTOR inhibitor in the low nanomolar range that is highly specific, as evidenced by its poor inhibition (IC50 > 5 μM) of a representative panel of protein kinases.33 Our studies revealed when the BRAFV600E cell lines VACO432 and RKO were treated with GDC-0879 at their relative IC50 (0.5μM and 1μM, respectively), cell viability was reduced by 50%. When 1nM of NVP-BEZ235 was added in combination with GDC-0879, the overall cell viability was further reduced to 29% and 43%, respectively, as compared to untreated cells (Figure 2B). Comparison of combination drug therapy to single agent treatment revealed a significant difference (p < 0.05) in both cell lines. Western blot analyses indicated that combination treatment is associated with decreased MAPK and PI3K/mTOR signaling in the BRAFV600E cell lines, while combination treatment has no effect on MAPK signaling in BRAF wild-type or KRAS mutant cells (Figure 2C). These findings suggest that PI3K/mTOR signaling provides resistance to BRAF inhibition, which can be abrogated by concurrent BRAF and PI3K/mTOR blockade.
Figure 2. Concomitant PI3K/mTOR blockade increases the effectiveness of in vitro BRAF inhibition.
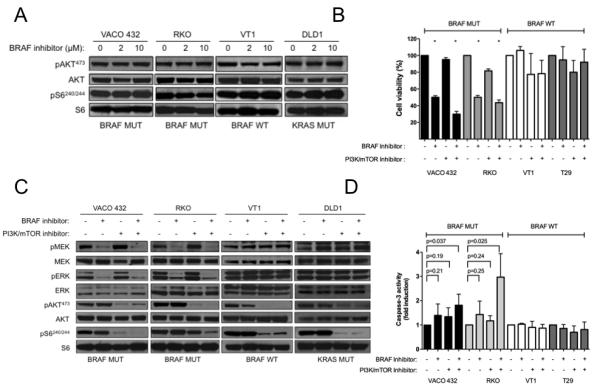
(A) PI3K/mTOR signaling in BRAF mutant (VACO432 and RKO), BRAF/KRAS wild-type (VT1), and KRAS mutant (DLD-1) cells was examined by western blot analysis for p-AKT Thr473 and p-S6 Ser240/244 after treatment with 0, 2, and 10 μM GDC-0879 for 2 hours. (B) Cell viability in BRAF mutant (VACO432 and RKO) and BRAF/KRAS wild-type (VT1 and T29) cells was examined after treatment with GDC-0879 (0.5μM and 1μM for VACO432/VT1 and RKO/T29, respectively), NVP-BEZ235 (1 and 10 nM for VACO432/VT1 and RKO/T29, respectively), and GDC-0879 + NVP-BEZ235 for 48 hours. (C) MAPK, PI3K, and mTOR signaling in BRAF mutant (VACO432 and RKO), BRAF/KRAS wild-type (VT1), and KRAS mutant (DLD-1) cells was examined by western blot for p-MEK, p-ERK, p-AKT, and p-S6 after treatment with 2μM GDC-0879 and 100nM NVP-BEZ235 for 2 hours. (D) Caspase-3 activity was assessed after treatment of VACO432 and RKO cells with 2μM GDC-0879 and 100nM NVP-BEZ235 for 16 hours. * P < .05.
Concurrent BRAF and PI3K/mTOR blockade results in induction of apoptosis
To assess the effects of combined PI3K/mTOR and BRAF inhibition on apoptosis, we examined caspase-3 activity after treatment of BRAFV600E (VACO432 and RKO) and BRAF/KRAS wild-type (VT1 and T29) cells with 2μM GDC-0879 and 100nM NVP-BEZ235 for 16 hours. This revealed a significant 1.8 and 3.0-fold induction of caspase-3 activity as compared to untreated cells after combination treatment in the BRAF mutants VACO432 and RKO, respectively (P < 0.05, Figure 2D). Comparison of combination drug therapy to single agent treatment revealed a significant difference (p < 0.05) in the RKO cell line. However, similar analysis of the RKO cell line revealed a trend towards increased apoptosis that was not statistically significant (GDC-0879 vs. combination, P < 0.00 and NVP-BEZ235 vs. combination, P < 0.001). These findings suggest that concurrent BRAF and PI3K/mTOR inhibition decreases viability through apoptotic induction.
Development of a GEMM for MSS BRAFV600E CRC
Pre-clinical drug studies are problematic in most CRC GEMMs, as they present with mainly small intestinal tumors, thereby precluding the use of optical colonoscopy to follow dynamic treatment responses. Because of this, we have described the development of multiple GEMMs for sporadic CRC based on surgical administration of AdCre to the distal colon of floxed mice.23 To develop a GEMM for MSS BRAFV600E CRC, we administered AdCre to mice bearing floxed Apc and latent Braf alleles (Apc-Braf) (Supplemental Figure 1).26,27 We performed this procedure in 192 mice, which resulted in distal colonic tumor formation in 178 animals (93%). Following AdCre injection, we used colonoscopy to monitor development of individual tumors, which revealed significantly faster growth in Apc-Braf than in mice bearing floxed Apc alleles alone (Apc) (P < .0001) (Figure 3A). Furthermore, we observed that the mean tumor multiplicities in Apc-Braf and Apc mice were 2.25 and 1.45, respectively (P < .0001, Figure 3B). MSI testing in ten individual colonic tumors using pyrosequencing of a microsatellite locus after PCR amplification for a specific mononucleotide repeat revealed no allelic size variations, demonstrating that these tumors are MSS (data not shown). Western blot and immunohistochemistry confirmed increased MAPK signaling in Apc-Braf tumors (Figure 3C). These results suggest that our strategy results in a reproducible, robust GEMM for MSS BRAFV600E CRC.
Figure 3. Colon-restricted activation of a latent BrafV600E allele results in a robust CRC GEMM.
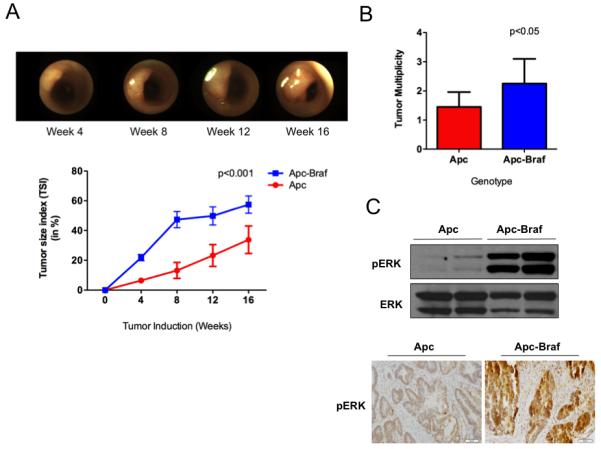
Following AdCre injection, colonoscopy was used to (A) serially examine individual tumors and derive growth curves (n = 8), and (B) determine tumor multiplicity. p-ERK was assessed by (C) western blot and immunohistochemistry.
Apc-Braf tumors recapitulate the BRAFV600E sessile serrated CRC pathway
While it has been demonstrated that intestinal expression of mutant BRAFV600E is sufficient to induce serrated epithelium and crypt hyperplasia, it does not reliably result in adenoma or carcinoma formation.34 Since human BRAFV600E mutant adenomas display evidence of Wnt activation, we sought to determine if tumors from our Apc-Braf GEMM would recapitulate the sessile serrated CRC pathway in humans. We examined 36 tumors from Apc-Braf mice at 8 to 24 weeks after AdCre infection and observed 1) discrete foci with architectural abnormalities of gland serration, gland branching, basal dilatation, lateral gland extension, and dystrophic goblet cells, which are consistent with human sessile serrated adenoma/polyp (SSA/P) and 2) SSA/P with cytologic dysplasia, which is characteristic of SSA progression in humans.35,36 We examined 20 tumors from mice at 25 or more weeks after AdCre infection and in 10 (50%) of these mice observed SSA/P with adenocarcinoma (Figure 4A). Of these, 16 tumors were flat or laterally spreading with central depression on endoscopic and gross examination (Figure 4B). Four of these tumors exhibited evidence of invasive adenocarcinoma (Figure 4C). Finally, we observed lesions with epithelial serration, mucin depletion in the upper polyp, and straight, mucin-depleted bases, which are consistent with hyperplastic polyps (Figure 4D). No serration was seen in 40 tumors from mice bearing only the modified Apc allele (Supplemental Figure 2). Taken together, these results suggest that our GEMM model is a robust surrogate for the human sessile serrated adenoma-carcinoma pathway.
Figure 4. Apc-Braf mice recapitulate the human BRAFV600E sessile serrated adenoma-carcinoma pathway.
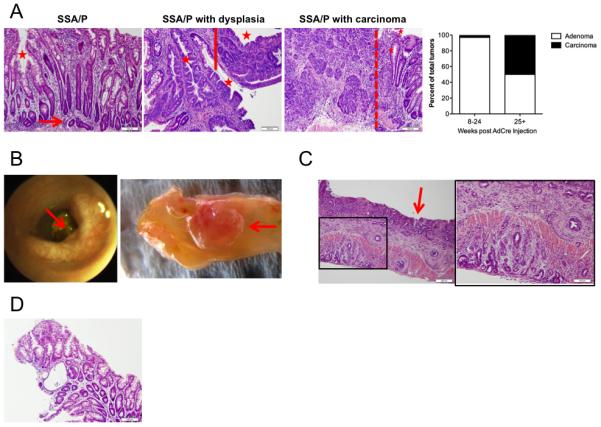
(A) Tumors have features of sessile serrated adenoma/polyp (SSA/P), including serrated glands (asterisk) and L-shaped basal glands (arrow); SSA/P with cytologic dysplasia (right of solid line); and SSA/P with adenocarcinoma (left of dotted line). The distribution of adenomas and carcinomas after AdCre administration is shown. Flat tumors with central depression (arrow) were identified on (B) endoscopy and gross examination. Representative flat tumor demonstrating (C) invasive adenocarcinoma. (D) Representative hyperplastic polyp with evidence of epithelial serration and mucin depletion.
In vivo BRAF blockade inhibits tumor growth in Apc-Braf mice
We next ascertained the effect of in vivo BRAF inhibition. After AdCre infection, we used colonoscopy to monitor tumor development and to randomize comparably-sized tumors for treatment with either control or 100mg/kg of GDC-0879 for 28 days. We monitored growth and/or regression of individual tumors during treatment (Figure 5A). Whereas the mean Tumor Size Index (TSI) in controls significantly increased during treatment (32.6% vs. 51.4%, P = 0.017), the mean TSI in the GDC-0879-treated cohort did not (27.5% vs. 27.7%, P = 0.95; Figure 5B). These results demonstrate that in vivo BRAF blockade results in stasis of BrafV600E tumors.
Figure 5. Concomitant in vivo BRAF and PI3K/mTOR inhibition results in tumor regression.
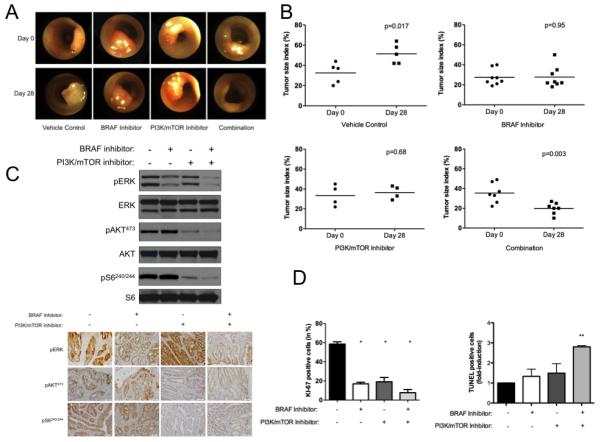
(A) Tumor-bearing Apc-Braf mice were treated with control, 100 mg/kg GDC-0879, 25 mg/kg NVP-BEZ235, or 100 mg/kg GDC-0879 + 25 mg/kg NVP-BEZ235 and examined by colonoscopy. (B) Serial imaging was used to assess the effects of drug treatment. MAPK and PI3K/mTOR tumor signaling was examined by (C) western blot and immunohistochemistry for p-ERK, p-AKT, and p-S6. The effect of concomitant BRAF and PI3K/mTOR inhibition on tumor and apoptosis was examined by (D) KI-67 immunohistochemistry and TUNEL assay. * P < .05, ** P < 0.01.
In vivo BRAF blockade results in inhibition of MAPK and sustained PI3K/mTOR signaling
We next ascertained if the resistance mechanisms we had identified in vitro would have in vivo significance in our GEMM for BRAFV600E CRC. To examine the effects of in vivo BRAF inhibition on MAPK and PI3K/mTOR signaling, we performed western blot analysis and immunohistochemistry for p-ERK, p-AKT, and p-S6 in Apc-Braf tumors. These studies revealed a decrease in p-ERK levels is seen in tumor lysates following treatment with GDC-0879, but sustained p-AKT and p-S6 levels (Figure 5C). These results demonstrate that in vivo BRAF inhibition results in MAPK blockade and sustained PI3K/mTOR signaling.
In vivo BRAF inhibition blocks proliferation but does not induce apoptosis
We next sought to determine whether the observed tumor stasis after in vivo BRAF inhibition derived from effects on tumor proliferation and/or induction of apoptosis. To assess the effects of in vivo BRAF inhibition on cellular proliferation, we performed immunohistochemistry for the proliferation marker KI-67, which revealed a 71% decrease (P < .05). To assess the effects of in vivo BRAF inhibition on cellular apoptosis in colonic tumors, we used an in situ TUNEL assay that revealed a non-significant 1.9-fold increase (P = 0.08, Figure 5D). These results suggest that in vivo BRAF blockade results in tumor stasis primarily through a decrease in tumor proliferation.
Concomitant in vivo BRAF and PI3K/mTOR blockade results in tumor regression
We next sought to examine if concomitant PI3K/mTOR blockade enhances the effects of in vivo BRAF inhibition. Following AdCre induction, we used optical colonoscopy to randomize comparablysized tumors for treatment with either control, 100mg/kg of GDC-879, 25 mg/kg of NVP-BEZ235, or both GDC-0879 and NVP-BEZ235 for 28 days (Figure 5A). We did not observe any in vivo effects on the histopathology of the normal mucosa or the overall health of the mice following either single or combination drug treatment. GDC-0879 (27.5% in pre-treatment vs. 27.7% in post-treatment; P = 0.95) and NVP-BEZ235 (33.7% in pre-treatment vs. 36.5% in post-treatment; P = 0.68) treated cohorts showed tumor stasis (Figure 5B). However, the combination GDC-0879 and NVP-BEZ235-treated cohort showed significant tumor regression (35.4% in pre-treatment vs. 19.8% in post-treatment; P = .003; Figure 5B). These results demonstrate that concomitant PI3K/mTOR blockade enhances the effects of in vivo BRAF inhibition.
Concomitant in vivo BRAF and PI3K/mTOR blockade results in inhibition of MAPK and PI3K/mTOR signaling
To examine the effects of in vivo BRAF and PI3K/mTOR inhibition on MAPK and PI3K/mTOR signaling, we performed western blot analyses that revealed decreased p-ERK, p-AKT, and p-S6 levels in tumor lysates following treatment. The continuous down regulation of p-S6 likely plays an important role in the impressive therapeutic responses seen in combination treated tumors. These findings were confirmed by subsequent immunohistochemistry analysis (Figure 5C). These results suggest that concomitant BRAF and PI3K/mTOR blockade induces in vivo tumor regression.
Concomitant in vivo BRAF and PI3K/mTOR inhibition blocks proliferation and induces apoptosis
We next sought to determine whether the observed colonic tumor regression after in vivo BRAF inhibition and PI3K/mTOR inhibition derives from changes in proliferation and/or apoptosis. To assess the effects of in vivo BRAF and PI3K/mTOR inhibition on proliferation, we used immunohistochemistry analysis for the proliferation marker KI-67, which revealed a 87% decrease (P < 0.01). To assess the effects of in vivo BRAF inhibition on apoptosis, we used an in situ TUNEL assay that revealed a statistically significant five-fold induction with concomitant BRAF and PI3K/mTOR blockade (P < 0.05, Figure 5D). These results suggest that the observed tumor regression after concomitant BRAF and PI3K/mTOR inhibition derives from a significant decrease in proliferation and induction of apoptosis.
Discussion
Due to the extremely poor prognosis for patients with BRAFV600E CRC, the development of novel therapies for this clinical segment is urgently needed.3, 4 The limited response of BRAFV600E CRC to vemurafenib suggests the presence of de novo or rapidly acquired resistance mechanisms.17 Besides the de novo resistance seen in BRAFV600E CRC towards single-agent BRAF-inhibition, the MAPK pathway can be reactivated by additional mechanisms. For example, two publications have reported it is the increased activity of phospho-EGFR that follows BRAF-inhibition in CRC that leads to reactivation of p-ERK.19,20 Similar findings have been observed in in vivo melanoma xenograft models of acquired resistance and in in vitro CRC systems.14,16 Additionally, elevated levels of RAS-GTP, CRAF, and pAKT have been found in melanoma that is resistant to BRAF-inhibition.37 Here, we demonstrate the requirement for concomitant targeting of BRAF and PI3K/mTOR for effective treatment of BRAFV600E CRC.
Robust GEMMs should recapitulate the appropriate histopathologic spectrum observed in humans. BRAFV600E CRC derives from the recently described serrated pathway associated with characteristic histologic features and Wnt activation.34 A recent BrafV600E GEM model based on the AcreERT system has been reported to have similar serrated features and subsequent activation of the Wnt pathway.38 However, as BrafV600E is activated along the entire gastrointestinal tract, these animals die prematurely from overwhelming tumor burden before the individual lesions evolve to advanced carcinoma. To develop a tractable model that is amenable to assessment of experimental therapeutics, we focally activated BrafV600E and inactivated Apc in the distal colon of floxed mice using AdCre, as we have previously described.23,24 In these mice, we observed early sessile serrated adenoma/polyp (SSA/P), SSA/P with cytologic dysplasia, SSA/P with adenocarcinoma, and invasive carcinoma. These findings demonstrate that we are able to recapitulate the histopathologic spectrum of the sessile serrated adenoma-carcinoma pathway seen in humans, making this a robust surrogate for BRAFV600E CRC.
Cancer GEMMs that are robust preclinical drug testing platforms should present with the following features: 1) tumors develop rapidly along a reproducible time line and 2) tumors can be continuously monitored throughout drug treatment. Because of the association between BRAF mutation and activation of the Wnt pathway in human CRC, we began with conditional Apc-Braf mice to accelerate tumorigenesis.36,38 To enable the use of optical colonoscopy for longitudinal tumor evaluation, we focally modify floxed genes in the distal colon using AdCre, as we have previously described.23,24 As such, these tumors reproducibly progress in the distal colon, permitting monitoring by optical colonoscopy throughout experimental drug treatments. Our GEMM fulfills the above requirements, making it a robust platform for evaluation of experimental therapeutics.
In both our in vitro and our in vivo data utilizing our novel BRAFV600E GEMM, we corroborate the findings seen in human patients: BRAF mutant tumors are not sensitive to single-agent BRAF inhibition. As tumor growth is modulated by complex, interdigitating signaling networks, blockade at a single node often results in compensation through a parallel pathway. Recently, publications have indicated that in the presence of a BRAF inhibitor, the MAPK pathway can be re-activated by the expression of RTKs.14,19 In contrast, the PI3K pathway shows sustained activity in 60% of treatment-naive BRAFV600E CRC, which may explain the ineffectiveness of BRAF inhibitors.22 Consequently, concurrent inhibition of both pathways may be required. For example, chronic BRAF inhibition has been suggested to confer drug resistance in BRAFV600E melanoma through recruitment of PI3K signaling, while simultaneous blockade of both signaling pathways results in effective treatment.27,29 Furthermore, the requirement for concomitant inhibition of the MAPK and PI3K/mTOR pathways has been shown in other cancer models.40-42
To examine if this feature were present in our model for BRAFV600E CRC, we interrogated PI3K/mTOR pathway signaling both in vitro and in vivo after BRAF inhibition, revealing the presence of p-AKT Thr473 and p-S6 Ser240/244 indicating activity through the PI3K/mTOR pathway (Figures 2A and 5C). While one BRAFV600E CRC cell line carries the activating PIK3CA mutation H1047R (RKO), the other (VACO432) does not. While VACO432 is heterozygous for a P124T mutation, this has to our knowledge not been characterized as an activating mutation. Taken together with the significant proportion of BRAFV600E CRC patients with sustained PI3K/mTOR activity,22 we believe that the need for combination therapy is independent of PIK3CA mutational status
Our subsequent in vitro and in vivo studies showed that combined BRAF and PI3K/mTOR inhibition is required for effective treatment of BRAFV600E CRC. While it has been suggested that NVP-BEZ235 is a more potent mTOR inhibitor than an PI3K inhibitor,43 inhibition of this particular signaling node seems to be critical to the successful treatment of BRAFV600E CRC. As we did not test this directly, we cannot say whether pure PI3K inhibitors or pure mTOR inhibitors will be as or more effective in combination with BRAF inhibition. However, the more potent mTOR inhibition seen by the down regulation of p-S6 with NVP-BEZ235 may suggest this to be a more important component. To our knowledge, we are the first to show a BRAF inhibitor-based combination therapy that effectively down regulates TORC1, as evidenced by down regulation of p-S6. Additionally, others have also reported the need for a combinational approach. It has been reported that combination treatment with a RAF inhibitor (RAF265) and a dual PI3K/mTOR inhibitor (BEZ) is effective in thyroid cancer.44 Future efforts are underway to further refine which targets along the PI3K/mTOR axis provide the strongest results. Collectively, these results suggest that patients with BRAFV600E CRC may benefit from such combination therapy.
To maximize tumor regression, robust strategies for induction of apoptosis are critical. Whereas BRAF inhibition alone is sufficient to induce apoptosis in melanoma,45 such treatment results in only growth arrest of BRAFV600E thyroid and colon cancers 44,46-49 with resistance to cell death.41 This suggests that differential apoptotic responses are one underlying mechanism of the disparate clinical outcomes seen in melanoma and CRC patients treated with BRAF inhibitors. In our experiments, the effects on apoptosis may have appeared more pronounced in RKO because it harbors a prototypic PIK3CA activating mutation (H1047R), whereas VACO432 contains a rare and uncharacterized PIK3CA mutation (P124T) that might possess lower intrinsic activity. Furthermore, we examined apoptosis after 16 hours, as this has been reported to be the time of maximal apoptotic induction by agents such as staurosporine. It is possible that examination at a different time point might further augment the differences in VACO432 cells to achieve statistical significance. Nonetheless, our data suggest that concomitant treatment of BRAFV600E CRC cell lines with NVP-BEZ235 may be important to tip the scales from inhibition of proliferation towards induction of apoptosis. Furthermore, concomitant BRAF and PI3K inhibition has been observed to increase the sensitivity of CRC to TRAIL treatment.34 This indicates the need for both growth inhibition along with apoptosis. Our studies show that concomitant BRAF and PI3K/mTOR inhibition results in both growth inhibition and an induction of apoptosis leading to in vivo tumor regression.
The approach to targeted therapy in BRAFV600E cancers has been shifting and is seen in the design of recent clinical trials. The identification of the need for BRAF-inhibitor based treatments to include additional inhibitors has led to trials that include a BRAF-inhibitor given in combination with additional drugs such as MEK-inhibitors, PI3K-specific-inhibitors, and monoclonal antibodies against the protein death receptor ligand-1 (PD-L1). Additionally, a recently opened clinical trial is assessing the efficacy of a BRAF inhibitor given along with both an EGFR inhibitor and a PI3K inhibitor. We believe the strength of this study is that we have shown that combination treatment of BRAFV600E CRC with a BRAF inhibitor and PI3K/mTOR inhibitor down regulates both p-ERK and p-S6, reduces cellular viability, and induces tumor regression in BRAFV600E GEMMs. It remains unknown whether EGFR inhibition in combination with BRAF inhibition is capable of down regulating TORC1 and achieving the same in vivo tumor regression.
In summary, the establishment of robust therapeutic strategies for BRAFV600E CRC is an unmet clinical need. Collectively, our findings provide the pre-clinical rationale for further examination of BRAF and PI3K/mTOR inhibitors in clinical trials for BRAFV600E CRC. Furthermore, our novel GEMM recapitulates the sessile serrated adenoma-carcinoma pathway in humans, making it a robust platform to interrogate BRAFV600E CRC biology and refine further combinatorial therapeutic regimens.
Supplementary Material
Statement of Translational Relevance.
BRAFV600E mutations are associated with poor clinical prognosis in colorectal cancer (CRC). Whereas selective BRAF inhibitors are effective for melanoma treatment, comparable efforts in CRC have been disappointing. Further understanding of potential resistance mechanisms to treatment is an unmet clinical need in CRC. Using colon-restricted delivery of adenovirus expressing cre recombinase to floxed mice, we developed a novel genetically engineered mouse model (GEMM) for BRAFV600E CRC that recapitulates the sessile serrated adenoma-carcinoma pathway in humans. In conjunction with in vitro cell line studies, we used this GEMM to identify continued PI3K/mTOR signaling as a potential resistance mechanism and to demonstrate the efficacy of concomitant BRAF and PI3K/mTOR blockade for therapy. Taken together, this GEMM provides a robust preclinical platform to interrogate BRAFV600E CRC biology and to refine further combinatorial therapeutic regimens and suggests that combined treatment with BRAF and PI3K/mTOR inhibitors should be explored in future clinical CRC trials.
Acknowledgements
We would like to thank Drs. Cory Abate-Shen, Alain Charest, Eric Collisson, Raju Kucherlapati, and Martin McMahon for critical review of this manuscript.
Financial Support: This research was supported by grants from the NIDDK (K08-DK078033) and NCI (U01-CA084301).
Footnotes
Present address: Oncology Research Unit, Pfizer Global Research and Development, San Diego, CA 92121
Conflicts of Interest: E. Martin is a current employee of Pfizer Global Research and Development.
REFERENCES
- 1.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nature Reviews Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. British Journal of Cancer. 2009;101:465–472. doi: 10.1038/sj.bjc.6605164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Research. 2005;65:6063–9. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 5.Tian S, Roepman P, Popovici V, Michaut M, Majewski I, Salazar R, et al. A robust genomic signature for the detection of colorectal cancer patients with microsatellite instability phenotype and high mutation frequency. Journal of Pathology. 2012;228:586–595. doi: 10.1002/path.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. Journal of Clinical Oncology. 2010;28:466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 7.Saridaki Z, Papadatos-Pastos D, Tzardi M, Mavroudis D, Bairaktari E, Arvanity H, et al. BRAF mutations, microsatellite instability status and cyclin D1 expression predict metastatic colorectal patients’ outcome. British Journal of Cancer. 2010;12:1762–1768. doi: 10.1038/sj.bjc.6605694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: A Cancer Journal for Clinicians. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 9.Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011;8:426–433. doi: 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- 10.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. New England Journal of Medicine. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solit DB, Rosen N. Resistance to BRAF Inhibition in Melanomas. New England Journal of Medicine. 2011;364:772–774. doi: 10.1056/NEJMcibr1013704. [DOI] [PubMed] [Google Scholar]
- 12.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang CC, Lai F, Thorne RF, Yang F, Liu H, Hersey P, et al. MEK-Independent Survival of B-RAFV600E Melanoma Cells Selected for Resistance to Apoptosis Induced by the RAF Inhibitor PLX4720. Clinical Cancer Research. 2011;17:721–730. doi: 10.1158/1078-0432.CCR-10-2225. [DOI] [PubMed] [Google Scholar]
- 16.Shi H, Kong X, Ribas A, Lo RS. Combinatorial Treatments That Overcome PDGFRβ-Driven Resistance of Melanoma Cells to V600EB-RAF Inhibition. Cancer Research. 2011;71:5067–5074. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao M, Tian F, Mariadason JT, Tsao CC, Lemos RJ, Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clinical Cancer Research. 2013;19:657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markowitz SD, Bertagnolli MM. Molecular Basis of Colorectal Cancer. New England Journal of Medicine. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discovery. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibiton through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 21.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 22.Baba Y, Nosho K, Shima K, Hayashi M, Meyerhardt JA, Chan AT, et al. Phosphorylated AKT expression is associated with PIK3CA mutation, low stage, and favorable outcome in 717 colorectal cancers. Cancer. 2011;117:1399–1408. doi: 10.1002/cncr.25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hung KE, Maricevich MA, Richard LG, Chen WY, Richardson MP, Kunin A, et al. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proceedings of the National Academy of Sciences. 2010;107:1565–1570. doi: 10.1073/pnas.0908682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roper J, Richardson MP, Wang WV, Richard LG, Chen W, Coffee EM, et al. The Dual PI3K/mTOR Inhibitor NVP-BEZ235 Induces Tumor Regression in a Genetically Engineered Mouse Model of PIK3CA Wild-Type Colorectal Cancer. PLoS ONE. 2011;6:e25132. doi: 10.1371/journal.pone.0025132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ, et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006;2:e146. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacher JW, Abdel Megid WM, Kent-First MG, Halberg RB. Use of mononucleotide repeat markers for detection of microsatellite instability in mouse tumors. Molecular carcinogenesis. 2005;44:285–92. doi: 10.1002/mc.20146. [DOI] [PubMed] [Google Scholar]
- 29.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 30.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KYJ, Artis DR, Schlessinger J, Su F, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoeflich KP, Herter S, Tien J, Wong L, Berry L, Chan J, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69:3042–51. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- 32.Oikonomou E, Koc M, Sourkova V, Andera L, Pintzas A. Selective BRAFV600E Inhibitor PLX4720, Requires TRAIL Assistance to Overcome Oncogenic PIK3CA Resistance. PLoS ONE. 2011;6:e21632. doi: 10.1371/journal.pone.0021632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maira S-M, Stauffer Fdr, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Molecular Cancer Therapeutics. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 34.Dale CS. Update on the serrated pathway to colorectal carcinoma. Human Pathology. 2011;42:1–10. doi: 10.1016/j.humpath.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Snover DC, Batts KP. Serrated Colorectal Neoplasia. Surgical Pathology Clinics. 2010;3:207–240. doi: 10.1016/j.path.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Fujita K, Yamamoto H, Matsumoto T, Hirahashi M, Gushima M, Kishimoto J, et al. Sessile serrated adenoma with early neoplastic progression: a clinicopathologic and molecular study. The American journal of surgical pathology. 2011;35:295–304. doi: 10.1097/PAS.0b013e318205df36. [DOI] [PubMed] [Google Scholar]
- 37.Su F, Bradley WD, Wang Q, Yang H, Xu L, Higgins B, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Research. 2012;72:969–978. doi: 10.1158/0008-5472.CAN-11-1875. [DOI] [PubMed] [Google Scholar]
- 38.Carragher LA, Snell KR, Giblett SM, Aldridge VS, Patel B, Cook SJ, et al. (V600E)Braf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16(INK4a) EMBO Mol Med. 2010;2:458–71. doi: 10.1002/emmm.201000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang HH, Walker F, Kiflemariam S, Whitehead RH, Williams D, Phillips WA, et al. Selective inhibition of proliferation in colorectal carcinoma cell lines expressing mutant APC or activated B-Raf. Int J Cancer. 2009;125:297–307. doi: 10.1002/ijc.24289. [DOI] [PubMed] [Google Scholar]
- 40.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–64. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of Clinical Investigation. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Molecular Cancer Therapeutics. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 44.Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clinical Cancer Research. 2011;17:6482–6489. doi: 10.1158/1078-0432.CCR-11-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Research. 2003;17:5198–5202. [PubMed] [Google Scholar]
- 46.Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Research. 2012;72:779–789. doi: 10.1158/0008-5472.CAN-11-2941. [DOI] [PubMed] [Google Scholar]
- 47.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proceedings of the National Academy of Sciences. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tap WD, Gong KW, Dering J, Tseng Y, Ginther C, Pauletti G, et al. Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with PLX4032 in malignant melanoma. Neoplasia. 2010;12:637–49. doi: 10.1593/neo.10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JT, Li L, Brafford PA, van den Eijnden M, Halloran MB, Sproesser K, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment cell & melanoma research. 2010;23:820–827. doi: 10.1111/j.1755-148X.2010.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.