

Summary
Lignin is the second most abundant constituent of the cell wall of vascular plants, where it protects cellulose towards hydrolytic attack by saprophytic and pathogenic microbes. Its removal represents a key step for carbon recycling in land ecosystems, as well as a central issue for industrial utilization of plant biomass. The lignin polymer is highly recalcitrant towards chemical and biological degradation due to its molecular architecture, where different non‐phenolic phenylpropanoid units form a complex three‐dimensional network linked by a variety of ether and carbon–carbon bonds. Ligninolytic microbes have developed a unique strategy to handle lignin degradation based on unspecific one‐electron oxidation of the benzenic rings in the different lignin substructures by extracellular haemperoxidases acting synergistically with peroxide‐generating oxidases. These peroxidases posses two outstanding characteristics: (i) they have unusually high redox potential due to haem pocket architecture that enables oxidation of non‐phenolic aromatic rings, and (ii) they are able to generate a protein oxidizer by electron transfer to the haem cofactor forming a catalytic tryptophanyl‐free radical at the protein surface, where it can interact with the bulky lignin polymer. The structure–function information currently available is being used to build tailor‐made peroxidases and other oxidoreductases as industrial biocatalysts.
Interest of microbial degradation of lignin
Lignin is a complex aromatic polymer, highly recalcitrant towards both chemical and biological degradation, characteristic of the cell wall of vascular plants (Fig. 1). Around 20% of the total carbon fixed by photosynthesis in land ecosystems is incorporated into lignin, being the second main constituent of plant biomass after cellulose. In addition of providing plant stems the rigidity required for growth on land, and waterproofing vascular tissues for sap circulation, a main role of lignin is to protect the cellulose polymer towards hydrolytic attack by most pathogen and saprophytic organisms. In spite of this, lignin‐degrading microbes evolved simultaneously with the colonization of land by vascular plants in the Palaeozoic era, around 400 million year ago (Taylor and Osborne, 1996). Microbial degradation of lignin (Martínez et al., 2005; Kersten and Cullen, 2007) represents a key step for closing the carbon cycle, since removal of the lignin barrier enabled the subsequent use of plant carbohydrates by other microorganisms.
Figure 1.
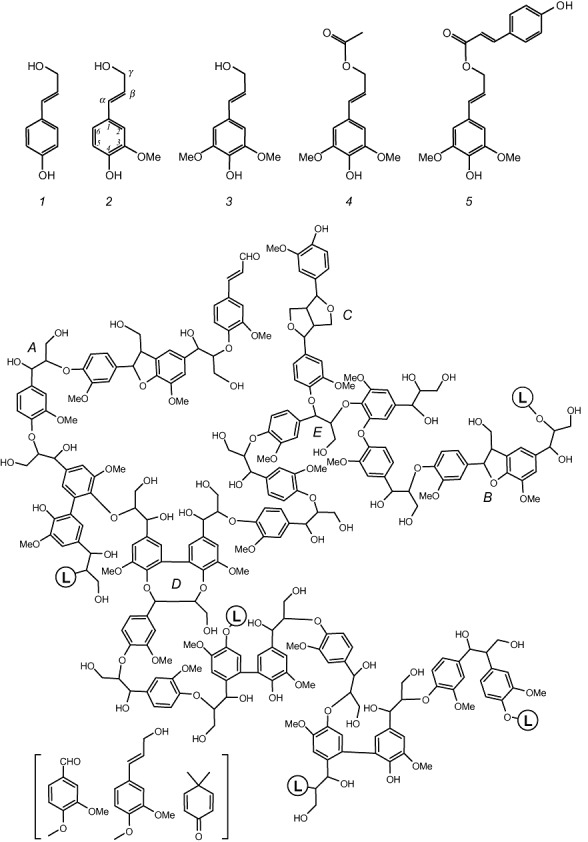
Three classical and two acylated lignin precursors or monolignols (top), and structural model for gymnosperm lignin (bottom). Gymnosperms produce the simplest lignin type formed only by guaiacyl units derived from coniferyl alcohol (2). In contrast, angiosperm lignin also include p‐hydroxyphenyl and sinapyl units derived from p‐coumaryl (1) and sinapyl (3) alcohols, as well as a variable amount of acylated lignin often derived from sinapyl alcohol γ‐esterified with acetic (4), p‐coumaric acid (5) or other organic acids (Ralph et al., 2004; Martínez et al., 2008). A variety of ether and carbon–carbon inter‐unit linkages are formed during monolignol radical polymerization resulting in β‐O‐4′ (A), phenylcoumaran (B), pinoresinol (C) and dibenzodioxocin (D) substructures, among others. Linkages to additional lignin chains are indicated (L‐containing circles). Other minor structures (in brackets) include vanillin, coniferyl alcohol and dimethylcyclohexadienone‐type units, the latter in new spirodienone substructures (Zhang et al., 2006) (courtesy of G. Gellerstedt).
Lignin removal is also a central aspect in industrial uses of cellulosic biomass, such as bioethanol production and manufacture of cellulose‐based chemicals and materials, including paper. In the plant cell wall, lignin concentrates in the middle lamella, its most external layer acting as a cementing agent between fibres (Fig. 2, top). Cellulose pulp manufacture basically consists in breaking down (chemically or mechanically) the middle lamella in such a way that wood fibres are individualized (Sixta, 2006). Although in lower concentration than plant carbohydrates (cellulose and hemicelluloses) lignin is also present in secondary wall, the thicker cell‐wall layer, where it is intimately associated to carbohydrates preventing their efficient hydrolysis in the production of bioethanol (Galbe and Zacchi, 2007). In the above industrial applications, biotechnology based on lignin‐degrading microbes and their enzymes can contribute to more efficient and environmentally sound use of renewable lignocellulosic feedstocks for sustainable production of materials, chemicals, biofuels and energy.
Figure 2.
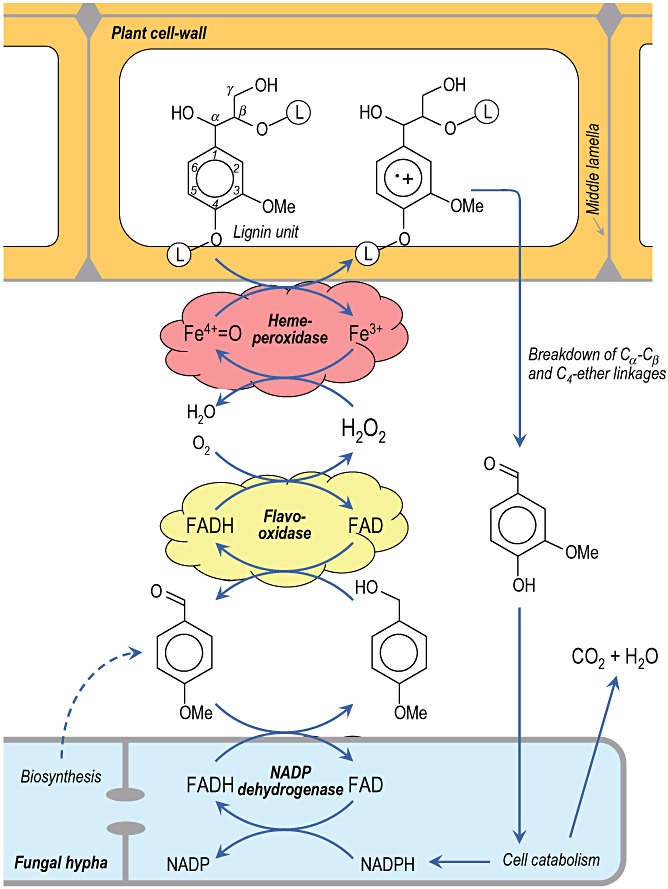
Pictorial scheme of the enzymatic degradation of plant cell‐wall lignin (L‐containing circles represent the remaining lignin polymer) by Pleurotus VP, with contribution of extracellular flavooxidases (such as AAO) generating hydrogen peroxide during redox cycling of non‐phenolic aromatic aldehydes (such as the fungal metabolite p‐anisaldehyde) with participation of intracellular aryl‐aldehyde dehydrogenase. Peroxidase one‐electron oxidation of lignin units (the key step in the degradative process) results in an unstable cation radical that experiences different reactions including breakdown of Cα–Cβ and C4–ether linkages releasing the corresponding aromatic aldehydes (vanillin in the case of guaiacyl units) that can be intracellularly mineralized. In the case of P. chrysosporium LiP, lignin attack requires the presence of veratryl alcohol, probably as an enzyme‐bound mediator, and hydrogen peroxide is mainly generated by glyoxal oxidase.
Chemical bases of lignin recalcitrance
Although lignin precursors, i.e. the three classical p‐hydroxycinnamyl alcohols and their recently reported acylated forms (Ralph et al., 2004; Martínez et al., 2008), are phenolic compounds (Fig. 1, top), the lignin polymer formed from these monolignols is basically non‐phenolic (Fig. 1, bottom). In the last step of lignin biosynthesis, plant peroxidases (and maybe also laccases) oxidize monolignol to their phenoxy radicals (Higuchi, 1997). Chemical coupling between the resonant forms of these radicals results in a variety of phenolic dimeric structures (dilignols) that can be enzymatically oxidized again, the process finally leading to lignin polymer formation. However, due to predominance of the corresponding radical forms and higher stability of the coupling products, ether linkages between the phenolic position (C4) and a side‐chain (or aromatic ring) carbon of the p‐hydroxyphenylpropenoid precursors (substructures A, B and D) are strongly predominant in the growing polymer. Due to the high frequency of these ether linkages, the aromatic lignin units are basically non‐phenolic. Moreover, in contrast to cellulose formed by linear (anhydroglucose) chains and hemicelluloses that include short branches on a main polysaccharidic backbone, the lignin polymerization mechanism (based on resonant radical coupling) results in a complex three‐dimensional network (Fig. 1, bottom) due to both chain branching and inter/intra‐chain coupling during polymerization, as shown in updated lignin models (Gellerstedt and Henriksson, 2008).
Due to its non‐phenolic aromatic nature, lignin units cannot be oxidized by low‐redox‐potential oxidoreductases, such as the plant peroxidases initiating the polymerization process. In fact, only a small group of highly specialized peroxidases secreted by ligninolytic fungi are able to degrade model compounds representing the main lignin substructures. The bulky nature of the heterogeneous lignin polymer forming a complex three‐dimensional network represents an additional limitation for biodegradation since the enzyme accessibility is strongly reduced. To overcome this difficulty, two main strategies have been developed by ligninolytic organisms based on: (i) presence of catalytic residues widely exposed at the surface of ligninolytic peroxidases, and (ii) use of redox mediators participating in the enzymatic attack.
Ligninolytic organisms and enzymes
The first lignin structural models were available in the 1970s (Nimz, 1974; Adler, 1977) and new substructures are still being identified using modern analytical techniques (Karhunen et al., 1995; Zhang et al., 2006; del Río et al., 2007). Before this date no contrasted information on lignin structure was available, preventing studies on its microbial or enzymatic degradation. Synthetic lignins and simple model compounds (incorporating radioactive labelling) were used to unravel the mechanisms of lignin attack by white‐rot basidiomycetes, the only organisms that are able to extensively mineralize lignin (Eriksson et al., 1990). The complexity shown by the first lignin models and the variety of compounds identified in early lignocellulose biodegradation studies (Chen and Chang, 1985) suggested a set of different degradative enzymes attacking the different lignin substructures and releasing different breakdown products. However, subsequent work using model compounds representative of the main substructures found in lignin revealed that lignin‐degrading organisms just adopted the opposite strategy. These studies, instead of revealing a variety of enzymes catalysing the different reactions observed, showed that the latter were the result of an unspecific oxidative attack on the benzenic rings of lignin units followed by different bond breakdown reactions due to the chemical instability of the cation radicals formed (Kirk and Farrell, 1987).
Moreover, using simple model compounds, it was possible to demonstrate that among the different oxidative enzymes produced by lignin‐degrading organisms, only a group of basidiomycete haemperoxidases could directly attack the non‐phenolic lignin network (Martínez, 2002; Hammel and Cullen, 2008). These enzymes include lignin peroxidase (LiP) initially described in Phanerochaete chrysosporium, the first basidiomycete whose genome was sequenced due to the interest on biological degradation of lignin (Martínez et al., 2004), and a versatile peroxidase (VP) more recently reported in Pleurotus and Bjerkandera species, the former genus including species being able to degrade lignin selectively (Martínez et al., 1994). VP is also able to oxidize Mn2+, as reported for P. chrysosporium manganese peroxidase (MnP). The Mn3+ resulting from the action of these two peroxidases oxidizes phenolic compounds but, in the presence of unsaturated lipids, it can also oxidize non‐phenolic lignin via peroxidation radicals (Bao et al., 1994). The molecular evolution of ligninolytic peroxidases has been shown by a recent study (Morgenstern et al., 2008).
Taking into account the unique characteristics of the microbial attack to lignin, compared with hydrolytic attack to other natural polymers including plant carbohydrates, the process was described as an ‘enzymatic combustion’ where enzymatically generated hydrogen peroxide oxidizes the lignin polymer in a reaction catalysed by the above‐mentioned high‐redox‐potential peroxidases (Kirk and Farrell, 1987) (Fig. 2). Simultaneously to the discovery of ligninolytic peroxidases, several peroxide‐generating oxidases were described in ligninolytic fungi, such as glyoxal oxidase (a copper radical enzyme), aryl‐alcohol oxidase (AAO) and pyranose‐2 oxidase (two flavoenzymes) (Martínez et al., 2005). In Pleurotus species continuous production of hydrogen peroxide during redox cycling of anisaldehyde, an extracellular metabolite (Gutiérrez et al., 1994) is produced involving mycelium‐associated aryl‐alcohol dehydrogenase (a NADPH dehydrogenase) working together with extracellular AAO (Guillén and Evans, 1994) (Fig. 2). In P. chrysosporium, glyoxal oxidase uses products from lignocellulose degradation as the enzyme‐reducing substrates for generating the hydrogen peroxide required by the ligninolytic peroxidases (Kersten and Cullen, 2007). The unstable cation radicals formed during peroxidase attack to lignin model compounds experience different chemical reactions including breakdown of Cα–Cβ and C4–ether linkages resulting in the release of aromatic aldehydes, one of the main products found during enzymatic depolymerization of lignin (Fig. 2), together with other reactions such as demethoxylation (methanol release) and aromatic‐ring cleavage (with formation of muconate‐type structures) (Martínez et al., 2005).
The involvement of ligninolytic peroxidases in wood lignin degradation has been recently supported by comparison of the genomes of P. chrysosporium (Martínez et al., 2004), a model white‐rot fungus characterized by its ability to remove wood lignin leaving a whitish cellulosic residue, and Postia placenta (Martinez et al., 2009), a model brown‐rot fungus characterized by its ability to remove wood polysaccharides leaving a brown lignin‐enriched residue. This comparison showed a large set of ligninolytic peroxidase genes in the genome of P. chrysosporium, in agreement with previous studies (Cullen, 1997). In contrast, the P. placenta genome includes genes of oxidases and other enzymes involved in cellulose attack via Fenton chemistry, but contains an unique peroxidase gene related to the low‐redox‐potential peroxidase of the non‐ligninolytic basidiomycete Coprinopsis cinerea (CIP), and completely lacks ligninolytic peroxidase genes (lip, vp or mnp).
A third type of microbial oxidoreductases, laccases and related multicopper oxidases are produced by most ligninolytic basidiomycetes (Baldrian, 2006) as well as by eubacteria and actinomycetes growing on lignocellulosic materials (Sharma et al., 2007). The low redox potential of laccases prevents their direct action on lignin. However, they can degrade this and other recalcitrant compounds in the presence of redox mediators, as discussed below. Laccases also play a variety of other physiological roles including mushroom morphogenesis, detoxification and humification, among others (Claus, 2004). Moreover, they seem involved in generation of lignocellulose‐degrading hydroxyl radical (via Fenton chemistry) in both white‐rot (Guillén et al., 2000) and at least some brown‐rot basidiomycetes, in agreement with the presence of laccase genes in the genome of P. placenta (http://genome.jgi‐psf.org/Pospl1/Pospl1.home.html).
General determinants of ligninolytic peroxidase structure and activity
All prokaryotic, fungal and plant haemperoxidases share a common folding and helical topology described in cytochrome c peroxidase (CCP), the first peroxidase whose crystal structure was reported (Poulos et al., 1980) and only more recently (Gajhede et al., 1997) in the best‐known peroxidase, horseradish peroxidase (HRP). It is noteworthy that LiP, an enzyme that was completely unknown until 1983 (Glenn et al., 1983; Tien and Kirk, 1983), was the second peroxidase whose crystal structure was reported (Piontek et al., 1993; Poulos et al., 1993). The rapid progresses of LiP structure–function studies, as well as the fact that two groups reported simultaneously its discovery and crystal structure, demonstrate the interest on high‐redox‐potential peroxidases.
As mentioned above, all peroxidases require hydrogen peroxide, or other hydroperoxides, to activate the haem cofactor yielding the so‐called compound‐I in a common catalytic cycle (Dunford, 1999) (Fig. 3). Compound‐I contains a reactive Fe4+ oxo complex with a cation radical at the porphyrin ring, formed by two‐electron oxidation of the Fe3+‐containing haem of the resting enzyme. One‐electron oxidation of one substrate molecule yields compound‐II, where the porphyrin cation radical has been reduced. The remaining Fe4+=O in compound‐II oxidizes a second substrate molecule, and the enzyme returns to its ferric resting state to initiate a new catalytic cycle.
Figure 3.
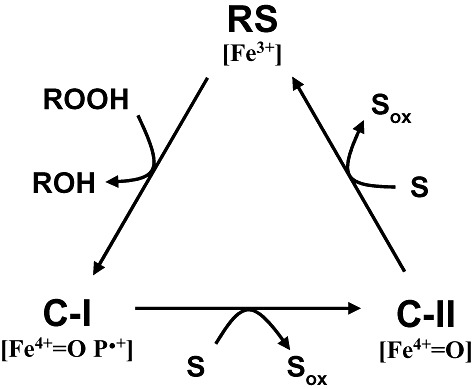
General catalytic cycle of peroxidases (Dunford, 1999). The cycle includes two‐electron oxidation of the enzyme resting state (RS, containing Fe3+) by hydroperoxide to yield compound‐I (C‐I; containing Fe4+‐oxo and porphyrin cation radical), whose reduction in two one‐electron steps results in the intermediate compound‐II (C‐II; containing Fe4+=O after porphyrin reduction) and then the resting form of the enzyme, with concomitant oxidation of two substrate molecules (S; which could be low‐redox‐potential phenols and dyes, or Mn2+ in the cases of MnP and VP).
The molecular structure of haemperoxidases includes two domains, probably originating from ancestral gene duplication, delimiting a central cavity where the haem cofactor is located (Li and Poulos, 1994; Banci, 1997). In most cases, the access of both the enzyme‐oxidizing (peroxide) and ‐reducing substrates to the haem cofactor is produced through a main access channel (Fig. 4, left). Taking into account the high reactivity of both compound‐I and compound‐II, this internal location of the cofactor most probably plays a dual role. First, it prevents unspecific reduction of the activated enzyme by a variety of reductants different from its specific target substrate. Second, it prevents intermolecular reaction resulting in oxidation of surface susceptible amino acids (e.g. tyrosine residues) leading to enzyme inactivation (e.g. by dimerization reactions). Oxidation of Mn2+ by basidiomycete MnP and VP is also produced through a specific access channel enabling entering of the cation to reach one of the haem propionates (Fig. 4, left). The edge of this channel includes three acidic residues that bind Mn2+ specifically enabling its high‐efficiency oxidation.
Figure 4.
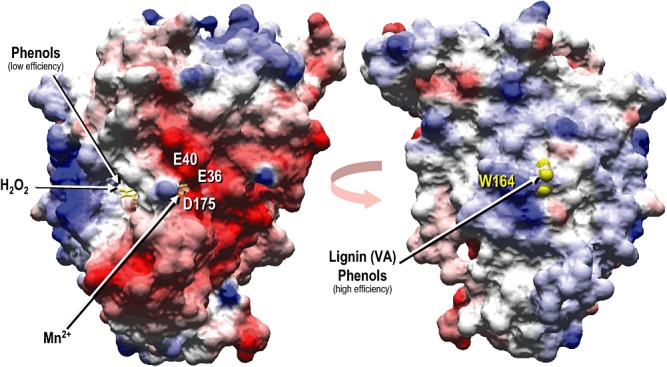
Two different views of the solvent access surface in a ligninolytic peroxidase (P. eryngii VP; PDB entry 2BOQ) revealing (left) the main haem access channel enabling hydrogen peroxide entrance for activation of the haem cofactor (in yellow) located in a central pocket (low‐redox‐potential phenols and dyes can also be oxidized at this channel albeit with low efficiency), and the Mn2+‐oxidation channel formed by three acidic residues (Glu‐36, Glu‐40 and Asp‐175); as well as an approximately 180° rotated view (right) of the same peroxidase showing the partially exposed side‐chain (yellow van der Waals spheres including hydrogen atoms) of the catalytic tryptophan (Trp‐164) involved in oxidation of high‐redox‐potential compounds, such as veratryl alcohol (VA) and lignin models, as well as in high‐efficiency oxidation of some phenols and dyes, by long‐range electron transfer (LRET) to the haem cofactor (surface colours correspond to electrostatic charge).
A detail of the haem environment and other neighbour amino acid residues in a ligninolytic peroxidase (Pleurotus eryngii VP) is shown in Fig. 5. These include two axial histidine residues, the so‐called proximal (His‐169) and distal (His‐47) histidines, and other conserved residues (Martínez, 2002). Proximal histidine acts as the fifth ligand of the haem iron together with the four nitrogens of the tetrapyrrolic macrocycle, while distal histidine together with a conserved arginine (VP Arg‐43) contributes to iron reaction with hydrogen peroxide in compound‐I formation (Hiner et al., 2002). Two aromatic residues are also highly conserved at the proximal (Phe‐186) and distal (Phe‐46) sides of the haem of many peroxidases, in CCP being two tryptophan residues one of them playing a direct role in catalysis as mentioned below. Two more residues at the proximal (Asp‐231) and distal (Asn‐78) sides of the haem establish hydrogen bonds with the two histidines. Two structural Ca2+ ions are located in conserved binding sites at the two peroxidase domains contributing to folding. The above‐mentioned Mn2+‐oxidation site of VP (and MnP) is shown at the right side of haem, being formed by three acidic residues positioning the cation near one of the haem propionates for direct electron transfer (Gold et al., 2000; Ruiz‐Dueñas et al., 2007). Finally, a tryptophan residue (Trp‐164) is shown at the left side of the haem, being involved in oxidation of high‐redox‐potential aromatic compounds (together with contiguous Leu‐165) as described in the next section. The VP structure also includes several molecules of water, including those completing the characteristic coordinations of Mn2+ and Ca2+ ions. Another water molecule (w71) could act as the sixth ligand of the haem Fe3+ at a position close to that occupied by the Fe4+=O oxygen of compound‐I and compound‐II, as shown for HRP (Berglund et al., 2002).
Figure 5.
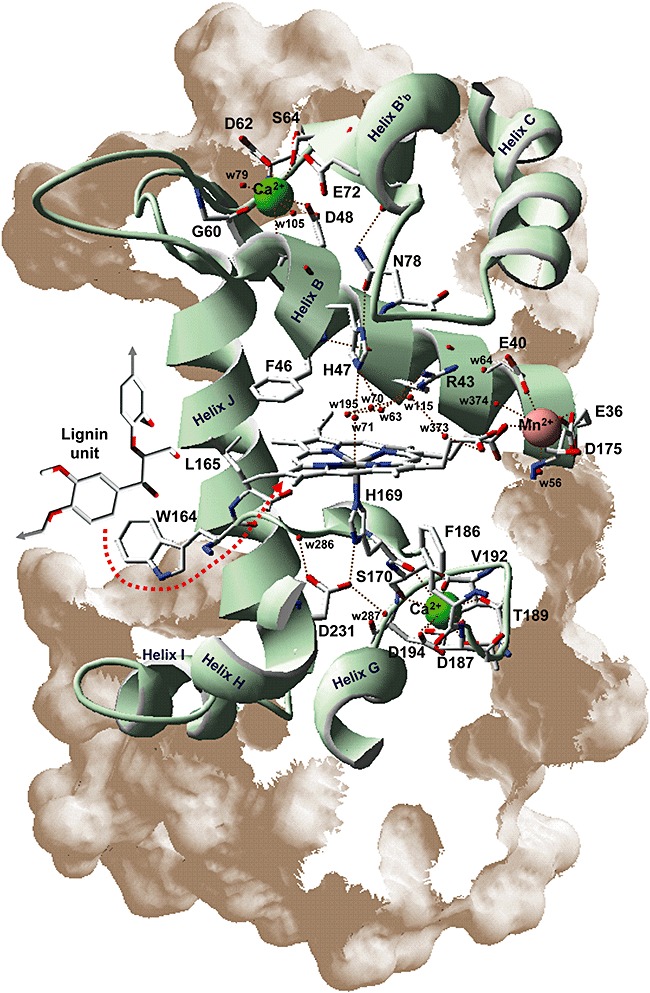
Details of haem environment and other structurally and catalytically relevant residues in P. eryngii VP. His‐169 (the fifth ligand of haem iron), Phe‐186 and Asp‐231 (corresponding to His‐176, Phe‐193 and Asp‐238 in LiP‐H8) are shown at the proximal side of the haem, while His‐47, Phe‐46, Arg‐43 and Asn‐78 (corresponding to His‐47, Phe‐46 and Arg‐43 in LiP‐H8) are shown at the distal side. Glu‐36, Glu‐40 and Asp‐175 (corresponding to Ala‐36, Glu‐40 and Asn‐182 in LiP‐H8) constitute the site of oxidation of Mn2+ (red van der Waals sphere) near the internal propionate of haem, while Trp‐164 (corresponding to Trp‐171 in LiP‐H8) is responsible for oxidation of lignin units and other aromatic compounds by LRET (red arrow) to the activated haem cofactor via Leu‐165 (corresponding to Leu‐172 in LiP‐H8). Finally, the ligands of the two structural Ca2+ ions (green spheres) are indicated at the proximal (Ser‐170, Asp‐187, Thr‐189, Val‐192 and Asp‐194) and distal (Asp‐48, Gly‐60, Asp‐62 and Ser‐64) sides (corresponding, respectively, to Ser‐177, Asp‐194, Thr‐196, Ile‐199 and Asp‐201; and Asp‐48, Gly‐66, Asp‐68 and Ser‐70 in LiP‐H8). Several water molecules are also shown including those completing Mn2+, Ca2+ and haem Fe3+ coordination.
It has been shown that the redox potential of peroxidase compound‐I and compound‐II depends of the haem environment characteristics, as shown by 1H‐NMR spectra of the cyanide adduct of the enzyme (Banci et al., 1991; 2003). These included, as one of the most important factors, the position and more or less marked imidazolate character of the proximal histidine side‐chain, whose N1 acts as the fifth ligand of the haem iron. The strength of this bond affects the electron deficiency of iron and, consequently, the reactivity of the activated enzyme. The characteristics of the proximal histidine side‐chain that are affected by H‐bonding to other residues (such as Asp‐231 in Fig. 5) (Banci et al., 1995) and the position of helix F where this residue is located (Piontek et al., 1993) affect the chemical shift of its Hε1 signal (which was found at about −8 p.p.m. in LiP, −16 p.p.m. in VP, −22 p.p.m. in CCP and −32 p.p.m. in HRP spectra) in agreement with the reported higher redox potential of ligninolytic peroxidases compared with plant peroxidases (Millis et al., 1989).
Direct peroxidase oxidation of lignin: long‐range electron transfer (LRET) mechanism
When the crystal structure of P. chrysosporium LiP was reported, it was assumed that this peroxidase would oxidize high‐redox‐potential aromatic compounds at the main haem access channel. In fact, veratryl alcohol was modelled at the crystal structure channel to determine the eventual contacts (Poulos et al., 1993). This is the classical peroxidase substrate oxidation site, as shown in plant HRP and in the low‐redox‐potential fungal CIP (Smith and Veitch, 1998). However, the main haem access channel of LiP is significantly narrower than those of CIP or HRP, and the hypothesis of long‐range electron transfer (LRET) oxidation of lignin at the protein surface, initially proposed by Du and colleagues (1992) and Schoemaker and colleagues (1994), was adopted by other authors.
Protein LRET requires the existence of an amino acid residue susceptible to form a stable radical (preferably a tyrosine, tryptophan or histidine residue) located at the protein surface and adequately connected to the haem cofactor for electron transfer. The above authors suggested two residues (Trp‐171 and His‐82) as the origin of two LRET pathways for oxidation of high‐redox‐potential aromatic compounds by LiP. Several years later a third pathway was proposed (initiating at His‐239) (Johjima et al., 1999). No pathway starting at a tyrosine residue has been proposed since all ligninolytic peroxidases cloned up to date (a total of near 50 sequences) are free of tyrosine residues (to prevent oxidative inactivation) with the only exception of a Trametes cervina LiP discussed below.
The same three putative LRET pathways were identified in P. eryngii VP, and their eventual operation was investigated by site‐directed mutagenesis by Pérez‐Boada and colleagues (2005). This study definitively showed that only the pathway starting at the exposed tryptophan (Trp‐164 of isoenzyme VPL) was operative for oxidation of high‐redox‐potential aromatic compounds including veratryl alcohol. The position of this residue at the vicinity of the haem cofactor is illustrated in Fig. 5, which also shows a leucine residue involved in the electron transfer; and the solvent exposed aromatic side‐chain of this tryptophan is shown in Fig. 4 (right). The catalytic role of a homologous tryptophan has also been shown in VP from other fungi (Tinoco et al., 2007; Tsukihara et al., 2008). Formation of the tryptophanyl‐free radical postulated by Du and colleagues (1992) was confirmed by low‐temperature electron paramagnetic resonance (EPR) of peroxide‐activated VP (Pogni et al., 2006).
The involvement of the homologous tryptophan residue of LiP (Trp‐171) in oxidation of veratryl alcohol and, more interestingly, of a tetrameric lignin model compound was also confirmed by mutagenesis (Doyle et al., 1998; Mester et al., 2001). Although the LiP tryptophanyl radical was not directly detected, as reported by VP EPR, indirect evidence for its presence in the activated enzyme was obtained by adduct formation (Blodig et al., 1999).
A difference between the two ligninolytic peroxidases concerns the surface environment of the catalytic tryptophan, which in LiP has a partial negative charge, whereas in VP some acidic residues are substituted by basic residues (Fig. 4, right). A noteworthy characteristic of VP is its ability to oxidize directly a series of high‐redox‐potential substrates that LiP is able to oxidize only in the presence of veratryl alcohol (see below). This fact is related to the catalytic tryptophan environment, and a charge inversion VP variant (R257D) was obtained that exhibited a LiP‐type behaviour during oxidation of high‐redox‐potential dyes (Ruiz‐Dueñas et al., 2008).
After demonstrating the involvement of a protein radical in substrate oxidation by ligninolytic peroxidases, an extended catalytic cycle has been proposed for VP (Fig. 6) that in general terms can be also applied to P. chrysosporium LiP. In this cycle, compound‐IB containing Fe4+=O and tryptophan radical, and compound‐IIB containing Fe3+ and tryptophan radical, are included together with normal compound‐I and compound‐II (containing Fe4+=O and porphyrin radical, and Fe4+=O respectively) that are now called compound‐IA and compound‐IIA. Compound‐IB and compound‐IIB (formed by LRET to the activated haem cofactor) represent a small percentage of the total activated enzyme (Pogni et al., 2006) being in equilibrium with the corresponding compound‐IA and compound‐IIA.
Figure 6.
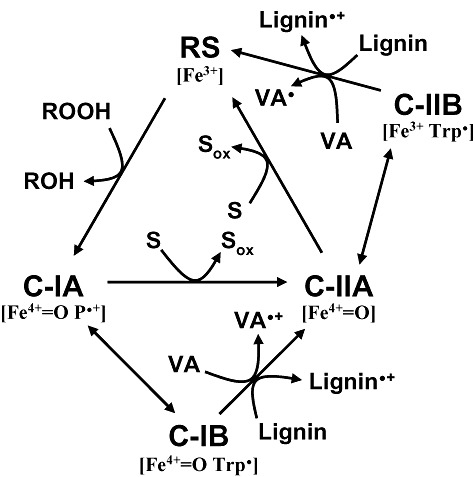
Extended catalytic cycle proposed for ligninolytic peroxidases (LiP and VP). In addition to normal compound‐I and compound‐II of Fig. 3 (now C‐IA and C‐IIA), C‐IB (containing Fe4+=O and tryptophan radical) and C‐IIB (containing Fe3+ and tryptophan radical) are included, being involved in oxidation of high‐redox‐potential compounds such as veratryl alcohol (VA) and lignin units to their corresponding cation radicals. C‐IB and C‐IIB are formed by LRET to the activated haem cofactor. Adapted from Pérez‐Boada and colleagues (2005).
A different LiP form lacking a catalytic tryptophan residue has been reported in T. cervina (Miki et al., 2006). This unique peroxidase presents an exposed tyrosine at a different position of the molecule that seems to play the same role of the above tryptophan. Interestingly, a plant peroxidase using a tyrosyl radical for oxidation of bulky phenols such as sinapyl alcohol (structure 3 in Fig. 1) has been recently reported (Sasaki et al., 2008).
Redox mediators in lignin degradation
The use of small chemical oxidizers acting as redox mediators represents a second alternative to overcome the difficulties associated to the limited access of the bulky lignin polymer to the activated cofactor of peroxidases and other oxidoreductases. Enzyme‐mediator systems have been extensively investigated in the case of laccases after the work of Bourbonnais and Paice (1990) reporting that synthetic mediators expanded the application potential of these low‐redox‐potential oxidoreductases enabling oxidation of non‐phenolic lignin model compounds. These mediators are low‐molecular‐mass compounds that: (i) form stable free radicals oxidizing compounds that the enzyme alone is not able to oxidize, and (ii) diffuse away from the enzyme and can easily penetrate the lignocellulose matrix. It has been recently shown that some phenolic lignin precursors or degradation products can be used as ‘natural’ laccase mediators in industrial processes, and suggested that some of them could play a similar role in nature (Camarero et al., 2005). However, the real significance of the laccase‐mediator systems in natural biodegradation of lignin is still to be demonstrated.
The existence of an exposed protein radical transferring electrons to the haem via a LRET pathway enables LiP direct oxidation of veratryl alcohol and lignin model compounds, including non‐phenolic tetramers (Mester et al., 2001). However, LiP requires the presence of veratryl alcohol or other compounds forming aromatic cation radicals, to oxidize polymeric lignin and high‐redox‐potential dyes (Harvey et al., 1986). This contrasts with the ‘veratryl alcohol‐independent’ activity of VP oxidizing different compounds including lignin (Heinfling et al., 1998a; Camarero et al., 2001). The role of veratryl alcohol cation radical as a real mediator in LiP reactions has been matter of controversy, taking into account the short half‐life of this species in aqueous media (Candeias and Harvey, 1995). However, the acidic environment surrounding LiP Trp‐171 could stabilize the cation radical that would act as an enzyme‐bound mediator (Khindaria et al., 1996).
Biotechnological interest and future trends
Microbial oxidoreductases, including both peroxidases and laccases, have been investigated for biotechnological application including paper pulp bleaching in chlorine‐free sequences (Paice et al., 1995; Bajpai, 2004; Sigoillot et al., 2005). Ligninolytic peroxidases, in contrast to laccases and low‐redox‐potential peroxidases that are used in several industrial sectors (e.g. textiles, detergents, food and beverages, etc.), are not still commercially available due to different reasons including the low levels of enzyme obtained from natural and recombinant hosts. Moreover, these oxidoreductases in most cases are not suitable biocatalysts as they are produced in nature. Industrial processes often require enzymes recognizing specific substrates or proceeding under extreme conditions (e.g. high hydrogen peroxide concentration or extreme pH and temperature).
Laccases have low redox potential and can degrade recalcitrant compounds only in the presence of redox mediators. However, the laccase‐mediator system has became extremely popular in biodegradation studies, as well as for processing and functionalization of lignocellulosic fibres (Riva, 2006; Widsten and Kandelbauer, 2008). In spite of ‘natural’ laccase mediators have been reported as potential substitutes of synthetic ‐N(OH)‐ compounds in different applications (Camarero et al., 2007; Cañas et al., 2007; Gutiérrez et al., 2007), economic issues related to the cost of the mediator, and environmental concerns related to the eventual release of toxic compounds during the enzymatic treatment are main drawbacks for the industrial implementation of this enzymatic system in the pulp and paper and other industrial sectors.
In contrast to laccases, some ligninolytic peroxidases do not require mediators to degrade high‐redox‐potential compounds. Indeed, they should be the enzymes of choice for removing lignin or transforming high‐redox‐potential aromatic compounds in different applications. The rapid acquisition of knowledge on the structure and function of these enzymes over the last years has been used to modulate their catalytic and operational properties using site‐directed mutagenesis in a variety of studies (Timofeevski et al., 1999; Reading and Aust, 2000; Celik et al., 2001; Mester and Tien, 2001; Feng et al., 2003). In those cases where the structural basis of the property to be improved is unknown, or too difficult to be predicted, directed evolution or saturation mutagenesis is the approach of choice (Cherry et al., 1999; Miyazaki‐Imamura et al., 2003; Ryu et al., 2008). Some future trends in the industrial use of basidiomycete peroxidases are discussed below.
Considerable efforts have been devoted during the last years to improve the expression of ligninolytic and other peroxidases in different fungal hosts using a variety of strategies. Among others, strong promoters, protease deficient strains, molecular chaperones and external sources of haem have been used with only partially successful results (Stewart et al., 1996; Conesa et al., 2000; 2002; Gu et al., 2003; Lú‐Chau et al., 2004; Wang et al., 2004; Eibes et al., 2009). New strategies based on converting low‐redox‐potential peroxidases, easily to express at levels of several grams per litre, into high‐redox‐potential peroxidases are being developed taking advantage of the structure–function knowledge accumulated. In this sense, CIP has been successfully modified and transformed into a LiP‐like enzyme by introducing a catalytic tryptophan, at the same time that the acidity of the local environment of this residue was increased (Smith and Doyle, 2006). Although the mutated enzyme was not so efficient as a true LiP, it is possible to predict that additional modifications will yield an enzyme with the expected catalytic activity.
Hydrogen peroxide inactivate all peroxidases after several cycles of catalysis as a consequence of the competition between productive and unproductive electron sources (including enzyme components) in a process described as a suicide inactivation (Valderrama et al., 2002). Different attempts aimed to improve hydrogen peroxide stability of ligninolytic peroxidases have been performed, mainly removing easily oxidizable and conformationally unstable amino acid residues located at the peroxide‐binding side of haem with very promising results (Miyazaki and Takahashi, 2001; Miyazaki‐Imamura et al., 2003). Recently, a novel peroxidase from Raphanus sativus has been identified and characterized as the only known case of a haemperoxidase intrinsically resistant to hydrogen peroxide (Gil‐Rodríguez et al., 2008). In‐depth analysis of its structure will give the first structural evidences of peroxide stability to be used to increase the peroxide stability of ligninolytic and other peroxidases.
pH and temperature inactivation of ligninolytic peroxidases is associated to the release of the two structural Ca2+ involved in stabilization of the molecular architecture (Sutherland and Aust, 1996; George et al., 1999; Lú‐Chau et al., 2004). It has been described that losing these ions causes hexacoordination of the haem iron preventing LiP and MnP activation by hydrogen peroxide, although more recently a decrease in redox potential has been suggested as the main inactivation cause in VP (Verdín et al., 2006). Some authors have succeeded stabilizing peroxidases by avoiding the lose of Ca2+ by adding extra disulfide bridges (Reading and Aust, 2000). Generation of the appropriate disulfide bridges could not only make these enzymes resistant to high pH and temperature, but it could also be used to increase their redox potential. Thermal inactivation has been also related to destabilizing interactions between adjacent acidic side‐chains. Mutations removing one of these interactions in CIP improved its thermal stability by 134‐fold (Cherry et al., 1999). Curiously these two residues are conserved in ligninolytic peroxidases (LiP, MnP and VP), and additional interactions between acidic residues can be observed in other regions of their molecular structures. It is expected that substitution of one amino acid residue of these acidic couples will promote enzyme stabilization towards high temperature, as described for CIP.
Among ligninolytic peroxidases, VP presents especial biotechnological interest due to different reasons including: (i) catalytic versatility by combination of different substrate oxidation sites, and (ii) ability to degrade some compounds of interest that LiP and MnP (as well as CIP) are not able to oxidize directly. Its catalytic versatility permits the application of VP in Mn3+‐mediated or Mn‐independent reactions on both low‐ and high‐redox‐potential substrates. The possibility to degrade directly a variety of recalcitrant compounds represents a considerable advantage compared with LiP since the cost of veratryl alcohol required as mediator can be saved. Among the different compounds of industrial and/or environmental interest that VP can transform, polycyclic aromatic hydrocarbons (Wang et al., 2003), phenolic and non‐phenolic aromatic pollutants (Rodríguez et al., 2004), pesticides (Dávila‐Vázquez et al., 2005) and a variety of industrial dyes (Heinfling et al., 1998b) can be cited (including, among others, Reactive Blue 38 and other azo dyes, Reactive Black 5 and other phthalocyanine dyes, anthracene and derivatives, benzo[a]pyrene, pyrene, 2,4‐dichlorophenol and pentachlorophenol). For some applications the use of VP in combination with redox mediators can also be considered (Tinoco et al., 2007). Among them, the use of VP to reoxidize Mn2+‐containing polyoxometalates is an interesting possibility (Marques et al., 2008), since these highly promising catalysts for environmentally friendly delignification are very difficult to be chemically reoxidized.
The promiscuity of ligninolytic fungi and their enzymes oxidizing aromatic xenobiotics and other recalcitrant compounds is due to the involvement of the lignin‐degrading enzymatic machinery in many of these reactions. To overcome the bulky nature and structural heterogeneity of the lignin polymer, these microorganisms have developed a highly unspecific extracellular system being able to subtract one electron directly from the benzenic rings of the different lignin units. High‐redox‐potential extracellular peroxidases, often forming catalytic radicals at the protein surface, are the key enzymes in this initial attack yielding partially oxidized products whose catabolism is finally completed intracellularly.
Acknowledgments
This work has been partially supported by the Spanish projects BIO2005‐03569 and BIO2008‐01533, and the BIORENEW (http://www.biorenew.org) EU‐contract (NMP2‐CT‐2006‐026456). The authors thank Göran Gellerstedt (KTH, Stockholm, Sweden) for the lignin structure model. María Morales, Yuta Miki, Susana Camarero and María Jesús Martínez from CIB (CSIC, Madrid, Spain), Ana Gutiérrez and José C. del Río from IRNAS (CSIC, Sevilla, Spain) and Francisco Guillén from University of Alcalá (Alcalá de Henares, Spain) are acknowledged for providing data and helpful discussions. F.J.R.‐D. thanks an EU‐project contract.
References
- Adler E. Lignin chemistry – past, present and future. Wood Sci Technol. 1977;11:169–218. [Google Scholar]
- Bajpai P. Biological bleaching of chemical pulps. Crit Rev Biotechnol. 2004;24:1–58. doi: 10.1080/07388550490465817. [DOI] [PubMed] [Google Scholar]
- Baldrian P. Fungal laccases – occurrence and properties. FEMS Microbiol Rev. 2006;30:215–242. doi: 10.1111/j.1574-4976.2005.00010.x. [DOI] [PubMed] [Google Scholar]
- Banci L. Structural properties of peroxidases. J Biotechnol. 1997;53:253–263. doi: 10.1016/s0168-1656(97)01677-5. [DOI] [PubMed] [Google Scholar]
- Banci L., Bertini I., Turano P., Tien M., Kirk T.K. Proton NMR investigation into the basis for the relatively high redox potential of lignin peroxidase. Proc Natl Acad Sci USA. 1991;88:6956–6960. doi: 10.1073/pnas.88.16.6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L., Bertini I., Pierattelli R., Tien M., Vila A.J. Factoring of the hyperfine shifts in the cyanide adduct of lignin peroxidase from P. chrysosporium. J Am Chem Soc. 1995;117:8659–8667. [Google Scholar]
- Banci L., Camarero S., Martínez A.T., Martínez M.J., Pérez‐Boada M., Pierattelli R. NMR study of Mn(II) binding by the new versatile peroxidase from the white‐rot fungus Pleurotus eryngii. J Biol Inorg Chem. 2003;8:751–760. doi: 10.1007/s00775-003-0476-1. et al. [DOI] [PubMed] [Google Scholar]
- Bao W.L., Fukushima Y., Jensen K.A., Moen M.A., Hammel K.E. Oxidative degradation of non‐phenolic lignin during lipid peroxidation by fungal manganese peroxidase. FEBS Lett. 1994;354:297–300. doi: 10.1016/0014-5793(94)01146-x. [DOI] [PubMed] [Google Scholar]
- Berglund G.I., Carlsson G.H., Smith A.T., Szoke H., Henriksen A., Hajdu J. The catalytic pathway of horseradish peroxidase at high resolution. Nature. 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- Blodig W., Smith A.T., Winterhalter K., Piontek K. Evidence from spin‐trapping for a transient radical on tryptophan residue 171 of lignin peroxidase. Arch Biochem Biophys. 1999;370:86–92. doi: 10.1006/abbi.1999.1365. [DOI] [PubMed] [Google Scholar]
- Bourbonnais R., Paice M.G. Oxidation of non‐phenolic substrates. An expanded role for laccase in lignin biodegradation. FEBS Lett. 1990;267:99–102. doi: 10.1016/0014-5793(90)80298-w. [DOI] [PubMed] [Google Scholar]
- Camarero S., Bocchini P., Galletti G.C., Martínez M.J., Martínez A.T. Compositional changes of wheat lignin by a fungal peroxidase analyzed by Pyrolysis‐GC/MS. J Anal Appl Pyrolysis. 2001;58/59:413–423. [Google Scholar]
- Camarero S., Ibarra D., Martínez M.J., Martínez A.T. Lignin‐derived compounds as efficient laccase mediators for decolorization of different types of recalcitrant dyes. Appl Environ Microbiol. 2005;71:1775–1784. doi: 10.1128/AEM.71.4.1775-1784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarero S., Ibarra D., Martínez A.T., Romero J., Gutiérrez A., Del Río J.C. Paper pulp delignification using laccase and natural mediators. Enzyme Microb Technol. 2007;40:1264–1271. [Google Scholar]
- Cañas A., Alcalde M., Plou F.J., Martínez M.J., Martínez A.T., Camarero S. Transformation of polycyclic aromatic hydrocarbons by laccase is strongly enhanced by phenolic compounds present in soil. Environ Sci Technol. 2007;41:2964–2971. doi: 10.1021/es062328j. [DOI] [PubMed] [Google Scholar]
- Candeias L.P., Harvey P.J. Lifetime and reactivity of the veratryl alcohol radical cation – implications for lignin peroxidase catalysis. J Biol Chem. 1995;270:16745–16748. doi: 10.1074/jbc.270.28.16745. [DOI] [PubMed] [Google Scholar]
- Celik A., Cullis P.M., Sutcliffe M.J., Sangar R., Raven E.L. Engineering the active site of ascorbate peroxidase. Eur J Biochem. 2001;268:78–85. doi: 10.1046/j.1432-1327.2001.01851.x. [DOI] [PubMed] [Google Scholar]
- Chen C.‐L., Chang H. Chemistry of lignin biodegradation. In: Higuchi T., editor. Academic Press; 1985. pp. 535–555. [Google Scholar]
- Cherry J.R., Lamsa M.H., Schneider P., Vind J., Svendsen A., Jones A. Directed evolution of a fungal peroxidase. Nat Biotechnol. 1999;17:379–384. doi: 10.1038/7939. et al. [DOI] [PubMed] [Google Scholar]
- Claus H. Laccases: structure reactions, distribution. Micron. 2004;35:93–96. doi: 10.1016/j.micron.2003.10.029. [DOI] [PubMed] [Google Scholar]
- Conesa A., Van Den Hondel C.A.M.J.J., Punt P.J. Studies on the production of fungal peroxidases in Aspergillus niger. Appl Environ Microbiol. 2000;66:3016–3023. doi: 10.1128/aem.66.7.3016-3023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A., Jeenes D., Archer D.B., VandenHondel C.A.M.J., Punt P.J. Calnexin overexpression increases manganese peroxidase production in Aspergillus niger. Appl Environ Microbiol. 2002;68:846–851. doi: 10.1128/AEM.68.2.846-851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen D. Recent advances on the molecular genetics of ligninolytic fungi. J Biotechnol. 1997;53:273–289. doi: 10.1016/s0168-1656(97)01684-2. [DOI] [PubMed] [Google Scholar]
- Dávila‐Vázquez G., Tinoco R., Pickard M.A., Vázquez‐Duhalt R. Transformation of halogenated pesticides by versatile peroxidase from Bjerkandera adusta. Enzyme Microb Technol. 2005;36:223–231. [Google Scholar]
- Doyle W.A., Blodig W., Veitch N.C., Piontek K., Smith A.T. Two substrate interaction sites in lignin peroxidase revealed by site‐directed mutagenesis. Biochemistry. 1998;37:15097–15105. doi: 10.1021/bi981633h. [DOI] [PubMed] [Google Scholar]
- Du P., Collins J.R., Loew G.H. Homology modeling of a heme protein, lignin peroxidase, from the crystal structure of cytochrome c peroxidase. Protein Eng. 1992;5:679–691. doi: 10.1093/protein/5.7.679. [DOI] [PubMed] [Google Scholar]
- Dunford H.B. Wiley‐VCH; 1999. [Google Scholar]
- Eibes G.M., Lú‐Chau T.A., Ruiz‐Dueñas F.J., Feijoo G., Martínez M.J., Martínez A.T. Effect of culture temperature on the heterologous expression of Pleurotus eryngii versatile peroxidase in Aspergillus hosts. Bioprocess Biosyst Eng. 2009;32:129–134. doi: 10.1007/s00449-008-0231-7. et al. [DOI] [PubMed] [Google Scholar]
- Eriksson K.‐E.L., Blanchette R.A., Ander P. Springer‐Verlag; 1990. [Google Scholar]
- Feng M.L., Tachikawa H., Wang X.T., Pfister T.D., Gengenbach A.J., Lu Y. Resonance Raman spectroscopy of cytochrome c peroxidase variants that mimic manganese peroxidase. J Biol Inorg Chem. 2003;8:699–706. doi: 10.1007/s00775-003-0460-9. [DOI] [PubMed] [Google Scholar]
- Gajhede M., Schuller D.J., Henriksen A., Smith A.T., Poulos T.L. Crystal structure of horseradish peroxidase C at 2.15 Å resolution. Nat Struct Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- Galbe M., Zacchi G. Pretreatment of lignocellulosic materials for efficient bioethanol production. Adv Biochem Engineer Biotechnol. 2007;108:41–65. doi: 10.1007/10_2007_070. [DOI] [PubMed] [Google Scholar]
- Gellerstedt G., Henriksson G. Lignins: major sources, structure and properties. In: Belgacem M., Gandini A., editors. Elsevier; 2008. pp. 201–224. [Google Scholar]
- George S.J., Kvaratskhelia M., Dilworth M.J., Thorneley R.N.F. Reversible alkaline inactivation of lignin peroxidase involves the release of both the distal and proximal site calcium ions and bishistidine co‐ordination of the haem. Biochem J. 1999;344:237–244. [PMC free article] [PubMed] [Google Scholar]
- Gil‐Rodríguez P., Ferreira‐Batista C., Vázquez‐Duhalt R., Valderrama B. A novel heme peroxidase from Raphanus sativus intrinsically resistant to hydrogen peroxide. Eng Life Sci. 2008;8:286–296. [Google Scholar]
- Glenn J.K., Morgan M.A., Mayfield M.B., Kuwahara M., Gold M.H. An extracellular H2O2‐requiring enzyme preparation involved in lignin biodegradation by the white rot basidiomycete Phanerochaete chrysosporium. Biochem Biophys Res Commun. 1983;114:1077–1083. doi: 10.1016/0006-291x(83)90672-1. [DOI] [PubMed] [Google Scholar]
- Gold M.H., Youngs H.L., Gelpke M.D. Manganese peroxidase. Met Ions Biol Syst. 2000;37:559–586. [PubMed] [Google Scholar]
- Gu L., Lajoie C., Kelly C. Expression of a Phanerochaete chrysosporium manganese peroxidase gene in the yeast Pichia pastoris. Biotechnol Prog. 2003;19:1403–1409. doi: 10.1021/bp025781h. [DOI] [PubMed] [Google Scholar]
- Guillén F., Evans C.S. Anisaldehyde and veratraldehyde acting as redox cycling agents for H2O2 production by Pleurotus eryngii. Appl Environ Microbiol. 1994;60:2811–2817. doi: 10.1128/aem.60.8.2811-2817.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillén F., Gómez‐Toribio V., Martínez M.J., Martínez A.T. Production of hydroxyl radical by the synergistic action of fungal laccase and aryl alcohol oxidase. Arch Biochem Biophys. 2000;383:142–147. doi: 10.1006/abbi.2000.2053. [DOI] [PubMed] [Google Scholar]
- Gutiérrez A., Caramelo L., Prieto A., Martínez M.J., Martínez A.T. Anisaldehyde production and aryl‐alcohol oxidase and dehydrogenase activities in ligninolytic fungi from the genus Pleurotus. Appl Environ Microbiol. 1994;60:1783–1788. doi: 10.1128/aem.60.6.1783-1788.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez A., Rencoret J., Ibarra D., Molina S., Camarero S., Romero J. Removal of lipophilic extractives from paper pulp by laccase and lignin‐derived phenols as natural mediators. Environ Sci Technol. 2007;41:4124–4129. doi: 10.1021/es062723+. et al. [DOI] [PubMed] [Google Scholar]
- Hammel K.E., Cullen D. Role of fungal peroxidases in biological ligninolysis. Curr Opin Plant Biol. 2008;11:349–355. doi: 10.1016/j.pbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Harvey P.J., Schoemaker H.E., Palmer J.M. Veratryl alcohol as a mediator and the role of radical cations in lignin biodegradation by Phanerochaete chrysosporium. FEBS Lett. 1986;195:242–246. [Google Scholar]
- Heinfling A., Ruiz‐Dueñas F.J., Martínez M.J., Bergbauer M., Szewzyk U., Martínez A.T. A study on reducing substrates of manganese‐oxidizing peroxidases from Pleurotus eryngii and Bjerkandera adusta. FEBS Lett. 1998a;428:141–146. doi: 10.1016/s0014-5793(98)00512-2. [DOI] [PubMed] [Google Scholar]
- Heinfling A., Martínez M.J., Martínez A.T., Bergbauer M., Szewzyk U. Transformation of industrial dyes by manganese peroxidase from Bjerkandera adusta and Pleurotus eryngii in a manganese‐independent reaction. Appl Environ Microbiol. 1998b;64:2788–2793. doi: 10.1128/aem.64.8.2788-2793.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi T. Springer, Verlag; 1997. [Google Scholar]
- Hiner A.N.P., Raven E.L., Thorneley R.N.F., García‐Canovas F., Rodríguez‐López J.N. Mechanisms of compound I formation in heme peroxidases. J Inorg Biochem. 2002;91:27–34. doi: 10.1016/s0162-0134(02)00390-2. [DOI] [PubMed] [Google Scholar]
- Johjima T., Itoh H., Kabuto M., Tokimura F., Nakagawa T., Wariishi H. Direct interaction of lignin and lignin peroxidase from Phanerochaete chrysosporium. Proc Natl Acad Sci USA. 1999;96:1989–1994. doi: 10.1073/pnas.96.5.1989. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhunen P., Rummakko P., Sipila J., Brunow G., Kilpeläinen I. Dibenzodioxocins – a novel type of linkage in softwood lignins. Tetrahedron Lett. 1995;36:169–170. [Google Scholar]
- Kersten P., Cullen D. Extracellular oxidative systems of the lignin‐degrading Basidiomycete Phanerochaete chrysosporium. Fungal Genet Biol. 2007;44:77–87. doi: 10.1016/j.fgb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Khindaria A., Yamazaki I., Aust S.D. Stabilization of the veratryl alcohol cation radical by lignin peroxidase. Biochemistry. 1996;35:6418–6424. doi: 10.1021/bi9601666. [DOI] [PubMed] [Google Scholar]
- Kirk T.K., Farrell R.L. Enzymatic ‘combustion’: the microbial degradation of lignin. Annu Rev Microbiol. 1987;41:465–505. doi: 10.1146/annurev.mi.41.100187.002341. [DOI] [PubMed] [Google Scholar]
- Li H.Y., Poulos T.L. Structural variation in heme enzymes: a comparative analysis of peroxidase and P450 crystal structures. Structure. 1994;2:461–464. doi: 10.1016/s0969-2126(00)00046-0. [DOI] [PubMed] [Google Scholar]
- Lú‐Chau T.A., Ruiz‐Dueñas F.J., Camarero S., Feijoo G., Martínez M.J., Lema J.M. Effect of pH on the stability of Pleurotus eryngii versatile peroxidase during heterologous production in Emericella nidulans. Bioprocess Biosyst Eng. 2004;26:287–293. doi: 10.1007/s00449-004-0365-1. et al. [DOI] [PubMed] [Google Scholar]
- Marques G., Gamelas J.A.F., Evtuguin D., Ruiz‐Dueñas F.J., Morales M., Del Río J.C. 2008. et al.) Versatile peroxidase reoxidation of Mn‐based polyoxomethalate in chlorine‐free bleaching of eucalypt pulp. Proceeding of the 8th International Peroxidase Symposium, Tampere, 2023 August.
- Martínez A.T. Molecular biology and structure–function of lignin‐degrading heme peroxidases. Enzyme Microb Technol. 2002;30:425–444. [Google Scholar]
- Martínez A.T., Camarero S., Guillén F., Gutiérrez A., Muñoz C., Varela E. Progress in biopulping of non‐woody materials: chemical, enzymatic and ultrastructural aspects of wheat‐straw delignification with ligninolytic fungi from the genus Pleurotus. FEMS Microbiol Rev. 1994;13:265–274. et al. [Google Scholar]
- Martínez A.T., Speranza M., Ruiz‐Dueñas F.J., Ferreira P., Camarero S., Guillén F. Biodegradation of lignocellulosics: microbiological, chemical and enzymatic aspects of fungal attack to lignin. Int Microbiol. 2005;8:195–204. et al. [PubMed] [Google Scholar]
- Martínez A.T., Rencoret J., Marques G., Gutiérrez A., Ibarra D., Jiménez‐Barbero J. Monolignol acylation and lignin structure in some nonwoody plants: a 2D NMR study. Phytochemistry. 2008;69:2831–2843. doi: 10.1016/j.phytochem.2008.09.005. et al. [DOI] [PubMed] [Google Scholar]
- Martínez D., Larrondo L.F., Putnam N., Gelpke M.D., Huang K., Chapman J. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol. 2004;22:695–700. doi: 10.1038/nbt967. et al. [DOI] [PubMed] [Google Scholar]
- Martinez D., Challacombe J., Morgenstern I., Hibbett D.S., Schmoll M., Kubicek C.P. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0809575106. et al. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester T., Tien M. Engineering of a manganese‐binding site in lignin peroxidase isozyme H8 from Phanerochaete chrysosporium. Biochem Biophys Res Commun. 2001;284:723–728. doi: 10.1006/bbrc.2001.5015. [DOI] [PubMed] [Google Scholar]
- Mester T., Ambert‐Balay K., Ciofi‐Baffoni S., Banci L., Jones A.D., Tien M. Oxidation of a tetrameric nonphenolic lignin model compound by lignin peroxidase. J Biol Chem. 2001;276:22985–22990. doi: 10.1074/jbc.M010739200. [DOI] [PubMed] [Google Scholar]
- Miki Y., Tanaka H., Nakamura M., Wariishi H. Isolation and characterization of a novel lignin peroxidase from the white‐rot basidiomycete Trametes cervina. J Fac Agr Kyushu Univ. 2006;51:99–104. [Google Scholar]
- Millis C.D., Cai D., Stankovich M.T., Tien M. Oxidation‐reduction potentials and ionization states of extracellular peroxidases from the lignin‐degrading fungus Phanerochaete chrysosporium. Biochemistry. 1989;28:8484–8489. doi: 10.1021/bi00447a032. [DOI] [PubMed] [Google Scholar]
- Miyazaki C., Takahashi H. Engineering of the H2O2‐binding pocket region of a recombinant manganese peroxidase to be resistant to H2O2. FEBS Lett. 2001;509:111–114. doi: 10.1016/s0014-5793(01)03127-1. [DOI] [PubMed] [Google Scholar]
- Miyazaki‐Imamura C., Oohira K., Kitagawa R., Nakano H., Yamane T., Takahashi H. Improvement of H2O2 stability of manganese peroxidase by combinatorial mutagenesis and high‐throughput screening using in vitro expression with protein disulfide isomerase. Protein Eng. 2003;16:423–428. doi: 10.1093/protein/gzg054. [DOI] [PubMed] [Google Scholar]
- Morgenstern I., Klopman S., Hibbett D.S. Molecular evolution and diversity of lignin degrading heme peroxidases in the agaricomycetes. J Mol Evol. 2008;66:243–257. doi: 10.1007/s00239-008-9079-3. [DOI] [PubMed] [Google Scholar]
- Nimz H. Beech lignin – proposal of a constitutional scheme. Angew Chem Int Ed Engl. 1974;13:313–321. [Google Scholar]
- Paice M.G., Bourbonnais R., Reid I.D., Archibald F.S., Jurasek L. Oxidative bleaching enzymes: a review. J Pulp Paper Sci. 1995;21:J280–J284. [Google Scholar]
- Pérez‐Boada M., Ruiz‐Dueñas F.J., Pogni R., Basosi R., Choinowski T., Martínez M.J. Versatile peroxidase oxidation of high redox potential aromatic compounds: site‐directed mutagenesis, spectroscopic and crystallographic investigations of three long‐range electron transfer pathways. J Mol Biol. 2005;354:385–402. doi: 10.1016/j.jmb.2005.09.047. et al. [DOI] [PubMed] [Google Scholar]
- Piontek K., Glumoff T., Winterhalter K. Low pH crystal structure of glycosylated lignin peroxidase from Phanerochaete chrysosporium at 2.5 Å resolution. FEBS Lett. 1993;315:119–124. doi: 10.1016/0014-5793(93)81146-q. [DOI] [PubMed] [Google Scholar]
- Pogni R., Baratto M.C., Teutloff C., Giansanti S., Ruiz‐Dueñas F.J., Choinowski T. A tryptophan neutral radical in the oxidized state of versatile peroxidase from Pleurotus eryngii: a combined multi‐frequency EPR and DFT study. J Biol Chem. 2006;281:9517–9526. doi: 10.1074/jbc.M510424200. et al. [DOI] [PubMed] [Google Scholar]
- Poulos T.L., Freer S.T., Alden R.A., Edwards S.L., Skogland U., Takio K. The crystal structure of cytochrome c peroxidase. J Biol Chem. 1980;255:575–580. et al. [PubMed] [Google Scholar]
- Poulos T.L., Edwards S.L., Wariishi H., Gold M.H. Crystallographic refinement of lignin peroxidase at 2 Å. J Biol Chem. 1993;268:4429–4440. doi: 10.2210/pdb1lga/pdb. [DOI] [PubMed] [Google Scholar]
- Ralph J., Lundquist K., Brunow G., Lu F., Kim H., Schatz P.F. Lignins: natural polymers from oxidative coupling of 4‐hydroxyphenylpropanoids. Phytochem Rev. 2004;3:29–60. et al. [Google Scholar]
- Reading N.S., Aust S.D. Engineering a disulfide bond in recombinant manganese peroxidase results in increased thermostability. Biotechnol Prog. 2000;16: 326–333. doi: 10.1021/bp0000151. [DOI] [PubMed] [Google Scholar]
- Del Río J.C., Marques G., Rencoret J., Martínez A.T., Gutiérrez A. Occurrence of naturally acetylated lignin units. J Agric Food Chem. 2007;55:5461–5468. doi: 10.1021/jf0705264. [DOI] [PubMed] [Google Scholar]
- Riva S. Laccases: blue enzymes for green chemistry. Trends Biotechnol. 2006;24:219–226. doi: 10.1016/j.tibtech.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Rodríguez E., Nuero O., Guillén F., Martínez A.T., Martínez M.J. Degradation of phenolic and non‐phenolic aromatic pollutants by four Pleurotus species: the role of laccase and versatile peroxidase. Soil Biol Biochem. 2004;36:909–916. [Google Scholar]
- Ruiz‐Dueñas F.J., Morales M., Pérez‐Boada M., Choinowski T., Martínez M.J., Piontek K. Manganese oxidation site in Pleurotus eryngii versatile peroxidase: a site‐directed mutagenesis, kinetic and crystallographic study. Biochemistry. 2007;46:66–77. doi: 10.1021/bi061542h. et al. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Dueñas F.J., Morales M., Mate M.J., Romero A., Martínez M.J., Smith A.T. Site‐directed mutagenesis of the catalytic tryptophan environment in Pleurotus eryngii versatile peroxidase. Biochemistry. 2008;47:1685–1695. doi: 10.1021/bi7020298. et al. [DOI] [PubMed] [Google Scholar]
- Ryu K., Hwang S.Y., Kim K.H., Kang J.H., Lee E.K. Functionality improvement of fungal lignin peroxidase by DNA shuffling for 2,4‐dichlorophenol degradability and H2O2 stability. J Biotechnol. 2008;133:110–115. doi: 10.1016/j.jbiotec.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Sasaki S., Nonaka D., Wariishi H., Tsutsumi Y., Kondo R. Role of Tyr residues on the protein surface of cationic cell‐wall‐peroxidase (CWPO‐C) from poplar: potential oxidation sites for oxidative polymerization of lignin. Phytochemistry. 2008;69:348–355. doi: 10.1016/j.phytochem.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Schoemaker H.E., Lundell T.K., Floris R., Glumoff T., Winterhalter K.H., Piontek K. Do carbohydrates play a role in lignin peroxidase cycle? Redox catalysis in the endergonic region of the driving force. Bioorganic Med Chem. 1994;2:509–519. doi: 10.1016/0968-0896(94)80021-9. [DOI] [PubMed] [Google Scholar]
- Sharma P., Goel R., Capalash N. Bacterial laccases. World J Microbiol Biotechnol. 2007;23:823–832. [Google Scholar]
- Sigoillot C., Camarero S., Vidal T., Record E., Asther M., Pérez‐Boada M. Comparison of different fungal enzymes for bleaching high‐quality paper pulps. J Biotechnol. 2005;115:333–343. doi: 10.1016/j.jbiotec.2004.09.006. et al. [DOI] [PubMed] [Google Scholar]
- Sixta H. Wiley‐VCH; 2006. [Google Scholar]
- Smith A.T., Doyle W.A. 2006.
- Smith A.T., Veitch N.C. Substrate binding and catalysis in heme peroxidases. Curr Opin Chem Biol. 1998;2:269–278. doi: 10.1016/s1367-5931(98)80069-0. [DOI] [PubMed] [Google Scholar]
- Stewart P., Whitwam R.E., Kersten P.J., Cullen D., Tien M. Efficient expression of a Phanerochaete chrysosporium manganese peroxidase gene in Aspergillus oryzae. Appl Environ Microbiol. 1996;62:860–864. doi: 10.1128/aem.62.3.860-864.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland G.R.J., Aust S.D. The effects of calcium on the thermal stability and activity of manganese peroxidase. Arch Biochem Biophys. 1996;332:128–134. doi: 10.1006/abbi.1996.0324. [DOI] [PubMed] [Google Scholar]
- Taylor T.N., Osborne J.M. The importance of fungi in shaping the paleoecosystem. Rev Paleobot Palyn. 1996;90:249–262. [Google Scholar]
- Tien M., Kirk T.K. Lignin‐degrading enzyme from the hymenomycete Phanerochaete chrysosporium Burds. Science. 1983;221:661–663. doi: 10.1126/science.221.4611.661. [DOI] [PubMed] [Google Scholar]
- Timofeevski S.L., Nie G., Reading N.S., Aust S.D. Engineering a functional hybrid of manganese peroxidase and lignin peroxidase. Plant Peroxidase Newsl. 1999;13:99–111. [Google Scholar]
- Tinoco R., Verdín J., Vázquez‐Duhalt R. Role of oxidizing mediators and tryptophan 172 in the decoloration of industrial dyes by the versatile peroxidase from Bjerkandera adusta. J Mol Catal B Enzym. 2007;46:1–7. [Google Scholar]
- Tsukihara T., Honda Y., Sakai R., Watanabe T., Watanabe T. Mechanism for oxidation of high‐molecular‐weight substrates by a fungal versatile peroxidase, MnP2. Appl Environ Microbiol. 2008;74:2873–2881. doi: 10.1128/AEM.02080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valderrama B., Ayala M., Vázquez‐Duhalt R. Suicide inactivation of peroxidases and the challenge of engineering more robust enzymes. Chem Biol. 2002;9:555–565. doi: 10.1016/s1074-5521(02)00149-7. [DOI] [PubMed] [Google Scholar]
- Verdín J., Pogni R., Baeza A., Baratto M.C., Basosi R., Vázquez‐Duhalt R. Mechanism of versatile peroxidase inactivation by Ca2+ depletion. Biophys Chem. 2006;121:163–170. doi: 10.1016/j.bpc.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Wang H.K., Lu F.P., Sun W.F., Du L.X. Heterologous expression of lignin peroxidase of Phanerochaete chrysosporium in Pichia methanolica. Biotechnol Lett. 2004;26:1569–1573. doi: 10.1023/B:BILE.0000045654.66689.b4. [DOI] [PubMed] [Google Scholar]
- Wang Y.X., Vázquez‐Duhalt R., Pickard M.A. Manganese‐lignin peroxidase hybrid from Bjerkandera adusta oxidizes polycyclic aromatic hydrocarbons more actively in the absence of manganese. Can J Microbiol. 2003;49:675–682. doi: 10.1139/w03-091. [DOI] [PubMed] [Google Scholar]
- Widsten P., Kandelbauer A. Laccase applications in the forest products industry: a review. Enzyme Microb Technol. 2008;42:293–307. [Google Scholar]
- Zhang L.M., Gellerstedt G., Ralph J., Lu F.C. NMR studies on the occurrence of spirodienone structures in lignins. J Wood Chem Technol. 2006;26:65–79. [Google Scholar]