

Summary
We developed a rapid detection method for Legionella pneumophila (Lp) by filtration, immunomagnetic separation, double fluorescent staining, and flow cytometry (IMS‐FCM method). The method requires 120 min and can discriminate ‘viable’ and ‘membrane‐damaged’ cells. The recovery is over 85% of spiked Lp SG 1 cells in 1 l of tap water and detection limits are around 50 and 15 cells per litre for total and viable Lp, respectively. The method was compared using water samples from house installations in a blind study with three environmental laboratories performing the ISO 11731 plating method. In 53% of the water samples from different taps and showers significantly higher concentrations of Lp were detected by flow cytometry. No correlation to the plate culture method was found. Since also ‘viable but not culturable’ (VNBC) cells are detected by our method, this result was expected. The IMS‐FCM method is limited by the specificity of the used antibodies; in the presented case they target Lp serogroups 1–12. This and the fact that no Lp‐containing amoebae are detected may explain why in 21% of all samples higher counts were observed using the plate culture method. Though the IMS‐FCM method is not yet fit to completely displace the established plating method (ISO 11731) for routine Lp monitoring, it has major advantages to plating and can quickly provide important insights into the ecology of this pathogen in water distribution systems.
Introduction
Members of the bacterial genus Legionella are waterborne pathogens and parasites of aquatic protozoa (Fields, 1996), which can be found in soil (Fliermans et al., 1981) and ubiquitously in water (Dufour et al., 2003). Legionella spp. (Ls) consists of more than 50 species and 65 serogroups (Fields et al., 2002). Ls are able to infect pulmonary cells through inhalation of aerosols (Gao et al., 1998) and thus present a health threat. Since their favoured growth temperatures lie in the range of 20–45°C, man‐made installation systems, such as showers, air conditioners, and cooling towers are of major concern (Fields et al., 2002). Legionella pneumophila (Lp) is the causative agent of 50–90% of all infections (Breiman and Butler, 1998; Yu et al., 2002; Yáñez et al., 2005; Pravinkumar et al., 2010), though most Ls are potentially human pathogens (Alli et al., 2003; Palusińska‐Szysz and Cendrowska‐Pinkosz, 2009). In 2011 the incidence recorded by the Swiss mandatory surveillance system was 3.2 cases/100 000 inhabitants (BAG, 2012). The incidence shows an upward trend with an average lethality of 6.6% (BAG, 2009).
The gold standard for the detection of Ls are presently plate culture methods, in Europe most prominently ISO 11731 (ISO, 2004), which take up to 10 days for a result. Repeatedly, ring trials revealed high inter‐laboratory volatility for the plating methods (Boulanger and Edelstein, 1995; Bentham, 2000; Napoli et al., 2009; Lucas et al., 2011). One of the reasons for this might be that Lp can enter the ‘viable‐but‐not‐cultivable’ (VBNC) state (Allegra, et al. 2008), which results in a major drawback for all cultivation‐dependent methods. Furthermore, when plating Ls from water the number detected is not proportional to the sampling volume, since the distribution of Ls appears to be not homogeneous in water samples (Schaefer, 2007). One reason for this may be amoeba carrying Ls (Oliver, 2010).
Ls detection using polymerase chain reaction (PCR) methods is frequently hampered by inhibitory compounds like humic acids, which are present in environmental samples (Palmer et al., 1993). Such factors lead to reduced sensitivity, false negative results and higher limits of detection (LOD) (Touron‐Bodilis et al., 2011). In most protocols not only cellular but also free and dead cell DNA is amplified, which leads to false‐positive amplification (Fields et al., 2002) and makes interpretation of data difficult (Krojgaard et al., 2011).
Faster methods for Lp‐detection based on immunofluorescent microscopy (IFM) were reported with variable recoveries of 25–75% (Edelstein, 1987; Palmer et al., 1993), where also the successful application of viability stains was demonstrated (Delgado‐Viscogliosi et al., 2005). Also, several studies employing fluorescent in situ hybridization (FISH) to DNA, mRNA or rRNA (Buchbinder et al., 2002; Declerck et al., 2003; Dutil et al., 2006; Ditommaso et al., 2010; Whiley et al., 2011) demonstrated the successful detection of total or viable Lp in environmental samples. However, low signal intensity, long overnight incubations (Hambsch et al., 2010) and low photostability of the dyes were reported (Declerck et al., 2003), leading to false negative results and, hence, only qualitative interpretation of results (Ditommaso et al., 2010). Despite recent advances in instrumentation and image analysis technology, all microscopy‐based methods are tedious, time‐consuming (Hambsch et al., 2010), require highly skilled laboratory personnel, and are known to generate variable results due to, e.g., viewer fatigue (Garcia and Shimizu, 1997). Consequently, the potential of these methods for routine use is considerably reduced.
Solid phase cytometry‐based assays address these issues by automated counting of stained cells collected on membrane filters (Aurell et al., 2004; Parthuisot et al., 2011). However, this technique is hampered by low sensitivity (Hambsch et al., 2010) and low throughput of a maximum of 15–20 samples per day (Aurell et al., 2004) due to the time‐consuming and often difficult manual validation of potentially identified targets (Aurell et al., 2004; Vanhee et al., 2009). Also the scanning procedure may fail completely due to low signal intensity (Hambsch et al., 2010). Furthermore, it was shown that the obscuration of organisms on a filter by debris and other particles may lead to false negative results (Keserue et al., 2011).
Flow cytometry (FCM) was already used 30 years ago for Lp detection. These first approaches, however, had very high LODs of 104 to 106 cells per litre (Tyndall et al., 1985; Lebaron et al., 1998). More recently, FCM‐based approaches reached detection limits as low as 500 cells per litre, but high background signals, not well discriminated cell clusters, high counts of false‐positives and lacking information about physiological state are some of the drawbacks of these methods (Füchslin et al., 2010; Taguri et al., 2011). Recently, the usefulness of propidium iodide (PI)‐based discrimination of live and dead Lp cells was demonstrated in combination with FCM (Allegra et al., 2008).
In this study, we developed an improved FCM‐based method to detect and quantify total and viable Lp cells from tap water samples. It includes initial concentrating steps by filtration, re‐suspension, followed by immunomagnetic separation (IMS) of Lp cells to purify the samples. Detection by FCM is based on two fluorescent parameters, i.e. green and blue fluorescence (Fig. 1), coupled to two different polyclonal antibodies and thus minimizing the risk for cross‐reactions. Membrane‐damaged and thus potentially dead cells are discriminated by an additional PI‐staining and the time to result is only around two hours.
Figure 1.
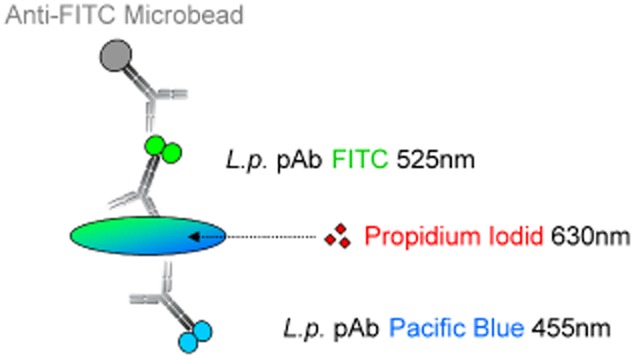
Labelling concept for Legionella pneumophila (Lp) serogroups 1–12. Lp cells are labelled with two polyclonal antibodies targeting Lp serogroups 1–12 conjugated to two different dyes and excited by two different lasers (405 and 488 nm). For the capture of cells by magnetic separation, a super paramagnetic particle is bound to the FITC molecule through a FITC‐specific antibody. Propidium iodide is employed for discrimination of membrane‐damaged cells.
Results
Spiking experiments
Since we demonstrated previously that Lp quantification by FCM and IFM agree very well (Füchslin et al., 2010), the spiking counts were determined by FCM. When applying our gating approach for spiked tap water samples it was possible to discriminate Lp cells very well from background signals in the main fluorescence dot plots, i.e., green versus blue fluorescence; the negative controls contained very few background noise events (Fig. 2A and B, and supporting data in Figs S1 and S2). Background noise events (false positives) for total and viable Lp were 26.0 ± 8.0 and 8.0 ± 2.5 events l−1 (n = 15), respectively.
Figure 2.

Examples for green versus blue fluorescence dot plots for different tap water samples. (A) shows quantification in spiked tap water and (B) is the corresponding negative control. (C–F) show different positive tap water samples with naturally occurring Lp cells. (D–F) indicate that environmental Lp cells may be smaller in some cases and display less fluorescence than spiked Lp cells grown in liquid culture. The complete FCM output for these examples can be found in Figs S1–S5, except for (C), which is from Fig. 6.
The average recovery from 1 l of spiked tap water for total and viable Lp was 85.4% ± 13.9% and 87.1% ± 8.4%, respectively (Fig. 3, Table S1). The two‐ sided Pearson r for both correlations is 0.9999. The method allowed to detect successfully cells at seeding levels of around 40 cells l−1. A simple approach to estimate the limit of detection (LOD) was applied and consisted of adding three times the blank (background noise) standard deviation to the very same value (MacDougall and Crummett, 1980; McNaught and Wilkinson, 1997), leading to a LOD of 49.88 and 15.41 cells l−1 for total and viable Lp respectively (P > 0.99). By doubling these values the determination limits (statistically safe qualitative determination) can be estimated to be around 100 and 30 cells l−1 and the quantification limits, calculated by tripling the LOD, were 150 and 45 cells l−1 for total and viable Lp (Deutsches Institut für Normung e.V., 2008). For simplicity and added statistical confidence we considered 50, 100 and 150 cells l−1 as detection, determination and quantification limit, respectively, for both viable and total Lp quantification. The standard deviations for the method above the quantification limit are on average 5.9% and 5.1% for total and viable Lp, respectively (Table S1).
Figure 3.
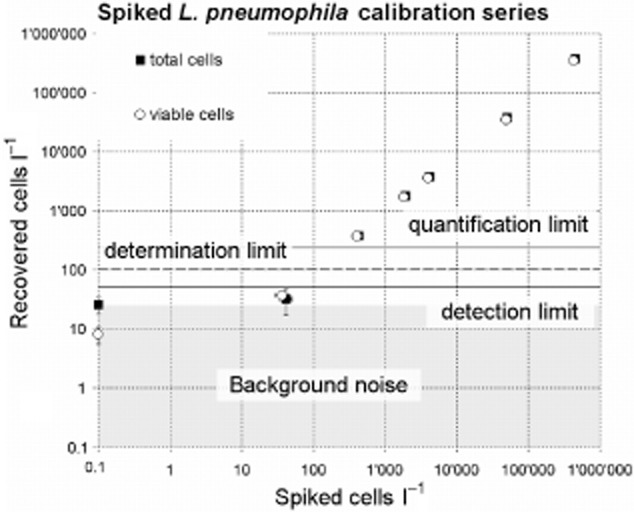
Calibration series of Lp spiked into 1 l tap water. Circles represent the viable Lp cells and squares the total Lp cells. Horizontal solid bold line, dotted line and solid thin line represent the estimated detection, determination and quantification limits. The actual data of this calibration series are presented in Table S1.
Detection of Lp in tap water samples
When detecting environmental Lp in water samples of contaminated distribution systems (Fig. 2C–F, Figs S3–S5), it was possible to discriminate well the Lp cluster from background signals. In contrast to the spiking experiments, strong background signals were often observed and environmental Lp cells sometimes appeared to be smaller than cultured ones, thus displaying less fluorescence intensity (Fig. 2D and E) than the latter.
When cells were heat‐killed at 90°C for 1 min, a shift of the Lp cells over the threshold level for red fluorescence was observed (Fig. S4). This is due to the properties of the PI dye that can enter only cells with a damaged membrane resulting in an elevated red fluorescence signal.
Comparison of Lp detection by FCM versus plating
For comparison, a total of 85 drinking water samples from different taps and showers were analysed by both plating and FCM. Laboratories 1 and 2 provided 45 samples and applied the ISO 11731 method (ISO, 2004), whereas Laboratory 3 provided 40 samples and did plate 1 ml of sample only. An overview of the results obtained is shown in Fig. 4.
Figure 4.
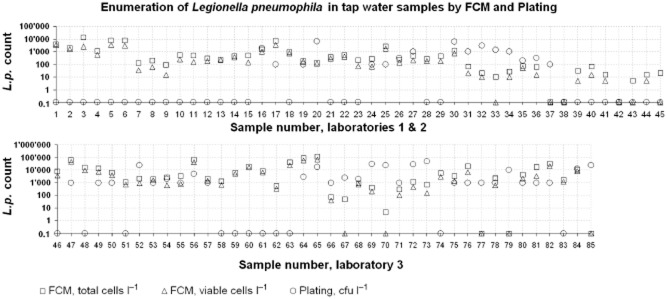
Comparison of tap water samples with naturally occurring Lp cells measured by plating and FCM. Laboratory 1 and 2 performed the complete ISO 11731 method whereas Laboratory 3 plated 1 ml only. Around 50% of the samples have significantly (> 0.5 log) higher total and viable FCM Lp counts than the plating.
As expected the FCM total Lp count was higher than that for viable Lp for each sample and usually both values were above the number of Lp colony‐forming units. No correlation was observed between plating and FCM (Fig. S7).
Positive or negative agreement of FCM and plating results were defined to be below or above 1000 counts l−1, respectively, since this value is recommended in Switzerland and other countries as a threshold level for initiating measures against Lp (BAG, 2009). Using this threshold, an agreement of 62% and 60% was obtained for total and viable Lp cells, respectively (Table 1). The Cohen's κ‐values were in the 0.2 range, indicating poor strength of agreement. Since both methods, FCM and plating, display false‐negative results, and high deviations of results obtained by plating are known from interlaboratory comparisons (Lucas et al., 2011), it seems not appropriate to consider plating as the reference method. Thus using this value as the ‘real’ Lp result for calculating specificity and sensitivity for the FCM approach would not lead to a correct assessment of this method.
Table 1.
Comparison of positive and negative results for FCM and plating of Lp.
| Culture | FCM total Lp | FCM viable Lp | ||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| Positive | 22 | 15 | 17 | 20 |
| Negative | 17 | 31 | 14 | 34 |
| Agreement | 62% | 60% | ||
| Cohen's κ | 0.239 ± 0.106 | 0.171 ± 0.107 | ||
The threshold discriminating between positive and negative results is 1000 counts per litre. This is the level of contamination recommended for intervention in several countries.
Quantification agreement
For comparing the quantification we took into consideration the FCM determination limit, the high plating deviations and detection limit. Thus, we defined the difference (Δ) of the plating and FCM results to be of significance if Δ > 0.5 log10 and Δ > 100 counts l−1, for laboratories 1 and 2. For laboratory 3 the definition for a significant difference of the result is Δ > 0.5 log10 and Δ > 1000 counts l−1 since this laboratory plated only 1 ml of sample and thus the detection limit was 1000 cells per litre. Applying this definition, plating gave significantly lower plate counts than the viable FCM Lp count for 22 (49%) of the samples from laboratory 1 and laboratory 2. For 8 (18%) samples of these laboratories the plating result was significantly higher. For laboratory 3 this ratio was 23 (58%) for significantly higher FCM and 10 (25%) for higher plate count results, respectively. Since the results (FCM/Plating ratio) of the samples from the three laboratories did not significantly differ (tested was lab 1 + 2 versus 3, two‐tailed; Mann–Whitney test: P = 0.31; independent samples Student's test: P = 0.16), they were combined in Table 2.
Table 2.
Agreement of quantification for Lp plating and FCM detection
| Plating versus | FCM versus plating quantification | |||
|---|---|---|---|---|
| FCM higher | Plate higher | Positive agreement | Negative agreement | |
| FCM total Lp | 45 | 16 | 15 | 9 |
| % | 53% | 19% | 18% | 11% |
| FCM viable Lp | 40 | 18 | 15 | 12 |
| % | 47% | 21% | 18% | 14% |
Quantification with a difference of > 0.5 log and > 100 counts for laboratories 1 and 2 and > 0.5 log and > 1000 counts for laboratory 3 is considered to not agree. These conventions were chosen since the plating volatility is generally very high and for results from laboratory 3 the detection limit is 1000 cfu l−1.
Statistical comparison of the methods
An overview of the total cell count (TCC) and the viable cell count (VCC) obtained by FCM for all samples is shown in Fig. 5. They were statistically compared with results of plating and of FCM detecting total and viable Lp (Table 3). As expected, viable and total cells in general, and total and viable Lp correlated very well. Furthermore, we encountered some correlation of TCC and VCC versus Lp plating (Pearson r = 0.425 and r = 0.357, respectively) (Table 3).
Figure 5.
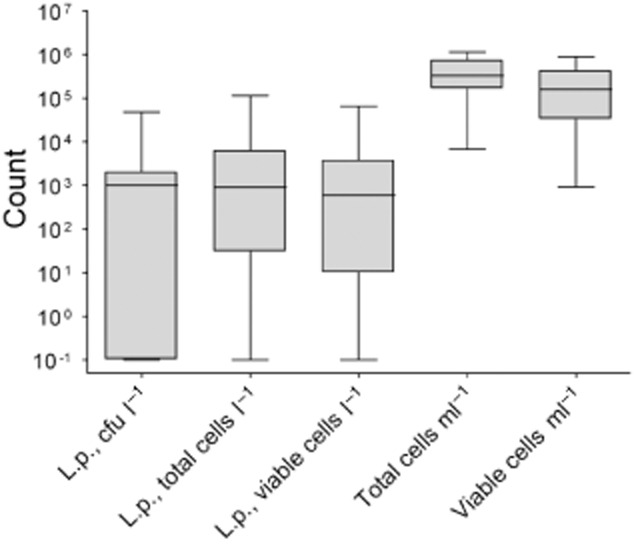
Box plots of the obtained concentrations for Lp detection by plating and FCM and total (TCC) and viable cell count (VCC). The box represents the first, second (median) and third quartiles, the whiskers represent the maximum and minimum values.
Table 3.
Pearson correlation matrix for all parameters measured
| Parameter | Lp, cfu l−1 | Lp, cells l−1 | Lp, viable cells l−1 | TCC per millilitre | ||||
|---|---|---|---|---|---|---|---|---|
| r | P | r | P | r | P | r | P | |
| Lp, cells l−1 | 0.052 | 0.693 | ||||||
| Lp, viable cells l−1 | 0.045 | 0.737 | 0.991 | < 0.001 | ||||
| TCC per millilitre | 0.425 | 0.001 | 0.007 | 0.955 | < 0.001 | 0.998 | ||
| VCC per millilitre | 0.357 | 0.005 | 0.094 | 0.480 | 0.070 | 0.596 | 0.905 | < 0.001 |
| Kolmogorov–Smirnova | < 0.001 | < 0.001 | < 0.001 | < 0.001 | ||||
| Shapiro–Wilk | < 0.001 | < 0.001 | < 0.001 | < 0.001 | ||||
All P‐values are two tailed.
Lilliefors significance correction.
Bold: correlation is significant at the < 0.01 level.
When comparing the values of Lp plating and FCM versus TCC and VCC, it appeared that most of the higher and alarming values (> 1000 Lp counts l−1) were found when TCC values were in the range from 108 to 109 cells l−1 (Fig. S8).
We compared the viable fraction of Lp and total bacteria and found the mean viable fraction of bacterial cells to be very similar, namely 57.3 ± 20.3% of the Lp and 56.8 ± 14.8% of the total bacteria were viable according to membrane integrity. The linearization function (y = −0.2255x + 69.699) displayed a poor fit and a negative slope (R2 = 0.096, Fig. S9) indicating a slightly negative correlation of these two parameters.
Discussion
We recently reported the suitability and good performance of similar IMS approaches for separation of Giardia and Cryptosporidium from tap and surface waters (Keserue et al., 2011; 2012) and separation of Lp from buffer (Füchslin et al., 2010). In this study we could greatly enhance the performance of the detection compared with the latter study. Accordingly, the presented method performed excellently in spiking experiments with tap water. Since our FCM procedure involves the combination of different parameters, a positive signal has to fulfil many conditions and thus, many background signals are eliminated and a high noise reduction is achieved. For both, total and viable Lp counts, recoveries exceeded on average 80% with standard deviations of around 10% and a low number of false positives. This shows that only a very small fraction of Lp cells possibly became damaged at the membrane level during our procedure and, therefore, this effect can be neglected for interpreting the results.
For about half of the tap water samples the counts measured by the plate culture method were significantly lower (Δ > 0.5 log) than FCM quantification. There are a number of reasonable explanations why the plating is in most cases lower than the FCM measurement. The typical high plating volatilities mentioned above might be one reason for this observation. In a proficiency testing scheme for US laboratories that culture Ls from tap water samples the cfu ml−1 was underestimated by 1.25 ± 0.78 logs on average (Lucas et al., 2011). Another important reason why cultivation methods may underestimate bacterial numbers is that ‘viable but not culturable’ (VNBC) state cells cannot be quantified by plating. It was shown that non‐culturable Ls cells can be detected by FCM and resuscitated by co‐culture with amoebae (Allegra et al., 2008). Furthermore, Allegra and colleagues demonstrated the development of resistance of Lp to high temperatures and the persistence of VNBC cells to a 30 min heat treatment at 70°C (Allegra et al., 2011). In addition, the used antibodies were not extensively tested against similar species and, therefore, cross‐reactions cannot be excluded. It was also reported decades ago that only a limited number of polyclonal antibodies to Lp is available and that most of them have not been tested vigorously for their sensitivity and specificity (Orrison et al., 1983); we are not aware that this has changed substantially since then. As we used two different antibodies and the binding of antibodies to a non‐target cell would need to be similarly strong and the distribution would have to resemble that for Lp in order to be quantified by our gating regime, we doubt that cross‐reactions did impair our overall measurement. Of course, this highly selective gating strategy bears the risk of missing some true positive cells; however, the spiking experiments revealed a high recovery, hence, this effect cannot be very pronounced.
A number of reasons can account for higher GVPC plate counts compared with FCM (∼ 20% of cases); these include: (i) the used antibodies bind only to Lp Sg 1–12, thus, if Lp belonging to the remaining serogroups (Lp Sg 13–16) were present they would not have been quantified. Since the commercial serotyping agglutination kits discriminate only between Lp 1, Lp 2–15 and Ls it was not possible to check for the presence of these serogroups. In South Korea, these rare serogroups were identified from water samples in 1.4–2.6% of cases (Lee et al., 2010). (ii) Similar colonies of other species may be quantified as Lp in the plating evaluation as not every colony is subjected to the agglutination tests. (iii) Also the before mentioned arithmetical inconsistency of plating different volumes may lead to overestimation, notably when plating only 1 ml (Schaefer, 2007). (iv) In samples with high iron oxide content, which is occasionally visible by a colouring of the filter membrane, an increased SCC signal of the target cells may result that might shift these signals outside the gating region and, thus, lead to false‐negative results. An acid treatment or magnetic separation before filtration might help to avoid this. Alternatively, a simpler gating strategy without using the SCC signal could circumvent this difficulty. (v) Free‐living amoebae (FLA) are abundant in drinking water systems and since Lp is an amoeba‐resisting microorganism (ARM) (Thomas and Ashbolt, 2010), Lp can replicate within the FLA and subsequently return into the water. Hsu and colleagues found 26.6% of Lp positive samples to contain FLA‐intracellular Lp in spring water (Hsu et al., 2011). Thus, the presence of Legionella‐containing amoebae, which will neither be destroyed nor quantified by the FCM assay, but may very well be detected by the plating approach, may lead to an underestimation of Lp cells by FCM.
We did not find a correlation of FCM Lp versus plating or TCC and VCC. Surprisingly though, we observed a weak correlation between TCC and VCC versus the LP plating.
Only very rarely we found plating and FCM Lp numbers to be above 1000 counts l−1 with TCC and VCC values up to 108 cells l−1. This may point to a need for future research to analyse correlation of these parameters and to estimate Lp growth potential relative to the total bacterial numbers (Fig. S8).
Furthermore, the results show that Lp viability does not correlate with total cell viability and thus Lp may profit from higher levels of other dead cells (Fig. S9). Results pointing towards necrotrophic growth were observed before (Temmerman et al., 2006).
The few results presented here already illustrate the great potential of our method to investigate and understand the behaviour of this pathogen in its environmental and man‐made habitats. A further optimization of the described method could help to overcome some of its present weaknesses. An improved method should include the possibility to destroy FLA and release the Lp cells present in these protozoa; it also should be based upon specific antibodies targeting the appropriate species and serogroups and should not be dependent on the SSC. The limitations of the antibodies caused by the limited range of serogroups may be overcome by using a pool of antibodies, e.g., either covering all Lp serogroups, or even all Ls. In the future antibody alternatives may provide improved binding properties and cost reduction (Stumpp and Amstutz, 2007; Chung et al., 2011).
The method developed and tested in the present study might not yet be fit to completely displace the plating methods used in laboratories for routine water analysis. However, it allows to quantify cultivable and VBNC state cells, thus offers the possibility to describe disinfection measures, to study physiological different viability states during disinfection processes, and to follow the survival and growth behaviour of Lp in water supply systems in more detail. This justifies studies for further improvement, which may finally lead to acceptance of this new approach to detecting and quantifying Legionella in water. When all the current drawbacks can be overcome, a fast, cost‐effective, potentially automatable and high throughput method will be available for routine application.
Experimental procedures
Legionella pneumophila cultivation
Legionella pneumophila SG1 (ATCC 33152, Oxoid, Hampshire, UK) was cultivated on buffered charcoal yeast extract (BCYE) agar plates (obtained ready‐poured from Oxoid, Pratteln, Switzerland) at 37°C. After at least 5 days of incubation, cells from one single colony were inoculated into 50 ml of a BCYE broth (10 g l−1 of yeast extract and 0.2 g l−1 activated charcoal (one tenth of the original concentration; Feeley et al., 1979; Edelstein, 1982) with one tenth of the original charcoal concentration of 0.2 g l−1 and BCYE growth supplement (Oxoid). The batch culture was incubated on a shaker at 150 r.p.m. for at least 3 days at 37°C. Then 100 μl of culture was transferred into 0.9 ml of sterile filtered phosphate‐buffered saline (PBS, 9.0 g l−1 NaCl, 1.62 g l−1 Na2HPO4·2H2O, 0.36 g l−1 KH2PO4, pH 7.2) and diluted as required for spiking experiments with the same buffer.
Spiking experiments
Tap water samples of 1 l used for spiking experiments were collected in 1 l screw‐cap glass bottles from a well‐frequented tap in the laboratory of the Federal Office of Public Health (FOPH, Bern, Liebefeld) having an outlet temperature above 55°C. Under these conditions, we found the samples to be virtually Ls‐free with both the plating (< 10 Ls l−1 with GVPC; BCYE with glycine, vancomycin, polymixin and cyclohexamide selective medium), and the FCM detection method. After the water temperature had dropped below 30°C, the water samples were spiked with 100 μl of PBS containing a defined amount of cells determined by FCM (see below).
Pre‐treatment of samples for method comparison experiments
Samples for Ls detection were obtained from three different routine laboratories that are regularly testing water samples for clients maintaining large inhabited facilities. Samples were primarily obtained from buildings where Lp were recently found, in order to obtain a larger number of presumably positive results.
Laboratory 1 performed the standard ISO 11731 (ISO, 2004) method. Samples were collected in sterile 2 l screw‐cap bottles and mixed after sampling. After the laboratory had withdrawn 1 l of the sample, the remaining litre was stored at 4°C and transported to the FOPH and FCM analysis was performed within 4–24 h after sampling.
Laboratory 2 performed the standard ISO 11731 method as well. Samples were collected in sterile 2 l screw‐cap bottles and mixed after sampling. After the laboratory had withdrawn 1 l for the ISO method, the remaining litre was filtered through a hydrophilic 0.22 μm polycarbonate track‐etch‐membrane filter (PCTE, Sterlitech Corporation, Kent, WA, USA) and suspended in 15 ml centrifugation tubes in 4–5 ml of PBST (PBS + 0.05% Tween 20; Fluka). These tubes were then sent cooled to the FOPH. FCM analysis was thus performed 24–48 h after sampling.
Laboratory 3 performed a shortened adaptation of ISO 11731, where simply 1 ml of sample is split into two 0.5 ml aliquots and plated directly onto two GVPC plates. The detection limit of this approach is 1000 cfu l−1. Samples were collected in sterile 1 l screw‐cap bottles and mixed after sampling. The remaining samples were transferred uncooled to the FOPH and further processed 6–12 h after sampling.
All laboratories confirmed up to 5 Lp colonies per plate by latex agglutination according to ISO 11731.
Filtration
After pre‐filtration of the water samples through a 30 μm nylon mesh filter (Millipore AG, Zug, Switzerland), water samples were subsequently vacuum‐filtered through a hydrophilic 0.22 μm pore size PCTE filter (Sterlitech). The filter was then carefully removed and re‐suspended in a 50 ml centrifugation tube, containing 3 ml of sterile PBST and 10 μl 15% BSA (bovine serum albumin, Sigma, Steinheim, Gemany) by strong vortexing.
Bacterial cell staining
The total flow cytometric bacterial cell concentration (TCC) and total number of Lp in pure cultures was determined as described by Hammes and colleagues (2008). Briefly, 1 ml of sample was stained with 10 μl SYBR Green I working solution (×10 000 stock solution diluted 100‐times in DMSO, Invitrogen, Zug, Switzerland) and incubated in the dark at 30°C for 15 min.
For the viable flow cytometric cell concentration (VCC) we added to 1 ml of sample 10 μl of PI solution (30 mM) dissolved in DMSO (Invitrogen) together with the SYBR Green as for the TCC (Berney et al., 2008).
Specific Lp antibody‐staining
To the re‐suspended sample (3 ml; 4–5 ml for laboratory 2) we added 1.2 μg of fluorescein isothiocyanate (FITC) conjugated polyclonal IgG rabbit antibodies (Cat. No. ab20818, Abcam, Cambridge, UK). After incubation for 20 min at ambient temperature in the dark, 10 μl of superparamagnetic anti‐fluorescein isothiocyanate (FITC) Microbeads (Miltenyi Biotech GmbH, Bergisch Gladbach, Germany) and 2 μg of anti‐Legionella pneumophila rabbit polyclonal antibody (Cat. No. GTX40943, Genetex, Irvine, CA, USA) conjugated to Pacific Blue was added. The conjugation of the antibody was performed using a Pacific Blue protein labelling kit according to the recommendations of the manufacturer (Cat. No. P30012, Molecular Probes, Basel, Switzerland). The assay was then incubated for 40 min at 4°C.
For the positive controls, cells spiked into PBS were stained in the same way and then counted by FCM as described later. This approach has shown excellent correlation and agreement to IFM and is, therefore, appropriate for preparation of the seeding aliquots (Füchslin et al., 2010).
Immunomagnetic separation (IMS)
Immunomagnetic separation using MACS™ MS columns (Miltenyi Biotech) was conducted according to the recommendations of the manufacturer. The whole sample (3–5 ml) was run through the column after placing the column in the separator magnet. Subsequently, the column was rinsed in succession with 3 ml of PBS, 3 ml of PBST and 3 ml PBS. Recovery of the collected cells was conducted after removal of the column from the magnet by flushing 1 ml PBS through the column into a 15 ml Falcon tube by firmly applying the provided plunger.
FCM detection
Flow cytometric detection was performed with a Partec CyFlow ML flow cytometer (Partec GmbH, Münster, Germany) equipped with a 200 mW blue solid‐state laser emitting light at 488 nm and a 110 mW violet diode‐laser emitting at 405 nm. The volumetric counting properties of the flow cytometer measure the events in 200 μl of sample volume and extrapolate the results to 1 ml of sample volume. Optical filters were adjusted to measure green fluorescence at 520 nm, red fluorescence at 630 nm and the sideward scatter (SSC) at 488 nm emitted from spot one, i.e., excited by the blue laser. For the second spot, excited by the violet laser, the optical filters were set to measure the sideward scatter at 405 nm and the blue fluorescence at 460 nm. The trigger was set on green fluorescence. Events were defined based on sideward scatters (488 and 405 nm), fluorescence emitted at 455 nm and 520 nm. The 630 nm fluorescence signal was used to discriminate PI‐positive and PI‐negative cells.
A typical result obtained with environmental Lp cells in tap water is shown in Fig. 2. Results were acquired by plotting green fluorescence against both sideward scatter (Fig. 6A and B), blue fluorescence against sideward scatter (Fig. 6D and E), blue versus red fluorescence (Fig. 6F), and green versus blue fluorescence (Fig. 6C). The Lp clusters were gated in Fig. 6A, B, D and E by gates R1–4 and combined to a multigate G1, requiring a valid event to lie inside all 4 gates. In order to discriminate viable from dead cells the gate R5 was set in Fig. 6F as a red fluorescence threshold for PI‐negative cells. A box plot and polygonal gate was chosen for better visualization as compared with a histogram with a range gate. Thus, for viable Lp quantification, gate G1 and R5 were combined as Gate G2.
Figure 6.
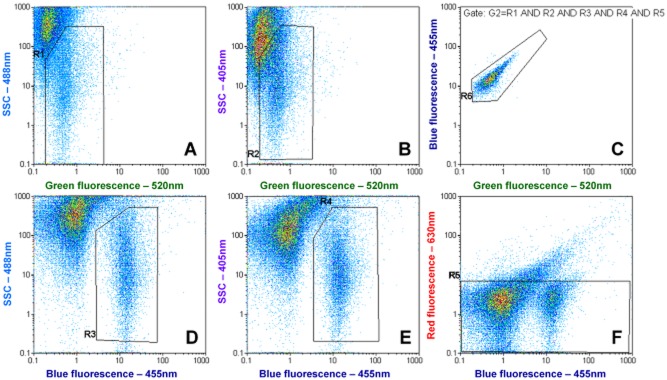
Typical example of the FCM dot plots for environmental detection of Lp cells in tap water. (A, B, D and E) show the main gates that are applied in combination to (C) to reduce background signals. (F) represents the membrane integrity discrimination approach; since membrane‐damaged Lp cells display a higher red fluorescence due to propidium iodide they move outside the gate and can be discriminated (Fig. S6). The counting is performed in (C).
For counting, gate R6 was then set around the Lp cluster on the main fluorescent parameters box plot, green versus blue fluorescence (Fig. 6C) and the gate G1, for total Lp or the gate G2 for viable Lp was applied.
The specific instrumental gain settings used for 520 nm, 455 nm, 630 nm, SSC 488 nm and SSC 405 nm channels were 300, 440, 500, 180 and 500, respectively. The speed rate was 3, implying a counting rate of less than 1000 events s−1.
The TCC and VCC were measured and gated as described previously (Berney et al., 2008; Hammes et al., 2008).
Acknowledgments
This work was supported by research grants of the Federal Office of Public Health (grant number FOPH 08.000086) and EAWAG. Thanks go to Dr A. Auckenthaler for initiating this study. We thank the laboratories participating in this study for their effort. Especially, we want to thank Dr Ralf Maibusch (Stadtlabor Bern), Dr Paul Svoboda and Dr Jill Engelmann (Kantonales Laboratorium Basel‐Land, Liestal), Frau Mathilde Mauerhofer (Bactlab AG, Bern‐Liebefeld), and Dr Guido Vogel and Dr Valentin Pflüger (Mabritec AG, Riehen).
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. FCM dot plots; complete view of Fig. 2A.
Fig. S2. FCM dot plots; complete view of Fig. 2B.
Fig. S3. FCM dot plots; complete view of Fig. 2D.
Fig. S4. FCM dot plots; complete view of Fig. 2E.
Fig. S5. FCM dot plots; complete view of Fig. 2F.
Fig. S6. Example dot plot for increase of red fluorescence in membrane‐damaged Lp cells.
Fig. S7. Correlation of Lp plating vs. flow cytometry for total and viable Lp.
Fig. S8. Correlations of FCM and plating Lp detection vs. total and viable cell count.
Fig. S9. Linear correlation of viable fraction of Lp and total cells measured with FCM.
Complete calibration data set for spiking experiments.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Allegra S., Berger F., Berthelot P., Grattard F., Pozzetto B., Riffard S. Use of flow cytometry to monitor Legionella viability. Appl Environ Microbiol. 2008;74:7813–7816. doi: 10.1128/AEM.01364-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegra S., Grattard F., Girardot F., Riffard S., Pozzetto B., Berthelot P. Longitudinal evaluation of the efficacy of heat treatment procedures against Legionella spp. in hospital water systems by using a flow cytometric assay. Appl Environ Microbiol. 2011;77:1268–1275. doi: 10.1128/AEM.02225-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alli O.A.T., von Zink S., Lackum N.K., Abu‐Kwaik Y. Comparative assessment of virulence traits in Legionella spp. Microbiology. 2003;149:631–641. doi: 10.1099/mic.0.25980-0. [DOI] [PubMed] [Google Scholar]
- Aurell H., Catala P., Farge P., Wallet F., Le Brun M., Helbig J.H. Rapid detection and enumeration of Legionella pneumophila in hot water systems by solid‐phase cytometry. Appl Environ Microbiol. 2004;70:1651–1657. doi: 10.1128/AEM.70.3.1651-1657.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAG. Bern, Switzerland: Bundesamt für Gesundheit; 2009. [Google Scholar]
- BAG. Bern, Switzerland: Bundesamt für Gesundheit; 2012. [Google Scholar]
- Bentham R.H. Routine sampling and the control of Legionella spp. in cooling tower water systems. Curr Microbiol. 2000;41:271–275. doi: 10.1007/s002840010133. [DOI] [PubMed] [Google Scholar]
- Berney M., Vital M., Hülshoff I., Weilenmann H.‐U., Egli T., Hammes F. Rapid, cultivation‐independent assessment of microbial viability in drinking water. Water Res. 2008;42:4010–4018. doi: 10.1016/j.watres.2008.07.017. [DOI] [PubMed] [Google Scholar]
- Boulanger C.A., Edelstein P.H. Precision and accuracy of recovery of Legionella pneumophila from seeded tap water by filtration and centrifugation. Appl Environ Microbiol. 1995;61:1805–1809. doi: 10.1128/aem.61.5.1805-1809.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman R.F., Butler J.C. Legionnaires' disease: clinical, epidemiological, and public health perspectives. Semin Respir Infect. 1998;13:84–89. [PubMed] [Google Scholar]
- Buchbinder S., Trebesius K., Heesemann J. Evaluation of detection of Legionella spp. in water samples by fluorescence in situ hybridization, PCR amplification and bacterial culture. Int J Med Microbiol. 2002;292:241–245. doi: 10.1078/1438-4221-00213. [DOI] [PubMed] [Google Scholar]
- Chung H.J., Reiner T., Budin G., Min C., Liong M., Issadore D. Ubiquitous detection of Gram‐positive bacteria with bioorthogonal magnetofluorescent nanoparticles. ACS Nano. 2011;5:8834–8841. doi: 10.1021/nn2029692. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declerck P., Verelst L., Duvivier L., Van Damme A., Ollevier F. A detection method for Legionella spp. in (cooling) water: fluorescent in situ hybridisation (FISH) on whole bacteria. Water Sci Technol. 2003;47:143–146. [PubMed] [Google Scholar]
- Delgado‐Viscogliosi P., Simonart T., Parent V., Marchand G., Dobbelaere M., Pierlot E. Rapid method for enumeration of viable Legionella pneumophila and other Legionella spp. in water. Appl Environ Microbiol. 2005;71:4086–4096. doi: 10.1128/AEM.71.7.4086-4096.2005. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsches Institut für Normung e.V. Berlin, Germany: DIN; 2008. [Google Scholar]
- Ditommaso S., Giacomuzzi M., Gentile M., Zotti C.M. Evaluation of the usefulness of a new direct immunofluorescence assay (ScanVIT‐Legionella) for monitoring hospital water systems contaminated with Legionella spp. Lett Appl Microbiol. 2010;50:341–346. doi: 10.1111/j.1472-765X.2010.02797.x. [DOI] [PubMed] [Google Scholar]
- Dufour A., Snozzi M., Köster W., editors. 1st edn. London, UK: International Water Association Publishing; 2003. [Google Scholar]
- Dutil S., Tessier S., Veillette M., Laflamme C., Mériaux A., Leduc A. Detection of Legionella spp. by fluorescent in situ hybridization in dental unit waterlines. J Appl Microbiol. 2006;100:955–963. doi: 10.1111/j.1365-2672.2006.02845.x. et al. [DOI] [PubMed] [Google Scholar]
- Edelstein P.H. Comparative study of selective media for isolation of Legionella pneumophila from potable water. J Clin Microbiol. 1982;16:697–699. doi: 10.1128/jcm.16.4.697-699.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein P.H. The laboratory diagnosis of Legionnaires' disease. Semin Respir Infect. 1987;2:235–241. [PubMed] [Google Scholar]
- Feeley J.C., Gibson R.J., Gorman G.W., Langford N.C., Rasheed J.K., Mackel D.C., Baine W.B. Charcoal‐yeast extract agar: primary isolation medium for Legionella pneumophila. J Clin Microbiol. 1979;10:437–441. doi: 10.1128/jcm.10.4.437-441.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields B.S. The molecular ecology of legionellae. Trends Microbiol. 1996;4:286–290. doi: 10.1016/0966-842x(96)10041-x. [DOI] [PubMed] [Google Scholar]
- Fields B.S., Benson R.F., Besser R.E. Legionella and Legionnaires' disease: 25 years of investigation. Clin Microbiol Rev. 2002;15:506–526. doi: 10.1128/CMR.15.3.506-526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliermans C.B., Cherry W.B., Orrison L.H., Smith S.J., Tison D.L., Pope D.H. Ecological distribution of Legionella pneumophila. Appl Environ Microbiol. 1981;41:9–16. doi: 10.1128/aem.41.1.9-16.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füchslin H.P., Kötzsch S., Keserue H.‐A., Egli T. Rapid and quantitative detection of Legionella pneumophila applying immunomagnetic separation and flow cytometry. Cytometry. 2010;77A:264–274. doi: 10.1002/cyto.a.20858. [DOI] [PubMed] [Google Scholar]
- Gao L.‐Y., Stone B.J., Brieland J.K., Kwaik Y.A. Different fates of Legionella pneumophila pmi and mil mutants within macrophages and alveolar epithelial cells. Microb Pathog. 1998;25:291–306. doi: 10.1006/mpat.1998.0237. [DOI] [PubMed] [Google Scholar]
- Garcia L.S., Shimizu R.Y. Evaluation of nine immunoassay kits (enzyme immunoassay and direct fluorescence) for detection of Giardia lamblia and Cryptosporidium parvum in human fecal specimens. J Clin Microbiol. 1997;35:1526–1529. doi: 10.1128/jcm.35.6.1526-1529.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambsch B., Hügler M., Eberhagen I., Beimfohr C., Thelen K. 2010. , and ) Report on automated quantification of FISH‐labelled bacteria. Techneau. [WWW document]. URL http://www.techneau.org/fileadmin/files/Publications/Publications/Deliverables/D3.4.17.pdf. [DOI] [PubMed]
- Hammes F., Berney M., Wang Y., Vital M., Köster O., Egli T. Flow‐cytometric total bacterial cell counts as a descriptive microbiological parameter for drinking water treatment processes. Water Res. 2008;42:269–277. doi: 10.1016/j.watres.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Hsu B.‐M., Huang C.‐C., Chen J.‐S., Chen N.‐H., Huang J.‐T. Comparison of potentially pathogenic free‐living amoeba hosts by Legionella spp. in substrate‐associated biofilms and floating biofilms from spring environments. Water Res. 2011;45:5171–5183. doi: 10.1016/j.watres.2011.07.019. [DOI] [PubMed] [Google Scholar]
- ISO. Geneva, Switzerland: International Organization for Standardization; 2004. ) ISO 11731. Water Quality Detection and Enumeration of Legionella. [Google Scholar]
- Keserue H.‐A., Füchslin H.P., Egli T. Rapid detection and enumeration of Giardia lamblia cysts in water samples by immunomagnetic separation and flow cytometric analysis. Appl Environ Microbiol. 2011;77:5420–5427. doi: 10.1128/AEM.00416-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keserue H.‐A., Füchslin H.P., Wittwer M., Nguyen‐Viet H., Nguyen T.T., Surinkul N. Comparison of rapid methods for detection of Giardia spp. and Cryptosporidium spp. (oo)cysts using transportable instrumentation in a field deployment. Env Sci Technol. 2012;46:8952–8959. doi: 10.1021/es301974m. et al. [DOI] [PubMed] [Google Scholar]
- Krojgaard L.H., Krogfelt K.A., Albrechtsen H.‐J., Uldum S.A. Detection of Legionella by quantitative‐polymerase chain reaction (qPCR) for monitoring and risk assessment. BMC Microbiol. 2011;11:254. doi: 10.1186/1471-2180-11-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebaron P., Parthuisot N., Catala P. Comparison of blue nucleic acid dyes for flow cytometric enumeration of bacteria in aquatic systems. Appl Environ Microbiol. 1998;64:1725–1730. doi: 10.1128/aem.64.5.1725-1730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.K., Shim J.I., Kim H.E., Yu J.Y., Kang Y.H. Distribution of Legionella species from environmental water sources of public facilities and genetic diversity of L. pneumophila serogroup 1 in South Korea. Appl Environ Microbiol. 2010;76:6547–6554. doi: 10.1128/AEM.00422-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C.E., Taylor T.H., Jr, Fields B.S. Accuracy and precision of Legionella isolation by US laboratories in the ELITE program pilot study. Water Res. 2011;45:4428–4436. doi: 10.1016/j.watres.2011.05.030. [DOI] [PubMed] [Google Scholar]
- MacDougall D., Crummett W.B. Guidelines for data acquisition and data quality evaluation in environmental chemistry. Anal Chem. 1980;52:2242–2249. [Google Scholar]
- McNaught A.D., Wilkinson A. Research Triangle Park, NC: IUPAC International Union of Pure and Applied Chemistry; 1997. [Google Scholar]
- Napoli C., Iatta R., Fasano F., Marsico T., Montagna M.T. Variable bacterial load of Legionella spp. in a hospital water system. Sci Total Environ. 2009;408:242–244. doi: 10.1016/j.scitotenv.2009.09.039. [DOI] [PubMed] [Google Scholar]
- Oliver J.D. Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol Rev. 2010;34:415–425. doi: 10.1111/j.1574-6976.2009.00200.x. [DOI] [PubMed] [Google Scholar]
- Orrison L.H., Bibb W.F., Cherry W.B., Thacker L. Determination of antigenic relationships among legionellae and non‐legionellae by direct fluorescent‐antibody and immunodiffusion tests. J Clin Microbiol. 1983;17:332–337. doi: 10.1128/jcm.17.2.332-337.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C.J., Tsai Y.L., Paszko‐Kolva C., Mayer C., Sangermano L.R. Detection of Legionella species in sewage and ocean water by polymerase chain reaction, direct fluorescent‐antibody, and plate culture methods. Appl Environ Microbiol. 1993;59:3618–3624. doi: 10.1128/aem.59.11.3618-3624.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palusińska‐Szysz M., Cendrowska‐Pinkosz M. Pathogenicity of the family Legionellaceae. Arch Immunol Ther Exp. 2009;57:279–290. doi: 10.1007/s00005-009-0035-8. [DOI] [PubMed] [Google Scholar]
- Parthuisot N., Binet M., Touron‐Bodilis A., Pougnard C., Lebaron P., Baudart J. Total and viable Legionella pneumophila cells in hot and natural waters as measured by immunofluorescence‐based assays and solid‐phase cytometry. Appl Environ Microbiol. 2011;77:6225–6232. doi: 10.1128/AEM.00393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravinkumar S.J., Edwards G., Lindsay D., Redmond S., Stirling J., House R. A cluster of Legionnaires' disease caused by Legionella longbeachae linked to potting compost in Scotland, 2008–2009. Euro Surveill. 2010;15:19496. doi: 10.2807/ese.15.08.19496-en. et al. [DOI] [PubMed] [Google Scholar]
- Schaefer B. Legionellenuntersuchung bei der Trinkwasseranalyse. Bundesgesundhbl Gesundheitsforsch Gesundheitsschutz. 2007;50:291–295. doi: 10.1007/s00103-007-0154-5. [DOI] [PubMed] [Google Scholar]
- Stumpp M.T., Amstutz P. DARPins: a true alternative to antibodies. Curr Opin Drug Discov Devel. 2007;10:153–159. [PubMed] [Google Scholar]
- Taguri T., Oda Y., Sugiyama K., Nishikawa T., Endo T., Izumiyama S. A rapid detection method using flow cytometry to monitor the risk of Legionella in bath water. J Microbiol Methods. 2011;86:25–32. doi: 10.1016/j.mimet.2011.03.012. et al. [DOI] [PubMed] [Google Scholar]
- Temmerman R., Vervaeren H., Noseda B., Boon N., Verstraete W. Necrotrophic growth of Legionella pneumophila. Appl Environ Microbiol. 2006;72:4323–4328. doi: 10.1128/AEM.00070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J.M., Ashbolt N.J. Do free‐living amoebae in treated drinking water systems present an emerging health risk? Environ Sci Technol. 2010;45:860–869. doi: 10.1021/es102876y. [DOI] [PubMed] [Google Scholar]
- Touron‐Bodilis A., Pougnard C., Frenkiel‐Lebossé H., Hallier‐Soulier S. Usefulness of real‐time PCR as a complementary tool to the monitoring of Legionella spp. and Legionella pneumophila by culture in industrial cooling systems. J Appl Microbiol. 2011;111:499–510. doi: 10.1111/j.1365-2672.2011.05063.x. [DOI] [PubMed] [Google Scholar]
- Tyndall R.L., Hand R.E., Jr, Mann R.C., Evans C., Jernigan R. Application of flow cytometry to detection and characterization of Legionella spp. Appl Environ Microbiol. 1985;49:852–857. doi: 10.1128/aem.49.4.852-857.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhee L.M.E., Nelis H.J., Coenye T. Detection and quantification of viable airborne bacteria and fungi using solid‐phase cytometry. Nat Protocols. 2009;4:224–231. doi: 10.1038/nprot.2008.228. [DOI] [PubMed] [Google Scholar]
- Whiley H., Taylor M., Bentham R.H. Detection of Legionella species in potting mixes using fluorescent in situ hybridisation (FISH) J Microbiol Methods. 2011;86:304–309. doi: 10.1016/j.mimet.2011.05.023. [DOI] [PubMed] [Google Scholar]
- Yáñez M.A., Carrasco‐Serrano C., Barberá V.M., Catalán V. Quantitative detection of Legionella pneumophila in water samples by immunomagnetic purification and real‐time PCR amplification of the dotA gene. Appl Environ Microbiol. 2005;71:3433–3441. doi: 10.1128/AEM.71.7.3433-3441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.L., Plouffe J.F., Pastoris M.C., Stout J.E., Schousboe M., Widmer A. Distribution of Legionella species and serogroups isolated by culture in patients with sporadic community‐acquired legionellosis: an international collaborative survey. J Infect Dis. 2002;186:127–128. doi: 10.1086/341087. et al. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. FCM dot plots; complete view of Fig. 2A.
Fig. S2. FCM dot plots; complete view of Fig. 2B.
Fig. S3. FCM dot plots; complete view of Fig. 2D.
Fig. S4. FCM dot plots; complete view of Fig. 2E.
Fig. S5. FCM dot plots; complete view of Fig. 2F.
Fig. S6. Example dot plot for increase of red fluorescence in membrane‐damaged Lp cells.
Fig. S7. Correlation of Lp plating vs. flow cytometry for total and viable Lp.
Fig. S8. Correlations of FCM and plating Lp detection vs. total and viable cell count.
Fig. S9. Linear correlation of viable fraction of Lp and total cells measured with FCM.
Complete calibration data set for spiking experiments.