

Abstract
A Type4b secretion system (T4bSS) is required for Legionella growth in alveolar macrophages. IcmQ associates with IcmR, binds to membranes and has a critical role in the T4bSS. We have now solved a crystal structure of IcmR-IcmQ to further our understanding of this complex. This structure revealed an amphipathic four-helix bundle, formed by IcmR and the N-terminal domain of IcmQ, which is linked to a novel C-terminal domain of IcmQ (Qc) by a linker helix. The Qc domain has structural homology with ADP ribosyltransferase domains in certain bacterial toxins and binds NAD+ with a Kd in the physiological range. Structural homology and molecular dynamics were used to identify an extended NAD+ binding site on Qc and the resulting model was tested by mutagenesis and binding assays. Based on the data, we suggest that IcmR-IcmQ binds to membranes where it may interact with or perhaps modify a protein in the T4bSS when NAD+ is bound.
Keywords: Legionella pneumophila, T4b secretion system, IcmQ, IcmR
Introduction
Legionella pneumophila is a Gram-negative pathogen of freshwater amoeba (Fields, 1996) that infects alveolar macrophages in humans exposed to contaminated aerosols. After internalization, L. pneumophila (Lp) evades the lysosome-endosome pathway and converts the vacuole into a safe haven for replication (Horwitz, 1983a; Horwitz, 1983b; Isberg et al., 2009). This process requires the translocation of protein effectors into the host cell by a Type IVb secretion system (T4bSS) (Christie and Vogel, 2000; Luo and Isberg, 2004; de Filipe et al., 2008; Ensminger and Isberg, 2009). Successive cycles of bacterial uptake, replication and cell lysis may cause a severe pneumonia known as Legionnaire’s disease (Horwitz, 1983a; Horwitz 1983b; Coers et al., 2000).
The T4bSS is encoded by roughly 26 dot/icm genes that are required for pathogenicity (Marra et al., 1992; Berger and Isberg, 1993; Segal and Shuman, 1997). Nineteen of the Dot/Icm genes share sequence homology to genes in bacterial conjugation systems, such as the R64 IncI and Col1b-P9 plasmids that export DNA (Vogel et al., 1998; Segal and Shuman, 1999). However, the primary job of the Dot/Icm translocase is to export protein effectors into the cytoplasm of host cells during infection (Vogel et al., 1998; Nagai et al., 2005). At least 240 proteins have carboxyl terminal signal sequences that allow translocation by the Dot/Icm system (Zhu et al., 2011). These effectors target a variety of host proteins that control secretory traffic, translation and cell survival, to promote the formation of a replication vacuole (see Isberg et al., 2009).
Seven Dot/Icm proteins have no obvious sequence homology with known protein families. IcmR and IcmQ are members of this novel group and may have a unique role in the T4bSS. IcmQ is also present in T4bSS of Rickettsia, Fluoribacter, Tatlockia, Bartonella and Coxiella. Conversely, the icmR gene is not well conserved in these genera although a putative IcmQ partner is present in all cases. These partners have been designated as FIR proteins (functional homologs of IcmR) (Feldman and Segal, 2004; Feldman et al., 2005). L. pneumophila strains harboring icmQ knockouts are killed by host cells, while those lacking a functional fir gene have a severe growth defect (Coers et al., 2000).
IcmR and IcmQ form a stable heterodimer and are localized to the Lp cytoplasm (Coers et al., 2000), where IcmR may act as a chaperone to prevent aggregation or non-specific interactions of IcmQ with other components (Duménil and Isberg, 2001; Duménil et al., 2004; Raychaudhury et al., 2009). Protease digestion has shown that the middle of IcmR (Rm) interacts with the N-terminal region of IcmQ (Qn; Figure 1A) (Duménil et al., 2004). These studies revealed the presence of a protease-resistant C-terminal domain of IcmQ (Qc). A previous crystal structure of interacting IcmR and IcmQ domains revealed a parallel four-helix bundle (Raychaudhury et al., 2009), in which Rm forms a helix-turn-helix motif that interacts with a similar motif in Qn through extensive hydrophobic contacts. The pattern of hydrophobic residues in IcmR is conserved in other FIR proteins, which suggests that FIR-IcmQ complexes in general will form a similar 4 helix bundle (Raychaudhury et al., 2009).
Figure 1.
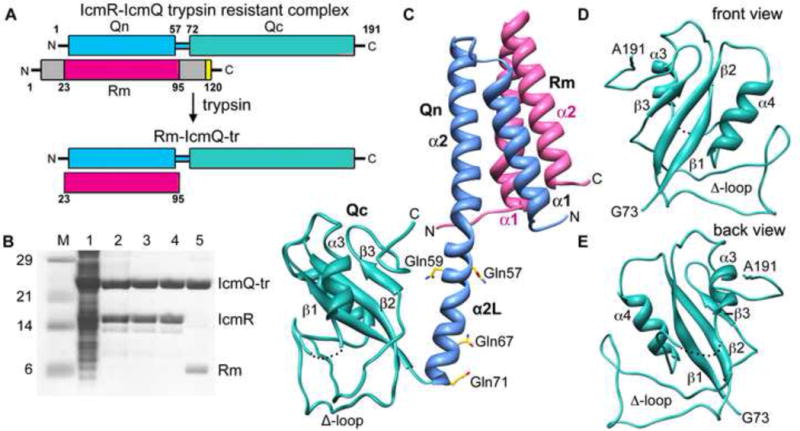
Overview, purification and structure of Rm-IcmQ.
A. A domain diagram for IcmR-IcmQ is shown. Trypsin treatment removes flexible N- and C-terminal regions of IcmR that may be unstructured, and yields the trypsin resistant (tr) Rm-IcmQ complex.
B. The purification of Rm-IcmQ-tr is shown on SDS-PAGE. Molecular weight markers (Lane M), high speed lysate (Lane 1), eluate from HisTrap HP (Lane 2), flow-through from HiTrap Q (Lane 3), eluate from HiTrap Sp (Lane 4), and Rm-IcmQ after trypsinization and purification (Lane 5) are shown.
C. A molecular model of Rm-IcmQ is shown. The middle region of IcmR (Rm, pink) interacts with the N-terminal domain of IcmQ (Qn, light blue) to form the Rm-Qn interacting domain, comprised of a 4-helix bundle. A linker helix (α2L) separates Rm-Qn from the C-terminal domain of IcmQ (Qc, green). Secondary structures are labeled, including the Δ-loop, and four residues mutated to glutamines (aa57, 59, 67 and 71) are also shown.
D. and E. Front and back views are shown of Qc.
See also Figures S1, S2 and S3.
To improve our understanding of this system we crystallized full-length IcmQ with Rm and solved the structure of this complex. The resulting structure revealed the Rm-Qn four-helix bundle (Raychaudhury et al., 2009), an extended linker helix and a novel C-terminal domain of IcmQ. The Qc domain shares considerable structural homology with NAD+ binding regions of secreted bacterial ADP ribosyltransferases (ADPRTs) and binding experiments demonstrated a Kd for NAD+ in the physiological range. Modeling and molecular dynamics were then used to generate a model of the Qc-NAD+ complex. This approach revealed seven conserved residues that form an extended binding site for NAD+ and targeted point mutations in three of these residues strongly reduced NAD+ binding. Finally, we show that moderately conserved arginines in the linker helix may help direct IcmQ to membranes. This may help target IcmR-IcmQ to the T4bSS, where the complex plays a critical role in the translocase.
Results
Structure of IcmR-IcmQ
IcmQ is comprised of an N-terminal domain (Qn; residues 1–57), a trypsin sensitive linker (residues 58–71) and a novel C-terminal domain (residues 72–191). In the current study, four basic linker residues (Lys57, Lys59, Arg67 and Arg71) were mutated to glutamines to yield a trypsin-resistant complex (denoted IcmR-IcmQ-tr; Figure 1A). IcmQ-tr was co-expressed with IcmR-6xHis, purified by pulling on the His-tagged complex, followed by HiTrap Q and HiTrap Sp columns (Figure 1B, lanes 1–4). To facilitate crystallization, IcmR-IcmQ-tr complexes were treated with protease to form trypsin-resistant complexes, denoted Rm-IcmQ-tr, and the protease was then removed with benzamidine agarose beads (Figure 1B, lane 5). Crystals of the Rm-IcmQ-tr complex diffracted to 2.4Å and were very sensitive to oxidation, which necessitated using fresh crystallization reagents. Leucine 119 of IcmQ and Leu84 in IcmR were mutated to methionine and crystals containing seleno-methionine labeled complexes were used to solve the structure with single-wavelength anomalous scattering.
After refinement, the final model consists of residues 28–86 of IcmR and residues 1–191 of IcmQ (see Table 1). The structure revealed a two-domain complex separated by an extended 18-residue α-helix (Figure 1C). This linker helix corresponds to a trypsin-sensitive region between Qn and Qc (Duménil et al., 2004) and contains four glutamine point mutations that were introduced to confer trypsin-resistance. Intriguingly, one of the mutations (Gln67) was involved in a crystal contact (Figure S1A). A structure of the IcmR-IcmQ interacting region (denoted Rm-Qn) was solved previously (Raychaudhury et al., 2009) and is present in the full complex (Figure 1C). In brief, two helices from each protein form a parallel four-helix bundle with hydrophobic residues buried in the interior. The trypsin sensitive linker between Qn and Qc forms an extension of helix α2 and has been denoted helix α2L (Figure 1C; residues 58–71). The Qc domain consists of residues 72–191 (Figures 1C–1E) and contains three β-strands, two α-helices and a loop shaped like the Greek letter Delta (residues 136–165). The Δ-loop is well-ordered due to extensive crystal contacts with a symmetry mate (Figures S1B, S1C) and forms van der Waals contacts with other regions of Qc (Figures 1C–1E).
Table 1.
Crystallographic and refinement statistics for Rm-IcmQ data sets
| Native | Selenomethionine derivative | |
|---|---|---|
| Space Group | P21212 | P21212 |
| Unit cell dimensions | 69 × 112 × 49 Å | 69 × 111 × 49 Å |
| Cell angles | 90°, 90°, 90° | 90°, 90°, 90° |
| Solvent content | 63% | 62% |
| Vm (Å3/Dalton) | 3.32 | 3.24 |
| Molecules / asu | 1 | 1 |
| Wavelength (Å) | 1.000 | 0.9789 |
| I/σ(total/highest) | 20.8 / 5.4 | 27.6 / 9.3 |
| Completeness | 99.1 (99.6) | 99.7 (99.4) |
| Rmerge (total/highest) | 0.054 / 0.341 | 0.077 / 0.306 |
| Refinement statistics for the Rm-IcmQ native data set | ||
| Resolution range (Å) | 19.75 – 2.4 | |
| Unique reflections | 15,065 | |
| Test set | 1,560 (10%) | |
| Rcryst | 0.225 | |
| Rfree | 0.259 | |
| Mean B-value (Å3) | 45.6 | |
| RMS Deviations | ||
| Bond length (Å) | 0.017 | |
| Bond angle (°) | 1.72 | |
| Total waters | 118 | |
| Residues modeled | IcmQ, 1–100, 104–191; IcmR, 28–86 | |
| Residues omitted | IcmQ, 101–103; IcmR, 1–27, 87–120 (removed by proteolysis) | |
| Ramachandran Plot | ||
| Core region | 228 residues, 94.6% | |
| Additional allowed regions | 13 residues, 5.4% | |
Molecular dynamics simulations of Rm-IcmQ suggest that the linker helix may be flexible. This flexibility would allow the Rm-Qn four-helix bundle to reorient relative to Qc (Figure S2A). These movements are hinge-like and centered primarily at Gly51 and Gly73 located at either end of helix α2L. Hence, Rm-Qn and Qc may come into close contact in solution (Figure S2B). The physiological relevance of this flexibility is not known. However, the B-factor distribution suggests that some regions of Qc may be intrinsically flexible relative to the Rm-Qn helix bundle (Figure S3). Higher B-factors could be due to a lack of stabilizing crystal contacts or the absence of a suitable ligand in the small loop-rich Qc domain. Finally, extensive contacts between the linker helix and an adjacent molecule may have stabilized the linker helix in a more extended conformation, allowing the Rm-IcmQ complex to be crystallized (Figure S1B, S1C).
An NAD+ binding fold in the C-terminal domain of IcmQ
Coordinates for the Qc domain were submitted to the DALI server (Holm et al., 2008) to identify proteins with a similar fold and the results suggested possible roles for IcmQ. Based on structural homology, a region of Qc was found to be similar to ADP-ribosyltransferase domains in certain bacterial toxins (Figures 2A–2C, highlighted in gold). These toxin ADPRTs hydrolyze NAD+ and transfer the ADP-ribose moiety to target proteins in host cells (Koch-Nolte et al., 2001; Holbourn et al., 2006). This post-translational modification may result in mis-regulation of the target protein. Interestingly, IcmQ is not believed to be exported by Legionella into the host cytoplasm and thus, may act within the bacterium. Structures with similar NAD+ binding domains include cholera toxin (Zhang et al., 1995; O’Neal et al., 2005) and diphtheria toxin (Bennett et al., 1994) (Figures 2B, 2C) with an overall sequence identity of 7–13% and an average RMSD of 2.9 Å between the appropriate toxin regions and Qc. The ADPRT domains contain a β-αα-ββ motif, and each toxin has a unique region inserted between β-strands 2 and 3. The equivalent insert in IcmQ corresponds to the Δ-loop (Figures 2A–2C, shown in grey). A binding pocket for the nicotinamide-ribose moiety in ADPRTs has been characterized as a “scorpion” motif with a loop or β-strand connected by a tight turn to an α-helix to form a hairpin-like structure (Lee et al., 2010). Comparison of Qc with NAD+-bound structures of cholera and diphtheria toxins revealed a similar hairpin in the three proteins (Figures 2D–2F).
Figure 2.
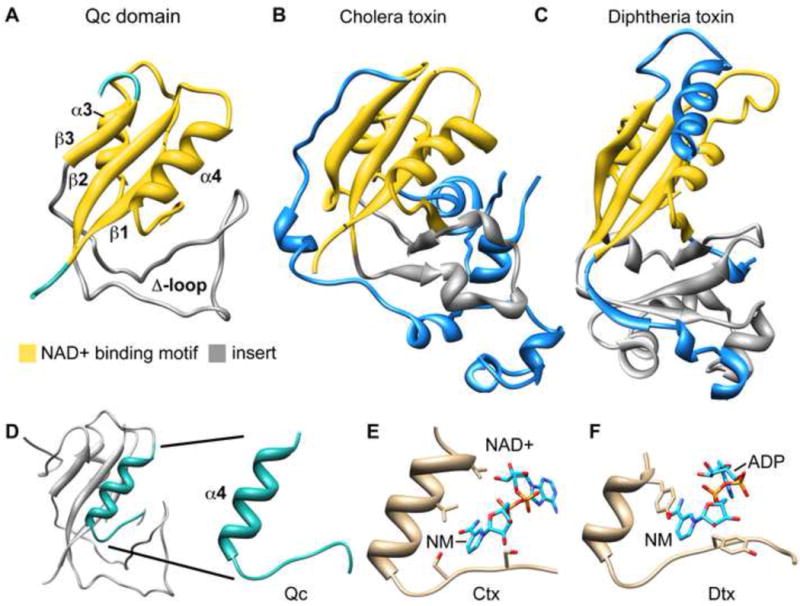
The Qc domain shares structural homology with ADP-ribosyltransferase domains (ADPRTs) of bacterial toxins.
A. The Qc domain is shown with a region of structural homology to ADPRTs colored in yellow. The inserted region between ß-strands 2 and 3 contains the Δ-loop colored in grey.
B. A similar color coded depiction is used for the ADPRT domain of cholera toxin (2A5F). Structures in panels A–C have been aligned on their ADPRT domains.
C. The ADPRT domain from diphtheria toxin is shown (1TOX).
D. The scorpion motif in Qc is highlighted.
E. The cholera toxin scorpion motif is shown with bound NAD+ (2A5F) and the nicotinamide ring is labeled (NM).
F. The diphtheria toxin scorpion motif is shown with bound NAD+ (1TOX).
The program BLAST (Altschul et al., 1990) was then used to identify 54 IcmQ orthologs in five bacterial genera (Legionella, Rickettsiella, Fluorobacter, Tatlockia and Coxiella) that have Type IVb secretion systems. IcmQ sequences were then aligned with T-Coffee to reveal conserved residues (Figure 3A) (Notredame et al., 2000). In total, seven residues that may form an NAD+ binding surface were mapped onto the scorpion motif and the surrounding region. These residues include Ser80, Tyr82, Arg106, Tyr109, Lys122, Glu128 and Asp151 (marked with black dots in Figure 3A). In addition, Pro107 and Ileu108 form part of a conserved RPIY motif that is located on the loop proximal to helix α4 in the scorpion motif (Figures 3A, 3B; shown in gold). Interestingly, IcmQ lacks a conserved glutamic acid residue that is usually adjacent to the scorpion motif in all known ADPRTs (Lee et al., 2010). The side chain carboxyl group of glutamate destabilizes the N-glycosidic bond between the ribose ring and nicotinamide, which facilitates an SN1 addition of the ribose-ADP moiety to a side chain of the target protein (Holbourn et al., 2006). We hypothesize that Asp151 in the Δ-loop could perform a similar role in IcmQ.
Figure 3.
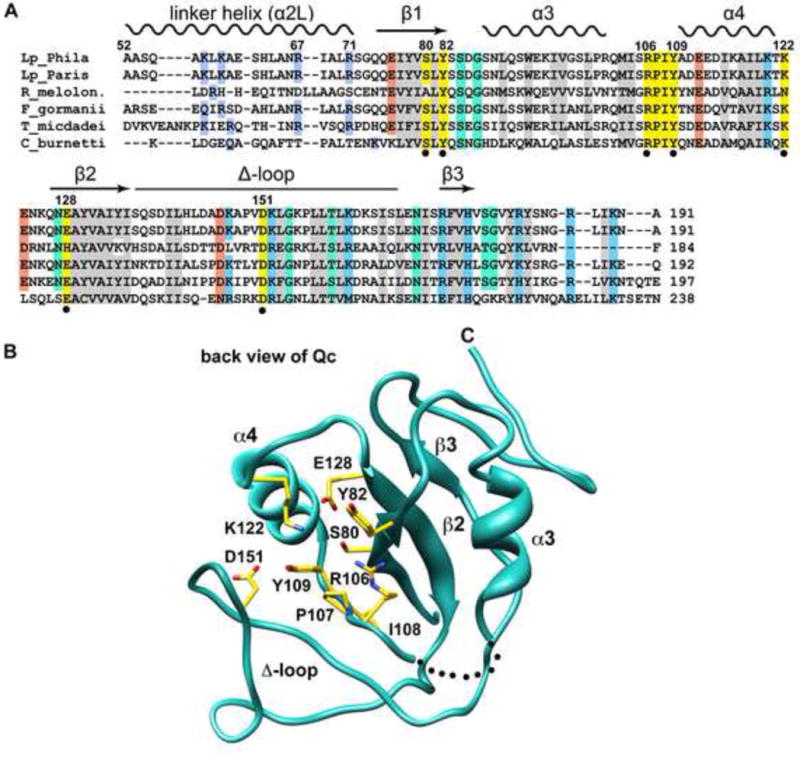
Conserved residues in the Qc domain may play a role in NAD+ binding.
A. A structural alignment is shown for IcmQ sequences from 5 genera. Similar hydrophobic residues are colored grey, positively charged residues are light blue, negatively charged residues are orange, and hydrophilic residues are green. Residues that may interact with NAD+ are colored yellow and are marked with a black dot.
B. A back view of the Qc domain is shown. Conserved residues in the vicinity of the scorpion motif are labeled and their side chains are colored yellow.
See also Figure S4.
NAD+ binding to IcmQ
The similarity between Qc and ADPRTs prompted us to test NAD+ binding by IcmQ. To this end, 15N labeled Qc with the linker helix (Qcl; residues 49/50 to 191) was probed using 2D NMR because of the high solubility of this domain. HSQC spectra were collected in a titration series with increasing ratios of NAD+ to Qcl and the resulting spectra were overlayed (Figure 4A, inset shown in 4B). Intriguingly, only a small number of peaks shifted, suggesting that a distinct and local conformational change may occur upon NAD+ binding. Similar experiments with NADH showed no detectable peak shifts with ~5 mM NADH (not shown). After plotting peak shifts as a function of NAD+ concentration we obtained an apparent Kd in the range of ~0.5–1 mM (Figure 4C). However, a non-specific binding component is present which made curve fitting difficult. To quantitate NAD+ binding, we carried out filter binding assays using 32P labeled NAD+. After subtraction of non-specific binding from the experimental data (Figure 4D red and blue curves), a well fit hyperbolic curve was obtained (Figure 4D, black triangles). Direct curve fitting and Lineweaver-Burk analysis gave a Kd of 0.4–0.6 mM (Figure 4E). Although this Kd indicates a relatively low affinity, the value falls in the middle of measured NAD+ concentrations in bacteria, which may vary depending upon the metabolic state of the cells (Grose et al., 2006). Hence, NAD+ binding by IcmQ may be physiologically-relevant.
Figure 4.
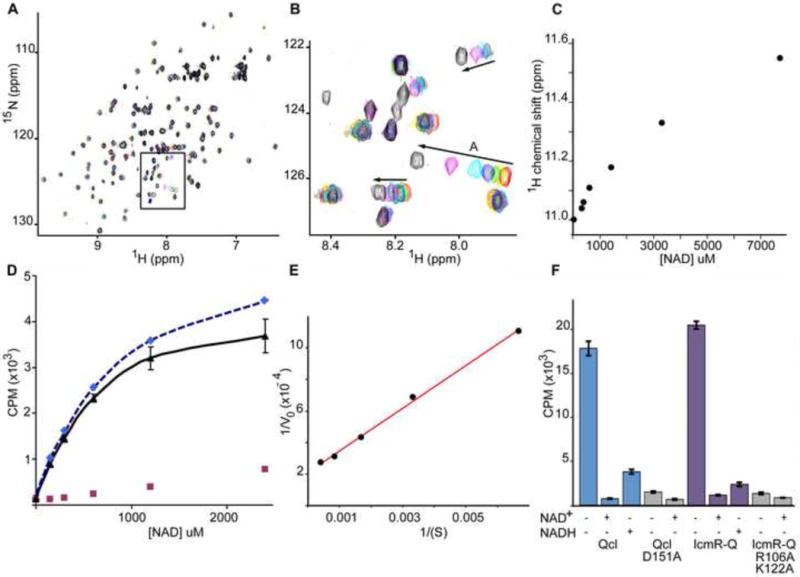
NAD+ binding by IcmQ.
A. A titration of Qcl with NAD+ was monitored with 15N-HSQC spectra. The original sample contained 330 uM Qcl (red peaks). NAD+ was added to final concentrations of 330 uM (yellow), 390 uM (green), 620 uM (blue), 1.4 mM (cyan), 3.3 mM (magenta) and 7.7 mM (black). A region of interest is boxed and shown in panel B.
B. Arrows indicate the direction of peak movements in response to increasing NAD+ concentration. All spectra were acquired at 20° C in 20 mM phosphate buffer, 100 mM NaCl at pH 7.5.
C. A plot is shown of peak shifts as a function of increasing NAD+ concentration for peak A. The apparent Kd for NAD+ binding is ~ 0.5–1 mM.
D. Fitted curves are shown for 32P labeled NAD+ bound to Qcl in filter binding assays. A non-specific component (red squares) was subtracted from the raw data (dashed blue fitted line) to give the corrected binding curve (black) with a calculated Kd of ~0.38 mM.
E. A Lineweaver-Burk plot is shown for the corrected data with a Kd of ~0.64 mM.
F. A histogram is shown for NAD+ binding to wild type, mutant Qcl and double mutant IcmR-IcmQ, either alone (−/−) or in the presence of excess unlabeled NAD+ or NADH. The mutant data are shaded in grey.
We then attempted to determine the structure of Rm-IcmQ with bound NAD+. Crystals of Rm-IcmQ soaked with NAD+ were stable up to a ligand concentration of 10 mM before cracking or dissolving. Unfortunately, the diffraction quality of these crystals was poor, so co-crystallization was attempted. Crystals with the usual morphology grew in the presence of 5 mM NAD+, but no density for the cofactor was found in the resulting maps. This could be due to the fact that the NAD+ binding motif (specifically the Δ-loop which contains D151A) may be involved in lattice contacts.
Therefore, computational methods were used to model NAD+ binding. First, a rough model with docked NAD+ was constructed based on toxin co-crystal structures. Molecular dynamics was then used to optimize the binding geometry (Figures 5A, 5B). The resulting model suggests a number of plausible interactions between NAD+ and conserved Qc residues, as highlighted in Figures 3 and 5. For example, Tyr82 and Tyr109 form ring-stacking interactions with the adenine and nicotinamide ring systems (Figures 5C–5F), while Arg106 and Lys122 may form hydrogen bonds with phosphate groups (Figure 5D). In addition, Ser80 and Glu128 form hydrogen bonds with suitable partners in the nicotinamide and ADP-ribose moiety, respectively (Figures 5C, 5E). Importantly, Asp151 may form hydrogen bonds with both 2′- and 3′- hydroxyl groups of the nicotinamide-ribose moiety (Figure 5F). This may destabilize the N-glycosidic bond with nicotinamide to facilitate an SN1 mechanism as suggested for other ADPRTs (Holbourn et al., 2006). Finally, the overall structures of apo- and NAD+-bound Qc molecules are quite similar, as suggested by NMR spectra. However, a small perturbation of the Δ-loop (grey model in Figure 5A) is required to position Asp151 to interact with bound NAD+. The Δ-loop forms extensive lattice contacts in the current crystal form. Thus, loop movements required for NAD+ binding may have precluded the formation of a liganded complex in crystal soaking experiments.
Figure 5.
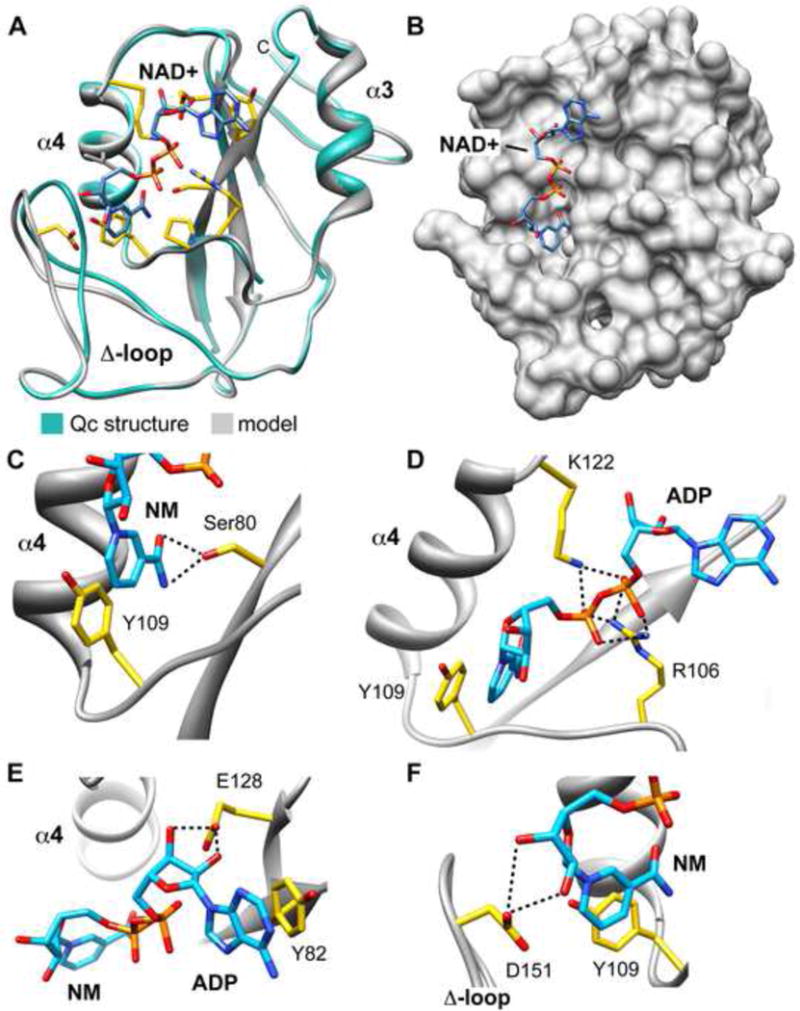
A model of NAD+ bound to Qc.
A. The structure of unliganded Qc (green) is super-imposed with the computational model of NAD+ bound to Qc (grey). Note movement of the Δ-loop and the extended conformation of NAD+ within the scorpion motif. In addition, the modeling restored connectivity of the α3–α4 loop.
B. Modeled NAD+ is shown in an extended conformation bound to a surface groove on Qc.
C. Possible molecular interactions between NAD+ and Qc are shown in panels C–F. Hydrogen bonds are depicted as black dashed lines. Hydrogen bonding interactions between Ser80 and nicotinamide in a small pocket are shown. In addition, a ring stacking interaction between nicotinamide and Tyr109 is present.
D. Hydrogen bonding between the phosphates of NAD+ and Arg106/Lys122 is shown.
E. Ring stacking between the adenine of NAD+ and Tyr82 is present, along with hydrogen bonding between the ribose moiety of ADP and Glu128.
F. Hydrogen bonding is present between Asp151 and hydroxyl groups of the ribose ring attached to the nicotinamide ring.
See also Figure S5.
To test the NAD+ binding model, a number of IcmQ mutants were prepared with altered residues in the predicted binding site including constructs with D151A, D151G, D151S and Y82A/Y109A mutations. When over-expressed in E. coli, the D151A, D151G and D151S mutations gave insoluble proteins, while Y82A/Y109A mutations were not expressed. However a double mutant of IcmQ (R106A/K122A) was prepared in complex with IcmR and the CD spectrum was similar to that obtained with wild type complexes (Figure S4). In addition, a soluble D151A mutant of Qcl had a normal CD spectrum (Figure S4). Wild type and mutant proteins were then tested in filter binding assays with 32P labeled NAD+. In this assay, we also performed competition experiments with cold (unlabeled) NAD+ and NADH (~50 mM) to test specificity. As shown in Figure 4F, only background binding of NAD+ was observed for the Qcl D151A mutant and the IcmQ R106A/K122A double mutant relative to wild type proteins. There was no further reduction in NAD+ binding after the addition of cold substrate. This supports our hypothesis that D151, R106 and K122 may play important roles in NAD+ binding.
We also found that NAD+ binding to wild type Qcl could be competed with a large excess of cold NAD+ or NADH (Figure 4F), unlike the situation with NMR binding experiments in which ~ 5 mM NADH did not cause peak shifts. A similar binding profile was observed for wild type IcmR-IcmQ complexes. The different NADH effects may be due to the very different protein concentrations used in NMR and filter binding experiments, coupled with the likely presence of a small amount of contaminating NAD+ in the large excess of NADH employed in filter binding, whereas a much lower concentration of NADH was used for the NMR titration. Alternatively, NADH at very high concentrations may bind to Qcl but its lifetime may preclude it from showing up on the NMR timescale. When taken together our data support the idea that Qcl and IcmQ may bind NAD+, whereas NADH binding, if present, may be very weak.
To investigate the possible role of NAD+ binding, we created an Lp02 strain in which the chromosomal copy of IcmQ was replaced with a D151A mutant. However, as observed in over-expression experiments, the IcmQ D151A mutant proved to be less stable than wild type protein, which resulted in much lower levels of IcmQ in the mutant. Presumably misfolded IcmQ was rapidly degraded leaving protein that was folded and more stable. This mismatch in IcmQ levels when compared to wild type Lp would have made interpretations difficult. Thus, we manipulated the expression of wild type and IcmQ D151A in separate Lp strains, so that these proteins were present in similar amounts. We estimate that the steady state level of IcmQ in these cells was ~5–10% of that present in wild type cells (Figure S5A, S5B). We then assayed infectivity with these matched Lp strains by monitoring the number of bacteria in replication vacuoles after exposing mouse macrophages to post-exponential bacteria. No discernible differences in replication efficiency were found under these conditions (Figure S5C). This indicates that IcmQ levels present in wild type Lp are at least 10X more than is needed for growth within mouse macrophages. Perhaps as little as 10–20 IcmQ molecules per bacterial cell can maintain an intracellular growth phenotype for Lp under these conditions. Additional studies are now needed to work out the role of NAD+ binding and IcmQ function in vivo.
Membrane interactions of IcmQ
In a previous study, IcmQ was shown to bind to membranes and permeabilize vesicles (Duménil et al., 2004). More recently, membrane binding and permeabilization were shown to reside in different domains of IcmQ. Thus, Qc with the linker helix (Qcl 49/50–191) directs membrane binding, while calcein efflux from pre-loaded vesicles is dependent on the N-terminal domain (Qn) (Raychaudhury et al., 2009).
We re-examined the ability of IcmQ to permeabilize membranes and asked how many molecules are required for this process. We reasoned that fewer IcmQ molecules would be required to form a defined membrane pore and release calcein from vesicles than would be required in a carpet model (Shai, 2002). In the latter case, massive protein binding to the outer leaflet would destabilize the membrane making it leaky. To study this process, preformed lipid vesicles of ~1 μ diameter were formed by extrusion with a 3:1 ratio of egg PC:DMPG and loaded with calcein dye during extrusion (Duménil et al., 2004). Increasing amounts of IcmQ were added to the vesicles and calcein efflux was followed by monitoring the increase in fluorescence of released dye. The resulting data are summarized in Table S1. Calcein release is maximal at ~50 nM IcmQ which corresponded to a calculated ratio of ~2800 molecules per vesicle. This high ratio suggests that membrane permeabilization by IcmQ may occur via the carpet mechanism. Since roughly 200 molecules of IcmQ may be present in each Legionella cell (Duménil and Isberg, 2001), we surmise that membrane binding may be the physiologically important property of IcmQ rather than bilayer permeabilization.
In previous studies, we showed that Qn(1–57) and Qc(72–191) did not bind to vesicles while Qcl(49/50–191) did bind to membranes (Duménil et al, 2004; Raychaudhury et al., 2009). This suggested that residues involved in membrane binding may be located between residues 49/50 and 71 in the α2L linker helix. To identify specific residues, IcmQ-tr with four basic residues in the linker helix mutated to glutamine was tested for membrane binding with a vesicle floatation assay (Duménil et al., 2004). Remarkably, IcmQ-tr was not able to associate efficiently with membranes (Figure 6A, right panel) when compared to wild type IcmQ (Figure 6A, left panel). In addition, IcmQ-tr did not release calcein from vesicles (Figure 6B, grey curve), presumably because this molecule is not able to bind membranes. These data suggest that basic residues in the linker helix of IcmQ-tr may be responsible for membrane binding. Two moderately conserved arginine residues in the linker helix (Arg67, Arg71; Figure S6A) are also present in Qn(1–72) which binds to membranes (Duménil et al., 2004). Thus, Arg67 and Arg71 may be required for membrane binding. To ensure that diminished membrane binding was not due to large structural changes introduced by linker mutations, CD spectra were obtained for IcmQ and IcmQ-tr and the resulting spectra were essentially identical (Figure S7). We also examined the dependence of membrane binding on acidic phospholipids and salt concentration. When phosphatidyl glycerol (DMPG) was omitted during vesicle formation we saw very little binding of IcmQ to membranes (Figure 6C). In addition, vesicle binding was greatly reduced in 250 mM NaCl with vesicles made with DMPG and egg PC (Figure 6D). Together, these data suggest that electrostatic interactions are important for membrane binding by the rather basic IcmQ molecule.
Figure 6.
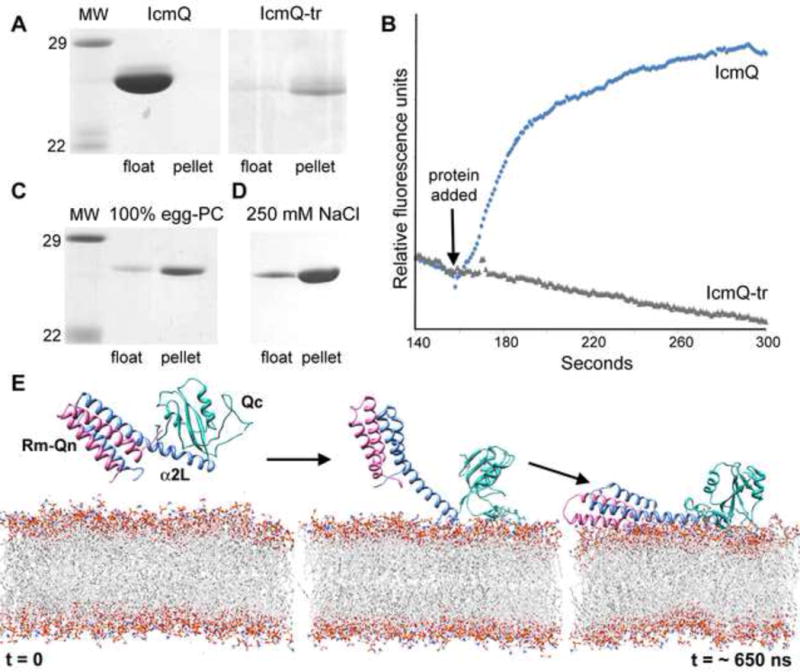
Membrane binding of IcmQ.
A. IcmQ binds efficiently to floated vesicles (left), while IcmQ-tr does not bind to membranes and is present in the pellet (right) in this qualitative assay.
B. IcmQ-tr (grey line) does not release calcein from vesicles, while IcmQ (blue line) is able to release trapped dye molecules as monitored by increased fluorescence.
C. IcmQ binding to vesicles is greatly diminished when membranes do not contain an acidic phospholipid.
D. Higher salt diminishes IcmQ binding to vesicles made with an acidic phospholipid (DMPG).
E. Molecular dynamics simulations show that Rm-IcmQ may bind to the membrane surface using electrostatic interactions.
See also Figures S6, S7 and S8.
We have shown previously that IcmR-IcmQ will interact with membranes (Raychaudhury et al., 2009). Thus, we used molecular dynamics simulations to model the interaction of Rm-IcmQ with a bilayer that contained POPG and POPE in the presence of water and cations, beginning from a state in which the protein was separated from the membrane by ~10–15Å (Figure 6E, left). During the simulations, basic residues near the end of the linker helix (Arg67 and Arg71) may target the complex to the membrane surface (Figure 6E, middle panel). In addition, the Δ-loop also interacts with the charged membrane surface. After ~650 ns, the entire Rm-Qn region and linker helix formed numerous contacts with the head group region (Figure 6E, right panel) and charged residues in contact with the membrane at this point are shown in Figure S6B. The electrostatic surface shows that the α2 linker helix is strikingly basic when compared to the rest of the complex, while charged residues in Rm are not well conserved. This charge distribution supports the idea that the basic linker helix is important in driving membrane association. Intriguingly, the spacing between the two membrane leaflets is perturbed when a large surface area of Rm-IcmQ is bound to the bilayer. A similar local disordering of lipids may lead to vesicle permeabilization when the bilayer is coated with IcmQ molecules with an exposed and destabilizing Qn domain. However, membranes are not permeabilized by IcmQ when Rm or IcmR are bound to Qn because the amphipathic helix-turn-turn helix motif of Qn is shielded from non-polar regions of the membrane, so that cracks in the bilayer may not occur (Duménil et al., 2004; Raychaudhury et al., 2009).
To examine whether membrane association of IcmQ is necessary for translocation activity, a strain of Lp01 was constructed with icmQ-tr replacing the chromosomal copy of icmQ (Lp01-IcmQ-tr). Legionellae with an active T4bSS are sensitive to NaCl concentrations greater than 0.1 M when grown on solid media, which may be due to functional translocases providing a conduit for salt entry into cells (Sadosky et al., 1993). Both Lp01 and Lp01-IcmQ-tr were unable to grow on media containing 0.1 M NaCl (not shown), indicating that assembled translocases are present in both strains. In addition, protein levels in Lp01-IcmQ-tr cells appeared to be normal (Figure S8A). Translocation assays were performed to examine the effect of membrane-deficient binding on IcmQ function. In these assays, translocation of an adenyl cyclase construct (CyaA-MavU) from Lp01-IcmQ-tr cells into macrophages was diminished by ~50% (Figure S8B), relative to the control. This data indicates that membrane binding may increase the targeting efficiency of IcmQ to the T4bSS, where it may interact with the translocase.
Discussion
An NAD+ binding module in IcmQ
IcmQ is an essential protein in the T4bSS of Legionella pneumophila, as icmQ deficient strains are killed by host cells after phagocytosis (Coers et al., 2000). In this paper, we report a crystal structure of Rm-IcmQ complexes that lack trypsin-sensitive N- and C-termini of IcmR. This analysis shows that ordered regions of IcmR-IcmQ are comprised of two domains separated by a trypsin sensitive linker helix. IcmR appears to be an integral part of the complex, as it chaperones the amphipathic helix-turn-helix motif of Qn and prevents IcmQ aggregation (Duménil and Isberg, 2001; Duménil et al., 2004; Raychaudhury et al., 2009). The β–αα–ββ motif in the C-terminal domain of IcmQ has structural homology with ADPRT domains of certain bacterial toxins (Koch-Nolte et al., 2001; Holbourn et al., 2006), and is consistent with IcmQ having a scorpion motif for NAD+ binding (Lee et al., 2010). We propose that Qc may represent a minimal NAD+ binding module because the Δ-loop between strands β2 and β3 is much smaller than similar inserts in cholera and diphtheria toxins.
Sequence identity between Qc and ADPRTs is very low (7–13%). However, we were able to identify residues in IcmQ orthologs from five bacterial genera that may be involved in NAD+ binding. We then demonstrated that Qc binds NAD+ with a Kd of ~0.4–0.6 mM. While this value reflects a rather weak affinity, the intracellular concentration of NAD+ in E. coli, a gram negative bacterium like Lp, is ~1–2 mM (Albe et al., 1990). After identifying the scorpion motif, we used structural homology and toxin-NAD+ co-crystal structures to create a model of NAD+ binding to Qc. This model was refined with molecular dynamics to provide a structure with reasonable stereochemistry in which NAD+ adopts an extended conformation on the surface of Qc. The analysis highlighted plausible roles for seven conserved residues, including two tyrosines that may be involved in ring stacking interactions, and five residues that could form hydrogen bonds and salt bridges with NAD+. In particular, Asp151 was predicted to form H-bond interactions with hydroxyl groups on the ribose attached to nicotinamide, while two basic residues (Arg 106/Lys122) may form a salt bridge network with phosphates that link adenine-ribose and ribose-nicotinamide moieties. The importance of Arg106, Lys122 and Asp151 was verified by a significant loss of NAD+ binding after mutation of respective residues to alanine. Importantly, the distribution of putative ligand binding residues on Qc differs from previously identified NAD+ binding motifs in ADPRTs (Holbourn et al., 2006).
Towards identifying the role of IcmR-IcmQ
Our work now sets the stage for further investigations into the function of IcmR-IcmQ in the T4bSS. The fact that IcmQ contains a novel NAD+ binding module suggests that this ligand may regulate IcmQ function. The concentration of NAD+ varies between 0.2 and 0.9 mM under anaerobic and aerobic conditions in Salmonella enterica (Grose et al., 2006). Similar fluctuations in the intra-cellular concentration of NAD+ may occur in Gram-negative Legionella. Hence, a Kd of 0.4 – 0.6 mM may be physiologically-relevant, as variations in the intra-cellular NAD+ concentration could influence the ratio of free and NAD+ bound IcmQ.
A recent study has shown that nicotinic acid (a precursor of NAD+) is an important regulator of Lp virulence and can up-regulate the transition from a replicating to a transmission phenotype (Edwards et al., 2013). Addition of 5 mM nicotinic acid to exponentially growing cultures induced a post-exponential phenotype with increased transcription of ~39 virulence genes including icmB/DotO, icmD/DotP, and icmX, along with components of the core translocase icmG/DotF, icmJ/DotN and icmO/DotL (Vincent et al., 2006). This in turn, promoted growth in macrophages and caused salt sensitivity for cell growth, which suggests that a functional T4bSS had been assembled (Edwards et al., 2013). Nicotinic acid also regulates virulence factors in Bordetella pertussis (Cotter and DiRita. 2000; Miller et al., 1989). These observations support the idea that the role of IcmR-IcmQ in the T4bSS may be regulated by NAD+ levels.
There are two possible roles for IcmQ binding of NAD+. First, IcmQ could be a specialized ADPRT that modifies T4bSS components. All known ADPRTs have a conserved glutamic acid that hydrogen bonds to hydroxyl groups of the ribose ring attached to nicotinamide, to destabilize the N-glycosidic bond (Koch-Nolte et al., 2001; Holbourn et al., 2006; Jørgensen et al., 2008). In our model, Asp151 in the Δ-loop may replace this conserved glutamate and a small rearrangement of the Δ-loop may facilitate NAD+ binding. In some ADPRTs, a glutamate to aspartate mutation leads to an inactive enzyme (Wilson et al., 1990; Bohmer et al., 1996). This inhibition may be due to the shorter side chain of aspartate, which weakens its interaction with bound NAD+. However, the Δ-loop in Qc is probably flexible, as our modeling shows that Asp151 can be brought into close proximity to the ribose ring. This acidic residue is important for NAD+ binding. However, our data show that the relatively unstable D151A variant has no impact on intracellular growth when compared to a matched wild type strain, which also has a greatly reduced expression of IcmQ. Trial assays with etheno-NAD+ and model substrates, such as histones, arginine or polylysine showed no ADPRT activity. In addition, no NAD+ hydrolase activity could be detected (not shown). Together, this data argues against an enzymatic role for Asp151 in IcmQ.
In a second scenario, NAD+ binding may stabilize the Qc domain, allowing IcmQ to function by interacting with a component of the T4bSS. This idea is supported by data which show that IcmQ stability is compromised by mutations that may block NAD+ binding. Hence, Qc could act as a sensor that allows IcmR-IcmQ to interact with a critical component of the T4bSS when intracellular NAD+ levels are sufficient for binding. In this model, low intracellular levels of NAD+ would lead to an increased turnover of IcmQ, and this may occur during growth conditions in which steady state levels of other virulence-associated proteins are similarly reduced. The ability of the D151A mutant to promote intracellular growth of Lp may reflect the fact that a small pool of the protein is able to fold properly and bind NAD+. This idea is compatible with our data which shows a significant, though not complete loss of NAD+ binding activity in vitro (~90%) by the D151A mutant.
The T4bSS must translocate protein effectors across 3 membranes but little is known about its overall structure (Figure 7). Functions have been assigned for some of the roughly 26 Dot and Icm proteins in the T4bSS (reviewed in Isberg et al. 2009). The core translocase is comprised of Dot C, D, F, G and H (Vincent et al., 2006; Nakano et al., 2010), while Dot U and IcmF may stabilize the assembly (Isberg et al., 2009). The trimeric Dot LMN complex binds ATP and may act as a gatekeeper to the translocation pore and coupling complex that interacts with IcmS-IcmW, a receptor for protein effectors (Vincent et al., 2012; Ninio et al., 2005). DotA is a candidate to interact with the host plasma membrane, because it forms an oligomeric ring when it is secreted in post-exponential cells (Nagai and Roy, 2001).
Figure 7.
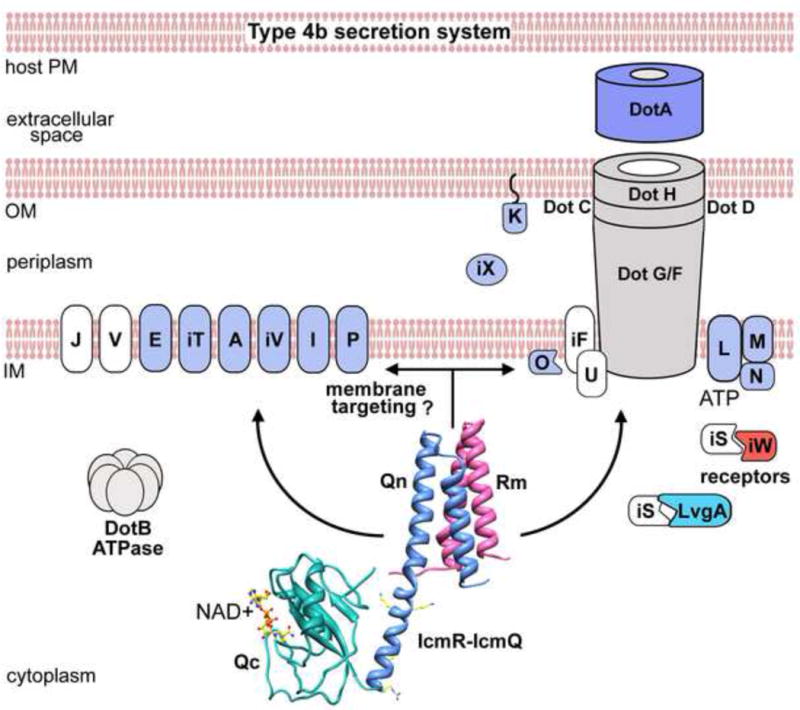
A model is shown of membrane localization and possible targeting of Rm-IcmQ to the T4bSS (see text for discussion). IcmQ function may be modulated by the intra-cellular concentration of NAD+ in response to changes in the metabolic state of the bacterium.
Against this backdrop, 7 Dot/Icm proteins may have novel functions in the T4bSS because they are not present in bacterial conjugation systems. These proteins include DotJ, DotU, DotV, IcmF, IcmS, IcmR and IcmQ (Komano et al., 2000; Sexton and Vogel, 2002). The IcmR-IcmQ complex binds to membranes that contain acidic phospholipids (Duménil et al., 2004 and this work). Membrane binding by IcmQ may be directed by two moderately conserved arginines in the linker helix. Notably, mutation of these arginines led to a significant loss of translocation activity (~50%) without affecting IcmQ expression in Legionella. We suggest that transient membrane binding may direct IcmR-IcmQ to the T4bSS more efficiently, where the complex could interact with (or perhaps modify) a target protein in the translocase (Figure 7). This membrane targeting may be particularly important given the relatively low number of IcmQ molecules that are present in each Lp cell (Duménil and Isberg, 2001). This work has now set the stage for studies to further define the roles of IcmR-IcmQ and NAD+ in the T4bSS.
Experimental Procedures
Protein preparation and crystallization
UniProt accession numbers for IcmQ and IcmR are A5IHF0 and Q5ZYC9 respectively. Expression clones of IcmR and IcmQ in the pQE-70 vector (Qiagen) with C-terminal 6xHis tags were provided by T. Montminy (Raychaudhury et al., 2009). To facilitate co-expression, IcmQ lacking a 6xHis tag was moved to the pET-24a vector. In order to generate a trypsin-resistant version of IcmQ, Lys57, Lys59, Arg67 and Arg71 were mutated to glutamines via PCR QuikChange reactions. For membrane binding experiments, these mutations were also prepared in a construct of IcmQ containing a C-terminal 6xHis-tag. A construct of icmQ for phasing the Rm-Q crystals was generated by mutating Leu119 to methionine using PCR mutagenesis. To create mutants for NAD+ binding, QuikChange PCR was used to mutate Asp151, Arg106 and Lys122, as well as Tyr82 and Tyr109 to alanines.
The Rm-IcmQ complex was prepared with BL21 (DE3) cells (Novagen) which were transformed with pQE-70 IcmR-6xHis and pET-24a IcmQ (TR). Cells were grown in the presence of 100 μg/ml ampicillin and 25 μg/ml kanamycin in 1L volumes of LB at 37°C to an OD600 of 1.0 and induced with 1 mM IPTG for 3 hours. Frozen cell pellets were thawed and resuspended in 50 mM Tris (pH 8.0), 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 mM PMSF, 1 mM benzamidine, 100 μM chymostatin, 1 μM leupeptin and 125 μM aprotinin. To promote lysis, cells were treated with lysozyme (0.3 mg/ml lysate, Sigma), deoxycholic acid (1.3 mg/ml) and DNase I (30 μg/ml, Sigma). High-speed lysate (100,000 × g) was bound to a HisTrap HP column (GE Biosciences) and washed extensively prior to elution with 350 mM imidazole in 20 mM Hepes pH 7.2, 1.0 M NaCl. Eluted protein was dialyzed into a buffer consisting of 20 mM Hepes pH 7.2, 50 mM NaCl and 1 mM DTT and further purified by ion-exchange chromatography using HiTrap Q and HiTrap Sp columns (GE Biosciences). The IcmR-IcmQ complex eluted from the HiTrap Sp column in 900 mM NaCl. Trypsin (1000:1 w/w, Sigma) was added to the complex for 2 hours, prior to protease removal by passage over agarose-linked benzamidine. The complex was then dialyzed into 20 mM Tris, pH 8.5, 50 mM NaCl and 1 mM DTT, concentrated to 10 mg/ml and stored until used for crystallization. To prepare selenomethionine-incorporated Rm-IcmQ, pET-24a IcmQ was replaced with pET-24a IcmQ (L119M) and pQE-70 IcmR-6xHis contained an L84M mutation, as described previously (Raychaudhury et al., 2009). Transformed cells were grown in M9 minimal media (supplemented with selenomethionine) designed to inhibit the methionine biosynthesis pathway (Doublié, 1997) and IcmR-IcmQ complexes were prepared. To make full-length IcmR-IcmQ, a similar purification was performed without trypsinization. Complexes were concentrated prior to experiments with a Centriprep. 15N-labeled Qcl protein (residues 49/50 to 191) for NMR studies was expressed in M9 media supplemented with 15NH4Cl and purified as described previously (Raychaudhury et al., 2009). Unlabelled Qcl and a D151A mutant of Qcl were also purified (Raychaudhury et al., 2009).
Crystals of native Rm-IcmQ were grown at 294 K in hanging drops at a concentration of 8 mg/ml in 24% PEG 1,500, 100 mM Hepes pH 7.5, 1 mM DTT and 1% ethylene glycol. Crystals of the selenomethionine-derivative Rm-IcmQ (L119M) crystals were grown at 294 K in hanging drops (concentration 8 mg/ml) in 24% PEG 1,500, 100 mM citrate pH 5.9, 1 mM DTT and 1% EG. To cryo-protect the crystals, equal volumes of buffer consisting of 24% PEG 1,500, 0.1 M Hepes pH 7.2 or citrate pH 5.9, 50 mM DTT and 30% ethylene glycol were added to crystallization drops. Crystals were then harvested using CryoLoops (Hampton Research), frozen in liquid nitrogen and stored until data collection.
Data collection and model building
Native and derivative datasets were collected under nitrogen gas flow at 90 K on beamline X25 at the NSLS in 1° oscillations using a ADSC Q315 CCD X-ray detector and processed with HKL-2000 (Otwinowsky and Minor, 1997). Initial attempts to phase the crystals using the Rm-Qn 4-helix bundle (3FXD, 3FXE; Raychaudhury et al., 2009) by molecular replacment were unsuccessful, as resulting electron density in the Qc region was poor. Suitable initial electron density maps were calculated using autoSHARP (Vonrhein et al., 2007) with a 3.0 Å selenomethionine derivative dataset. This revealed three major peaks containing seleniums from three selenomethionine residues, including M1 and L119M from IcmQ and L84M in IcmR. Phases were improved and extended to 2.4 Å with SOLOMON (Abrahams and Leslie, 1996) with the native dataset, and the model was built using COOT (Emsley and Cowtan, 2004). For refinement, 10% of the data was excluded for calculation of R-free. The model was refined with PHENIX (Adams et al., 2010) using iterative manual modifications, as well as “omit maps.” The final model converged to an R-factor of 22.5% and an R-free of 25.9%. The Ramachandran plot revealed 94.6% and 5.4% of the residues in the preferred and allowed regions respectively. Data collection and refinement statistics are summarized in Table 1. All molecular figures were made with Chimera (Pettersen et al., 2004).
Computational modeling of the Qc-NAD+ complex and membrane binding by Rm-IcmQ
The NAD+-bound Qc was generated by starting from a rough model and then undertaking multiple, successive rounds of refinement with molecular dynamics simulations using NAMD (Phillips et al., 2005). Simulations ranged from 50 to 500 ps in length, with varying levels of restraints applied to optimize interactions between NAD+ and Qc. In the final step, NAD+ was freed completely and found to remain bound for 50 ps. With the exception of a portion of the Δ-loop, backbone atoms of Qc were restrained to their crystallographic positions during refinement. For the 50-ns simulation of IcmR bound to IcmQ without a bound ligand, a system containing the two proteins in a water box was constructed and ionized with K+ and Cl− ions to a concentration of 100 mM. Solvation was sufficient to maintain a minimum distance of 30 Å between periodic copies of the proteins. The total system size was 57,000 atoms. For membrane-binding simulations, a bilayer was constructed with POPG and POPE (1:2 ratio) to mimic the negative charge density of a bacterial membrane. Rm-IcmQ was initially placed in a random orientation ~10–15 Å distant from the membrane surface. The system contained Ca2+ and Cl−1 ions at a concentration of ~100 mM and the final system size was 130,000 atoms. The first 50 ns of equilibration was carried out using NAMD on conventional supercomputers, with the remaining 550 ns carried out using the special-purpose MD machine Anton (Shaw et al., 2009).
Additional Methods are provided in the Supplement.
Accession numbers
Coordinates for Rm-IcmQ and structure factors have been deposited in the Protein Data Bank (4EYY).
Supplementary Material
Highlights.
A crystal structure was determined of the ordered regions in an IcmR-IcmQ complex.
The C-terminal domain of IcmQ contains an NAD+ binding module with a scorpion motif.
IcmQ may be targeted to the T4bSS by binding to membranes.
IcmQ in the T4bSS of Legionella may act as a sensor of bacterial metabolic status.
Acknowledgments
The authors would like to thank Jon Vural for assistance with NMR. Anton computer time was provided by the National Resource for Biomedical Supercomputing and the Pittsburgh Supercomputing Center through Grant RC2GM093307 from the NIH, using a machine donated by D.E. Shaw Research. J.C. Gumbart was supported by grant K22-A1100927 from the NIH. R. Isberg is a member of the HHMI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D Biol Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen GVB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX, a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albe KR, Butler MH, Wright BE. Cellular concentrations of enzymes and their substrates. J Theor Biol. 1990;143:163–195. doi: 10.1016/s0022-5193(05)80266-8. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Meyer EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Choe S, Eisenberg D. Refined structure of dimeric diphtheria toxin at 2.0 Å resolution. Prot Sci. 1994;3:1444–1463. doi: 10.1002/pro.5560030911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger KH, Isberg RR. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol. 1993;7:7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Bohmer J, Jung M, Sehr P, Fritz G, Popoff M, Just I, Aktories K. Active site mutation of the C3-like ADP-ribosyltransferase from Clostridium limosum-Analysis of Glutamic Acid 174. Biochemistry. 1996;35:282–289. doi: 10.1021/bi951784+. [DOI] [PubMed] [Google Scholar]
- Christie PJ, Vogel JP. Bacterial type IV secretion, conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 2000;8:354–360. doi: 10.1016/s0966-842x(00)01792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coers J, Kagan JC, Matthews M, Nagai H, Zuckerman D, Roy CR. Identification of Icm protein complexes that play distinct roles in the biogenesis of an organelle permissive for Legionella pneumophila intracellular growth. Mol Microbiol. 2000;38:719–736. doi: 10.1046/j.1365-2958.2000.02176.x. [DOI] [PubMed] [Google Scholar]
- Cotter PA, DiRita VJ. Bacterial virulence gene regulation: an evolutionary perspective. Annu Rev Microbiol. 2000;54:519–565. doi: 10.1146/annurev.micro.54.1.519. [DOI] [PubMed] [Google Scholar]
- de Filipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 2008;4:e1000117. doi: 10.1371/journal.ppat.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S. Preparation of Selenomethionyl Proteins for Phase Determination. Methods Enzymol. 1997;276:523–550. [PubMed] [Google Scholar]
- Duménil G, Isberg RR. The Legionella pneumophila IcmR protein exhibits chaperone activity for IcmQ by preventing its participation in high-molecular weight complexes. Mol Microbiol. 2001;40:1113–1127. doi: 10.1046/j.1365-2958.2001.02454.x. [DOI] [PubMed] [Google Scholar]
- Duménil G, Montminy TP, Tang M, Isberg RR. IcmR-regulated membrane insertion and efflux by the Legionella pneumophila IcmQ protein. J Biol Chem. 2004;279:4686–4695. doi: 10.1074/jbc.M309908200. [DOI] [PubMed] [Google Scholar]
- Edwards RL, Bryan A, Jules M, Harada K, Buchrieser C, Swanson MS. Nicotinic acid modulates Legionella pneumophila gene expression and induces virulence traits. Infect Immun. 2013 doi: 10.1128/IAI.00999-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot, model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ensminger AW, Isberg RR. Legionella pneumophila Dot/Icm translocated substrates, a sum of parts. Curr Op in Microbio. 2009;12:67–73. doi: 10.1016/j.mib.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M, Segal G. A specific genomic location within the icm/dot pathogenesis region of different Legionella species encodes functionally similar but nonhomologous virulence proteins. Infect Immun. 2004;72:4503–4511. doi: 10.1128/IAI.72.8.4503-4511.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M, Zusman T, Hagag S, Segal G. Coevolution between nonhomologous but functionally similar proteins and their conserved partners in the Legionella pathogenesis system. PNAS. 2005;102:12206–12211. doi: 10.1073/pnas.0501850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields BS. The molecular ecology of Legionellae. Trends in Microbiol. 1996;4:286–290. doi: 10.1016/0966-842x(96)10041-x. [DOI] [PubMed] [Google Scholar]
- Grose JH, Joss L, Velick SF, Roth JR. Evidence that feedback inhibition of NAD kinase controls responses to oxidative stress. Proc Natl Acad Sci U S A. 2006;103:7601–7606. doi: 10.1073/pnas.0602494103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbourn KP, Shone CC, Acharya KR. A family of killer toxins, Exploring the mechanisms of ADP-ribosylating toxins. FASEB. 2006;273:4579–4593. doi: 10.1111/j.1742-4658.2006.05442.x. [DOI] [PubMed] [Google Scholar]
- Holm L, Kääriäinen S, Rosenström P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz MA. Cell-mediated immunity in Legionnaires’ Disease. J Clin Invest. 1983a;71:1686–1697. doi: 10.1172/JCI110923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz MA. The Legionnaires’ Disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J Exp Med. 1983b;158:2108–2126. doi: 10.1084/jem.158.6.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR, O’Connor TJ, Heidtman M. The Legionella pneumophila replication vacuole, making a cosy niche inside host cells. Nat Rev Microbiol. 2009;7:13–24. doi: 10.1038/nrmicro1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen R, Wang Y, Visschedyk D, Merrill AR. The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO reports. 2008;9:802–809. doi: 10.1038/embor.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz SM, Hashemi S. Electron microscopic examination of the inflammatory response to Legionella pneumophila in guinea pigs. Lab Invest. 1982;46:24–32. [PubMed] [Google Scholar]
- Koch-Nolte F, Reche P, Haag F, Bazan F. ADP-ribosyltransferases, plastic tools for inactivating protein and small molecular weight targets. J Biotech. 2001;92:81–87. doi: 10.1016/s0168-1656(01)00356-x. [DOI] [PubMed] [Google Scholar]
- Komano T, Yoshida T, Narahara K, Furuya N. The transfer region of IncI1 plasmid R64: similarities between R64 tra and legionella icm/dot genes. Molecular Microbiology. 2000;35:1348–1359. doi: 10.1046/j.1365-2958.2000.01769.x. [DOI] [PubMed] [Google Scholar]
- Lee YM, Babu CS, Chen YC, Milcic M, Qu Y, Lim C. Conserved structural motif for recognizing nicotinamide adenine dinucleotide in poly(ADP-ribose) polymerases and ADP-ribosylating toxins, implications for structure based drug design. J Med Chem. 2010;53:4038–4049. doi: 10.1021/jm1001106. [DOI] [PubMed] [Google Scholar]
- Luo ZQ, Isberg RR. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. PNAS. 2004;101:841–846. doi: 10.1073/pnas.0304916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra A, Blander SJ, Horwitz MA, Shuman HA. Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. PNAS. 1992;89:9607–11. doi: 10.1073/pnas.89.20.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JF, Roy CR, Falkow S. Analysis of Bordetella pertussis virulence gene regulation by use of transcriptional fusions in Escherichia coli. J Bacteriol. 1989;171:6345–6348. doi: 10.1128/jb.171.11.6345-6348.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey JH. Silver stain for proteins in polyacrylamide gels, a modified procedure with enhanced uniform sensitivity. Anal Biochem. 1981;117:307–310. doi: 10.1016/0003-2697(81)90783-1. [DOI] [PubMed] [Google Scholar]
- Nakano N, Kubori T, Kinoshita M, Imada K, Nagai H. Crystal structure of Legionella DotD, insights into the relationship between type IVB and type II/III secretion systems. PLoS Pathog. 2010;6(10):e1001129. doi: 10.1371/journal.ppat.1001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H, Roy CR. The DotA protein from Legionella pneumophila is secreted by a novel process that requires the Dot/Icm transporter. EMBO J. 2001;20:5962–5970. doi: 10.1093/emboj/20.21.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. PNAS. 2005;102:826–831. doi: 10.1073/pnas.0406239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio S, Zuckman-Cholon DM, Cambronne ED, Roy CR. The Legionella IcmS-IcmW protein complex is important for Dot/Icm-mediated protein translocation. Mol Microbiol. 2005;55:912–926. doi: 10.1111/j.1365-2958.2004.04435.x. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee, A Novel Method for Fast and Accurate Multiple Sequence Alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- O’Neal CJ, Jobling MG, Holmes RK, Hol WGJ. Structural basis for the activation of cholera toxin by human ARF6-GTP. Science. 2005;309:1093–1096. doi: 10.1126/science.1113398. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzym. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera, A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhury S, Farelli JD, Montminy TP, Matthews M, Ménétret JF, Duménil G, Roy CR, Head JF, Isberg RR, Akey CW. Structure and function of interacting IcmR-IcmQ domains from a type IVb secretion system in Legionella pneumophila. Structure. 2009;17:590–601. doi: 10.1016/j.str.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadosky AB, Wiater LA, Shuman HA. Identification of Legionella pneumophila genes required for growth wi thin and killing of human macrophages. Infection and Immunity. 1993;61(12):5361–5373. doi: 10.1128/iai.61.12.5361-5373.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal G, Shuman HA. Characterization of a new region required for both proper trafficking of the LCV and subsequent intracellular growth of L. pneumophila. Infect Immun. 1997;65:5057–5066. doi: 10.1128/iai.65.12.5057-5066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal G, Shuman HA. Possible origin of the Legionella pneumophila virulence genes and their relation to Coxiella burnetii. Mol Microbiol. 1999;33:667–672. doi: 10.1046/j.1365-2958.1999.01511.x. [DOI] [PubMed] [Google Scholar]
- Sexton JA, Vogel JP. Type IVB secretion by intracellular pathogens. Traffic. 2002;3:178–185. doi: 10.1034/j.1600-0854.2002.030303.x. [DOI] [PubMed] [Google Scholar]
- Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, et al. Millisecond-scale molecular dynamics simulations on Anton Proceedings of the ACM/IEEE Conference on Supercomputing (SC09) ACM Press; New York: 2009. [Google Scholar]
- Friedman JR, Jeong KC, Buford EC, Miller JL, Vogel JP. Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol Microbiol. 2006;62:1278–1291. doi: 10.1111/j.1365-2958.2006.05446.x. [DOI] [PubMed] [Google Scholar]
- Vincent CD, Friedman JR, Jeong KC, Sutherland MC, Vogel JP. Identification of the DotL coupling protein subcomplex of the Legionella Dot/Icm type IV secretion system. Mol Microbiol. 2012;85:378–391. doi: 10.1111/j.1365-2958.2012.08118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JP, Andrews HL, Wong S, Isberg R. Conjugative transfer by the virulence system of Legionella pneumophila. Science. 1998;279:873–876. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- Wilson BA, Reich KA, Weinstein BR, Collier RJ. Active-site mutations of diphtheria toxin: effects of replacing glutamic acid-148 with aspartic acid, glutamine or serine. Biochemistry. 1990;29:8643–8651. doi: 10.1021/bi00489a021. [DOI] [PubMed] [Google Scholar]
- Zhang RG, Scott DL, Westbrook ML, Nance S, Spangler BD, Shipley GG, Wesbrook EM. The three-dimensional crystal structure of cholera toxin. J Mol Biol. 1995;251:563–573. doi: 10.1006/jmbi.1995.0456. [DOI] [PubMed] [Google Scholar]
- Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One. 2011;6(3):e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.