

Abstract
The oxidative modification hypothesis of atherosclerosis, which assigns to oxidized low-density lipoproteins (LDLs) a crucial role in atherosclerosis initiation and progression, is still debated. This review examines the role played by oxidized LDLs in atherogenesis taking into account data derived by studies based on molecular and clinical approaches. Experimental data carried out in cellular lines and animal models of atherosclerosis support the proatherogenic role of oxidized LDLs: (a) through chemotactic and proliferating actions on monocytes/macrophages, inciting their transformation into foam cells; (b) through stimulation of smooth muscle cells (SMCs) recruitment and proliferation in the tunica intima; (c) through eliciting endothelial cells, SMCs, and macrophages apoptosis with ensuing necrotic core development. Moreover, most of the experimental data on atherosclerosis-prone animals benefiting from antioxidant treatment points towards a link between oxidative stress and atherosclerosis. The evidence coming from cohort studies demonstrating an association between oxidized LDLs and cardiovascular events, notwithstanding some discrepancies, seems to point towards a role of oxidized LDLs in atherosclerotic plaque development and destabilization. Finally, the results of randomized clinical trials employing antioxidants completed up to date, despite demonstrating no benefits in healthy populations, suggest a benefit in high-risk patients. In conclusion, available data seem to validate the oxidative modification hypothesis of atherosclerosis, although additional proofs are still needed.
1. Introduction
Recent postulates on atherosclerosis designate the appearance of qualitative changes on endothelial cells, triggered by “irritative” stimuli (e.g., hypertension, dyslipidemia, and cigarette smoking), as an early pathogenic event [1]. This process occurs at specific segments of the arterial tree, mainly branching points and bifurcations, characterized by disturbed laminar blood flow, probably owing to differences in arteries regional development [2] and to the loss of the atheroprotective effect of laminar shear stress [3]. In this setting, the endothelium expresses adhesion and chemotactic molecules and acquires an increased permeability to macromolecules, which modifies the composition of the subendothelial extracellular matrix. Hence, the entry of low-density lipoprotein (LDL) particles in the arterial wall followed by their retention through the binding of apolipoprotein B100 to proteoglycans of the extracellular matrix [4] is held to be a key-initiating factor in early atherogenesis [5]. The LDL particles trapped in the subintimal extracellular matrix are mildly oxidized by resident vascular cells [6]. They retain the capability of binding to the LDL receptor [6, 7] and to exert their proatherogenic effects [8–10], including stimulation of the resident vascular cells to produce monocyte chemotactic protein-1, granulocyte, and macrophage colony-stimulating factors. These molecules promote monocytes recruitment and their differentiation into macrophages, which are able to further promote the oxidation of LDLs [11] through myeloperoxidase and reactive oxygen species. Completely oxidized LDLs, characterized by an increased apolipoprotein B100 negative charge, are recognized by scavenger receptors on macrophages and internalized to form foam cells [12], the hallmark of the atherosclerotic lesion. Furthermore, macrophages play a key role in atherogenesis through their proinflammatory action, which involves the production of interleukin-1β and tumor necrosis factor (Figure 1).
Figure 1.
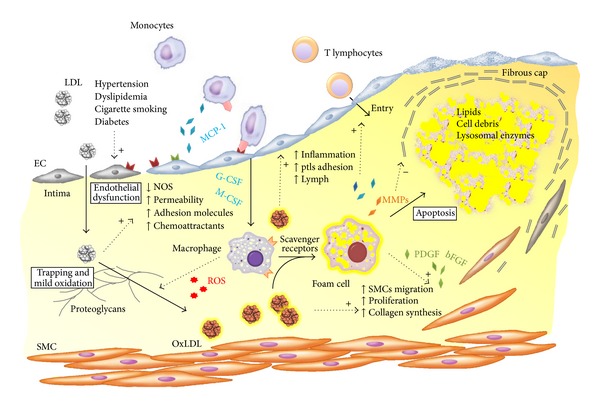
Putative pathway of oxidized low-density lipoprotein (oxLDL) in the atherogenetic process according to the oxidative hypothesis of atherosclerosis.
Other main effectors in the development of atherosclerotic lesions are smooth muscle cells (SMCs), which are recruited from the tunica media to the subendothelial space, where they proliferate in response to mediators such as the platelet-derived growth factor. SMCs residing in the tunica intima produce extracellular matrix molecules, for example, interstitial collagen and elastin, and build the fibrous cap that overlies the growing atherosclerotic plaque. The latter entails macrophage-derived foam cells, cellular debris, and extracellular lipids, which are inefficiently cleared due to defective efferocytosis and thereby form the so-called necrotic core of the plaque [13].
The atherosclerotic plaque becomes clinically manifest when it reaches an advanced stage due to its blood flow-limiting effects or its destabilization with ensuing thrombosis. Unfortunately, the latter complication, which is responsible for ischemic events, is not strictly related to the degree of stenosis at angiography [14, 15] as its occurrence stands mostly on the cellular features of the plaque and particularly on the thickness of the overlying fibrous cap [16, 17]. In fact, atherosclerotic plaques prone to rupture are characterized by accumulation of inflammatory cells, mostly at the shoulder regions. These cells degrade collagen through release of collagenolytic enzymes, mainly matrix metalloproteinases (MMPs), and also reduce its synthesis by inducing SMCs apoptosis [18].
Many excellent reviews on the current knowledge of atherosclerosis are available, but few are focused on oxidized LDLs. Hence, this review examines the role played by oxidized LDLs in atherogenesis taking into account data derived by studies based on molecular and clinical approaches.
2. Evidence Linking Oxidized LDLs to Atherosclerosis
The oxidative modification hypothesis designates the oxidative change of LDLs as a crucial, if not mandatory, step in atherogenesis [19]. This theory originated from studies demonstrating that LDLs modified by endothelial cells, transformation entailing an oxidation process [20], could be internalized and accumulated avidly by macrophages [21, 22], leading to foam cell formation, although these cells could also be generated from macrophages internalizing native LDLs from the medium through micropinocytosis [23], or by uptake of aggregated LDLs or LDL immune complexes.
Several potential mechanisms can explain how LDL oxidative modification occurs within the arterial wall in vivo. A major role has been proposed for the 12/15-lipoxygenase [24, 25] because (1) it is expressed in atherosclerotic plaques but not in normal vessels [26] and (2) its inhibition was associated with decreased oxidation of LDLs [27] and reduced atherosclerosis in animal models [25, 28, 29]. Myeloperoxidase, a heme enzyme secreted by neutrophils and monocytes/macrophages, is another suggested agent. It was found in human atherosclerotic plaques [30] and modifies LDLs, thus increasing their affinity for CD36 and SR-A [31, 32], the scavenger receptors mediating the uptake of oxidized LDLs by macrophages. Nitric oxide synthase (NOS) and nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase are other putative players as their products nitric oxide and superoxide anion, respectively, can combine to form the strong oxidizing agent peroxynitrite.
Native LDLs are internalized by macrophages at a pace too low to account for foam cells formation [33] owing to LDL receptor downregulation. Oxidative modification of LDLs increases their uptake by macrophages [20], via scavenger receptors. The latter are not downregulated in response to increased intracellular cholesterol, which explains why foam cells formation is held to occur with oxidized LDLs and not with native LDLs.
Besides contributing to the formation of lipid-laden macrophages, oxidized LDLs exhibit a wide array of biological properties, which are deemed to promote atherosclerosis.
Oxidized LDLs exert chemotactic activity for monocytes [34], stimulate their binding to endothelial cells [35] by inducing the expression of intercellular adhesion molecule-1 and vascular-cell adhesion molecule-1 [36], are mitogenic for macrophages [37], and promote their trapping in the intima, while limiting their egress from the arterial wall [38]. Hence, oxLDL is key for recruitment, activation, and proliferation of monocytes/macrophages in the arterial wall.
Oxidized LDLs increase the expression of growth factors, such as platelet-derived growth factor (PDGF) and basic fibroblast growth factor (FGF) by endothelial cells and macrophages. The former stimulates migration of SMCs [39–41], while the latter induces SMCs proliferation [42].
Oxidized LDLs stimulate collagen production by SMCs [43], thus contributing to the fibrous cap lining the atherosclerotic plaque and the expansion of the lesion size. However, they could also promote fibrous cap thinning by increasing secretion of matrix metalloproteinase 1 [44] and matrix metalloproteinase 9, decreasing production of the tissue inhibitor of metalloproteinase 1 [45], and inducing SMCs apoptosis [46]. Therefore, they can contribute to the occurrence of vulnerable plaques [16, 17]. Hence, taken together, this evidence involved oxidized LDLs in the progression of the atherosclerotic plaque and the development of its complications.
Oxidized LDLs are cytotoxic to vascular cells [47, 48] and promote their apoptosis [49, 50] with ensuing release in the subendothelial space of lipids and lysosomal enzymes, enhancing the progression of the atherosclerotic plaque [47] and the production of the necrotic core.
Oxidized LDLs stimulate platelet adhesion and aggregation, by decreasing endothelial production of nitric oxide, increasing prostacyclin production [51, 52], and stimulating the synthesis of prostaglandins and prostaglandin precursors [53]. Moreover, they decrease the secretion of the tissue-type plasminogen activator and increase that of plasminogen activator inhibitor-1 followed by a reduction of the fibrinolytic activity of endothelium [54–56]. Ultimately, they may determine vasoconstriction by inhibiting nitric oxide [57] and increasing endothelin production [58]. Taken together, these findings may explain the thrombotic complications of advanced atherosclerotic plaques.
3. In Vivo Models Supporting the Oxidized LDL Role in Atherosclerosis
Several studies were carried out in vivo in animal models where either a modulation of oxidative stress or manipulation of the scavenger receptor was undertaken, in order to prove the role of oxidized LDLs in the pathogenesis of atherosclerosis.
In an animal model of increased oxidative stress obtained through the overexpression of 15-lipoxygenase in the vascular wall, larger atherosclerotic lesions were found in LDL receptor-deficient mice [59]. However, a decreased atherosclerosis in cholesterol-fed rabbits and WHHL rabbits whose macrophages overexpressed human 15-lipoxygenase was also reported [60]. Animal models of reduced oxidative stress, instead, were obtained through knockout of oxidative stress-related genes or increasing the antioxidants: in three different knockout mouse models for 12/15-lipoxygenase, a decreased severity of atherosclerosis was seen [25, 61–64]. However, in apoE-deficient mice, the knocking out of the macrophage-specific 12/15-lipoxygenase increased the extension of atherosclerotic lesions [65].
The knockout in atherosclerosis-prone mice models of either SR-A or CD36 scavenger receptors, accounting for almost 90% of macrophage oxidized LDLs uptake [66], was demonstrated to be efficacious in decreasing the atherosclerotic burden [67, 68]. However, these results were not confirmed in another mice model with a CD36 and SR-A double knockout [69].
The results of these studies proved to be highly contradictory, due to the different animal models used, the different genetic background, and the unexpected consequences of gene deletions [70].
Finally, in spite of these conflicting data, support to the oxidative theory comes from extensive literature on the treatment of atherosclerosis-prone animals with antioxidants (reviewed by Witztum and Steinberg [71]). Most of these studies were carried out with probucol and demonstrated a protection from atherosclerotic lesions with the exception of the murine models, possibly secondary to a peculiar toxicity of this molecule in mice. In fact, in apoE-deficient mice fed with vitamin E, decreased atherosclerosis, paralleled with a decrease of aortic wall, plasma, and urinary F2 isoprostanes, a marker of oxidative stress, was observed [72].
4. Human Findings Supporting the Oxidized LDLs Role in Atherosclerosis
There is a wealth of literature on the association between oxidized LDLs and cardiovascular events. An important premise needs to be made beforehand, however, in that oxidation of LDLs induces immunogenic epitopes in their particles [73] with ensuing generation of antibodies against them (oxLDL Abs). Since these autoantibodies are detectable in the sera of the majority of patients with advanced atherosclerotic lesions [74], they can be viewed as in vivo markers of LDL oxidation. Oxidized LDLs and their involvement in atherosclerosis can therefore be assessed by two ways: (1) using murine monoclonal antibodies directed toward different oxidized LDLs epitopes and (2) determining the immunogenic response to oxidized LDLs. Both approaches have advantages and pitfalls, as reviewed in detail by Tsimikas [75].
Human studies on the association of oxidized LDLs with atherosclerosis or cardiovascular events have been highly conflicting (for rev. [76]). Some cross-sectional studies suggested a direct association of oxidized LDLs or oxLDL Abs with atherosclerosis in different vascular beds [74, 77, 78], whereas others found no association with coronary atherosclerotic burden in coronary artery disease patients [79–81]. Owing to these contradictory results, we focused on cohort studies, which are more solid and less prone to serendipitous findings, because of a lower chance for selection and recall bias [82].
Among the twenty-two cohort studies reporting on cardiovascular events, fourteen were positive [81, 83–94] (Table 1) and eight were negative [80, 95–101] (Table 2). Due to potential publication bias, the preponderance of positive results clearly does not provide a proof of the strength of the association [102]. However, it is important to highlight that three [95, 99, 100] out of eight negative studies were completed in healthy people. This carries a limitation in that the robustness of cohort studies depends on the assumption that the control group—in this case those exposed to low levels of oxidized LDLs—has features as close as possible to the group exposed to high levels of oxidized LDLs [103, 104]. Theoretically, this goal can be better accomplished in selected populations made of patients with similar risk profile, rather than in studies recruiting healthy persons. Among these negative studies, the first one reported on cardiovascular events in a large cohort of more than three thousand elderly patients who had 420 cardiovascular events after 5 years of followup [95]. Oxidized LDLs were predictive of cardiovascular events only if a multivariate analysis was not adjusted for the presence of metabolic syndrome. In the second study, which enrolled almost three thousand healthy subjects, malonyldialdehyde-LDL autoantibodies were not associated with cardiovascular events [99]. In the latter, similarly performed in a healthy population, malonyldialdehyde- and Cu-LDL autoantibodies and oxidized LDLs were not predictive of progression of carotid atherosclerosis [100].
Table 1.
Cohort studies demonstrating an association between oxidized low-density lipoprotein measurement and cardiovascular events.
| Oxidative oxLDL test | Population under study | CV endpoints | Number of events | Followup | Findings | Reference |
|---|---|---|---|---|---|---|
| OxLDL Abs 4E06 | 326 men | IMT | na | 3 years | OxLDL predicted IMT and carotid plaque progression | Wallenfeldt et al. [88] |
|
| ||||||
| OxLDL Abs 4E06 | 765 subjects | CV events | 77 CV events | 5 years | OxLDL predicted CV events | Tsimikas et al. [90] |
|
| ||||||
| OxLDL Abs 4E06 | 765 subjects | CV events | 82 CV events | 10 years | OxLDL predicted CV events | Kiechl et al. [92] |
|
| ||||||
| OxPL/apoB, AutoAbs MDA-/Cu-oxLDL | 765 subjects | CV events | 138 CV events | 15 years | OxPL/apoB predicted CV events and stroke; AutoAbs predicted CV events, stroke, and ACS | Tsimikas et al. [94] |
|
| ||||||
| MDA-LDL | 907 NIDDM | CV events, MI | 152 CV events, 43 MI | 3.7 years | MDA-LDL predicted MI and CV events | Lopes-Virella et al. [93] |
|
| ||||||
| AutoAbs Cu-oxLDL | 249 ESRD | CV mortality | 74 CV deaths | 63 months | AutoAbs predicted CV mortality | Shoji et al. [84] |
|
| ||||||
| AutoAbs Cu-oxLDL | 94 ESRD on hemodialysis | Total mortality | 32 deaths | 24 months | AutoAbs predicted mortality | Bays et al. [85] |
|
| ||||||
| AutoAbs Cu-oxLDL | 94 ESRD on hemodialysis | CV mortality | 33 CV deaths | 4 years | AutoAbs predicted CV mortality | Bays et al. [86] |
|
| ||||||
| OxLDL Abs DLH3 | 246 pts with coronary angiography | CV events: cardiac death, MI, PTCA, and CABG | 76 CV events | 38 months | OxLDL predicted CV events | Shimada et al. [87] |
|
| ||||||
| AutoAbs MDA-oxLDL | 734 IHD pts | CV mortality, MI, ACS, and CV events | 65 CV deaths, 153 CV events | 7.2 years | OxLDL predicted CV death and events | Maiolino et al. [81] |
|
| ||||||
| AutoAbs Cu-oxLDL | 74 PTCA pts, 14 ctr | Restenosis | 34 restenosis | 6 months | AutoAbs predicted restenosis | Lee et al. [107] |
|
| ||||||
| OxLDL Abs DLH3 | 102 primary PTCA pts, 86 ctr | Restenosis | 25 restenosis | 6 months | OxLDL predicted restenosis | Naruko et al. [89] |
|
| ||||||
| OxLDL Abs 4E06 | 433 ACS pts | CV death, MI | 17 CV deaths, 57 MI | 2 years | OxLDL predicted MI | Johnston et al. [91] |
|
| ||||||
| OxLDL Abs FOH1a/DLH3 | 84 CHF pts (EF < 45%), 18 ctr | CV death, CV hospitalization, and fatal arrhythmia | 26 CV events | 780 days | OxLDL predicted CV events | Tsutsui et al. [83] |
Abs: antibodies; ACS: acute coronary syndrome; AutoAbs: autoantibodies; CABG: coronary artery by-pass surgery; CHF: congestive heart failure; Crt: controls; CV: cardiovascular; ESRD: end-stage renal disease; IHD: ischemic heart disease; IMT: intima-media thickness; MI: myocardial infarction; oxLDL: oxidized low-density lipoproteins; OxPL/apoB: oxidized phospholipids on apolipoprotein B-100; PTCA: percutaneous transluminal coronary angioplasty; pts: patients.
Table 2.
Cohort studies demonstrating no association between oxidized low-density lipoprotein measurement and cardiovascular events.
| Oxidative oxLDL test | Population under study | CV endpoints | Number of events | Followup | Findings | Reference |
|---|---|---|---|---|---|---|
| OxLDL Abs 4E06 | 3033 elderly | CV events | 418 IHD, 120 MI | 3 years | OxLDL did not predict CV events at MV analysis | Holvoet et al. [95] |
|
| ||||||
| AutoAbs MDA-oxLDL | 2619 subjects | IHD (angina, ACS, and IHD death); CV events (IHD + TIA/stroke) | 151 IHD, 234 CV events | 8 years | AutoAbs did not predict CV events | Wilson et al. [99] |
|
| ||||||
| OxLDL Abs 4E06, AutoAbs MDA-/Cu-oxLDL | 919 subjects | Carotid atherosclerosis progression | na | 5 years | AutoAbs and oxLDL did not predict CV events | Mayr et al. [100] |
|
| ||||||
| AutoAbs Cu-oxLDL | 92 NIDDM, 80 ctr | CV events | 34 CV events, 15 CV deaths | 10 years | AutoAbs did not predict CV events | Uusitupa et al. [96] |
|
| ||||||
| OxLDL Abs 4E06 | 69 ESRD on hemodialysis, 33 ctr | CV events | 18 CV events | 43 months | OxLDL did not predict CV events | Lee et al. [101] |
|
| ||||||
| AutoAbs Cu-oxLDL | 415 IHD | CV events | 35 CV deaths/MI, 33 PTCA/CABG | 5 years | AutoAbs did not predict CV events | Erkkil et al. [97] |
|
| ||||||
| OxLDL Abs 4E06 | 687 PTCA pts | Restenosis, CV events | 135 restenosis, 181 CV events | 1 year | OxLDL did not predict CV events | Braun et al. [98] |
Abs: antibodies; AutoAbs: autoantibodies; CABG: coronary artery by-pass surgery; CHF: congestive heart failure; Crt: controls; CV: cardiovascular; ESRD: end-stage renal disease; IHD: ischemic heart disease; IMT: intima-media thickness; MI: myocardial infarction; NIDDM: noninsulin dependent diabetes mellitus; oxLDL: oxidized low-density lipoproteins; PTCA: percutaneous transluminal coronary angioplasty; pts: patients.
The lack of association between oxidized LDLs and cardiovascular events, possibly due to lack of statistical power, was also reported in two small cohorts of high-risk end-stage renal disease [101] and diabetes mellitus patients [96].
Other negative studies enrolling patients with coronary heart disease [80, 97, 98] were either too small [80, 97] and with a number of cardiovascular events too low to allow detection of any effect of oxidized LDLs or had an endpoint not appropriate to study atherosclerosis because most of the cardiovascular events were coronary artery restenosis (75% of total events) [98]. Moreover, it is worth highlighting that the negative study published by Tsimikas et al. [80] was on the same cohort where an association between coronary artery atherosclerosis and oxidized LDLs was demonstrated [78].
Among the positive studies four out of fourteen were carried out in a healthy cohort [88, 90, 92, 94], thus exposing them to the same considerations expressed above. Moreover, it has to be pointed out that three of these studies were completed in the same cohort, the Brunick study, at different time points of follow up, that is, 5 [90], 10 [92], and 15 years [94], and all demonstrated a predictive value of oxidized LDLs on cardiovascular events, contradicting the results on carotid artery atherosclerosis [100] on the same population.
Other studies demonstrating an association between oxidized LDLs and cardiovascular events enrolled small cohorts of either high-risk patients [83–86] or coronary artery disease patients [87, 89, 105]. Therefore these positive results could be secondary to serendipitous findings, as suggested by the opposite results on similar cohorts of end-stage renal failure patients where high oxLDL Abs titer was associated to either low [84] or high [86] cardiovascular mortality.
Two studies reported an association of oxidized LDLs with cardiovascular events in diabetics [93] and acute coronary syndrome patients [91]. Finally, using a prospective cohort study design and an unequivocal definition of the coronary artery disease phenotype, we reported the association of oxLDL Abs with cardiovascular mortality and cardiovascular events in more than 700 coronary artery disease patients [81].
In conclusion, most cohort studies reported an association between oxidized LDLs and cardiovascular events or mortality, in particular those including either a very high-risk population, that is, with end-stage renal disease and diabetes, or coronary artery disease patients. However, despite being an appealing hypothesis, the oxidation theory of atherosclerosis is not conclusively corroborated by observational studies, which have conflicting results, probably owing to the enrolment selection criteria and low statistical power.
5. Clinical Trials on Antioxidants and the Oxidized LDL Hypothesis
The oxidative theory of atherosclerosis would be conclusively proven by the beneficial effects of oxidative stress decrease on cardiovascular events. Therefore, the analysis of controlled randomized trials on antioxidant therapy in this setting is crucial. The first report on efficacy of antioxidants on cardiovascular events in coronary artery disease patients [106] was later confirmed by further studies [107–110] (Table 3), but numerous subsequent randomized clinical trials failed to prove any benefit [111–126] (Table 4). Moreover, meta-analyses on this issue are discordant [127, 128].
Table 3.
Randomized controlled trials demonstrating a beneficial effect of antioxidant therapy.
| Source | Patients | Inclusion criteria | Antioxidant agent | Dose | Route | Endpoints | Followup | Events |
|---|---|---|---|---|---|---|---|---|
| CHAOS [106] | 2002 | Angiographically demonstrated CAD | Vit E | 400/800 IU | PO | CV death + MI; nonfatal MI | 510 d | CV death: 27 vit E, 23 pl; nonfatal MI: 14 vit E, 41 pl |
| WHS [107] | 39876 | Healthy women | Vit E | 600 IU q48 h | PO | Composite endpoint (CV death, MI, and stroke) | 10.1 y | CV events: Vit E 482, pl 517; CV death: Vit E 106, pl 140; MI: Vit E 196, pl 195 |
| SPACE [108] | 196 | Hemodialysis CV disease pts | Vit E | 800 IU | PO | Composite endpoint (MI, ACS, PAD, and stroke) | 519 d | Composite endpoint: Vit E 15, pl 33; CV death: vit E 9, pl 15; nonfatal MI: vit E 8, pl 18 |
| Tepel et al. [109] | 134 | Hemodialysis CV disease pts | Acetylcysteine | 1200 mg | PO | Composite endpoint (CV death, MI, PTCA/CABG, PAD, and stroke) | 14.5 m | Composite endpoint: acetylcysteine 18, pl 33 |
| Milman et al. [110] | 1434 | Diabetes mellitus Hp 2-2 genotype | Vit E | 400 IU | PO | Composite endpoint (CV death, MI, and stroke) | 18 m | Composite endpoint: Vit E 16, pl 33 |
CAD: coronary artery disease; CV: cardiovascular; d: days; DM: diabetes mellitus; HR: hazard ratio; HTN: arterial hypertension; m: months; MI: myocardial infarction; MLD: minimal luminal diameter; na: not available; PAD: peripheral artery disease, pl: placebo; PO: per os; pts: patients; RF: risk factor; vit: vitamin; y: years.
Table 4.
Randomized controlled trials demonstrating no effect of antioxidant therapy.
| Source | Number of patients | Inclusion criteria | Antioxidant agent | Dose | Route | Endpoints | Followup | Events |
|---|---|---|---|---|---|---|---|---|
| Virtamo et al. [111] | 27271 | Male smokers | Vit E, beta-carotene | 50 mg, 20 mg | PO | Major coronary events (CV death, MI) | 6.1 y | CV events: Vit E 519, beta-carotene 547, Vit E + beta-carotene 511, and pl 534; CV death: Vit E 212, beta-carotene 235, Vit E + beta-carotene 222, and pl 238; non-fatal MI: Vit E 307, beta-carotene 312, Vit E + beta-carotene 289, and pl 296 |
|
| ||||||||
| Rapola et al. [112] | 1862 | Previous MI | Vit E, beta-carotene | 50 mg, 20 mg | PO | Major coronary events (CV death, MI) | 5.3 y | CV events: Vit E 94, beta-carotene 113, Vit E + beta-carotene 123, and pl 94 |
|
| ||||||||
| HATS [113] | 80 | CAD | Vit E/C, beta-carotene, and selenium | 800 IU, 1 g, 25 mg, and 100 g | PO | Composite endpoint (CV death, MI, and PTCA/CABG) | 38 m | CV events: antiox 9, pl 9; CV death: antiox 0, pl 1; nonfatal MI: antiox 1, pl 4 |
|
| ||||||||
| PHS II [114] | 14641 | Male physicians | Vit E/C | 400 IU 500 mg |
PO | Composite endpoint (CV death, MI, and stroke) | 8 y | CV events: Vit E 620, pl 625; Vit C 619, pl 626; CV death: Vit E 258, pl 251; Vit C 256, pl 253; MI: Vit E 240, pl 271; Vit C 260, pl 251 |
|
| ||||||||
| WACS [115] | 8171 | High CV risk women | Vit E/C, beta-carotene | 600 IU q48 h, 500 mg, and 50 mg q48 h | PO | Composite endpoint (CV death, MI, PTCA/CABG, and stroke) | 9.4 y | CV events: Vit E 708, pl 742; Vit C 731, pl 719; beta-carotene 731, pl 719; CV death: Vit E 193, pl 202; Vit C 206, pl 189; beta-carotene 211, pl 184; MI: Vit E 131, pl 143; Vit C 140, pl 134; beta-carotene 135, pl 139 |
|
| ||||||||
| PPP [116] | 4495 | Subjects ≥ 1 RF | Vit E | 300 mg | PO | Composite endpoint (CV death, MI, and stroke) | 3.6 y | CV events: Vit E 56, pl 53; CV death: Vit E 22, pl 26; MI: Vit E 22, pl 25 |
|
| ||||||||
| GISSI-prevenzione [117] | 5660 | Recent MI | Vit E | 300 mg | PO | Composite endpoint (CV death, MI, and stroke) | 3.5 y | CV events: Vit E 371, pl 414; CV death: Vit E 155, pl 193; MI: Vit E 22, pl 25 |
|
| ||||||||
| Greenberg et al. [118] | 1720 | Skin cancer | beta-carotene | 50 mg | PO | CV death | 4.3 y | CV death: beta-carotene 68, pl 59 |
|
| ||||||||
| PHS [119] | 22071 | Male physicians | beta-carotene | 50 mg q48 | PO | Malignant neoplasm; composite endpoint (CV death, MI, and stroke) | 12 y | CV events: beta-carotene 967, pl 972; CV death: beta-carotene 338, pl 313; MI: beta-carotene 468, pl 489 |
|
| ||||||||
| SUVIMAX [120] | 13017 | Adult subjects | Vit E/C, beta-carotene, selenium, and zinc | 30 mg, 120 mg, 6 mg, 100 g, and 20 mg | PO | CV ischemic events | 7.5 y | CV events: antiox 134, pl 137 |
|
| ||||||||
| HPS [121] | 20536 | CAD, PAD, DM, and HTN | Vit E/C, beta-carotene | 600 mg, 250 mg, 20 mg | PO | Composite endpoint (CV death, and MI) | 5 y | CV death: antiox 878, pl 840; MI: antiox 1063, pl 1047; CV events: antiox 2306, pl 2312 |
|
| ||||||||
| HOPE [122] | 9541 | CV disease or DM + additional CV RF | Vit E | 400 IU | PO | Composite endpoint (CV death, MI, and stroke) | 7 y | CV events: Vit E 1022, pl 985; CV death: Vit E 482, pl 475; MI: Vit E 724, pl 686 |
|
| ||||||||
| Mark et al. [123] | 3318 | Esophageal dysplasia | Vit E/C, beta-carotene | 60 IU, 180 mg, and 15 mg | PO | CV death | 6 y | CV death: antiox 22, pl 35 |
|
| ||||||||
| CARET [124] | 1845 | Exposure to asbestos or smoke | Vit E/A | 15/30 mg, 25000 IU | PO | Lung cancer incidence | 5.5 y | CV death: HR 1.26 (0.99–1.61) |
|
| ||||||||
| WAVE [125] | 213 | Postmenopausal women with CAD | Vit E/C | 400 IU, 500 mg | PO | Change in MLD | 2.8 y | CV events: antiox 10, pl 5; CV death: antiox 4, pl 2; nonfatal MI: antiox 3, pl 1 |
|
| ||||||||
| HOPE [126] | 9541 | CV disease or DM + additional CV RF | Vit E | 400 IU | PO | Composite endpoint (CV death, MI, stroke) | 4.5 y | CV events: Vit E 772, pl 739; CV death: Vit E 342, pl 328; MI: Vit E 532, pl 524 |
CAD: coronary artery disease; CV: cardiovascular; d: days; DM: diabetes mellitus; HR: hazard ratio; HTN: arterial hypertension; m: months; MI: myocardial infarction; MLD: minimal luminal diameter; na: not available; PAD: peripheral artery disease, pl: placebo; PO: per os; pts: patients; RF: risk factor; vit: vitamin; y: years.
An in depth analysis of these studies, however, highlighted that most of the negative studies were completed in either healthy or high-risk individuals, whereas results of clinical trials completed in patients with cardiovascular disease demonstrated the benefit conferred by antioxidants in some cases [106, 108, 109], with notable exceptions [112, 113, 117, 125]. The fact that treatment was likely given to the wrong patients, that is, with very low risk profile, can explain the failure of antioxidants trials in preventing cardiovascular events in the aforementioned negative reports [129].
Moreover, as in cohort studies, positive effects of antioxidants were witnessed in randomized trials enrolling very high-risk populations, as end-stage renal disease patients in hemodialysis, characterized by an increased oxidative stress, possibly secondary to hemolysis and hemoglobin-induced LDL oxidation [130, 131]. In these patients, with vitamin E supplementation, as tested in the SPACE trial randomizing patients to vitamin E or placebo [108], cardiovascular events were reduced by 54% and myocardial infarction by 70%. Accordingly, the potent antioxidant N-acetylcysteine showed a significant 40% decrease in the combined primary endpoint of cardiovascular events in another study [109]. After these rewarding results, we proposed the use of vitamin E coated dialysis membrane in these patients, which effectively reduces oxidative stress markers [132, 133]. Finally, in another high-risk population of diabetics carrying the haptoglobin 2-2 genotype, which is associated with inferior antioxidant protection [134], vitamin E was able to reduce the primary composite end point of cardiovascular death, nonfatal myocardial infarction, or stroke [110], even on top of statin therapy [135].
Thus, most controlled randomized trials involving the use of antioxidants provided negative results. However, administration of antioxidants to patients with known cardiovascular disease or with a very high-risk profile proved to be beneficial in a nontrivial number of studies.
6. Conclusions
Evidence supports on a molecular ground the oxidative hypothesis of atherosclerosis. The translation of experimental evidence in humans with studies aimed at the demonstration of the association of oxidative stress with cardiovascular events proved to be difficult and resulted in contrasting findings, particularly with administration of antioxidant therapy. However, the selection of patients either at higher risk or with cardiovascular disease provided much rewarding outcomes with numerous positive studies. It seems therefore that although this theory still needs further proofs to be definitely clarified, data available so far strengthen the pivotal role for oxidative stress in atherosclerosis.
References
- 1.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 2.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(6):1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 3.Gimbrone MA, Jr., Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Annals of the New York Academy of Sciences. 2000;902:230–240. doi: 10.1111/j.1749-6632.2000.tb06318.x. [DOI] [PubMed] [Google Scholar]
- 4.Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 5.Skålén K, Gustafsson M, Knutsen Rydberg E, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417(6890):750–754. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 6.Navab M, Berliner JA, Watson AD, et al. The Yin and Yang of oxidation in the development of the fatty streak: a review based on the 1994 George Lyman Duff memorial lecture. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(7):831–842. doi: 10.1161/01.atv.16.7.831. [DOI] [PubMed] [Google Scholar]
- 7.Berliner JA, Territo MC, Sevanian A, et al. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. Journal of Clinical Investigation. 1990;85(4):1260–1266. doi: 10.1172/JCI114562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi S-H, Harkewicz R, Lee JH, et al. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circulation Research. 2009;104(12):1355–1363. doi: 10.1161/CIRCRESAHA.108.192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(6):1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- 10.Bae YS, Lee JH, Choi SH, et al. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circulation Research. 2009;104(2):210–218. doi: 10.1161/CIRCRESAHA.108.181040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parhami F, Fang ZT, Fogelman AM, Andalibi A, Territo MC, Berliner JA. Minimally modified low density lipoprotein-induced inflammatory responses in endothelial cells are mediated by cyclic adenosine monophosphate. Journal of Clinical Investigation. 1993;92(1):471–478. doi: 10.1172/JCI116590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: recognition by receptors for acetylated low density lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(10 I):6499–6503. doi: 10.1073/pnas.78.10.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature Reviews Immunology. 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ambrose JA, Tannenbaum MA, Alexopoulos D, et al. Angiographic progression of coronary artery disease and the development of myocardial infarction. Journal of the American College of Cardiology. 1988;12(1):56–62. doi: 10.1016/0735-1097(88)90356-7. [DOI] [PubMed] [Google Scholar]
- 15.Little WC, Constantinescu M, Applegate RJ, et al. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild-to-moderate coronary artery disease? Circulation. 1988;78(5 I):1157–1166. doi: 10.1161/01.cir.78.5.1157. [DOI] [PubMed] [Google Scholar]
- 16.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(5):1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 17.Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. New England Journal of Medicine. 2011;364(3):226–235. doi: 10.1056/NEJMoa1002358. [DOI] [PubMed] [Google Scholar]
- 18.Libby P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. Journal of Lipid Research. 2009;50:S352–S357. doi: 10.1194/jlr.R800099-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol: modifications of low-density lipoprotein that increase its atherogenicity. New England Journal of Medicine. 1989;320(14):915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 20.Steinbrecher UP, Parthasarathy S, Leake DS. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(12 I):3883–3887. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of biologically modified low density lipoprotein. Arteriosclerosis. 1983;3(2):149–159. doi: 10.1161/01.atv.3.2.149. [DOI] [PubMed] [Google Scholar]
- 22.Heinecke JW, Rosen H, Chait A. Iron and copper promote modification of low density lipoprotein by human arterial smooth muscle cells in culture. Journal of Clinical Investigation. 1984;74(5):1890–1894. doi: 10.1172/JCI111609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruth HS, Jones NL, Huang W, et al. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. Journal of Biological Chemistry. 2005;280(3):2352–2360. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- 24.Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104(4):503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 25.Cyrus T, Witztum JL, Rader DJ, et al. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. Journal of Clinical Investigation. 1999;103(11):1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yla-Herttuala S, Rosenfeld ME, Parthasarathy S, et al. Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage-rich areas of atherosclerotic lesions. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(18):6959–6963. doi: 10.1073/pnas.87.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rankin SM, Parthasarathy S, Steinberg D. Evidence for a dominant role of lipoxygenase(s) in the oxidation of LDL by mouse peritoneal macrophages. Journal of Lipid Research. 1991;32(3):449–456. [PubMed] [Google Scholar]
- 28.Sendobry SM, Cornicelli JA, Welch K, et al. Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. British Journal of Pharmacology. 1997;120(7):1199–1206. doi: 10.1038/sj.bjp.0701007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bocan TMA, Rosebury WS, Mueller SB, et al. A specific 15-lipoxygenase inhibitor limits the progression and monocyte-macrophage enrichment of hypercholesterolemia-induced atherosclerosis in the rabbit. Atherosclerosis. 1998;136:203–216. doi: 10.1016/s0021-9150(97)00204-9. [DOI] [PubMed] [Google Scholar]
- 30.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. Journal of Clinical Investigation. 1994;94(1):437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Podrez EA, Febbraio M, Sheibani N, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. Journal of Clinical Investigation. 2000;105:1095–1108. doi: 10.1172/JCI8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Nicholls SJ, Rodriguez ER, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nature Medicine. 2007;13(10):1176–1184. doi: 10.1038/nm1637. [DOI] [PubMed] [Google Scholar]
- 33.Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(1):333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinn MT, Parthasarathy S, Steinberg D. Lysophosphatidylcholine: a chemotactic factor for human monocytes and its potential role in atherogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(8):2805–2809. doi: 10.1073/pnas.85.8.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frostegard J, Haegerstrand A, Gidlund M, Nilsson J. Biologically modified LDL increases the adhesive properties of endothelial cells. Atherosclerosis. 1991;90(2-3):119–126. doi: 10.1016/0021-9150(91)90106-d. [DOI] [PubMed] [Google Scholar]
- 36.Cominacini L, Garbin U, Pasini AF, et al. Antioxidants inhibit the expression of intercellular cell adhesion molecule-1 and vascular cell adhesion molecule-1 induced by oxidized LDL on human umbilical vein endothelial cells. Free Radical Biology and Medicine. 1996;22(1-2):117–127. doi: 10.1016/s0891-5849(96)00271-7. [DOI] [PubMed] [Google Scholar]
- 37.Yui S, Sasaki T, Miyazaki A, Horiuchi S, Yamazaki M. Induction of murine macrophage growth by modified LDLs. Arteriosclerosis and Thrombosis. 1993;13(3):331–337. doi: 10.1161/01.atv.13.3.331. [DOI] [PubMed] [Google Scholar]
- 38.Quinn MT, Parthasarathy S, Fong LG, Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(9):2995–2998. doi: 10.1073/pnas.84.9.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stiko-Rahm A, Hultgardh-Nilsson A, Regnstrom J, Hamsten A, Nilsson J. Native and oxidized LDL enhances production of PDGF AA and the surface expression of PDGF receptors in cultured human smooth muscle cells. Arteriosclerosis and Thrombosis. 1992;12(9):1099–1109. doi: 10.1161/01.atv.12.9.1099. [DOI] [PubMed] [Google Scholar]
- 40.Kohno M, Yokokawa K, Yasunari K, et al. Induction by lysophosphatidylcholine, a major phospholipid component of atherogenic lipoproteins, of human coronary artery smooth muscle cell migration. Circulation. 1998;98(4):353–359. doi: 10.1161/01.cir.98.4.353. [DOI] [PubMed] [Google Scholar]
- 41.Kim JG, Taylor WR, Parthasarathy S. Demonstration of the presence of lipid peroxide-modified proteins in human atherosclerotic lesions using a novel lipid peroxide-modified anti-peptide antibody. Atherosclerosis. 1999;143(2):335–340. doi: 10.1016/s0021-9150(98)00320-7. [DOI] [PubMed] [Google Scholar]
- 42.Lindner V, Lappi DA, Baird A, Majack RA, Reidy MA. Role of basic fibroblast growth factor in vascular lesion formation. Circulation Research. 1991;68(1):106–113. doi: 10.1161/01.res.68.1.106. [DOI] [PubMed] [Google Scholar]
- 43.Jimi S, Saku K, Uesugi N, Sakata N, Takebayashi S. Oxidized low density lipoprotein stimulates collagen production in cultured arterial smooth muscle cells. Atherosclerosis. 1995;116(1):15–26. doi: 10.1016/0021-9150(95)05515-x. [DOI] [PubMed] [Google Scholar]
- 44.Rajavashisth TB, Liao JK, Galis ZS, et al. Inflammatory cytokines and oxidized low density lipoproteins increase endothelial cell expression of membrane type 1-matrix metalloproteinase. Journal of Biological Chemistry. 1999;274(17):11924–11929. doi: 10.1074/jbc.274.17.11924. [DOI] [PubMed] [Google Scholar]
- 45.Xu X-P, Meisel SR, Ong JM, et al. Oxidized low-density lipoprotein regulates matrix metalloproteinase-9 and its tissue inhibitor in human monocyte-derived macrophages. Circulation. 1999;99(8):993–998. doi: 10.1161/01.cir.99.8.993. [DOI] [PubMed] [Google Scholar]
- 46.Loidl A, Claus R, Ingolic E, Deigner H-P, Hermetter A. Role of ceramide in activation of stress-associated MAP kinases by minimally modified LDL in vascular smooth muscle cells. Biochimica et Biophysica Acta. 2004;1690(2):150–158. doi: 10.1016/j.bbadis.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Schwartz CJ, Valente AJ, Sprague EA, Kelley JL, Nerem RM. The pathogenesis of atherosclerosis: an overview. Clinical Cardiology. 1991;14(2):1–16. doi: 10.1002/clc.4960141302. [DOI] [PubMed] [Google Scholar]
- 48.Cathcart MK, Morel DW, Chisolm GM., III Monocytes and neutrophils oxidize low density lipoprotein making it cytotoxic. Journal of Leukocyte Biology. 1985;38(2):341–350. doi: 10.1002/jlb.38.2.341. [DOI] [PubMed] [Google Scholar]
- 49.Sata M, Walsh K. Oxidized LDL activates Fas-mediated endothelial cell apoptosis. Journal of Clinical Investigation. 1998;102(9):1682–1689. doi: 10.1172/JCI3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardwick SJ, Hegyi L, Clare K, et al. Apoptosis in human monocyte-macrophages exposed to oxidized low density lipoprotein. Journal of Pathology. 1996;179:294–302. doi: 10.1002/(SICI)1096-9896(199607)179:3<294::AID-PATH590>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 51.Thorin E, Hamilton CA, Dominiczak MH, Reid JL. Chronic exposure of cultured bovine endothelial cells to oxidized LDL abolishes prostacyclin release. Arteriosclerosis and Thrombosis. 1994;14(3):453–459. doi: 10.1161/01.atv.14.3.453. [DOI] [PubMed] [Google Scholar]
- 52.Li L-X, Chen J-X, Liao D-F, Yu L. Probucol inhibits oxidized-low density lipoprotein-induced adhesion of monocytes to endothelial cells by reducing P-selectin synthesis in vitro. Endothelium. 1998;6(1):1–8. doi: 10.3109/10623329809053400. [DOI] [PubMed] [Google Scholar]
- 53.Armstrong DA. Oxidized LDL, ceroid, and prostaglandin metabolism in human atherosclerosis. Medical Hypotheses. 1992;38(3):244–248. doi: 10.1016/0306-9877(92)90103-j. [DOI] [PubMed] [Google Scholar]
- 54.Kugiyama K, Sakamoto T, Misumi I, et al. Transferable lipids in oxidized low-density lipoprotein stimulate plasminogen activator inhibitor-1 and inhibit tissue-type plasminogen activator release from endothelial cells. Circulation Research. 1993;73(2):335–343. doi: 10.1161/01.res.73.2.335. [DOI] [PubMed] [Google Scholar]
- 55.Gräfe M, Auch-Schwelk W, Hertel H, et al. Human cardiac microvascular and macrovascular endothelial cells respond differently to oxidatively modified LDL. Atherosclerosis. 1998;137(1):87–95. doi: 10.1016/s0021-9150(97)00258-x. [DOI] [PubMed] [Google Scholar]
- 56.Allison BA, Nilsson L, Karpe F, Hamsten A, Eriksson P. Effects of native, triglyceride-enriched, and oxidatively modified LDL on plasminogen activator inhibitor-1 expression in human endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(5):1354–1360. doi: 10.1161/01.atv.19.5.1354. [DOI] [PubMed] [Google Scholar]
- 57.Liao JK, Wee Soo Shin WSS, Wen Yee Lee WYL, Clark SL. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. Journal of Biological Chemistry. 1995;270(1):319–324. doi: 10.1074/jbc.270.1.319. [DOI] [PubMed] [Google Scholar]
- 58.Boulanger CM, Tanner FC, Bea M-L, Hahn AWA, Werner A, Luscher TF. Oxidized low density lipoproteins induce mRNA expression and release of endothelin from human and porcine endothelium. Circulation Research. 1992;70(6):1191–1197. doi: 10.1161/01.res.70.6.1191. [DOI] [PubMed] [Google Scholar]
- 59.Harats D, Shaish A, George J, et al. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(9):2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 60.Shen J, Herderick E, Cornhill JF, et al. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. Journal of Clinical Investigation. 1996;98(10):2201–2208. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cyrus T, Praticò D, Zhao L, et al. Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein e-deficient mice. Circulation. 2001;103(18):2277–2282. doi: 10.1161/01.cir.103.18.2277. [DOI] [PubMed] [Google Scholar]
- 62.George J, Afek A, Shaish A, et al. 12/15-Lipoxygenase gene disruption attenuates atherogenesis in LDL receptor-deficient mice. Circulation. 2001;104(14):1646–1650. doi: 10.1161/hc3901.095772. [DOI] [PubMed] [Google Scholar]
- 63.Zhao L, Cuff CA, Moss E, et al. Selective interleukin-12 synthesis defect in 12/15-lipoxygenase-deficient macrophages associated with reduced atherosclerosis in a mouse model of familial hypercholesterolemia. Journal of Biological Chemistry. 2002;277(38):35350–35356. doi: 10.1074/jbc.M205738200. [DOI] [PubMed] [Google Scholar]
- 64.Huo H. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–2031. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- 65.Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB Journal. 2008;22(10):3595–3606. doi: 10.1096/fj.08-112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kunjathoor VV, Febbraio M, Podrez EA, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. Journal of Biological Chemistry. 2002;277(51):49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 67.Febbraio M, Podrez EA, Smith JD, et al. Targeted disruption of the class B, scavenger receptor CD36 protects against atherosclerotic lesion development in mice. Journal of Clinical Investigation. 2000;105(8):1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suzuki H, Kurihara Y, Takeya M, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386(6622):292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 69.Moore KJ, Kunjathoor VV, Koehn SL, et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. Journal of Clinical Investigation. 2005;115(8):2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Witztum JL. You are right too! Journal of Clinical Investigation. 2005;115(8):2072–2075. doi: 10.1172/JCI26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends in Cardiovascular Medicine. 2001;11(3-4):93–102. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- 72.Praticò D, Tangirala RK, Rader DJ, Rokach J, Fitzgerald GA. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nature Medicine. 1998;4(10):1189–1192. doi: 10.1038/2685. [DOI] [PubMed] [Google Scholar]
- 73.Erkkilä AT, Närvänen O, Lehto S, Uusitupa MIJ, Ylä-Herttuala S. Autoantibodies against oxidized low-density lipoprotein and cardiolipin in patients with coronary heart disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(1):204–209. doi: 10.1161/01.atv.20.1.204. [DOI] [PubMed] [Google Scholar]
- 74.Inoue T, Uchida T, Kamishirado H, Takayanagi K, Hayashi T, Morooka S. Clinical significance of antibody against oxidized low density lipoprotein in patients with atherosclerotic coronary artery disease. Journal of the American College of Cardiology. 2001;37(3):775–779. doi: 10.1016/s0735-1097(00)01199-2. [DOI] [PubMed] [Google Scholar]
- 75.Tsimikas S. Measures of oxidative stress. Clinics in Laboratory Medicine. 2006;26(3):571–590. doi: 10.1016/j.cll.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 76.Strobel NA, Fassett RG, Marsh SA, Coombes JS. Oxidative stress biomarkers as predictors of cardiovascular disease. International Journal of Cardiology. 2011;147(2):191–201. doi: 10.1016/j.ijcard.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 77.Nishi K, Itabe H, Uno M, et al. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(10):1649–1654. doi: 10.1161/01.atv.0000033829.14012.18. [DOI] [PubMed] [Google Scholar]
- 78.Tsimikas S, Brilakis ES, Miller ER, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. New England Journal of Medicine. 2005;353(1):46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- 79.Rossi GP, Cesari M, De Toni R, et al. Antibodies to oxidized low-density lipoproteins and angiographically assessed coronary artery disease in white patients. Circulation. 2003;108(20):2467–2472. doi: 10.1161/01.CIR.0000097122.19430.48. [DOI] [PubMed] [Google Scholar]
- 80.Tsimikas S, Brilakis ES, Lennon RJ, et al. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. Journal of Lipid Research. 2007;48(2):425–433. doi: 10.1194/jlr.M600361-JLR200. [DOI] [PubMed] [Google Scholar]
- 81.Maiolino G, Pedon L, Cesari M, et al. Antibodies to malondialdehyde oxidized low-density lipoproteins predict long term cardiovascular mortality in high risk patients. International Journal of Cardiology. 2013 doi: 10.1016/j.ijcard.2012.09.165. [DOI] [PubMed] [Google Scholar]
- 82.Röhrig B, Du Prel J-B, Wachtlin D, Blettner M. Types of study in medical research—part 3 of a series on evaluation of scientific publications. Deutsches Arzteblatt. 2009;106(15):262–268. doi: 10.3238/arztebl.2009.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsutsui T, Tsutamoto T, Wada A, et al. Plasma oxidized low-density lipoprotein as a prognostic predictor in patients with chronic congestive heart failure. Journal of the American College of Cardiology. 2002;39(6):957–962. doi: 10.1016/s0735-1097(02)01721-7. [DOI] [PubMed] [Google Scholar]
- 84.Shoji T, Fukumoto M, Kimoto E, et al. Antibody to oxidized low-density lipoprotein and cardiovascular mortality in end-stage renal disease. Kidney International. 2002;62(6):2230–2237. doi: 10.1046/j.1523-1755.2002.00692.x. [DOI] [PubMed] [Google Scholar]
- 85.Bayés B, Cruz Pastor M, Bonal J, et al. Homocysteine, C-reactive protein, lipid peroxidation and mortality in haemodialysis patients. Nephrology Dialysis Transplantation. 2003;18(1):106–112. doi: 10.1093/ndt/18.1.106. [DOI] [PubMed] [Google Scholar]
- 86.Bayés B, Pastor MC, Bonal J, Foraster A, Romero R. Oxidative stress, inflammation and cardiovascular mortality in haemodialysis—role of seniority and intravenous ferrotherapy: analysis at 4 years of follow-up. Nephrology Dialysis Transplantation. 2006;21(4):984–990. doi: 10.1093/ndt/gfi294. [DOI] [PubMed] [Google Scholar]
- 87.Shimada K, Mokuno H, Matsunaga E, et al. Circulating oxidized low-density lipoprotein is an independent predictor for cardiac event in patients with coronary artery disease. Atherosclerosis. 2004;174(2):343–347. doi: 10.1016/j.atherosclerosis.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 88.Wallenfeldt K, Fagerberg B, Wikstrand J, Hulthe J. Oxidized low-density lipoprotein in plasma is a prognostic marker of subclinical atherosclerosis development in clinically healthy men. Journal of Internal Medicine. 2004;256(5):413–420. doi: 10.1111/j.1365-2796.2004.01402.x. [DOI] [PubMed] [Google Scholar]
- 89.Naruko T, Ueda M, Ehara S, et al. Persistent high levels of plasma oxidized low-density lipoprotein after acute myocardial infarction predict stent restenosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(4):877–883. doi: 10.1161/01.ATV.0000209886.31510.7f. [DOI] [PubMed] [Google Scholar]
- 90.Tsimikas S, Kiechl S, Willeit J, et al. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: five-year prospective results from the bruneck study. Journal of the American College of Cardiology. 2006;47(11):2219–2228. doi: 10.1016/j.jacc.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 91.Johnston N, Jernberg T, Lagerqvist B, Siegbahn A, Wallentin L. Oxidized low-density lipoprotein as a predictor of outcome in patients with unstable coronary artery disease. International Journal of Cardiology. 2006;113(2):167–173. doi: 10.1016/j.ijcard.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 92.Kiechl S, Willeit J, Mayr M, et al. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 Activity, and 10-year cardiovascular outcomes: prospective results from the bruneck study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(8):1788–1795. doi: 10.1161/ATVBAHA.107.145805. [DOI] [PubMed] [Google Scholar]
- 93.Lopes-Virella MF, Hunt KJ, Baker NL, et al. The levels of MDA-LDL in circulating immune complexes predict myocardial infarction in the VADT study. Atherosclerosis. 2012;224:526–531. doi: 10.1016/j.atherosclerosis.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsimikas S, Willeit P, Willeit J, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. Journal of the American College of Cardiology. 2012;60:2218–2229. doi: 10.1016/j.jacc.2012.08.979. [DOI] [PubMed] [Google Scholar]
- 95.Holvoet P, Kritchevsky SB, Tracy RP, et al. The metabolic syndrome, circulating oxidized LDL, and risk of myocardial infarction in well-functioning elderly people in the health, aging, and body composition cohort. Diabetes. 2004;53(4):1068–1073. doi: 10.2337/diabetes.53.4.1068. [DOI] [PubMed] [Google Scholar]
- 96.Uusitupa MIJ, Niskanen L, Luoma J, et al. Autoantibodies against oxidized LDL do not predict atherosclerotic vascular disease in non-insulin-dependent diabetes mellitus. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(10):1236–1242. doi: 10.1161/01.atv.16.10.1236. [DOI] [PubMed] [Google Scholar]
- 97.Erkkilä AT, Närvänen O, Lehto S, Uusitupa MIJ, Ylä-Herttuala S. Antibodies against oxidized LDL and cardiolipin and mortality in patients with coronary heart disease. Atherosclerosis. 2005;183(1):157–162. doi: 10.1016/j.atherosclerosis.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 98.Braun S, Ndrepepa G, Von Beckerath N, et al. Lack of association between circulating levels of plasma oxidized low-density lipoproteins and clinical outcome after coronary stenting. American Heart Journal. 2005;150(3):550–556. doi: 10.1016/j.ahj.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 99.Wilson PWF, Ben-Yehuda O, McNamara J, Massaro J, Witztum J, Reaven PD. Autoantibodies to oxidized LDL and cardiovascular risk. The Framingham Offspring Study. Atherosclerosis. 2006;189(2):364–368. doi: 10.1016/j.atherosclerosis.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 100.Mayr M, Kiechl S, Tsimikas S, et al. Oxidized low-density lipoprotein autoantibodies, chronic infections, and carotid atherosclerosis in a population-based study. Journal of the American College of Cardiology. 2006;47(12):2436–2443. doi: 10.1016/j.jacc.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 101.Lee YK, Lee DH, Kim JK, et al. Lysophosphatidylcholine, oxidized low-density lipoprotein and cardiovascular disease in korean hemodialysis patients: analysis at 5 years of follow-up. Journal of Korean Medical Science. 2013;28:268–273. doi: 10.3346/jkms.2013.28.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions. version 5.1.0 . The Cochrane Collaboration; 2011. [Google Scholar]
- 103.Grimes DA, Schulz KF. Bias and causal associations in observational research. The Lancet. 2002;359(9302):248–252. doi: 10.1016/S0140-6736(02)07451-2. [DOI] [PubMed] [Google Scholar]
- 104.Rochon PA, Gurwitz JH, Sykora K, et al. Reader’s guide to critical appraisal of cohort studies—1. Role and design. British Medical Journal. 2005;330(7496):895–897. doi: 10.1136/bmj.330.7496.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.George J, Harats D, Bakshi E, et al. Anti-oxidized low density lipoprotein antibody determination as a predictor of restenosis following percutaneous transluminal coronary angioplasty. Immunology Letters. 1999;68(2-3):263–266. doi: 10.1016/s0165-2478(99)00050-4. [DOI] [PubMed] [Google Scholar]
- 106.Stephens NG, Parsons A, Schofield PM, et al. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS) The Lancet. 1996;347(9004):781–786. doi: 10.1016/s0140-6736(96)90866-1. [DOI] [PubMed] [Google Scholar]
- 107.Lee I-M, Cook NR, Gaziano JM, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer. The women’s health study: a randomized controlled trial. Journal of the American Medical Association. 2005;294(1):56–65. doi: 10.1001/jama.294.1.56. [DOI] [PubMed] [Google Scholar]
- 108.Boaz M, Smetana S, Weinstein T, et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. The Lancet. 2000;356(9237):1213–1218. doi: 10.1016/s0140-6736(00)02783-5. [DOI] [PubMed] [Google Scholar]
- 109.Tepel M, Van der Giet M, Statz M, Jankowski J, Zidek W. The antioxidant acetylcysteine reduces cardiovascular events in patients with end-stage renal failure: a randomized, controlled trial. Circulation. 2003;107(7):992–995. doi: 10.1161/01.cir.0000050628.11305.30. [DOI] [PubMed] [Google Scholar]
- 110.Milman U, Blum S, Shapira C, et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype: a prospective double-blinded clinical trial. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(2):341–347. doi: 10.1161/ATVBAHA.107.153965. [DOI] [PubMed] [Google Scholar]
- 111.Virtamo J, Rapola JM, Ripatti S, et al. Effect of vitamin E and beta carotene on the incidence of primary nonfatal myocardial infarction and fatal coronary heart disease. Archives of Internal Medicine. 1998;158(6):668–675. doi: 10.1001/archinte.158.6.668. [DOI] [PubMed] [Google Scholar]
- 112.Rapola JM, Virtamo J, Ripatti S, et al. Randomised trial of α-tocopherol and β-carotene supplements on incidence of major coronary events in men with previous myocardial infarction. The Lancet. 1997;349(9067):1715–1720. doi: 10.1016/S0140-6736(97)01234-8. [DOI] [PubMed] [Google Scholar]
- 113.Brown BG, Zhao X-Q, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. New England Journal of Medicine. 2001;345(22):1583–1592. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- 114.Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the physicians’ health study II randomized controlled trial. Journal of the American Medical Association. 2008;300(18):2123–2133. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cook NR, Albert CM, Gaziano JM, et al. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the women’s antioxidant cardiovascular study. Archives of Internal Medicine. 2007;167(15):1610–1618. doi: 10.1001/archinte.167.15.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.de Gaetano G, Collaborative Group of the Primary Prevention Project Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. collaborative group of the primary prevention project. The Lancet. 2001;357:89–95. doi: 10.1016/s0140-6736(00)03539-x. [DOI] [PubMed] [Google Scholar]
- 117.GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-prevenzione trial. gruppo italiano per lo studio della sopravvivenza nell'infarto miocardico. The Lancet. 1999;354:447–455. [PubMed] [Google Scholar]
- 118.Greenberg ER, Baron JA, Karagas MR, et al. Mortality associated with low plasma concentration of beta carotene and the effect of oral supplementation. Journal of the American Medical Association. 1996;275(9):699–703. doi: 10.1001/jama.1996.03530330043027. [DOI] [PubMed] [Google Scholar]
- 119.Hennekens CH, Buring JE, Manson JE, et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. New England Journal of Medicine. 1996;334(18):1145–1149. doi: 10.1056/NEJM199605023341801. [DOI] [PubMed] [Google Scholar]
- 120.Hercberg S, Galan P, Preziosi P, et al. The SU.VI.MAX study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Archives of Internal Medicine. 2004;164(21):2335–2342. doi: 10.1001/archinte.164.21.2335. [DOI] [PubMed] [Google Scholar]
- 121.Collins R, Armitage J, Parish S, Sleight P, Peto R. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20 536 high-risk individuals: a randomised placebo-controlled trial. The Lancet. 2002;360(9326):23–33. doi: 10.1016/S0140-6736(02)09328-5. [DOI] [PubMed] [Google Scholar]
- 122.Lonn E. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. Journal of the American Medical Association. 2005;293(11):1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 123.Mark SD, Wang W, Fraumeni JF, Jr., et al. Lowered risks of hypertension and cerebrovascular disease after vitamin/mineral supplementation: the Linxian nutrition intervention trial. American Journal of Epidemiology. 1996;143(7):658–664. doi: 10.1093/oxfordjournals.aje.a008798. [DOI] [PubMed] [Google Scholar]
- 124.Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. New England Journal of Medicine. 1996;334(18):1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 125.Waters DD, Alderman EL, Hsia J, et al. Effects of hormone replacement therapy and antioxidant vitamin supplements on coronary atherosclerosis in postmenopausal women: a randomized controlled trial. Journal of the American Medical Association. 2002;288(19):2432–2440. doi: 10.1001/jama.288.19.2432. [DOI] [PubMed] [Google Scholar]
- 126.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. the heart outcomes prevention evaluation study investigators. New England Journal of Medicine. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 127.Ye Z, Song H. Antioxidant vitamins intake and the risk of coronary heart disease: meta-analysis of cohort studies. European Journal of Cardiovascular Prevention and Rehabilitation. 2008;15(1):26–34. doi: 10.1097/HJR.0b013e3282f11f95. [DOI] [PubMed] [Google Scholar]
- 128.Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. The Lancet. 2004;361:2017–2023. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- 129.Steinhubl SR. Why have antioxidants failed in clinical trials? American Journal of Cardiology. 2008;101(10):S14–S19. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 130.Ziouzenkova O, Asatryan L, Tetta C, Wratten ML, Hwang J, Sevanian A. Oxidative stress during ex vivo hemodialysis of blood is decreased by a novel hemolipodialysis procedure utilizing antioxidants. Free Radical Biology and Medicine. 2002;33(2):248–258. doi: 10.1016/s0891-5849(02)00875-4. [DOI] [PubMed] [Google Scholar]
- 131.Sevanian A, Asatryan L. LDL modification during hemodialysis markers for oxidative stress. Contributions to Nephrology. 2002;137:386–395. [PubMed] [Google Scholar]
- 132.Calò LA, Naso A, Pagnin E, et al. Vitamin E-coated dialyzers reduce oxidative stress related proteins and markers in hemodialysis—a molecular biological approach. Clinical Nephrology. 2004;62(5):355–361. doi: 10.5414/cnp62355. [DOI] [PubMed] [Google Scholar]
- 133.Calò LA, Naso A, D’Angelo A, et al. Molecular biology-based assessment of vitamin E-coated dialyzer effects on oxidative stress, inflammation, and vascular remodeling. Artificial Organs. 2011;35(2):E33–E39. doi: 10.1111/j.1525-1594.2010.01125.x. [DOI] [PubMed] [Google Scholar]
- 134.Levy AP. Application of pharmacogenomics in the prevention of diabetic cardiovascular disease: mechanistic basis and clinical evidence for utilization of the haptoglobin genotype in determining benefit from antioxidant therapy. Pharmacology and Therapeutics. 2006;112(2):501–512. doi: 10.1016/j.pharmthera.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 135.Blum S, Milman U, Shapira C, et al. Dual therapy with statins and antioxidants is superior to statins alone in decreasing the risk of cardiovascular disease in a subgroup of middle-aged individuals with both diabetes mellitus and the haptoglobin 2-2 genotype. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(3):e18–e20. doi: 10.1161/ATVBAHA.107.159905. [DOI] [PubMed] [Google Scholar]