

Significance
As people live longer, the prevalence and economic impact of neurodegenerative diseases rise. No cures or effective treatments exist for any of these fatal disorders, so identifying potential therapeutics that extend survival in animal models is vital. Many neurodegenerative illnesses have been shown to be caused by the accumulation of self-propagating misfolded proteins—the hallmark of prion diseases. We report the efficacy of 2-aminothiazoles, which were identified in cell-based screens as antiprion compounds, in extending the lives of prion-infected animals. Efficacy was limited by the development of drug-resistant prions, which is likely to have important implications for creating therapeutics in many different neurodegenerative diseases.
Keywords: drug discovery, antiprion therapeutics, bioluminescence imaging
Abstract
There is not a single pharmaceutical that halts or even slows any neurodegenerative disease. Mounting evidence shows that prions cause many neurodegenerative diseases, and arguably, scrapie and Creutzfeldt–Jakob disease prions represent the best therapeutic targets. We report here that the previously identified 2-aminothiazoles IND24 and IND81 doubled the survival times of scrapie-infected, wild-type mice. However, mice infected with Rocky Mountain Laboratory (RML) prions, a scrapie-derived strain, and treated with IND24 eventually exhibited neurological dysfunction and died. We serially passaged their brain homogenates in mice and cultured cells. We found that the prion strain isolated from IND24-treated mice, designated RML[IND24], emerged during a single passage in treated mice. Although RML prions infect both the N2a and CAD5 cell lines, RML[IND24] prions could only infect CAD5 cells. When passaged in CAD5 cells, the prions remained resistant to high concentrations of IND24. However, one passage of RML[IND24] prions in untreated mice restored susceptibility to IND24 in CAD5 cells. Although IND24 treatment extended the lives of mice propagating different prion strains, including RML, another scrapie-derived prion strain ME7, and chronic wasting disease, it was ineffective in slowing propagation of Creutzfeldt–Jakob disease prions in transgenic mice. Our studies demonstrate that prion strains can acquire resistance upon exposure to IND24 that is lost upon passage in mice in the absence of IND24. These data suggest that monotherapy can select for resistance, thus intermittent therapy with mixtures of antiprion compounds may be required to slow or stop neurodegeneration.
Mounting evidence argues that many, if not all neurodegenerative illnesses are caused by prions, including such common disorders as Alzheimer’s and Parkinson diseases (1, 2). At present, there is not a single medication that halts or even slows neurodegeneration, which is invariably fatal. Perhaps the most well understood prion diseases are those caused by the aberrantly folded prion protein (PrP) (3, 4). In humans, PrP prions cause kuru, Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker disease and fatal insomnia; in animals, they include scrapie in sheep, bovine spongiform encephalopathy, transmissible mink encephalopathy, and chronic wasting disease (CWD).
Prions are composed of proteins with an alternative conformation that is self-propagating. PrP prions consist of a disease-causing isoform designated PrPSc, which is derived from the normal cellular isoform, PrPC. The conversion of PrPC into PrPSc occurs posttranslationally by a poorly understood process. In fungi, prion formation depends on several different chaperones, but no similar auxiliary proteins have been identified in mammalian prion formation despite evidence for their existence (5).
For many years, the existence of distinct strains of prions was used as an argument against infectious proteins devoid of nucleic acid. Studies of PrPSc in two transmissible mink encephalopathy strains demonstrated differences in the properties of the protein (6), while two human mutant PrPSc prions induced different strains composed of wild-type prions on passage in transgenic (Tg) mice (7). The number of different PrPSc conformers that encipher different prion strain-specific properties was initially thought to be relatively few but that view has changed with the recognition that many different strains of synthetic prions can be created de novo and passaged in fungi and mammals (8, 9).
The antimalarial compound quinacrine was found to reduce prion accumulation in cell culture (10, 11). However, when mice were inoculated with Rocky Mountain Laboratory (RML) prions, a mouse-adapted scrapie strain, and treated with quinacrine, there was no extension in survival and drug-resistant prions emerged (12, 13). When Weissmann and co-workers added swainsonine (swa) to RML-infected cultured cells, they were able to isolate swa-resistant and swa-dependent RML strains (14). Because swa is neurotoxic, its efficacy as a therapeutic for prion disease could not be evaluated.
Unlike quinacrine, treatment with Compound B or anle138b substantially extended the survival of prion-infected mice (15–17). Prions from the brains of mice treated with Compound B showed different ratios of mono- to diglycosylated PrPSc compared with the inoculum (15), and had a different conformational stability (17), but it was not determined whether the resulting prions were resistant to further treatment with Compound B. Importantly, Compound B contains a hydrazone functionality likely under physiologic conditions to produce an aryl hydrazine, a toxic metabolite and carcinogen (18), limiting its utility as a therapeutic (17). Treatment with anle138b decreased PrPSc accumulation at early timepoints and changed the size of PrPSc aggregates, but the resulting prions did not show evidence of altered strain properties. One potential problem with anle138b is the presence of a methylenedioxyphenol group, which has been shown to produce neurotoxic or hepatotoxic effects (19).
In the work reported here, we describe the oral administration of two therapeutic lead compounds, IND24 and IND81, in wild-type (WT) mice or Tg mice that were infected with mouse-adapted scrapie, CWD or sporadic (s)CJD prions. Although treatment of RML-infected WT mice with both IND24 and IND81 was successful in extending survival up to twofold, the mice eventually exhibited neurologic dysfunction and died.
To investigate why IND24 treatment did not cure the prion-infected mice, we serially passaged brain homogenates in mice and cultured cells. An IND24-resistant prion strain, designated RML[IND24], repeatedly emerged during a single passage in mice treated with IND24. Whereas RML[IND24] was unable to infect cultured mouse neuroblastoma (N2a) cells, which can be infected with RML prions, it could infect CAD5 cells, a cell line reported to support infection with additional prion strains (20). Even after several passages in CAD5 cells in the absence of IND24, the prions remained resistant to IND24 at high concentrations. However, one passage of RML[IND24] prions in untreated mice rendered them susceptible to IND24 in CAD5 cells. Although IND24 was also efficacious in mice propagating another mouse-adapted scrapie strain (ME7) and against CWD prions, it did not extend the incubation times for sCJD prions in Tg mice. Our results demonstrate that prion strains can acquire IND24-resistance upon exposure to IND24, and efficacy is highly strain-dependent. These results have broad implications for the development of therapeutics for prion diseases and argue that intermittent therapy with mixtures of antiprion drugs is likely to be required to stop neurodegeneration.
Results
Prion Strain Nomenclature.
To distinguish between prion strains following treatment, serial passage, and infection into cells, we introduce the following nomenclature. Treatments in mice are appended in brackets to the prion strain name: for example, prions from mice infected with the RML prion strain and treated with IND24 are denoted RML[IND24]. Analogously, prions from RML-infected, vehicle-treated mice are denoted RML[V] (Fig. 1). When prions are used to infect cultured cells, the name of the cell line is appended to the prion strain–treatment, e.g., RML[IND24]-CAD5. Treatments in subsequent passages in mice are separated by semicolons. As such, prions from mice infected with RML[IND24] prions and then treated with vehicle are denoted RML[IND24;V] (Fig. 1).
Fig. 1.
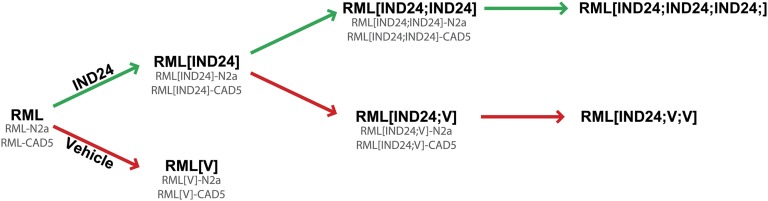
Prion strain nomenclature. Schematic of serial passages and treatment in mice, using the example of mice infected with the RML prion strain, treated with IND24 (green arrows) or vehicle (V; red arrows). Nomenclature for cells infected with each strain is denoted by appending the cell line name, as shown in gray.
2-Aminothiazoles Extended Survival.
The 2-aminothiazole (2-AMT) scaffold was identified in a screen for antiprion compounds in cell culture (21) and optimized by medicinal chemistry and pharmacokinetic studies (22, 23). Two compounds, IND24 and IND81, were selected for efficacy studies in RML-infected Tg(Gfap-luc)/FVB mice (24) expressing WT mouse PrPC. The mice were dosed with 210 mg⋅kg−1⋅d−1 of either compound from 1 d postinoculation (dpi), until they showed signs of neurologic dysfunction. Treatment with IND24 extended mean survival to 204 ± 5 dpi compared with 118 ± 1 dpi with vehicle alone (Fig. 2A and Table 1). Treatment with IND81 extended mean survival to 193 ± 3 dpi.
Fig. 2.

Effects of IND24 and IND81 treatment on RML-infected Tg(Gfap-luc)/FVB mice. (A) Kaplan–Meier survival curves for infected mice treated with vehicle (red; n = 12), IND24 (solid green; n = 12) or IND81 (dashed green; n = 11). This color scheme also applies to B–E and N–O. (B) Mean brain bioluminescence signal for mice in A, measured approximately every week; error bars represent SEM. (C) Immunoblot of PK-resistant PrP from the brains of terminal RML-infected mice treated with vehicle (RML[V]), IND24 (RML[IND24]) or IND81 (RML[IND81]). PrPSc was detected with HuM-P Fab conjugated to HRP. IND81-treated samples were loaded at 15× the protein level of other samples. Molecular weight markers of migrated protein standards represent 30 and 20 kDa. (D) Average ratio of monoglycosylated to diglycosylated PrPSc in mice treated with vehicle (n = 7), IND24 (n = 7), or IND81 (n = 9), as indicated. Error bars represent SD. ***P < 0.001, n.s., not significant, Student’s t test. (E) Conformational stability of prions from IND24-treated (n = 6), IND81-treated (n = 6), and vehicle-treated (n = 6) RML-infected mice. Samples were exposed to increasing concentrations of GdnHCl (0–4 M, as indicated) then subjected to PK digestion. Points represent average percentage of PK-resistant PrP remaining at each concentration of GdnHCl, and error bars represent SD. (F–M) Neuropathologic analysis of brain sections from RML[V] (Left) and RML[IND24] (Right) mice. (F and G) PrPSc immunohistochemistry of brainstem sections, stained using the R2 monoclonal antibody. (Scale bars in F and G: 100 μm.) (H–M) H&E stain of tissue sections from the brainstem (H and I), lateral cortex (J and K), and cerebellum (L and M). (Scale bars in H–M; 50 μm.) (N) Quantification of PrPSc deposition in multiple brain regions from RML-infected Tg(Gfap-luc)/FVB mice treated with IND24 (n = 12) or vehicle (n = 5), using the R2 monoclonal antibody. BF, basal forebrain; Bs, brainstem; CbG, cerebellum-granule cell layer; CbM, cerebellum-molecular cell layer; Cd, caudate nucleus; Hp, hippocampus; NC-L, neocortex-lateral; NC-M, neocortex-medial; Th, thalamus; Sp, septum. Error bars represent SEM. Measurements were made in intensity units (IU). (O) Quantification of vacuolation from multiple brain regions of mice treated with vehicle (n = 5) and IND24 (n = 14). Abbreviations as in N. Error bars represent SEM.
Table 1.
Incubation periods for prion-inoculated Tg(Gfap-luc)/FVB mice treated with 2-AMTs
| Inoculum | Treatment | Initiate dosing, dpi | Mean incubation period ± SEM, d | n/n0* | Survival index ± SEM |
| RML | Vehicle | — | 118 ± 1 | 12/12 | 100† |
| IND24 | 1 | 204 ± 5 | 12/12 | 173 ± 4 | |
| IND24 | 60 | 211 ± 1 | 6/6 | 179 ± 1 | |
| RML | IND81 | 1 | 193 ± 3 | 11/11 | 164 ± 3 |
| RML[IND24]‡ | Vehicle | — | 187 ± 13 | 9/9 | 158 ± 13 |
| IND24 | 1 | 166 ± 5 | 8/8 | 141 ± 5 | |
| RML[IND24]‡ | Vehicle | — | 188 ± 0 | 3/3 | 159 ± 1 |
| IND24 | 1 | 165 ± 5 | 5/5 | 140 ± 5 | |
| RML[IND24;IND24] | IND24 | 1 | 158 ± 0 | 5/5§ | 134 ± 1 |
| RML[IND24;V] | Vehicle | — | 113 ± 5 | 7/7 | 96 ± 5 |
| ME7 | Vehicle | — | 126 ± 2 | 5/5 | 100† |
| IND24 | 1 | 214 ± 4 | 9/9 | 170 ± 4 | |
| IND24 | 60 | 158 ± 5 | 7/7 | 125 ± 5 | |
| ME7[IND24] | Vehicle | — | 123 ± 1 | 8/8 | 98 ± 2 |
| IND24 | 1 | 183 ± 3 | 6/6 | 145 ± 3 |
n, number of ill mice; n0, number of inoculated mice.
Vehicle-treated control used to calculate survival index.
Inocula from two independent treatment experiments.
All mice showed clinical signs of prion disease, but were also suffering from an intercurrent peritonitis when euthanized.
Incubation time is dependent on the prion strain, titer, route of inoculation, and host model. Due to the small differences observed in disease onset within cohorts of prion-infected mice, extension in survival can appear highly significant regardless of whether these differences fall within the range of interexperimental variability (25). Moreover, many statistical tests fail to account for the magnitude of any survival extension. To enable the direct comparison of prion inoculation experiments and provide a quantitative metric for extension in survival, we calculated the survival index: the mean survival time of compound-treated mice divided by the mean survival time of the vehicle-treated control mice on first passage, multiplied by 100. For example, a survival index of 200 shows that treatment extended mean survival time twofold. Continuous treatment with IND24 or IND81 in WT mice resulted in survival indices of 173 ± 4 and 164 ± 3, respectively (Table 1).
Bioluminescence Imaging Predicted Efficacy.
Using Tg(Gfap-luc)/FVB mice, we previously showed that the luciferase reporter under the control of the glial fibrillary acidic protein (GFAP) promoter (24) could function as a surrogate for PrPSc prion production (26). Vehicle treatment of prion-infected mice resulted in an increase in the bioluminescence imaging (BLI) signal at ∼60 dpi that plateaued after ∼75 dpi (Fig. 2B). The BLI signal in both IND24- and IND81-treated mice did not begin to increase until ∼125 dpi and plateaued at ∼150 dpi (Fig. 2B). Thus, the suppressed BLI signal predicted efficacy beginning at 60 dpi.
2-AMT Treatment Altered the Characteristics of Prions in Brains of Mice.
In addition to survival time, a battery of metrics was used to characterize prion strains, including molecular weight of proteinase K (PK)-resistant PrP, as determined by gel electrophoresis; glycoform ratio, which is PK-resistant mono- to diglycosylated PrP; conformational stability, defined as the resistance to denaturation by guanidine hydrochloride (GdnHCl); spatial distribution of PrPSc and vacuolation in the brain; the ability to infect different cell lines; and drug resistance measured by clearance of PrPSc in infected cells, determined by gel electrophoresis.
PK-resistant PrPSc was present, but accumulation was variable in the brains of all ill mice and consistently lower in IND81-treated animals (Fig. 2C; IND81-treated samples loaded at 15× the concentration of other samples). There was no difference in molecular weight of PK-resistant PrP in treated samples. When comparing the relative intensities of the mono- and diglycosylated PrPSc glycoforms, RML[V] was characterized by a ∼1.6-fold stronger monoglycosylated band. This ratio was significantly different from RML[IND24] and RML[IND81], which exhibited more equal levels of mono- and diglycosylated PrPSc (Fig. 2 C and D and Table S1; P < 0.001, Student’s t test). We also observed a shift in PrPSc conformational stability for RML[IND24] and RML[IND81], with respective [GdnHCl]1/2 values of 1.8 ± 0.3 M and 1.6 ± 0.2 M, compared with 1.5 ± 0.1 M for RML[V]; this increased conformational stability was statistically significant for RML[IND24] (P = 0.04, Student’s t test) but not for RML[IND81] (Fig. 2E). Because IND24 administration consistently led to longer survival times compared with IND81, IND24 was used for subsequent experiments.
Neuropathological studies of IND24-treated animals showed that PrPSc deposition was both variable and less uniform compared with vehicle-treated controls (Fig. 2 F, G, and N). A significant increase in PrPSc levels was observed in the septum (P = 0.03), basal forebrain (P = 0.05), brainstem (P = 0.004), and cerebellum (P = 0.001) in RML[IND24] (n = 12) mice compared with RML[V] (n = 5) mice (Student’s t test with Bonferroni correction; Fig. 2 F, G, and N). Treatment with IND24 also changed the distribution of vacuolation: it was decreased in the cortex and brainstem (Fig. 2 H–K) of RML[IND24] mice and increased in the cerebellum compared with RML[V] mice (Fig. 2 L and M). Quantification of vacuolation over 10 brain regions showed significant differences between RML[V] (n = 5) and RML[IND24] (n = 14) mice in the caudate nucleus (P = 0.001), brainstem (P = 0.03), and the granule cell layer of the cerebellum (P = 0.01, Student’s t test with Bonferroni correction; Fig. 2O). These distinct biochemical and neuropathological differences between RML[V] and RML[IND24] suggested that a unique prion strain emerged with 2-AMT treatment.
RML[IND24] Was Resistant to IND24 in Cell Culture.
We then sought to determine the infectability of RML[IND24] prions in cultured cells, and subsequent resistance to IND24. The infection of N2a cell cultures with RML or RML[V] was successful: five of six attempts showed persistent prion infection (Table S2). In contrast, only one of eight attempts using RML[IND24] produced a persistent prion infection of N2a cells. When RML[IND24]-N2a or RML[V]-N2a cells were treated with 0.5 µM IND24 for 5 d, most of the PrPSc was cleared (Fig. 3 A and B), analogous to RML-N2a cells (23). When the infected N2a cells were analyzed biochemically by immunoblotting and quantification of the PrP glycoforms, we found similar PrP patterns regardless of the source of RML prion infection (Fig. S1A). These results suggest that N2a cells cannot propagate the RML[IND24] strain, and that the one successful infection was likely the result of a residual population of IND24-sensitive prions in the inoculum.
Fig. 3.

Infection of cultured cells with IND24-treated prions demonstrates IND24-resistance. PK-resistant PrPSc in N2a (A and B) and CAD5 (C and D) cells infected with RML[V] (A and C) and RML[IND24] (B and D) treated with 0–20 μM IND24, as indicated. PrPSc was assayed by immunoblot using HRP-conjugated HuM-D13 Fab. Images are representative of three replicates. Molecular weight markers of migrated protein standards represent 30 and 20 kDa. (E) Average ratio of monoglycosylated to diglycosylated PrPSc in CAD5 cells infected with RML[V] (n = 6) or RML[IND24] (n = 6). Error bars represent SD. ***P < 0.001, Student’s t test.
To determine whether our observations were limited to N2a cells, we repeated these studies with CAD5 cells, a neuronal mouse cell line reported to be permissive to infection with a wider range of prion strains compared with N2a cells (20). Infection of the CAD5 cells was successful in six of six attempts with prions in brain homogenates from either RML[V]- or RML[IND24]-infected mice (Table S2). Whereas IND24 eliminated most PrPSc in RML[V]-CAD5 cells (Fig. 3C), it was ineffective in RML[IND24]-CAD5 cells, even with 20 µM of IND24 (Fig. 3D). Quantification of the PrP glycoforms in CAD5 cells showed that cells infected with RML[V] had a predominantly monoglycosylated band, whereas RML[IND24] infection resulted in a significantly lower mono-/diglycosylated PrP ratio (P < 0.001, Student’s t test), similar to their respective inocula (compare Figs. 3E and 2D). These results indicate that RML[IND24] prions possessed an altered cell infection profile and were IND24-resistant in CAD5 cells, consistent with a strain change as a result of treatment.
IND24 Treatment in Mice Was Required to Maintain IND24 Resistance.
To characterize further the RML[IND24] prions, brain homogenates from RML[IND24] mice were passaged in Tg(Gfap-luc)/FVB mice that received either IND24 or vehicle. The mean survival time for the second passage in the presence of IND24 was 166 ± 5 dpi and 165 ± 5 dpi in two independent replicate experiments (Fig. 4A and Table 1). This survival time was ∼40 d shorter than that found on the initial passage of RML prions in mice treated with IND24 or IND81 (Fig. 2A; P < 0.001, Log-rank test).
Fig. 4.

Serial passage of prions from IND24-treated mice. (A) Kaplan–Meier survival curves for passage of RML[IND24] and RML[IND24;IND24] into Tg(Gfap-luc)/FVB mice treated with IND24. Mice were inoculated with either RML[IND24] (green, n = 8; and light green, n = 5) or RML[IND24;IND24] (dotted green, n = 5). (B) Mean brain bioluminescence signal for mice in A, measured approximately every week; error bars represent SEM. Color scheme as in A. (C) PK-resistant PrPSc in CAD5 cells infected with RML[IND24;IND24] treated with 0–20 µM IND24, as indicated. PrPSc was assayed by immunoblot using HRP-conjugated HuM-D13 Fab. Images are representative of three replicates. Molecular weight markers of migrated protein standards represent 30 and 20 kDa. (D) Kaplan–Meier survival curves for passage of RML[IND24] or RML[IND24;V] into Tg(Gfap-luc)/FVB mice treated with vehicle. Mice were inoculated with either RML[IND24] (red, n = 8; and light red, n = 3) or RML[IND24;V] (dotted red, n = 7). (E) Mean brain bioluminescence signal for mice in D, measured approximately every week; error bars represent SEM. Color scheme as in D. (F–K) PK-resistant PrPSc in CAD5 cells (F) and N2a cells (G) infected with RML[IND24;V] then treated with 0–4 µM IND24, as indicated. (H–K) PK-resistant PrPSc in CAD5 cells infected with RML (H and J) and RML[IND24;IND24] (I and K) treated with 0–10 µM Compound B (H and I) or quinacrine (J and K). In F–K, PrPSc was assayed by immunoblot using HRP-conjugated HuM-D13 Fab. Images are representative of three replicates. Molecular weight markers of migrated protein standards represent 30 and 20 kDa.
Consistent with this shortening of the incubation time, the BLI signal increased rapidly beginning at ∼60 dpi and plateaued at ∼90 dpi (Fig. 4B). The BLI profiles of the animals in the two independent experiments were virtually superimposable. The prions in these mice were designated RML[IND24;IND24] and they showed the same ratio of mono-/diglycosylated PrP as RML[IND24] (Fig. S1B).
RML[IND24;IND24] prions were unable to infect N2a cells (no infections in six attempts), but did infect CAD5 cells (six of six; Table S2). The RML[IND24;IND24]-CAD5 prions were resistant to IND24 at concentrations as high as 20 µM (Fig. 4C). Immunoblotting showed that the PrP glycoform ratios of RML[IND24;IND24]-CAD5 were similar to RML[IND24]-CAD5 (Fig. S1C). These results indicate that RML[IND24;IND24] maintained the characteristics of RML[IND24], including IND24-resistance, when passaged in the presence of IND24. Further serial passage of RML[IND24;IND24] in the presence of IND24, giving RML[IND24;IND24;IND24], resulted in a mean survival time of 158 ± 0 dpi and an indistinguishable BLI profile to RML[IND24;IND24] (Fig. 4 A and B).
RML[IND24] prions passaged in the absence of IND24 resulted in incubation periods of 187 ± 13 dpi and 188 ± 0 dpi in two independent experiments (Table 1 and Fig. 4D). The BLI profiles of the animals in the two independent experiments were nearly identical: the BLI signal increased rapidly beginning at ∼125 dpi and continued to rise until the mice were euthanized at ∼190 dpi (Fig. 4E). In contrast to the small difference in mean survival times between RML[IND24;V] and RML[IND24;IND24], the BLI profiles argue that the strains are distinct (compare solid lines in Fig. 4 B and E). Consistent with the BLI data, the mono-/diglycosylated ratio of RML[IND24;V] was intermediate between RML[IND24] and RML[V] (Fig. S1B and Table S1).
In contrast to RML[IND24;IND24], RML[IND24;V] was able to infect both N2a (six of six) and CAD5 cells (six of six) (Table S2). Whereas RML[IND24;IND24]-CAD5 was highly IND24-resistant (Fig. 4C), both RML[IND24;V]-N2a and RML[IND24;V]-CAD5 showed substantially reduced PrPSc levels with all IND24 doses tested (Fig. 4 F and G). Similar to RML[IND24;V] in mice, glycoform ratios for RML[IND24;V]-CAD5 were between RML[V]-CAD5 and RML[IND24]-CAD5 (Fig. S1C). Taken together, these results argue that during the passage of RML[IND24] in vehicle-treated mice, there was a progressive reversion of the RML[IND24] phenotype to RML[V]. A second passage of RML[IND24;V] into Tg(Gfap-luc)/FVB mice administered vehicle resulted in a mean survival time of 113 ± 5 dpi, and a BLI profile and glycoform ratio indistinguishable from the parent RML strain (compare dotted lines in Figs. 4 D and E and 2 A and B; Fig. S1B, Table 1, and Table S1).
Cell Infection Experiments Demonstrated Multidrug Resistance.
Replication of the IND24-resistant prion strain in cells allowed us to challenge this strain with additional antiprion compounds. RML-CAD5 or RML[IND24;IND24]-CAD5 were treated with 0–10 µM Compound B or 0–10 µM quinacrine for 5 d, and PrPSc was assayed by immunoblot (Fig. 4 H–K). Cells infected with RML proved highly susceptible to both Compound B (Fig. 4H) and quinacrine (Fig. 4J), consistent with previous reports (11, 15). Compound B treatment up to 10 µM was ineffective against the IND24-resistant strain (Fig. 4I), and the effective dose of quinacrine against RML[IND24;IND24] (Fig. 4K) was ∼5× higher than for RML-infected CAD5 cells. These results demonstrated the cross-resistance of the RML[IND24;IND24] strain to compounds with distinct chemical structures.
IND24 Was Effective Against ME7 Prion Infection.
To determine whether IND24 treatment was effective against other PrP prion strains, we treated Tg(Gfap-luc)/FVB mice infected with ME7 prions, an independently derived mouse-adapted sheep scrapie strain (27). ME7-infected Tg(Gfap-luc)/FVB mice were dosed with IND24 at 210 mg⋅kg−1⋅d−1 from 1 dpi and continued until mice showed signs of neurologic dysfunction. Mean survival times were 214 ± 4 dpi for IND24-treated mice and 126 ± 2 dpi for vehicle-treated mice, giving a survival index of 170 ± 4 (Table 1), similar to Tg(Gfap-luc)/FVB mice inoculated with RML prions.
Because IND24 treatment altered the strain characteristics of RML, we examined whether or not the ME7 strain characteristics also changed upon treatment with IND24. Based on the molecular weight of PK-resistant PrPSc, glycoform ratios, and stability to GdnHCl denaturation, ME7[IND24] and ME7[V] were indistinguishable (Fig. 5 A–C). Although PrPSc deposition and vacuolation in most regions showed no difference between IND24- and vehicle-treated, ME7-inoculated mice, we found a significant decrease in PrPSc deposition in the septum (P = 0.003) and the caudate nucleus (P = 0.002, Student’s t test with Bonferroni correction; Fig. S2A), and in vacuolation in the septum (Fig. S2B; P < 0.001) of IND24-treated animals.
Fig. 5.

Effects of IND24 treatment on ME7 prion infection. (A) PK-resistant PrP from the brains of terminal animals infected with ME7 prions and treated with vehicle (red) or IND24 (green), as indicated. PrPSc was probed using HRP-conjugated HuM-P Fab. Molecular weight markers of migrated protein standards represent 30 and 20 kDa. This color scheme applies to B, C, and H. (B) Average ratio of monoglycosylated to diglycosylated PrPSc in ME7-infected Tg(Gfap-luc)/FVB mice treated with vehicle (n = 4) or IND24 (n = 5). Error bars represent SD. n.s. = not significant. (C) Conformational stability of ME7 prions from IND24-treated (n = 4) and vehicle-treated (n = 4) Tg(Gfap-luc)/FVB mice. Samples were exposed to increasing concentrations of GdnHCl (0–4 M, as indicated), then subjected to PK digestion. Points represent average percentage of PK-resistant PrP remaining at each concentration of GdnHCl, and error bars represent SD. (D–G) PK-resistant PrP in CAD5 cells (D and E) and LD9 cells (F and G) infected with ME7 (D and F) or ME7[IND24] (E and G), and then treated with 0–20 µM of IND24, as indicated. PK-resistant PrP was assayed by immunoblot using HRP-conjugated HuM-D13 Fab. Images are representative of three replicates. Molecular weight markers of migrated protein standards represent 30 and 20 kDa. (H) Mean survival of mice infected with RML or ME7, then treated with vehicle and/or IND24, initiated at the timepoints indicated. Number of animals per experiment are shown. Error bars represent the SEM. ***P < 0.001, n.s. = not significant, Log-rank test.
Because ME7 does not infect N2a cells (28) but does infect CAD5 cells and a mouse fibroblast cell line termed “LD9” (20), we attempted to infect these cell lines with ME7, ME7[V], or ME7[IND24]. All infection attempts for ME7 (n = 5), ME7[V] (n = 3), ME7[IND24] (n = 6) were successful for CAD5 and LD9 cells (Table S2). IND24 treatment reduced PrPSc similarly in both ME7- and ME7[IND24]-infected CAD5 and LD9 cells (Fig. 5 D–G). Glycoform ratios showed no significant differences between cells infected with ME7[V] or ME7[IND24] within each cell line (Fig. S2C).
Efficacy of Delayed Treatment Was Strain-Dependent.
To test the effects of delayed treatment on survival time, we treated RML- and ME7-infected animals with IND24 starting at 60 dpi. The survival of RML-infected animals with delayed treatment was extended to 211 ± 1 dpi, a survival index of 179 ± 1, indistinguishable from treatment starting from 1 dpi (Fig. 5H and Table 1). Conversely, ME7-infected animals treated from 60 dpi survived 158 ± 5 dpi, giving a survival index of 125 ± 5, a significant decrease compared with treatment starting at day 1 (P < 0.001, Log-rank test; Fig. 5H and Table 1). These results suggest that RML and ME7 prions propagate with different kinetics.
IND24 Was Ineffective Against Human sCJD Prions.
To determine whether IND24 might extend survival in Tg mouse models, IND24 was administered at 210 mg⋅kg−1⋅d−1 beginning at 1 dpi. Tg4053 mice overexpressing WT mouse PrP (29) were inoculated with RML prions and treated with IND24, increasing survival from 51 ± 3 dpi to 112 ± 4 dpi, producing a survival index of 220 ± 14 (Fig. 6A and Table 2). This result demonstrates that IND24 is effective against transgene-expressed PrP.
Fig. 6.

Effects of IND24 treatment in Tg mouse models of prion disease. Kaplan–Meier survival curves of mice treated with vehicle (red) or IND24 (green) in (A) Tg4053 mice infected with RML (IND24: n = 9, vehicle: n = 8); (B) Tg1014 mice infected with RML (IND24: n = 9, vehicle: n = 4); (C) Tg1014 mice infected with sCJD(MM1) (IND24: n = 9, vehicle: n = 7); (D) Tg2669 mice infected with sCJD(MM1) (IND24: n = 7, vehicle: n = 9); (E and F) Tg1014 mice in E (IND24: n = 6, vehicle: n = 8) and Tg2669 mice in F (IND24: n = 8, vehicle: n = 9) infected with a 1:1,000 dilution of sCJD(MM1); (G) Tg152 mice infected with sCJD(VV2) (IND24: n = 9, vehicle: n = 9); and (H) Tg(ElkPrP)12584 mice infected with CWD(Elk) (IND24: n = 6, vehicle: n = 9).
Table 2.
Incubation periods for prion-inoculated transgenic mice treated from day 1 with IND24
| Inoculum | Host* | Treatment | Mean incubation period ± SEM, d | n/n0† | Survival index ± SEM |
| RML | Tg4053 | Vehicle | 51 ± 3 | 8/8 | 100‡ |
| IND24 | 112 ± 4 | 9/9 | 220 ± 14 | ||
| RML | Tg1014 | Vehicle | 107 ± 5 | 4/4 | 100‡ |
| IND24 | 217 ± 8 | 9/9 | 203 ± 12 | ||
| sCJD(MM1) | Tg1014 | Vehicle | 78 ± 1 | 7/7 | 100‡ |
| IND24 | 73 ± 2 | 9/9 | 94 ± 2 | ||
| sCJD(MM1) | Tg2669+/+ | Vehicle | 143 ± 2 | 9/9 | 100‡ |
| IND24 | 144 ± 2 | 6/7 | 101 ± 2 | ||
| sCJD(MM1) low dose | Tg1014 | Vehicle | 113 ± 4 | 8/8 | 100‡ |
| IND24 | 115 ± 5 | 6/6 | 102 ± 5 | ||
| sCJD(MM1) low dose | Tg2669+/+ | Vehicle | 176 ± 3 | 9/9 | 100‡ |
| IND24 | 182 ± 4 | 8/8 | 103 ± 3 | ||
| sCJD(VV2) | Tg152+/+ | Vehicle | 193 ± 2 | 9/9 | 100‡ |
| IND24 | 185 ± 9 | 9/9 | 96 ± 5 | ||
| CWD(Elk) | Tg12584+/+ | Vehicle | 108 ± 3 | 9/9 | 100‡ |
| IND24 | 237 ± 0 | 6/6 | 219 ± 3 |
Homozygous transgenes are denoted +/+.
n, number of ill mice; n0, number of inoculated mice.
Vehicle-treated control used to calculate survival index.
Next, we treated Tg1014 mice expressing a chimeric human/mouse PrP (30), which are capable of replicating both mouse and human prions. Following RML-infection, IND24 treatment produced a survival index of 203 ± 12 (Fig. 6B and Table 2). However, when we inoculated sCJD(MM1) prions (the most common human prion strain) into Tg1014 mice, mean survival times were 73 ± 2 dpi and 78 ± 1 dpi for IND24-treated and vehicle-treated Tg1014 mice, respectively, giving a survival index of 94 ± 2 (Fig. 6C and Table 2), indicating that efficacy is dependent on the inoculating strain, not the PrP sequence of the host.
We then infected Tg2669 mice, expressing WT human PrP with the M129 polymorphism, with sCJD(MM1) prions and treated them with IND24. The mean survival times of these mice were 144 ± 2 dpi and 143 ± 2 dpi for IND24 and vehicle treatments, respectively, giving a survival index of 101 ± 2 (Fig. 6D and Table 2). To test the possibility that IND24 treatment could have a limited effect masked by the high prion titer in the inoculum, we treated Tg1014 and Tg2669 mice infected with a 1:1,000 dilution of the original sCJD(MM1) inoculum. Again, we observed no increase in survival in IND24-treated animals compared with vehicle-treated mice: respective survival indexes were 102 ± 5 and 103 ± 3 for Tg1014 and Tg2669 mice (Fig. 6 E and F and Table 2). Finally, we inoculated sCJD(VV2) prions (the second most common human prion strain) into Tg(HuPrP,V129)152 mice that express WT human PrP with the V129 polymorphism (31, 32). IND24 treatment failed to produce any extension in lifespan: the survival index of treated mice was 96 ± 5 (Fig. 6G and Table 2). Together, these results indicate that IND24 treatment had no effect on the survival of Tg mice infected with the two most common human CJD prion strains.
IND24 Treatment Prolonged the Lives of Animals Infected with CWD.
To assess whether IND24 antiprion efficacy was limited to mouse-adapted scrapie strains, we inoculated Tg mice expressing elk PrPC with brain homogenate from an elk with CWD, and treated them with IND24. Vehicle-treated mice survived 108 ± 3 dpi, but mice treated with IND24 at 210 mg⋅kg−1⋅d−1, starting the day after inoculation, survived 237 ± 0 dpi, giving a survival index of 219 ± 3 (Fig. 6H and Table 2).
Discussion
The lack of any drugs that halt or even slow neurodegeneration is becoming a crisis. In developed and developing countries, people over 65 y of age represent the fastest growing segment of these populations. For most neurodegenerative diseases, the greatest risk factor is age. In Alzheimer’s disease, the risk increases from 5% at age 65, to 20% at age 75, and to 40% at age 85 (33).
We began with an unbiased screening of compounds to identify those that substantially lower PrPSc levels. Among such compounds were several 2-AMTs (21). Several dozen analogs were subsequently synthesized to optimize the potency as well as the drug-like properties of these 2-AMTs (22, 23). In the studies reported here, two 2-AMTs significantly extended the lives of mice infected with mouse-adapted scrapie or with CWD prions (Figs. 2A, 5H, and 6H) but the mice eventually showed signs of progressive neurologic dysfunction, which required them to be euthanized.
To understand why mice treated with 2-AMTs eventually succumbed to CNS disease, we serially passaged the prions from RML-infected mice that received the AMT IND24, termed RML[IND24], into mice and cultured cells. The ability of cultured CAD5 cells to propagate both IND24-sensitive and -resistant RML prions demonstrated the emergence of an IND24-resistant prion strain, hence the treated mice eventually developed neurodegeneration. However, the mechanism by which RML prions acquired IND24 resistance remains unknown. Two passages of RML[IND24] prions in mice receiving only vehicle seemed to restore the original properties of RML prions and render them susceptible to IND24.
While screening and medicinal chemistry optimization resulting in IND24 and IND81 were performed in RML-infected N2a cells (21–23), IND24-resistant prions were not able to propagate in N2a cells, thereby making the cells unable to predict the emergence of resistance. CAD5 cells were similarly sensitive to IND24 following RML infection, so even though they are able to propagate the IND24-resistant strain, they would not have been more predictive of resistance than N2a cells. Although IND24 treatment of ME7-infected CAD5 and LD9 cells resulted in minimal reductions in PrPSc levels, it did prolong the survival of ME7-infected mice from 126 dpi to 214 dpi (a survival index of 170), similar to IND24 treatment of RML-infected mice. Biochemical analysis and infection experiments indicated that the resulting ME7[IND24] prion strain seems largely unaltered. Taken together, these observations imply that the extension in survival by IND24 treatment of ME7 infection may be caused by impaired propagation of prions rather than through strain adaptation. These results underscore the limitations of cell culture for predicting the results of animal studies, but raise the possibility that some compounds with limited efficacy in cells may prove more effective in animals.
Despite the efficacy against RML and ME7 prion strains, IND24 treatment of multiple Tg mouse lines inoculated with the two most common CJD prion strains did not lead to any extensions in survival, highlighting the need to validate therapeutics intended for human use in the most relevant animal models. This resistance seems to be an inherent property of both the MM1 and VV2 CJD strains, but the lack of a cell culture system for the propagation of human prions limits this interrogation. Although the results in humanized mice were disappointing, IND24 was effective in extending the lifespan of mice infected with CWD. This extension marks the successful treatment of a naturally occurring prion strain and demonstrates the validity of cell-based drug discovery for identifying compounds to treat neurodegenerative disorders.
We have generated additional compounds based on the 2-AMT and other chemical scaffolds that are more potent than IND24 and IND81 (34–36); whether these compounds will show efficacy against human prions remains to be determined. However, the rapid acquisition of IND24 resistance and the strain-specific efficacy of the compounds, combined with the observation that some CJD patients may harbor more than one prion strain (reviewed in ref. 37), indicate that a single therapeutic agent may not be sufficient to treat individuals with prion diseases.
Some investigators have argued for the strain selection mechanism to explain prion mutagenesis (38, 39). Our previous studies on serial passage of variant CJD and synthetic prions in Tg mice led to similar conclusions: the selection pressure was for more rapid prion formation, i.e., shorter survival times (9, 30, 40). It is unknown whether IND24 selected for a subpopulation of prions in the RML inoculum that replicates more slowly or induced a conformational change. The RML[IND24] prions in brain homogenates prepared from ill mice remained resistant to IND24 in CAD5 cells for more than 20 passages. These observations suggest that IND24 resistance was unlikely to require that the compound be bound to the RML[IND24] prions. Despite all of the data presented here, the mechanisms by which IND24 prolonged incubation times and led to resistance remain to be established.
The studies reported here have broad implications for the development of pharmacotherapeutics for many neurodegenerative diseases. Deciphering the mechanisms of drug resistance and building mixtures of medications to treat these dreaded disorders would seem paramount in view of the widening spectrum of disease thought to be caused by prions. Importantly, the frequency of IND24-resistant prions emerging appears to approach 100% (Tables 1 and 2). All of the animals receiving RML, ME7, or CWD prions showed a doubling of the incubation time when treated with IND24 or IND81, but none survived longer, indicating that efficacy of these 2-AMTs was limited. Our findings portend the emergence of a discipline where drug resistance is governed by the “laws” of conformational mutagenesis of protein structure instead of nucleic acid changes that give rise to amino acid substitutions.
Materials and Methods
Materials.
CAD5 and LD9 cells were a gift from Charles Weissmann. Minimal essential medium (MEM) with Earle’s salts; Dulbecco’s Modified Eagle Medium (DMEM) high glucose 1× with 4.5 g/L d-glucose and l-glutamine and without sodium pyruvate; Opti-MEM; FBS; penicillin-streptomycin (pen-strep; each at 10,000 units per mL); GlutaMAX; DTT (DTT; 0.5 M 10×); 4× loading buffer and PK were purchased from Invitrogen. Enzyme-free cell dissociation buffer was purchased from Millipore. IND24 and IND81 were synthesized by ChemVeda.
Animal Husbandry and Tissue Preparation.
All protocols were approved by the University of California San Francisco Animal Care and Use Committee. All mice were bred in our facility. Tg(Gfap-luc)/FVB and Tg4053 mice were on the FVB/N genetic background; Tg1014, Tg2669, Tg152, and Tg12584 mice were on the FVB/Prnp0/0 background. Female mice were used for all experiments, due to problems observed with long-term dosing of male mice (17). Mice were inoculated intracerebrally with 30 µL of 1% brain homogenate containing RML, ME7, sCJD(MM1), sCJD(VV2), or CWD prions, unless otherwise stated, when 30 µL of 0.001% sCJD(MM1) brain homogenate (generated by serial tenfold dilution), was used. RML and ME7 prions were serially passaged multiple times in WT CD1 mice. sCJD(MM1) and sCJD(VV2) inocula were prepared from histopathologically confirmed patients, in which the PrP ORF sequence had been confirmed free of mutation, and were previously reported (32). The CWD isolate used has previously been reported as isolate Elk1 (41). Animals were observed every day for signs of neurologic disease. Mice were diagnosed with prion disease when they exhibited three or more neurologic signs characteristic of prion disease, as described (42). Upon diagnosis, mice were euthanized and their brains collected. The left hemisphere was snap-frozen on dry ice, then stored at −80 °C for biochemical analysis; the right hemisphere was fixed for pathological analysis.
Tg Mouse Models.
Tg mice expressing mouse PrP, chimeric human (Hu)/mouse PrP, HuPrP with the V129 polymorphism, and ElkPrP, denoted Tg4053, Tg1014, Tg152, and Tg12584 respectively, have been previously reported (29–32, 41). Hemizygous Tg4053 and Tg1014 mice, and homozygous Tg152 and Tg12584, the latter designated with “+/+”, were used for experiments. For Tg2669 mice expressing human PrP with the M129 polymorphism, a construct was amplified by PCR from the plasmid pcDNA3.HuPrP using the following primers: 5′-CTATATGTCGACACCATGAACTGGGCACCCGCA-3′ (forward) and CTATATGTCGACCTAGGGCCGCAGCAGCCCCA-3′ (reverse). Following digestion with SalI, the PCR product was inserted into SalI-digested cos. Tet cosmid vector, which drives neuronal expression of proteins using the hamster Prnp promoter. Preparation of the vector and microinjection were performed as previously reported (43). Three lines of Tg mice were obtained, one of which (Tg2669) was selected for further analysis. Tg2669 mice were maintained on an FVB/Prnp0/0 knockout background. Expression levels of HuPrP in the brains of hemizygous Tg2669 mice were determined to be four to five times higher than that of WT FVB mice. The validity of the transgene sequence in Tg2669 mice was verified by DNA sequencing. Homozygous Tg2669+/+ mice were generated by intercrossing hemizygous mice and confirmed by backcrossing.
Long-Term Dosing of Compounds.
Dosing was performed as previously reported (17). Briefly, compounds were dissolved in 100% (vol/vol) PEG400 and stored at −4 °C until use. Samples were diluted to make final dosing concentrations of 1.25% (vol/vol) PEG400 in the initial RML infection studies, and 0.125% (vol/vol) PEG400 in all other studies in a complete rodent liquid diet (Bio Serv, product no. F1256SP). For vehicle-treated mice, pure PEG400 was diluted appropriately into the previously prepared rodent liquid diet. The amount of food served was based on the number of animals in each cage and replaced by new batches every 2–4 d. All dosing was performed at 210 mg⋅kg−1⋅d−1.
Bioluminescence Imaging.
Tg(Gfap-luc) mice were a generous gift from Caliper Life Sciences, from which a homozygous line was generated (44). Mice were imaged weekly using an IVIS Lumina imaging system (Caliper Life Sciences). In brief, mice were injected intraperitoneally with a solution of d-luciferin potassium salt (Gold Biotechnology) in PBS, pH 7.4 (Invitrogen), receiving 50 mg d-luciferin per kilogram body weight. The mice were then anesthetized using an isoflurane–oxygen gas mix and imaged after 10 min for 30–60 s under anesthesia. The bioluminescence signal was quantified from images using Living Image 4.2 software (Caliper).
Conformational Stability Assay.
Samples (10 µL of 20% brain homogenate) were incubated with GdnHCl, in increments of 0.5 M from 0 to 4 M, at 22 °C for 2 h. The samples were subsequently diluted with lysis buffer to a final concentration of 0.4 M of GdnHCl. PK was added at a final concentration of 50 µg/mL and the samples were incubated at 37 °C for 1 h. Phenylmethylsulfonyl fluoride (final concentration of 1 mM) was added to stop the PK digestion. Samples were ultracentrifuged at 100,000 × g for 1 h, and the pellets resuspended in loading buffer. The samples were subsequently analyzed by immunoblot.
Immunoblotting.
For immunoblot analysis, brain samples were digested with 20 µg/mL PK at 37 °C for 1 h. PrPSc pellets from 500 µL of PK-digested samples were collected and resuspended with SDS sample running buffer and 0.1 M DTT. Samples were heated to 100 °C for 10 min and run in a 4–12% Tris⋅glycine SDS gel (Invitrogen). The gel was transferred to PVDF membrane using an iBlot (Invitrogen) and the membrane was blocked with 5% milk for 30 min. The membranes were subsequently incubated overnight with horseradish peroxidase (HRP)-conjugated HuM-D13 or HuM-P Fabs and washed 3× with Tris-buffered saline with Tween-20 for 10 min before developing with an enhanced chemiluminescence reagent (Invitrogen).
Bands on immunoblots were quantified using ImageJ 1.46r (45). Total intensity for an area encompassing each band was measured, and average background was subtracted. Normalized intensities were totaled for each lane. For the conformational stability assay, a ratio was calculated comparing each lane to the control (0 M GdnHCl), and these ratios were averaged across multiple samples. For glycoform proportions, a ratio was calculated for each normalized band intensity compared with the sum of the three isoforms, and these ratios were averaged across multiple samples.
Pathology.
Brains were immersion-fixed in 10% (vol/vol) buffered formalin. Fixed samples were paraffin-embedded, sectioned, and then stained with H&E, or processed by immunohistochemistry for PrP with the R2 monoclonal antibody, as described previously (46). Images were taken at 20× and 40× magnifications using the SpotFlex camera and program on the Leica DM-IRB microscope. Neuropathological analyses were performed on an average of 5–11 mice to qualify for statistical calculations. PrPSc concentrations were determined by a visual assessment of the intensity of immunohistological staining, multiplied by the area of the brain region occupied by staining, and divided by the cross-sectional area of the brain region. The intensity of immunostaining was given a value of 1 for barely detectable, 2 for mild, 3 for moderate, and 4 for strongly intensive. Vacuolation in a brain region was estimated by the percent of an area occupied by vacuoles (vacuolation score), multiplied by the fraction of a brain region containing vacuoles, and divided by the cross-sectional area of the brain region. PrPSc accumulation and vacuolation were evaluated by a single rater (S.J.D.).
Cell Culture.
Cells were infected with RML or ME7 prions as previously described (47). All cell lines were maintained at 37 °C in their respective media in 100-mm plates and fed with fresh media every 2–3 d. N2a cells were maintained in DMEM supplemented with 1% GlutaMAX, 100 units pen-strep and 10% FBS; LD9 cells maintained in MEM supplemented with 90 units pen-strep and 9% FBS; CAD5 cells maintained in Opti-MEM supplemented with 90 units pen-strep and 9% FBS. Dividing cells were plated at 10% confluency and split 1:10 when they become confluent. Cells were incubated with IND24 at the doses indicated over 5 d with refeeding. IND24 was diluted in DMSO, with 50-µL final volume in 10 mL of media. At the end of the 5-d treatment period, lysis buffer (100 mM Tris⋅HCl, pH 8.0; 150 mM NaCl; 0.5% Nonidet P-40; 0.5% sodium deoxycholate) was added to cells, and protein concentrations were measured using a bicinchoninic acid protein assay kit (Fisher). Protein extracts were normalized to 1 mg/mL total protein with lysis buffer before PK digestion. Lysates were digested with 20 µg/mL PK at 37 °C for 1 h. The reaction was stopped with phenylmethylsulfonyl fluoride (2 mM final concentration). The samples were ultracentrifuged at 100,000 × g for 1 h, resuspended in loading buffer, and subjected to immunoblot analysis.
Statistical Analysis.
All calculations were performed with Stata 11 (Stata Corp.), or Prism 5 (GraphPad Software Inc.).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants AG002132, AG010770, AG031220, and AG021601, as well as by gifts from the Sherman Fairchild Foundation, Fight for Mike Homer Program, and Rainwater Charitable Foundation. We also thank Dr. Charles Weissmann (Scripps Research Institute, Jupiter, FL) for the gift of CAD5 and LD9 cells; Sunny Grillo, Manuel Elepano, and the staff at the Hunter’s Point animal facility for bioluminescence imaging and animal management; and Ms. Hang Nguyen for expert editorial assistance.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317164110/-/DCSupplemental.
References
- 1.Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336(6088):1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prusiner SB. Shattuck lecture—neurodegenerative diseases and prions. N Engl J Med. 2001;344(20):1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 4.Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64(6):783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 5.Perrier V, et al. Dominant-negative inhibition of prion replication in transgenic mice. Proc Natl Acad Sci USA. 2002;99(20):13079–13084. doi: 10.1073/pnas.182425299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68(12):7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Telling GC, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274(5295):2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442(7102):585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 9.Ghaemmaghami S, et al. Conformational transformation and selection of synthetic prion strains. J Mol Biol. 2011;413(3):527–542. doi: 10.1016/j.jmb.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol. 2000;74(10):4894–4897. doi: 10.1128/jvi.74.10.4894-4897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korth C, May BCH, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA. 2001;98(17):9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barret A, et al. Evaluation of quinacrine treatment for prion diseases. J Virol. 2003;77(15):8462–8469. doi: 10.1128/JVI.77.15.8462-8469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghaemmaghami S, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5(11):e1000673. doi: 10.1371/journal.ppat.1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oelschlegel AM, Weissmann C. Acquisition of drug resistance and dependence by prions. PLoS Pathog. 2013;9(2):e1003158. doi: 10.1371/journal.ppat.1003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawasaki Y, et al. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol. 2007;81(23):12889–12898. doi: 10.1128/JVI.01563-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner J, et al. Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson's disease. Acta Neuropathol. 2013;125(6):795–813. doi: 10.1007/s00401-013-1114-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu D, et al. Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J Pharmacol Exp Ther. 2013 doi: 10.1124/jpet.113.205799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwu JR, et al. Aminyl and iminyl radicals from arylhydrazones in the photo-induced DNA cleavage. Bioorg Med Chem. 2004;12(10):2509–2515. doi: 10.1016/j.bmc.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 19.Murray M. Mechanisms of inhibitory and regulatory effects of methylenedioxyphenyl compounds on cytochrome P450-dependent drug oxidation. Curr Drug Metab. 2000;1(1):67–84. doi: 10.2174/1389200003339270. [DOI] [PubMed] [Google Scholar]
- 20.Mahal SP, et al. Prion strain discrimination in cell culture: The cell panel assay. Proc Natl Acad Sci USA. 2007;104(52):20908–20913. doi: 10.1073/pnas.0710054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghaemmaghami S, May BCH, Renslo AR, Prusiner SB. Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol. 2010;84(7):3408–3412. doi: 10.1128/JVI.02145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gallardo-Godoy A, et al. 2-Aminothiazoles as therapeutic leads for prion diseases. J Med Chem. 2011;54(4):1010–1021. doi: 10.1021/jm101250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silber BM, et al. Pharmacokinetics and metabolism of 2-aminothiazoles with antiprion activity in mice. Pharm Res. 2013;30(4):932–950. doi: 10.1007/s11095-012-0912-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu L, et al. Non-invasive imaging of GFAP expression after neuronal damage in mice. Neurosci Lett. 2004;367(2):210–212. doi: 10.1016/j.neulet.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 25.Tamgüney G, et al. Genes contributing to prion pathogenesis. J Gen Virol. 2008;89(Pt 7):1777–1788. doi: 10.1099/vir.0.2008/001255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamgüney G, et al. Measuring prions by bioluminescence imaging. Proc Natl Acad Sci USA. 2009;106(35):15002–15006. doi: 10.1073/pnas.0907339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zlotnik I. Spread of scrapie by contact in mice. J Comp Pathol. 1968;78(1):19–22. doi: 10.1016/0021-9975(68)90108-4. [DOI] [PubMed] [Google Scholar]
- 28.Bosque PJ, Prusiner SB. Cultured cell sublines highly susceptible to prion infection. J Virol. 2000;74(9):4377–4386. doi: 10.1128/jvi.74.9.4377-4386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson GA, et al. Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci USA. 1994;91(12):5690–5694. doi: 10.1073/pnas.91.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giles K, et al. Human prion strain selection in transgenic mice. Ann Neurol. 2010;68(2):151–161. doi: 10.1002/ana.22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Telling GC, et al. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA. 1994;91(21):9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giles K, et al. Identification of I137M and other mutations that modulate incubation periods for two human prion strains. J Virol. 2012;86(11):6033–6041. doi: 10.1128/JVI.07027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Arch Neurol. 2003;60(8):1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 34.Li Z, et al. 2-aminothiazoles with improved pharmacotherapeutic properties for treatment of prion disease. ChemMedChem. 2013;8(5):847–857. doi: 10.1002/cmdc.201300007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, et al. Discovery and preliminary structure–activity relationship of arylpiperazines as novel, brain-penetrant antiprion compounds. ACS Med Chem Lett. 2013;4:397–401. doi: 10.1021/ml300472n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, et al. Towards optimization of arylamides as novel, potent, and brain-penetrant antiprion lead compounds. ACS Med Chem Lett. 2013;4(7):647–650. doi: 10.1021/ml300454k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parchi P, et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: An updated classification. Acta Neuropathol. 2009;118(5):659–671. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327(5967):869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weissmann C, Li J, Mahal SP, Browning S. Prions on the move. EMBO Rep. 2011;12(11):1109–1117. doi: 10.1038/embor.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghaemmaghami S, et al. Convergent replication of mouse synthetic prion strains. Am J Pathol. 2013;182(3):866–874. doi: 10.1016/j.ajpath.2012.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tamgüney G, et al. Transmission of elk and deer prions to transgenic mice. J Virol. 2006;80(18):9104–9114. doi: 10.1128/JVI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlson GA, et al. Linkage of prion protein and scrapie incubation time genes. Cell. 1986;46(4):503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 43.Scott MR, Köhler R, Foster D, Prusiner SB. Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci. 1992;1(8):986–997. doi: 10.1002/pro.5560010804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stöhr J, et al. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci USA. 2012;109(27):11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11(7):36–42. [Google Scholar]
- 46.Muramoto T, et al. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an alpha-helix. Nat Med. 1997;3(7):750–755. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]
- 47.Butler DA, et al. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol. 1988;62(5):1558–1564. doi: 10.1128/jvi.62.5.1558-1564.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.