

Summary
Acute lymphoblastic leukaemia (ALL) is seen in both children and adults, but its incidence peaks between ages 2 and 5 years. The causation of ALL is considered to be multi-factorial, including exogenous or endogenous exposures, genetic susceptibility, and chance. The survival rate of paediatric ALL has improved to approximately 90% in recent trials with risk stratification by biologic features of leukaemic cells and response to therapy, therapy modification based on patient pharmacodynamics and pharmacogenomics, and improved supportive care. However, innovative approaches are needed to further improve survival while reducing adverse effects. While most children can be cured, the prognosis of infants and adults with ALL remains poor. Recent genome-wide profiling of germline and leukaemic cell DNA has identified novel submicroscopic structural genetic alterations and sequence mutations that contribute to leukaemogenesis, define new ALL subtypes, influence responsiveness to treatment, and may provide novel prognostic markers and therapeutic targets for personalized medicine.
Introduction
An estimated 6000 new cases (3400 male and 2600 female) of acute lymphoblastic leukaemia (ALL) are diagnosed annually in the US.1 Patients are predominantly children; approximately 60% of cases occur at age <20 years.2–5 The survival rate of childhood ALL is approaching 90% (appendix, figure 1),4,6 although the treatment of infants and adults needs improvement.5,7 Here we review recent advances in the epidemiology, pathobiology, and clinical management of ALL.
Epidemiology
ALL, like cancer in general, is likely to arise from interactions between exogenous or endogenous exposures, genetic (inherited) susceptibility, and chance (figure 1). These factors account for the approximately 1 in 2000 risk of childhood (0–15 years) ALL. The challenge is to identify the relevant exposures and inherited genetic variants and to decipher how and when they contribute to the multi-step natural history of ALL from its initiation (usually in utero) through its largely covert evolution to overt disease.8 The task is complicated by the relative rarity of ALL and the existence of biologically distinct subtypes that may not share common aetiological mechanisms.9 For example, ALL in infants (<12 months) is usually associated with MLL gene rearrangement, and the remarkably high concordance rate in monozygotic twins (approaching 100% in those with a single or monochorionic placenta) suggests that leukaemogenesis is largely complete at birth.10 In contrast, non-MLL-rearranged B-ALL has a peak incidence between 2 and 5 years and a concordance rate of 10–15%, suggesting that, although initiation in utero is common, other “promotional” exposures are probably required for the later emergence of disease.10
Figure 1.
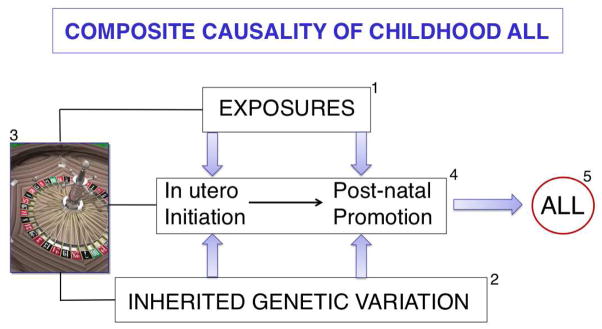
Composite causality of childhood ALL. 1) Exogenous (eg, infection) and endogenous (eg, inflammation, oxidative stress) exposures, 2) normal allelic variation in inherited genes, and 3) chance all play roles in 4) the covert natural history of childhood ALL,8,10,11 leading ultimately to 5) overt disease and clinical diagnosis. Cancer causation is riddled with chance,157 for example, incidental “external” exposure, incidental damage to a relevant oncogene in a relevant cell (stem or progenitor cell), and chance events at conception involving parental gene shuffling and recombination.
Exposures contributing to ALL
The topic of exposures and their role remains contentious. Epidemiological and case-control studies have found more than twenty candidate exposures that contribute to childhood ALL,11 but very few are based on reproducibly significant data or are biologically plausible. Some are of public concern, especially ionising and non-ionising (eg, electromagnetic field [EMF]) radiation. Ionising radiation is an established causal exposure for childhood ALL, as evidenced by the impact of the 1945 atomic bombs in Japan12 and by the modestly but significantly elevated risk caused by X-ray pelvimetry during pregnancy.13 These exposures are not currently relevant, although some argue that background or natural radiation could be significant.14 EMF exposures (eg, power lines) have been particularly controversial, and concern or confusion may be exacerbated by uncritical news reporting. Meta-analysis suggests a modestly elevated risk at high levels (>0.2μT),15 but the reliability of this finding is uncertain. It is impossible to prove that EMF never causes ALL, but at most, it might be involved in only a small minority of cases. Further, its credibility as a causal (promotional, late stage?) exposure is weakened by the lack of any known biological mechanism or credible modelling in vitro or in vivo.
Infection was the first suggested causal exposure for childhood ALL16 and remains the strongest candidate. Two specific hypotheses have been proposed, often referred to by their eponymous titles, and both are supported by epidemiological data (table 1).11,17,18 Both postulate that ALL results from an abnormal response to a common infection. The hypotheses differ in detail but are not mutually exclusive as explanations of rare time/space clusters of leukaemia19,20 or of ALL in the general community.11 There is no evidence to date of a unique or single transforming virus in ALL,11 as is the case of leukaemia in some animal species.21 Instead, it is likely that ALL is promoted indirectly by an abnormal or dysregulated immune response to one or more common infections (viral or bacterial) in a susceptible individual. Influenza viruses are plausible candidates.22 Susceptible children would be defined as having minimal prior exposure to infection during infancy and have a persistent in utero-generated pre-leukaemic clone11 plus a variable degree of genetic susceptibility, as described below. It is important to continue exploring the possible biological mechanisms of infectious promotion of ALL,23 as they could eventually lead to prophylactic interventions.
Table 1.
Infectious hypotheses for childhood ALL
| Hypotheses | Concepts | Timing | Agent | Evidence | |
|---|---|---|---|---|---|
| The Kinlen “population mixing” hypothesis17,19 | Unusual demographic mixing of susceptible and infected individuals | Perinatal? | Single novel virus? | Increased incidence (transient, 2x) in multiple situations of population mixing or clusters19 | |
| The Greaves “delayed infection” hypothesis11,18 | Delayed exposure to common infections in childhood under-exposed as infants | Later “promotional” or triggering event11 | One or (more probably) several common infections (bacterial or viral) | Reduced risk of ALL from day care attendance in infancy140,141 | |
Inherited susceptibility
There is very little evidence of inherited predisposition to ALL via highly penetrant mutations in children or adults.24 The high concordance in identical twin children has a non-genetic explanation (blood cell chimaerism).10 Infants born with constitutive trisomy 21 or Down’s syndrome are, however, at substantially elevated risk of ALL (~40-fold at age 0–4 years) and acute myeloid leukaemia (AML).25 The apparent absence of familial clustering of ALL or of greatly elevated sibling risk (maximum 2-fold) does not, however, argue against inherited susceptibility. Previous attempts to identify inherited genetic susceptibility to ALL have used a candidate gene approach. These studies found interesting potential candidates in, for example, folate metabolism and the immune response, but most were statistically underpowered or not consistently reproducible.26 More recent genome-wide association studies (GWAS) of childhood ALL compare the whole genome (usually remission blood-derived DNA) in a large series of patients to that in an ethnically matched control group, focusing on single-nucleotide polymorphisms in DNA sequences (with ~80% genome coverage).27–29 These studies demand hundreds or thousands of patients and controls, and, given the thousands of pan-genome markers being compared, a robust result is generally regarded as requiring a P value <10−7. They also require validation in a second, independent series of patients and independent confirmation by another group of researchers. To date, common allelic variants in four genes (IKZF1, ARID5B, CEBPE, and CDKN2A) have been significantly and consistently associated with childhood ALL (appendix, table 1).27–29 It is likely that variants in other genes have odds ratios (or impact) of <1.2, but these can be identified only in very large cooperative studies (3000–5000 cases). The gene variants have additive effects, so that an individual inheriting one copy of a variant will have risk elevated at ~50% above the norm while one who inherits all four variants in homozygous form (or double dose) would have an approximately10-fold increase in the risk of ALL. The overall conclusion from these studies is that the risk of childhood ALL is influenced by co-inheritance of multiple low-risk variants. The lower incidence of B-ALL with high hyperdiploidy in African-American children in the US may at least partially reflect lower prevalence of the ARID5B risk allele (appendix, table 1).30 Inherited allelic variation may also affect response to treatment.31
A striking finding in these GWAS is the nature of the four genes implicated (appendix, table 1). Their products do not impinge directly on potential exposure pathways, such as immune or liver detoxification pathways, but rather are key regulators of blood cell development, proliferation, and differentiation. Further, acquired or somatic mutants of each of these genes have been detected in cases of ALL. This fact suggests that the inherited gene variants contribute to the intrinsic vulnerability of stem or precursor blood cells to transforming events either in utero at initiation and/or with subsequent post-natal promotion and clonal evolution (figure 1). There is suggestive evidence that the risk-conferring gene variants have lowered expression of their product,27 but their functional aspects remain to be explored. These data provide valuable new insights into the aetiology of childhood ALL, but, at the moment, they do not carry strong enough predictive value to merit screening of the whole population of children.
Pathobiology
Genetic basis of ALL
High-resolution profiling of genetic alterations has transformed our understanding of the genetic basis of ALL. It has been known for several decades that the majority of childhood ALL cases harbour gross chromosomal alterations (figure 2).32 In B-ALL, these include high hyperdiploidy with non-random gain of at least five chromosomes (including X, 4, 6, 10, 14, 17, 18, and 21), hypodiploidy with fewer than 44 chromosomes, and recurring translocations, including t(12;21)(p13;q22), encoding ETV6-RUNX1 (TEL-AML1); t(1;19)(q23;p13), encoding TCF3-PBX1 (E2A-PBX1); t(9;22)(q34;q11), encoding BCR-ABL1; MLL rearrangement involving 11q23 with a wide range of partner genes; and rearrangement of MYC into antigen receptor gene loci. Dysregulation of TAL1, TLX1, TLX3, and LYL1, particularly by rearrangement into T cell antigen receptor loci, is common in T-ALL. These alterations are of key importance in both the pathogenesis and clinical management of ALL (figure 3). Many chromosomal rearrangements disrupt genes that regulate normal hematopoiesis and lymphoid development (eg, RUNX1 and ETV6), activate oncogenes (eg, MYC), or constitutively activate tyrosine kinases (eg, ABL1). Several of these alterations are significantly associated with outcome, particularly in B-ALL, and are used in risk stratification. Notably, high hyperdiploidy and ETV6-RUNX1 are associated with favorable outcome, whereas low hypodiploidy, and MLL rearrangement (especially in infants and adults) are associated with a dismal prognosis, in both children and adults.
Figure 2.

Cytogenetic and molecular genetic abnormalities in childhood ALL. ALL with rearrangement of CRLF2 but without the BCR-ABL1-like transcriptional profile rarely presents with other classifying karyotypic alterations but may be seen with high hyperdiploidy. The dicentric cases may have a range of heterogeneous translocations, including classifying translocations (eg, ETV6-RUNX1). iAMP21, intrachromosomal amplification of chromosome 21.
Figure 3.

Schema of genetic pathogenesis of B-ALL at diagnosis and relapse. HSC, hematopoietic stem cell; RAG, recombinase activating gene.
However, many of these alterations alone do not induce leukaemia in experimental models, and many cases of ALL lack a gross chromosomal alteration, indicating that additional submicroscopic genetic alterations contribute to leukaemogenesis. High-resolution microarray profiling of DNA copy number alterations (deletions and gains) and sequencing have identified several novel structural genetic alterations and sequence mutations that define new subtypes of ALL, contribute to leukaemogenesis, and influence treatment responsiveness; in several cases, these are being explored as novel prognostic markers and therapeutic targets.33–38 Importantly, many ALL subtypes are characterized by distinct constellations of structural genetic alterations that together drive establishment of the leukaemic clone.
Submicroscopic genetic alterations in ALL
More than 50 regions of recurring DNA copy number alteration have been identified in ALL.33,34,39–43 Deletions are more common than amplification and typically are focal, often involving only a single gene. The nature and frequency of these alterations are associated with cell lineage (B or T) and cytogenetic ALL subtype. Notably, MLL-rearranged ALL, which is typically aggressive and arises early in life, harbors <1 additional genetic alteration per case, whereas ETV6-RUNX1 and BCR-ABL1 leukaemias manifest later in childhood and typically have at least 6–8 additional genetic alterations. Many of the involved genes encode regulators of lymphoid development, cell cycle, tumor suppressors, and lymphoid signaling molecules (figure 3, table 2). Most commonly altered are transcriptional regulators of B lymphoid development (eg, PAX5, IZKF1, and EBF1) in more than two-thirds of B-ALL cases and the CDKN2A/CDKN2B loci encoding the INK4/ARF tumor suppressors in more than 80% of T-ALL cases. While our understanding of sequence mutations in ALL is incomplete, existing data show that several genes are altered by multiple mechanisms, including deletion or amplification, sequence mutation, and translocation. The nature of genetic alteration is highly gene-dependent; for example, structural alterations are more common than sequence mutations in PAX5 and IKZF1 in B-ALL, whereas several genes (eg, WT1, PHF6, and NOTCH1 in T-ALL) are more commonly targeted by sequence alteration.
Table 2.
Key genetic alterations in B-ALL
| Gene | Alteration | Frequency | Pathway and consequences of alteration | Clinical relevance | References |
|---|---|---|---|---|---|
| PAX5 | Focal deletions, translocations, sequence mutations | 31.7% of B-ALL | Transcription factor required for B-lymphoid development. Mutations impair DNA binding and transcriptional activation. | 33,34,39 | |
|
| |||||
| IKZF1 | Focal deletions or sequence mutations | 15% of all paediatric B-ALL | Transcription factor required for development of HSC to lymphoid precursors. Deletions and mutations result in loss of function or dominant negative isoforms. | 33 | |
| More than 80% of BCR-ABL1 ALL and 66% of CML in lymphoid blast crisis | Associated with poor outcome | 34,46,142 | |||
| One-third of high-risk BCR-ABL1-negative ALL | Tripling in cumulative incidence of relapse | 47,48,143 | |||
| Inherited variants | Increased risk of ALL | 27,28 | |||
|
| |||||
| JAK1/2 | Pseudokinase and kinase domain mutations | 18–35% of DS-ALL and 10.7% of High-risk BCR-ABL1-negative ALL | Mutations result in constitutive JAK-STAT activation. Transforms mouse Ba/F3-EpoR hematopoetic cell line. | 35,144–146 | |
|
| |||||
| CRLF2 | Rearrangement as IGH@-CRLF2 or P2RY8-CRLF2 resulting in overexpression | 5–16% of paediatric and adult B-ALL and >50% of DS-ALL | Associated with mutant JAK in up to 50% of cases. CRLF2 mutations and JAK mutations cotransforming in Ba/F3 cells and results in constitutive STAT activation. | 36,37,49,50 | |
| 14% of paediatric high-risk ALL | Associated with IKZF1 alteration and JAK mutations | Associated with poor outcome | 51,147 | ||
|
| |||||
| CREBBP | Focal deletion and sequence mutations | 19% of relapsed ALL. Also mutated in non-Hodgkin lymphoma | Mutations result in impaired histone acetylation and transcriptional regulation. | Mutations selected for at relapse, and associated with glucocorticoid resistance. | 38,148 |
CML, chronic myeloid leukaemia; DS, Down syndrome; HSC, hematopoietic stem cells.
Several genetic alterations have well-established roles in leukaemogenesis, such as activating mutations in NOTCH1; however, the roles of many other genes remain unknown, in part because of the paucity of mouse models that faithfully recapitulate human ALL. However, several alterations are known to disrupt the activity of the encoded proteins in vitro or to result in dominant negative activity, and recent studies have shown that loss-of-function mutations in Pax544 and Ikzf145 accelerate the onset of B-ALL in mouse models.
Prognostic genetic alterations in ALL
As cytogenetic alterations alone do not accurately predict the risk of relapse, there is great interest in the relation between novel genetic alterations and ALL outcome. The most consistent association is that of IKZF1 alterations and poor outcome (table 2). IKZF1 encodes IKAROS, the founding member of a family of pleiotropic zinc finger-containing transcription factors, and is required for lymphoid lineage development (figure 3). IKZF1 is altered in 15% of B-ALL cases and is a hallmark of two high-risk ALL subtypes: BCR-ABL1 lymphoid leukaemia (either de novo or chronic myeloid leukaemia in lymphoid blast crisis)34,46 and a new subtype termed “BCR-ABL1-like ALL”, described below.47,48
IKZF1 alterations include loss-of-function deletions of the entire IKZF1 locus, focal deletions resulting in expression of dominant negative IKZF1 alleles, and less commonly, sequence mutations. Alteration of IKZF1 is associated with an increased risk of treatment failure and relapse in both BCR-ABL1-positive and -negative ALL; hence, there is considerable interest in testing for IKZF1 alterations at diagnosis, although current treatment protocols do not consider IKZF1 status in risk stratification.
Novel subtypes of ALL
Rearrangement of CRLF2
As many as 8% of childhood ALL cases have CRLF2 rearrangement at the pseudoautosomal region 1 (PAR1) of Xp/Yp (figure 2 and table 2). CRLF2 encodes cytokine receptor-like factor 2, the receptor for thymic stromal lymphopoietin (TSLP).36,37 The arrangement occurs either as a rearrangement of CRLF2 into the immunoglobulin heavy chain locus at 14q32, or as a focal deletion immediately upstream of CRLF2 that results in expression of a novel fusion, P2RY8-CRLF2. Both events dysregulate CRLF2 expression, resulting in increased expression by lymphoblasts that may be detected by diagnostic immunophenotyping. Less common is a p.Phe232Cys mutation that results in receptor dimerization.49 CRLF2 rearrangement is particularly common (>50% of cases) in ALL associated with Down syndrome (DS-ALL). In both DS and non-DS ALL, approximately 50% of cases with CRLF2-rearrangement harbor concomitant activating mutations in the Janus kinase genes JAK1 or JAK2 (table 2).36,37,50 Particularly in non-DS-ALL, CRLF2 and JAK alterations are also associated with deleterious IKZF1 alterations and poor outcome.51 The JAK mutations are located either at or near the active site of the JAK kinase domain or in the pseudokinase domains of JAK1/2, most commonly at p.Arg683 of JAK2. Importantly, JAK2 mutations common in ALL are distinct from those observed in myeloproliferative neoplasms.52 Co-expression of CRLF2 and JAK mutant alleles transforms model cell lines and activates downstream Jak-Stat signaling, suggesting that these alterations are co-transforming in B-ALL.37 In DS-ALL, alterations of IKZF1, but not of CRLF2 or JAK2, were associated with a worse prognosis.53
BCR-ABL1-like ALL
Ten to 12 percent of B-ALL cases exhibit a gene expression profile similar to that of BCR-ABL1 ALL but are BCR-ABL1-negative, commonly show IKZF1 alteration, and have a poor outcome (figure 2).47,48 As many as 50% of BCR-ABL1-like cases have CRLF2 rearrangements and JAK mutations. Next-generation sequencing, including transcriptome and whole-genome sequencing, has shown that the remaining cases harbour a diverse range of rearrangements, deletions, and sequence mutations that activate cytokine receptor and kinase signaling (eg, those involving ABL1, EPOR, IL7R, JAK2, and PDGFRB). Several of these were shown to be transforming in vitro and to activate kinase signaling in primary leukaemic cells.54
Intrachromosomal amplification of chromosome 21 (iAMP21) occurs in approximately 2% of BALL cases (figure 2).55 This entity was originally defined by fluorescence in-situ hybridization (FISH) as gain of at least three copies of RUNX1.56 Subsequent FISH and microarray studies showed that the region of amplification is typically large and complex and is often accompanied by deletion of the subtelomeric regions of chromosome 21.57 The functional consequences of iAMP21 are largely unknown, but identification is important as it is associated with poor outcome on standard-risk regimens.55,58
Genetics of relapse and clonal heterogeneity in ALL
Cytogenetic59 and genomic profiling of samples collected at diagnosis and relapse has shown that the majority of ALL cases exhibit substantial changes in the nature of genetic alterations during the disease course and that relapse often arises from the emergence of a minor subclone with genetic alterations distinct from those of the predominant clone at diagnosis (figure 3).60–62 In most cases, the relapse clone shares lesions with the predominant clone at diagnosis, indicating a common “ancestral” or pre-leukaemic origin, but other lesions are discordant. Sensitive assays specific for individual alterations demonstrate that the relapse clone is often present at a low level at diagnosis, suggesting that the alterations that emerge at relapse confer resistance to therapy. Moreover, several of the alterations that most commonly emerge at relapse are those also associated with poor treatment outcome when present at diagnosis, such as deletions of IKZF1 and CDKN2A/CDKN2B. Less commonly, the relapse clone appears identical to or completely dissimilar to that at diagnosis. Similar findings were observed in candidate gene sequencing studies of relapsed ALL samples, which identified enrichment of loss-of-function mutations of the transcriptional coactivator and acetyltransferase CREBBP (encoding CREB-binding protein) and of TP53 (table 2, figure 3).38,63,64
Genome sequencing of ALL
Recent studies have shown that next-generation sequencing approaches are required to comprehensively identify the genetic alterations in leukaemia. Simultaneous sequencing of hundreds of thousands of nucleic acids (“massively parallel” sequencing) may be used to identify sequence mutations and structural variants in the encoding portion of the genome (“exome” sequencing), the transcriptome (mRNA sequencing), or the entire genome. A recent study sequenced 120 candidate genes and pathways targeted by DNA copy number alterations in 187 high-risk B-ALL cases and identified a high frequency of alterations targeting B lymphoid development (68%), the TP53/RB1 tumour suppressor pathway (54%), Ras signalling (50%), and Janus kinases (11%), as well as recurring mutations in genes including ETV6, TBL1XR1, CREBBP, MUC4, ASMTL, and ADARB2.65
Early T-cell precursor ALL (ETP-ALL) is an aggressive leukaemia characterized by an immature immunophenotype reminiscent of the murine thymic early T cell precursor,66 aberrant expression of myeloid and stem cell markers, a distinct gene expression profile, and dismal outcome.67 Whole genome sequencing of 12 ETP-ALL cases and mutational recurrence testing in an additional 94 ETP and non-ETP T-ALL cases showed that three pathways commonly mutated in AML were mutated at high frequency in ETP-ALL.68 These were inactivating mutations targeting haematopoietic and lymphoid development (including GATA3, ETV6, RUNX1 and IKZF1), mutations driving aberrant cytokine receptor and Ras signaling (NRAS, KRAS, FLT3, JAK1, JAK3 and IL7R), and deleterious mutations in chromatin-modifying genes, most notably components of the polycomb repressor complex 2 (PRC2) (EZH2, EED, and SUZ12). PRC2 normally mediates trimethylation of lysine 27 of histone 3 (H3K27), resulting in transcriptional repression; thus, these mutations are predicted to de-repress transcription. These results extend recent findings of PHF6 mutations in T-ALL which were obtained by sequencing X chromosome genes and explained increased incidence of T-ALL in males.69
These findings provide compelling evidence that whole-genome sequencing of the entire spectrum of ALL subtypes is required to identify all genetic alterations contributing to leukaemogenesis. In addition, studies investigating the nature of non-coding genetic mutations and the interaction of genetic, epigenetic, and transcriptomic factors in ALL will be of great interest.
Diagnosis
Morphological identification of lymphoblasts by microscopy and immunophenotypic determination of lineage commitment and developmental stage by flow cytometry are essential for correct diagnosis of ALL.2 Chromosomal analysis still plays an important role in the initial cytogenetic work-up. RT-PCR, FISH/multiplex ligation-dependent probe amplification, and flow cytometry are used to identify leukaemia-specific translocations, submicroscopic chromosomal abnormalities, and cellular DNA content, respectively. After genome-wide analysis becomes time- and cost-effective, it may replace many current diagnostic techniques. Appendix table 2 lists the tests that have prognostic and therapeutic implications.
Risk assignment
Clinical and biological factors
Age (infant or ≥10 years old), presenting leukocyte count (≥50×109/L), race (Hispanic or black), male sex, and T-cell immunophenotype have been considered adverse clinical prognostic factors in children, although their effect is diminished by contemporary risk-adapted therapy and improved supportive care.2–6 Infants with MLL rearrangement, especially those < 6 months old with a leukocyte count >300×109/L at diagnosis, still have a dismal prognosis.7 Cytogenetic and molecular risk factors have been discussed above.
Racial/ethnic differences in prognosis have been linked not only to socioeconomic factors but also to differences in genomic alterations.51,70,71 For example, germline single-nucleotide polymorphisms of PDE4B70 and ARID5B71 were shown to be associated with Native American genetic ancestry, and somatic CRLF2 rearrangements in ALL blasts51 were overrepresented in children from a Hispanic ethnic background; these alterations were found to contribute to inferior outcomes in Hispanics. Adverse prognosis conferred by genetic ancestry was mitigated by adding a course of delayed intensification therapy.70
Adolescents and adults have a greater prevalence of biologically high-risk leukaemia (eg, BCR-ABL1 and MLL rearrangement), a low incidence of favorable subtypes (eg, ETV6-RUNX1 and hyperdiploidy), and poorer adherence and tolerance to therapy.72 Older age (especially ≥60 years) and high presenting leukocyte count are also poor prognostic factors in this population. Recent studies showed that they had better outcomes when treated on paediatric rather than adult regimens.72–76 Typically, paediatric regimens provide higher doses of non-myelosuppressive drugs, early and frequent intrathecal therapy, reinduction and long maintenance phases, and strict oversight of adherence. Appendix figure 2 shows ALL survival according to age group.
Response to therapy
Early treatment response is predictive of the risk of relapse and is used to assign patients to subsequent risk-adapted therapy.77 Methods that track residual leukaemic cells by flow cytometry (detecting aberrant immunophenotypes) and by PCR amplification (detecting leukaemia-specific immunoglobulin and T-cell receptor genes or fusion transcripts) allow the recognition of ALL cells present at levels well below those detectable by microscopic morphologic assessment, ie, minimal residual disease (MRD). MRD is currently the most powerful prognostic indicator in childhood and adult ALL, even in patients with low-risk features at presentation.6,78–82 The kinetics of MRD clearance in response to identical remission-induction chemotherapy differed between B- and T-ALL; negative MRD on day 33 (after administration of 4 drugs) was the strongest prognostic factor in B-ALL,79 while negative MRD on day 78 (after 7 drugs) was also predictive in T-ALL, regardless of positive MRD on day 33.80
PCR is typically more sensitive than flow cytometry for measurement of MRD (~0.001% vs ~0.01%), and PCR-measurable low levels of MRD (0.001 to < 0.01%) after remission-induction therapy showed prognostic significance in childhood ALL.83 However, flow cytometry is faster, generally less expensive, and applicable to a larger proportion of patients,77 allowing early tailoring of therapy.81 The sensitivity of flow cytometry can be improved by using multi-color combinations of additional leukaemia-associated markers identified from differently expressed genes in ALL cells, yielding a detection threshold of ~0.001%.84
Treatment
Treatment of ALL typically spans 2–2.5 years, comprising 3 phases: remission-induction, intensification (or consolidation), and continuation (or maintenance).2 Most of the drugs used were developed before 1970. However, their dosage and schedule of administration in combination chemotherapy have been optimized on the basis of leukaemic-cell biological features, response to therapy (MRD), and patient pharmacodynamic and pharmacogenomic findings, resulting in the current high survival rate. Central nervous system (CNS)-directed therapy is administered to prevent relapse caused by leukaemia cells sequestered in this sanctuary site. Allogeneic haematopoietic stem-cell transplantation is considered for patients at very high risk. This section will focus on the most important advances in ALL treatment over the past 5 years.
Remission-induction therapy
Four to 6 weeks of remission-induction treatment eradicates the initial leukaemic cell burden and restores normal haematopoiesis in 96–99% of children and 78–92% of adults.2–5 The chemotherapy agents typically include a glucocorticoid (prednisone or dexamethasone), vincristine, and asparaginase, with or without anthracycline. This regimen appears to be sufficient for standard-risk ALL if intensified post-remission treatment is given. Patients at high or very high risk receive four or more drugs.
Prednisone (or prednisolone) has traditionally been used in ALL treatment, but dexamethasone is increasingly considered.85 However, the optimal doses and bioequivalence of these drugs are unclear. In prospective randomized trials, dexamethasone provided better control of CNS leukaemia and, at a prednisone-to-dexamethasone dose ratio <7, yielded better event-free survival, especially in children with T-ALL who responded well to prednisone pre-phase treatment and children age <10 years with B-ALL.86–89 However, when a higher dose of prednisolone (dose ratio >7) was used, the two drugs showed no difference in efficacy.90,91 Glucocorticoid treatment is associated with adverse effects, including infection, osteonecrosis, fracture, psychosis, and myopathy, whose incidence is generally higher with dexamethasone than with prednisone. Thus, high-dose dexamethasone (eg, 10 mg/m2/day) is not recommended for adolescent B-ALL.92
Three preparations of asparaginase are currently available: Escherichia coli-derived, Erwinia caratovora-derived, and a monoethoxypolyethylene glycol succinimidyl conjugate of E. coli L-asparaginase (PEG-asparaginase).93 As these formulations have different half-lives (PEG-asparaginase > E. coli > Erwinia), it is crucial to maintain asparagine depletion by optimizing the dose intensity and schedule of the asparaginase used. PEG-asparaginase has largely replaced the native product, as it provides at least 2 weeks of therapeutic activity after a single dose and less frequently induces antibodies.94,95 Native E. coli and PEG-asparaginase activity were reported to be inversely related to anti-E. coli asparaginase antibody titers, although PEG-asparaginase was inhibited only at high antibody titers.96 Therefore, PEG-asparaginase may be considered when antibody titers are low to intermediate, and Erwinia asparaginase should be considered when titers are high. A significant pharmacokinetic interaction has been observed between glucocorticoid and asparaginase.97,98 Higher systemic exposure to asparaginase was found to be associated with higher exposure to dexamethasone, presumably because of impaired hepatic synthesis of proteins involved in dexamethasone clearance. Thus, anti-asparaginase antibodies can reduce exposure to both drugs and may increase the risk of relapse.98
Patients with BCR-ABL1-positive ALL have been considered to have a poor prognosis but benefit from early administration of a tyrosine kinase inhibitor (eg, imatinib, dasatinib). When this agent is added to multiagent chemotherapy, complete remission rates are >90% and event-free survival is superior to that of historical controls.99,100 Unlike imatinib, dasatinib targets both ABL1 and Src kinases; it also has more potent activity against BCR-ABL1, is active against imatinib-resistant BCR-ABL1 (except for T315I mutation), and has better CNS penetration.100–102
Intensification (consolidation) therapy
Intensification (consolidation) therapy is administered after remission-induction to eradicate residual leukaemic cells.2,3 This phase commonly uses high-dose (ie, 1–8g/m2) methotrexate (MTX) with mercaptopurine, frequent pulses of vincristine and glucocorticoid, uninterrupted asparaginase for 20–30 weeks, and reinduction therapy with agents similar to those used during remission-induction.
The accumulation of the active MTX metabolites, MTX polyglutamates (MTXPG1–7), in leukaemic cells is associated with anti-leukaemic activity, the results of which can be affected by somatic and germline genetic factors, dose and duration of MTX administration, and leucovorin rescue. Functional enzyme and somatic genetic studies show that MTXPG1–7 accumulation varies widely among ALL subtypes, being low in TCF3-PBX1 ALL, T-ALL, and ETV6-RUNX1 ALL and high in hyperdiploid B-ALL, especially with gain of chromosome 18 or 10; therefore, the former group may benefit from higher MTX doses.103–105 Germline single-nucleotide polymorphisms of the organic anion transporter polypeptide SLCO1B1 were found to be associated with high MTX clearance.106,107 In patients with high-risk ALL, high-dose MTX (5 g/m2, 4 doses, every 14 days) plus mercaptopurine was more effective than escalating-dose MTX (initial dose 100 mg/m2, increasing by 50 mg/m2, 5 doses, every 10 days) plus PEG-asparaginase, without increased acute toxicity.108 The duration of an effective serum MTX level is also important; accumulation of MTXPG1–7 was less with 4-hour infusions of high-dose MTX than with 24-hour infusions.109 Leucovorin rescue is required after high-dose MTX administration; however, its excessive use can counteract the anti-leukaemic effects of MTX and increase the risk of relapse.110
Reinduction therapy has proven to be a crucial element of ALL protocols. Intensified reinduction therapy with vincristine and asparaginase improved the outcome of patients with high-risk ALL.111 However, an identical second reinduction cycle did not improve the outcome of patients with high-risk ALL and a rapid marrow response to 7 days of induction therapy or of those with standard-risk ALL, suggesting that residual leukaemia clones after a course of reinduction therapy may represent intrinsic drug resistance.111,112 It is not clear whether this second reinduction cycle offers a benefit to patients with high-risk ALL and a slow early response, in the context of contemporary therapy. Osteonecrosis frequently occurs after reinduction therapy, especially in children 10 years and older. Alternative-week (10 mg/m2/day on days 0–6 and 14–20, 2 courses) rather than continuous (on days 0–20, 1 course) administration of dexamethasone significantly reduced osteonecrosis despite a higher cumulative dose.113
Hematopoietic stem cell transplantation and cellular therapy
Allogeneic haematopoietic stem cell transplantation (HSCT) is considered for children with very high-risk ALL and/or persistent disease.114 Contemporary HSCT protocols with high-resolution HLA typing, case-based conditioning, and improved supportive care have reduced relapse-related mortality, regimen-related toxicity, and infection.115,116 Further, survival is comparable regardless of stem cell source (matched related, matched unrelated, cord blood, or haploidentical donor).116–118
In view of the ongoing development of disease detection and frontline therapies, the indications for allogeneic HSCT should be reassessed continuously. A level of MRD ≥10−4 before HSCT is strongly associated with relapse, and new strategies are needed to reduce the disease burden before and/or after HSCT.119,120 Patients with BCR-ABL1-positive ALL who obtain remission after multi-agent chemotherapy with ABL1 kinase inhibitors and young children (age <6 years) with B-ALL in delayed remission after induction failure can be treated without HSCT.99,100,121 The benefit of HSCT for infants with ALL is controversial; the role of HSCT, if any, is limited to a small high-risk group.122,123 Although many adult centers have considered HSCT during first complete remission a key element of therapy, treatment with paediatric-based regimens will decrease its use.5,72
Continuation therapy
Continuation therapy typically lasts 2 years or longer and comprises mainly daily mercaptopurine and weekly methotrexate with or without pulses of vincristine and dexamethasone.
Mercaptopurine and thioguanine are structural analogs of hypoxanthine and guanine, respectively, and inhibit de novo purine synthesis. Although thioguanine requires fewer steps to form the active metabolite thioguanine nucleotides and has greater in vitro cytotoxicity to lymphoblasts, randomized studies have not consistently shown a benefit of thioguanine in event-free survival124,125 or overall survival,126 and protracted doses •40 mg/m2/day were associated with death during remission, veno-occlusive disease, portal hypertension, and thrombocytopenia.124–126 Thus, mercaptopurine is preferred for continuation therapy. Thiopurine methyltransferase (TPMT) catalyzes S-methylation of thiopurines to inactive methylated metabolites. Patients with homozygous or heterozygous TPMT deficiency experience moderate to profound myelosuppression when treated with thiopurines.127 They may also develop secondary malignancy, especially at higher doses (eg, mercaptopurine 75 mg/m2/day).128 Further, an adherence rate <95% to planned mercaptopurine doses is associated with relapse.129 Therefore, uninterrupted, pharmacogenetics-based mercaptopurine dosing is important.2 After thioguanine nucleotides are incorporated into DNA, DNA mismatch repair enzymes exert cytotoxicity. Deficiency of such enzymes (eg, MSH2) renders leukaemic cells thiopurine-resistant.130 MSH2 expression was low or undetectable in approximately 11% of children with newly diagnosed ALL due to partial or complete somatic deletion of genes that regulate MSH2 degradation (FRAP1, HERC1, PRKCZ, and PIK3C2B). These children experienced a high incidence of relapse.130
CNS-directed therapy
Control of CNS disease is a key component of ALL therapy. Prophylactic cranial irradiation (12–18 Gy) effectively controls disease, but its use has recently been reduced or eliminated to prevent acute neurotoxicity, neurocognitive deficits, endocrinopathies, secondary malignancies, and excess late mortality.131 The St. Jude Total XV6 and Dutch Childhood Oncology Group ALL-9132 protocols replaced cranial irradiation with triple intrathecal chemotherapy (methotrexate, hydrocortisone, and cytarabine) for all newly diagnosed patients; the 5-year cumulative risk of isolated CNS relapse was 2.7% and 2.6%, respectively, within the range achieved by prophylactic cranial irradiation (1.5–4.5%). Patients at high-risk of CNS relapse, comprising those with any CNS involvement (including leukemic-cell contamination by traumatic spinal tap) and/or T-ALL,6 should be treated with intensified intrathecal therapy during early remission-induction. Cranial irradiation can be reserved only for salvage treatment, as the retrieval rate is high for patients with an isolated CNS relapse who did not receive irradiation with initial treatment.6
In a randomized study in standard-risk ALL, triple intrathecal treatment reduced the frequency of CNS relapse compared with single-agent intrathecal methotrexate, but was associated with increased risk of bone marrow and testicular relapse, possibly due to less intensive systemic therapy.133 In St. Jude Total XV, not only excellent CNS outcomes but also excellent overall outcomes (5-year event-free survival, 85.6%; overall survival, 93.5%) were achieved.6 As CNS and hematologic relapses are competing events, systemic chemotherapy with high-dose methotrexate, intensive asparaginase, and dexamethasone, plus risk-based early intensive intrathecal chemotherapy, play a substantial role in preventing CNS relapse.131
Remaining questions and future directions
There remain subsets of ALL that carry an adverse prognosis. Further intensification of current regimens is unlikely to dramatically improve survival but is certain to increase short- and long-term adverse effects. Reduction of treatment intensity should be sought for patients at low risk. Studies of chronic health complications in the growing number of long-term adult survivors will help to refine therapy to reduce the toxicity of treatment, while functional genomics and proteomics will provide a deeper understanding of the epidemiology and pathogenesis of individual cases, allowing targeted “personalized medicine” (table 3). Although the majority of epidemiology and pathobiology studies have been developed for paediatric ALL, these should be expanded to adult patients.
Table 3.
Targeted antileukaemic drugs in current clinical trials
| Class | Agent | Target | Indication |
|---|---|---|---|
| Purine nucleoside analogue | |||
| Clofarabine149 | Ribonucleotide reductase; DNA polymerase; mitochondria | All ALL | |
| Nelarabine150 | Ribonucleotide reductase; DNA synthesis | T-ALL | |
| Forodesine | Purine nucleoside phosphorylase | T-ALL | |
| Vinca alkaloid | |||
| Vincristine sulfate liposome151 | Tubulin | All ALL | |
| Kinase inhibitor | |||
| ABL1 kinase inhibitor | |||
| Dasatinib;100 Nilotinib;152 Imatinib;99 Ponatinib | ABL1 kinase; platelet-derived growth factor receptor B | BCR-ABL1-positive ALL; BCR-ABL1-like ALL (eg, NUP214-ABL1) | |
| Aurora kinase inhibitor | |||
| MLN8237 | Aurora A kinase | BCR-ABL1-positive ALL | |
| Janus kinase (JAK) inhibitor | |||
| Ruxolitinib; TG101348; CYT387 | JAK | JAK-mutated ALL; BCR-ABL1-like ALL (eg, BCR-JAK2; mutated IL7R) | |
| Tyrosine kinase inhibitor | |||
| Lestaurtinib; Midostaurin; Sorafenib; Quizartinib; Tandutinib; Sunitinib | FMS-like tyrosine kinase 3 | MLL-rearranged ALL; hyperdiploid ALL | |
| Other molecular or signaling inhibitor | |||
| Proteasome inhibitor | |||
| Bortezomib153 | Ubiquitin-proteasome pathway | All ALL | |
| Mammalian target of rapamycin (mTOR) inhibitor | |||
| Sirolimus; Temsirolimus; Everolimus | mTOR | All ALL | |
| Farnesyltransferase inhibitor | |||
| Tipifarnib; Lonafarnib | Ras, lamin A | All ALL | |
| γ-Secreatase inhibitor | |||
| RO4929097 | γ-Secretase | T-ALL | |
| Angiogenesis inhibitor | |||
| Bevacizumab | Vascular endothelial growth factor | All ALL | |
| Apoptosis inducer | |||
| Obatoclax; Oblimersen | Bcl-2 | All ALL | |
| Chemokine receptor (CXCR4) antagonist | |||
| Plerixafor | CXCL12 (SDF1)/CXCR4 axis | All ALL | |
| Epigenetic therapy | |||
| DNA methyltransferase inhibitor | |||
| Azacitidine; Decitabine | DNA methyltransferase | All ALL | |
| Histone methyltransferase inhibitor | |||
| EPZ-5676 | DOT1L | MLL-rearranged ALL | |
| Histone deacetylase inhibitor | |||
| Vorinostat; Panobinostat; Depsipeptide; Valproic acid | Histone deacetylase | All ALL | |
| Immune therapy | |||
| Monoclonal antibody | |||
| Blinatumomab138 | CD19 (engages CD3 T cells) | CD19-positive ALL | |
| SAR3419 | CD19 | CD19-positive ALL | |
| DT2219ARL | CD19 and 22 | CD19/CD22-positive ALL | |
| Rituximab137 | CD20 | CD20-positive ALL | |
| Epratuzumab;154 Moxetumomab; Inotuzumab ozogamicin155 | CD22 | CD22-positive ALL | |
| Alemtuzumab156 | CD52 | CD52-positive ALL | |
| Cellular therapy | |||
| Natural killer cells | Killer immunoglobulin-like receptor (KIR)-ligand | Donor KIR-recipient ligand mismatch | |
| T cells with CD19-specific chimeric antigen receptor | CD19 | CD19-positive ALL | |
Pharmacologic inhibitors of Janus kinases, such as ruxolitinib, are being explored in cases of childhood ALL harboring CRLF2 and JAK alterations.35 Detailed preclinical studies in BCR-ABL1-like ALL showed that the ABL1/PDGFRB inhibitors imatinib and dasatinib are effective against ALL cells with NUP214-ABL1 and that JAK inhibitors are effective against those with BCR-JAK2 or mutated IL7R; therefore, cases harboring these alterations may be candidates for targeted therapy.54 DNA and histone methyltransferase inhibitors and histone deacetylase inhibitors may reactivate silenced tumor-suppressor genes or augment sensitivity to concomitant chemotherapy. These epigenetic agents may be considered for infants with MLL-rearranged ALL, in whom aberrant DNA and histone methylation is frequently observed,134,135 and for patients with CREBBP mutations that encode histone acetyltransferase CREB-binding protein.38 Mutations in ETP-ALL are also observed in AML, suggesting that AML-directed therapies or agents that target JAK signaling may be beneficial.68 Monoclonal antibodies to surface antigens such as CD19, CD20, CD22, and CD52 have been used in unconjugated form (eg, rituximab and epratuzumab), conjugated to immunotoxins or chemotherapeutic agents (moxetumomab and inotuzumab ozogamicin), or in the form of a bispecific antibody (blinatumomab).136 The incorporation of rituximab into the hyper-CVAD regimen (fractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone) appears to improve outcome for younger adults (<60 years) with CD20-positive, BCR-ABL1-negative B-ALL.137 Blinatumomab, a bispecific, single-chain antibody to CD19 and CD3∊, recruits and activates CD3 effector cytotoxic T cells and is cytotoxic to CD19-expressing target cells bound to the other arm of the antibody; it has shown activity against relapsed/refractory ALL in an adult phase II study.138
Emerging evidence shows that newly diagnosed ALL comprises multiple subclones and that chemoresistance is frequently driven by subpopulations harboring genetic alterations that confer resistance.60–62 Thus, efforts should be made to identify patients at high risk of relapse by using highly sensitive methods to detect these subpopulations at diagnosis, and therapy should target these subpopulations to augment the efficacy of the current therapeutic regimen while reducing the intensity.
Search strategy and selection criteria
We searched Medline and PubMed for articles published between January 2007 and November 2012, using the keywords “acute lymphoblastic leukaemia”, “acute lymphocytic leukaemia”, and “acute lymphoid leukaemia”. Additional information was obtained from abstracts presented to the American Society of Hematology and the American Society of Clinical Oncology. We focused on publications from the past 5 years, but did not exclude commonly referenced and highly regarded older publications. We also searched the reference lists of articles identified by this search strategy and selected those we judged relevant. Review articles and book chapters are cited to provide readers with more details and more references than this Seminar can provide. Our reference list was modified on the basis of comments from peer reviewers.
Supplementary Material
Acknowledgments
Supported in part by grants from the National Institutes of Health (CA21765) (HI and CGM) and Leukaemia and Lymphoma Research UK (MG), and by the American Lebanese Syrian Associated Charities (ALSAC) (HI and CGM). CGM is a Pew Scholar in the Biomedical Sciences and a St Baldrick’s Scholar. These sponsors had no role in writing the Seminar. The authors thank Prof. Ching-Hon Pui, MD, for critical review and Sharon Naron, ELS, for editing the manuscript.
Footnotes
Contributors
All authors contributed to the writing of this paper and have seen and approved the final submitted version.
Conflicts of interest statement
HI receives unrelated research support from Bayer/Onyx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–43. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 3.Stanulla M, Schrappe M. Treatment of childhood acute lymphoblastic leukemia. Semin Hematol. 2009;46:52–63. doi: 10.1053/j.seminhematol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Hunger SP, Lu X, Devidas M, et al. Improved Survival for Children and Adolescents With Acute Lymphoblastic Leukemia Between 1990 and 2005: A Report From the Children’s Oncology Group. J Clin Oncol. 2012 doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011;29:532–43. doi: 10.1200/JCO.2010.30.1382. [DOI] [PubMed] [Google Scholar]
- 6.Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730–41. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pieters R, Schrappe M, De Lorenzo P, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370:240–50. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 8.Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nature Rev Cancer. 2003;3:639–49. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 9.Greaves MF. Aetiology of acute leukaemia. Lancet. 1997;349:344–49. doi: 10.1016/s0140-6736(96)09412-3. [DOI] [PubMed] [Google Scholar]
- 10.Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. 2003;102:2321–33. doi: 10.1182/blood-2002-12-3817. [DOI] [PubMed] [Google Scholar]
- 11.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6:193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 12.Preston DL, Kusumi S, Tomonaga M, et al. Cancer incidence in atomic bomb survivors. Part III: Leukemia, lymphoma and multiple myeloma, 1950–1987. Radiat Res. 1994;137 (Suppl):S68–S97. [PubMed] [Google Scholar]
- 13.Doll R, Wakeford R. Risk of childhood cancer from fetal irradiation. Br J Radiol. 1997;70:130–39. doi: 10.1259/bjr.70.830.9135438. [DOI] [PubMed] [Google Scholar]
- 14.Kendall G, Little MP, Wakeford R. Numbers and proportions of leukemias in young people and adults induced by radiation of natural origin. Leuk Res. 2011;35:1039–43. doi: 10.1016/j.leukres.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schüz J. Exposure to extremely low-frequency magnetic fields and the risk of childhood cancer: update of the epidemiological evidence. Progr Biophysics Mol Biol. 2011;107:339–42. doi: 10.1016/j.pbiomolbio.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Ward G. The infective theory of acute leukaemia. Br J Child Dis. 1917;14:10–20. [Google Scholar]
- 17.Kinlen L. Evidence for an infective cause of childhood leukaemia: comparison of a Scottish New Town with nuclear reprocessing sites in Britain. Lancet. 1988;ii:1323–27. doi: 10.1016/s0140-6736(88)90867-7. [DOI] [PubMed] [Google Scholar]
- 18.Greaves MF. Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia. 1988;2:120–25. [PubMed] [Google Scholar]
- 19.Kinlen LJ. Epidemiological evidence for an infective basis in childhood leukaemia. Br J Cancer. 1995;71:1–5. doi: 10.1038/bjc.1995.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinlen L, Doll R. Population mixing and childhood leukaemia: Fallon and other US clusters. Br J Cancer. 2004;91:1–3. doi: 10.1038/sj.bjc.6601982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldman JM, Jarrett O, editors. Mechanisms of viral leukaemogenesis. Edinburgh: Churchill Livingstone; 1984. [Google Scholar]
- 22.Kroll ME, Draper GJ, Stiller CA, Murphy MFG. Childhood leukemia incidence in Britain, 1974–2000: time trends and possible relation to influenza epidemics. J Natl Cancer Inst. 2006;98:417–20. doi: 10.1093/jnci/djj095. [DOI] [PubMed] [Google Scholar]
- 23.Ford AM, Palmi C, Bueno C, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGFβ pathway in early B lineage progenitor cells. J Clin Invest. 2009;119:826–36. doi: 10.1172/JCI36428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor GM, Birch JM. The hereditary basis of human leukemia. In: Henderson ES, Lister TA, Greaves MF, editors. Leukemia. 6. Philadelphia: WB Saunders; 1996. pp. 210–45. [Google Scholar]
- 25.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355:165–69. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 26.Vijayakrishnan J, Houlston RS. Candidate gene association studies and risk of childhood acute lymphoblastic leukemia: a systematic review and meta-analysis. Haematologica. 2010;95:1405–14. doi: 10.3324/haematol.2010.022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11. 2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1006–10. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trevino LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1001–05. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21. 3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42:492–94. doi: 10.1038/ng.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang W, Trevino LR, Yang JJ, et al. ARID5B SNP rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and contributes to racial differences in leukemia incidence. Leukemia. 2010;24:894–96. doi: 10.1038/leu.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JJ, Cheng C, Yang W, et al. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. J Am Med Assoc. 2009;301:393–403. doi: 10.1001/jama.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison CJ. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br J Haematol. 2009;144:147–56. doi: 10.1111/j.1365-2141.2008.07417.x. [DOI] [PubMed] [Google Scholar]
- 33.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 34.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–4. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 35.Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106:9414–8. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–98. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- 37.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–6. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuiper RP, Schoenmakers EF, van Reijmersdal SV, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–66. doi: 10.1038/sj.leu.2404691. [DOI] [PubMed] [Google Scholar]
- 40.Kawamata N, Ogawa S, Zimmermann M, et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood. 2008;111:776–84. doi: 10.1182/blood-2007-05-088310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strefford JC, Worley H, Barber K, et al. Genome complexity in acute lymphoblastic leukemia is revealed by array-based comparative genomic hybridization. Oncogene. 2007;26:4306–18. doi: 10.1038/sj.onc.1210190. [DOI] [PubMed] [Google Scholar]
- 42.Van Vlierberghe P, Homminga I, Zuurbier L, et al. Cooperative genetic defects in TLX3 rearranged pediatric T-ALL. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2008;22:762–70. doi: 10.1038/sj.leu.2405082. [DOI] [PubMed] [Google Scholar]
- 43.van Vlierberghe P, Meijerink JP, Lee C, et al. A new recurrent 9q34 duplication in pediatric T-cell acute lymphoblastic leukemia. Leukemia. 2006;20:1245–53. doi: 10.1038/sj.leu.2404247. [DOI] [PubMed] [Google Scholar]
- 44.Miller C, Mullighan CG, Su X, Ma J, Wang M, Zhang J, Williams RT, Downing JR. Pax5 haploinsufficiency cooperates with BCR-ABL1 to induce acute lymphoblastic leukemia. ASH Annual Meeting Abstracts. 2008;112:293s. [Google Scholar]
- 45.Virely C, Moulin S, Cobaleda C, et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia. 2010;24:1200–4. doi: 10.1038/leu.2010.63. [DOI] [PubMed] [Google Scholar]
- 46.Iacobucci I, Storlazzi CT, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP) Blood. 2009;114:2159–67. doi: 10.1182/blood-2008-08-173963. [DOI] [PubMed] [Google Scholar]
- 47.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. N Engl J Med. 2009;360:470–80. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–34. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010;107:252–57. doi: 10.1073/pnas.0911726107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hertzberg L, Vendramini E, Ganmore I, et al. Down syndrome acute lymphoblastic leukemia: a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the iBFM Study Group. Blood. 2010;115:1006–17. doi: 10.1182/blood-2009-08-235408. [DOI] [PubMed] [Google Scholar]
- 51.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–21. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112:2190–8. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buitenkamp TD, Pieters R, Gallimore NE, et al. Outcome in children with Down’s syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia. 2012;26:2204–11. doi: 10.1038/leu.2012.84. [DOI] [PubMed] [Google Scholar]
- 54.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–66. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moorman AV, Ensor HM, Richards SM, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11:429–38. doi: 10.1016/S1470-2045(10)70066-8. [DOI] [PubMed] [Google Scholar]
- 56.Harewood L, Robinson H, Harris R, et al. Amplification of AML1 on a duplicated chromosome 21 in acute lymphoblastic leukemia: a study of 20 cases. Leukemia. 2003;17:547–53. doi: 10.1038/sj.leu.2402849. [DOI] [PubMed] [Google Scholar]
- 57.Robinson HM, Harrison CJ, Moorman AV, Chudoba I, Strefford JC. Intrachromosomal amplification of chromosome 21 (iAMP21) may arise from a breakage-fusion-bridge cycle. Genes Chromosomes Cancer. 2007;46:318–26. doi: 10.1002/gcc.20412. [DOI] [PubMed] [Google Scholar]
- 58.Attarbaschi A, Mann G, Panzer-Grumayer R, et al. Minimal residual disease values discriminate between low and high relapse risk in children with B-cell precursor acute lymphoblastic leukemia and an intrachromosomal amplification of chromosome 21: the Austrian and German acute lymphoblastic leukemia Berlin-Frankfurt-Munster (ALL-BFM) trials. J Clin Oncol. 2008;26:3046–50. doi: 10.1200/JCO.2008.16.1117. [DOI] [PubMed] [Google Scholar]
- 59.Raimondi SC, Pui CH, Head DR, Rivera GK, Behm FG. Cytogenetically different leukemic clones at relapse of childhood acute lymphoblastic leukemia. Blood. 1993;82:576–80. [PubMed] [Google Scholar]
- 60.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang JJ, Bhojwani D, Yang W, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112:4178–83. doi: 10.1182/blood-2008-06-165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawamata N, Ogawa S, Seeger K, et al. Molecular allelokaryotyping of relapsed pediatric acute lymphoblastic leukemia. Int J Oncol. 2009;34:1603–12. doi: 10.3892/ijo_00000290. [DOI] [PubMed] [Google Scholar]
- 63.Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185–93. doi: 10.1200/JCO.2011.34.8144. [DOI] [PubMed] [Google Scholar]
- 64.Inthal A, Zeitlhofer P, Zeginigg M, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012 doi: 10.1038/leu.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118:3080–7. doi: 10.1182/blood-2011-03-341412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol. 2008;8:9–21. doi: 10.1038/nri2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–56. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Vlierberghe P, Palomero T, Khiabanian H, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42:338–42. doi: 10.1038/ng.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang JJ, Cheng C, Devidas M, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet. 2011;43:237–41. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu H, Cheng C, Devidas M, et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2012;30:751–7. doi: 10.1200/JCO.2011.38.0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schafer ES, Hunger SP. Optimal therapy for acute lymphoblastic leukemia in adolescents and young adults. Nat Rev Clin Oncol. 2011;8:417–24. doi: 10.1038/nrclinonc.2011.77. [DOI] [PubMed] [Google Scholar]
- 73.Boissel N, Auclerc MF, Lheritier V, et al. Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol. 2003;21:774–80. doi: 10.1200/JCO.2003.02.053. [DOI] [PubMed] [Google Scholar]
- 74.de Bont JM, Holt B, Dekker AW, van der Does-van den Berg A, Sonneveld P, Pieters R. Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia. 2004;18:2032–5. doi: 10.1038/sj.leu.2403538. [DOI] [PubMed] [Google Scholar]
- 75.Stock W, La M, Sanford B, et al. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood. 2008;112:1646–54. doi: 10.1182/blood-2008-01-130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pui CH, Pei D, Campana D, et al. Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol. 2011;29:386–91. doi: 10.1200/JCO.2010.32.0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Campana D. Minimal residual disease monitoring in childhood acute lymphoblastic leukemia. Curr Opin Hematol. 2012 doi: 10.1097/MOH.0b013e3283543d5c. [DOI] [PubMed] [Google Scholar]
- 78.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111:5477–85. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115:3206–14. doi: 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 80.Schrappe M, Valsecchi MG, Bartram CR, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118:2077–84. doi: 10.1182/blood-2011-03-338707. [DOI] [PubMed] [Google Scholar]
- 81.Basso G, Veltroni M, Valsecchi MG, et al. Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol. 2009;27:5168–74. doi: 10.1200/JCO.2008.20.8934. [DOI] [PubMed] [Google Scholar]
- 82.Gokbuget N, Kneba M, Raff T, et al. Adults with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012 doi: 10.1182/blood-2011-09-377713. [DOI] [PubMed] [Google Scholar]
- 83.Stow P, Key L, Chen X, et al. Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood. 2010;115:4657–63. doi: 10.1182/blood-2009-11-253435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coustan-Smith E, Song G, Clark C, et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2011;117:6267–76. doi: 10.1182/blood-2010-12-324004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Inaba H, Pui CH. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010;11:1096–106. doi: 10.1016/S1470-2045(10)70114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bostrom BC, Sensel MR, Sather HN, et al. Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Blood. 2003;101:3809–17. doi: 10.1182/blood-2002-08-2454. [DOI] [PubMed] [Google Scholar]
- 87.Mitchell CD, Richards SM, Kinsey SE, Lilleyman J, Vora A, Eden TO. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol. 2005;129:734–45. doi: 10.1111/j.1365-2141.2005.05509.x. [DOI] [PubMed] [Google Scholar]
- 88.Schrappe M, Zimmermann M, Moricke A, et al. Dexamethasone in induction can eliminate one third of all relapses in childhood acute lymphoblastic leukemia (ALL): results of an international randomized trial in 3655 patients (Trial AIEOP-BFM ALL 2000) ASH Annual Meeting Abstracts. 2008;112:7s. [Google Scholar]
- 89.Winick N, Salzer W, Devidas M, et al. Dexamethasone (DEX) versus prednisone (PRED) during induction for children with high-risk acute lymphoblastic leukemia (HR-ALL): A report from the Children’s Oncology Group Study AALL0232. J Clin Oncol. 2011;29:586s. [Google Scholar]
- 90.Igarashi S, Manabe A, Ohara A, et al. No advantage of dexamethasone over prednisolone for the outcome of standard- and intermediate-risk childhood acute lymphoblastic leukemia in the Tokyo Children’s Cancer Study Group L95-14 protocol. J Clin Oncol. 2005;23:6489–98. doi: 10.1200/JCO.2005.01.982. [DOI] [PubMed] [Google Scholar]
- 91.Bertrand Y, Suciu S, Benoit Y, et al. Dexamethasone(DEX)(6mg/sm/d) and prednisolone(PRED)(60mg/sm/d) in induction therapy of childhood ALL are equally effective: results of the 2nd interim analysis of EORTC Trial 58951. ASH Annual Meeting Abstracts. 2008;112:8s. [Google Scholar]
- 92.Pui CH, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120:1165–74. doi: 10.1182/blood-2012-05-378943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pieters R, Hunger SP, Boos J, et al. L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer. 2011;117:238–49. doi: 10.1002/cncr.25489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Silverman LB, Supko JG, Stevenson KE, et al. Intravenous PEG-asparaginase during remission induction in children and adolescents with newly diagnosed acute lymphoblastic leukemia. Blood. 2010;115:1351–3. doi: 10.1182/blood-2009-09-245951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Silverman LB, Gelber RD, Dalton VK, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood. 2001;97:1211–8. doi: 10.1182/blood.v97.5.1211. [DOI] [PubMed] [Google Scholar]
- 96.Willer A, Gerss J, Konig T, et al. Anti-Escherichia coli asparaginase antibody levels determine the activity of second-line treatment with pegylated E coli asparaginase: a retrospective analysis within the ALL-BFM trials. Blood. 2011;118:5774–82. doi: 10.1182/blood-2011-07-367904. [DOI] [PubMed] [Google Scholar]
- 97.Yang L, Panetta JC, Cai X, et al. Asparaginase may influence dexamethasone pharmacokinetics in acute lymphoblastic leukemia. J Clin Oncol. 2008;26:1932–9. doi: 10.1200/JCO.2007.13.8404. [DOI] [PubMed] [Google Scholar]
- 98.Kawedia JD, Liu C, Pei D, et al. Dexamethasone exposure and asparaginase antibodies affect relapse risk in acute lymphoblastic leukemia. Blood. 2012;119:1658–64. doi: 10.1182/blood-2011-09-381731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol. 2009;27:5175–81. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ravandi F, O’Brien S, Thomas D, et al. First report of phase 2 study of dasatinib with hyper-CVAD for the frontline treatment of patients with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia. Blood. 2010;116:2070–7. doi: 10.1182/blood-2009-12-261586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ottmann O, Dombret H, Martinelli G, et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: interim results of a phase 2 study. Blood. 2007;110:2309–15. doi: 10.1182/blood-2007-02-073528. [DOI] [PubMed] [Google Scholar]
- 102.Porkka K, Koskenvesa P, Lundan T, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112:1005–12. doi: 10.1182/blood-2008-02-140665. [DOI] [PubMed] [Google Scholar]
- 103.Whitehead VM, Vuchich MJ, Lauer SJ, et al. Accumulation of high levels of methotrexate polyglutamates in lymphoblasts from children with hyperdiploid (greater than 50 chromosomes) B-lineage acute lymphoblastic leukemia: a Pediatric Oncology Group study. Blood. 1992;80:1316–23. [PubMed] [Google Scholar]
- 104.Rots MG, Pieters R, Peters GJ, et al. Role of folylpolyglutamate synthetase and folylpolyglutamate hydrolase in methotrexate accumulation and polyglutamylation in childhood leukemia. Blood. 1999;93:1677–83. [PubMed] [Google Scholar]
- 105.French D, Yang W, Cheng C, et al. Acquired variation outweighs inherited variation in whole genome analysis of methotrexate polyglutamate accumulation in leukemia. Blood. 2009;113:4512–20. doi: 10.1182/blood-2008-07-172106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trevino LR, Shimasaki N, Yang W, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27:5972–8. doi: 10.1200/JCO.2008.20.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ramsey LB, Bruun GH, Yang W, et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012;22:1–8. doi: 10.1101/gr.129668.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Larsen E, Salzer WL, Devidas M, Nachman JB, Raetz EA, Loh ML, Heerema NA, Carroll AJ, Gastier-Foster JM, Borowitz MJ, Wood BL, Willman CL, Winick NJ, Hunger S, Carroll WL. Comparison of high-dose methotrexate (HD-MTX) with Capizzi methotrexate plus asparaginase (C-MTX/ASNase) in children and young adults with high-risk acute lymphoblastic leukemia (HR-ALL): A report from the Children’s Oncology Group Study AALL0232. J Clin Oncol. 2011:29. [Google Scholar]
- 109.Mikkelsen TS, Sparreboom A, Cheng C, et al. Shortening infusion time for high-dose methotrexate alters antileukemic effects: a randomized prospective clinical trial. J Clin Oncol. 2011;29:1771–8. doi: 10.1200/JCO.2010.32.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Skarby TV, Anderson H, Heldrup J, Kanerva JA, Seidel H, Schmiegelow K. High leucovorin doses during high-dose methotrexate treatment may reduce the cure rate in childhood acute lymphoblastic leukemia. Leukemia. 2006;20:1955–62. doi: 10.1038/sj.leu.2404404. [DOI] [PubMed] [Google Scholar]
- 111.Seibel NL, Steinherz PG, Sather HN, et al. Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2008;111:2548–55. doi: 10.1182/blood-2007-02-070342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Matloub Y, Bostrom BC, Hunger SP, et al. Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118:243–51. doi: 10.1182/blood-2010-12-322909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mattano LA, Jr, Devidas M, Nachman JB, et al. Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol. 2012;13:906–15. doi: 10.1016/S1470-2045(12)70274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Balduzzi A, Valsecchi MG, Uderzo C, et al. Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet. 2005;366:635–42. doi: 10.1016/S0140-6736(05)66998-X. [DOI] [PubMed] [Google Scholar]
- 115.Marks DI, Wang T, Perez WS, et al. The outcome of full-intensity and reduced-intensity conditioning matched sibling or unrelated donor transplantation in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia in first and second complete remission. Blood. 2010;116:366–74. doi: 10.1182/blood-2010-01-264077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leung W, Campana D, Yang J, et al. High success rate of hematopoietic cell transplantation regardless of donor source in children with very high-risk leukemia. Blood. 2011;118:223–30. doi: 10.1182/blood-2011-01-333070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Eapen M, Rubinstein P, Zhang MJ, et al. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet. 2007;369:1947–54. doi: 10.1016/S0140-6736(07)60915-5. [DOI] [PubMed] [Google Scholar]
- 118.Eapen M, Rocha V, Sanz G, et al. Effect of graft source on unrelated donor haemopoietic stem-cell transplantation in adults with acute leukaemia: a retrospective analysis. Lancet Oncol. 2010;11:653–60. doi: 10.1016/S1470-2045(10)70127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bader P, Kreyenberg H, Henze GH, et al. Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol. 2009;27:377–84. doi: 10.1200/JCO.2008.17.6065. [DOI] [PubMed] [Google Scholar]
- 120.Leung W, Pui CH, Coustan-Smith E, et al. Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood. 2012 doi: 10.1182/blood-2012-02-409813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schrappe M, Hunger SP, Pui CH, et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med. 2012;366:1371–81. doi: 10.1056/NEJMoa1110169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dreyer ZE, Dinndorf PA, Camitta B, et al. Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: a report from the Children’s Oncology Group. J Clin Oncol. 2011;29:214–22. doi: 10.1200/JCO.2009.26.8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mann G, Attarbaschi A, Schrappe M, et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood. 2010;116:2644–50. doi: 10.1182/blood-2010-03-273532. [DOI] [PubMed] [Google Scholar]
- 124.Harms DO, Gobel U, Spaar HJ, et al. Thioguanine offers no advantage over mercaptopurine in maintenance treatment of childhood ALL: results of the randomized trial COALL-92. Blood. 2003;102:2736–40. doi: 10.1182/blood-2002-08-2372. [DOI] [PubMed] [Google Scholar]
- 125.Vora A, Mitchell CD, Lennard L, et al. Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet. 2006;368:1339–48. doi: 10.1016/S0140-6736(06)69558-5. [DOI] [PubMed] [Google Scholar]
- 126.Stork LC, Matloub Y, Broxson E, et al. Oral 6-mercaptopurine versus oral 6-thioguanine and veno-occlusive disease in children with standard-risk acute lymphoblastic leukemia: report of the Children’s Oncology Group CCG-1952 clinical trial. Blood. 2010;115:2740–8. doi: 10.1182/blood-2009-07-230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89:387–91. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schmiegelow K, Al-Modhwahi I, Andersen MK, et al. Methotrexate/6-mercaptopurine maintenance therapy influences the risk of a second malignant neoplasm after childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Blood. 2009;113:6077–84. doi: 10.1182/blood-2008-11-187880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bhatia S, Landier W, Shangguan M, et al. Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:2094–101. doi: 10.1200/JCO.2011.38.9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Diouf B, Cheng Q, Krynetskaia NF, et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17:1298–303. doi: 10.1038/nm.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pui CH, Howard SC. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol. 2008;9:257–68. doi: 10.1016/S1470-2045(08)70070-6. [DOI] [PubMed] [Google Scholar]
- 132.Veerman AJ, Kamps WA, van den Berg H, et al. Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997–2004) Lancet Oncol. 2009;10:957–66. doi: 10.1016/S1470-2045(09)70228-1. [DOI] [PubMed] [Google Scholar]
- 133.Matloub Y, Lindemulder S, Gaynon PS, et al. Intrathecal triple therapy decreases central nervous system relapse but fails to improve event-free survival when compared with intrathecal methotrexate: results of the Children’s Cancer Group (CCG) 1952 study for standard-risk acute lymphoblastic leukemia, reported by the Children’s Oncology Group. Blood. 2006;108:1165–73. doi: 10.1182/blood-2005-12-011809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stumpel DJ, Schneider P, van Roon EH, et al. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood. 2009;114:5490–8. doi: 10.1182/blood-2009-06-227660. [DOI] [PubMed] [Google Scholar]
- 135.Bernt KM, Zhu N, Sinha AU, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hoelzer D. Novel antibody-based therapies for acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2011;2011:243–9. doi: 10.1182/asheducation-2011.1.243. [DOI] [PubMed] [Google Scholar]
- 137.Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28:3880–9. doi: 10.1200/JCO.2009.26.9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Topp MS, Kufer P, Gokbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29:2493–8. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- 139.Anderson RM, May RM. Immunisation and herd immunity. Lancet. 1990;335:641–45. doi: 10.1016/0140-6736(90)90420-a. [DOI] [PubMed] [Google Scholar]
- 140.Gilham C, Peto J, Simpson J, et al. Day care in infancy and risk of childhood acute lymphoblastic leukaemia: findings from a UK case-control study. Br Med J. 2005;330:1294–97. doi: 10.1136/bmj.38428.521042.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Urayama KY, Buffler PA, Gallagher ER, Ayoob JM, Ma X. A meta-analysis of the association between day-care attendance and childhood acute lymphoblastic leukaemia. Int J Epidemiol. 2010;39:718–32. doi: 10.1093/ije/dyp378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. 2009;27:5202–7. doi: 10.1200/JCO.2008.21.6408. [DOI] [PubMed] [Google Scholar]
- 143.Kuiper RP, Waanders E, van der Velden VH, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24:1258–64. doi: 10.1038/leu.2010.87. [DOI] [PubMed] [Google Scholar]
- 144.Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372:1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 145.Gaikwad A, Rye CL, Devidas M, et al. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol. 2009;144:930–2. doi: 10.1111/j.1365-2141.2008.07552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kearney L, Gonzalez De Castro D, Yeung J, et al. A specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukaemia. Blood. 2008;113:646–8. doi: 10.1182/blood-2008-08-170928. [DOI] [PubMed] [Google Scholar]
- 147.Cario G, Zimmermann M, Romey R, et al. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood. 2010;115:5393–7. doi: 10.1182/blood-2009-11-256131. [DOI] [PubMed] [Google Scholar]
- 148.Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hijiya N, Thomson B, Isakoff MS, et al. Phase 2 trial of clofarabine in combination with etoposide and cyclophosphamide in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. Blood. 2011;118:6043–9. doi: 10.1182/blood-2011-08-374710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dunsmore KP, Devidas M, Linda SB, et al. Pilot study of nelarabine in combination with intensive chemotherapy in high-risk T-cell acute lymphoblastic leukemia: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:2753–9. doi: 10.1200/JCO.2011.40.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.O’Brien S, Schiller G, Lister J, et al. High-Dose Vincristine Sulfate Liposome Injection for Advanced, Relapsed, and Refractory Adult Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia. J Clin Oncol. 2012 doi: 10.1200/JCO.2012.46.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ottmann OG, Larson RA, Kantarjian HM, et al. Phase II study of nilotinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoblastic leukemia. Leukemia. 2012 doi: 10.1038/leu.2012.324. [DOI] [PubMed] [Google Scholar]
- 153.Messinger YH, Gaynon PS, Sposto R, et al. Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood. 2012;120:285–90. doi: 10.1182/blood-2012-04-418640. [DOI] [PubMed] [Google Scholar]
- 154.Raetz EA, Cairo MS, Borowitz MJ, et al. Chemoimmunotherapy reinduction with epratuzumab in children with acute lymphoblastic leukemia in marrow relapse: a Children’s Oncology Group Pilot Study. J Clin Oncol. 2008;26:3756–62. doi: 10.1200/JCO.2007.15.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kantarjian H, Thomas D, Jorgensen J, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13:403–11. doi: 10.1016/S1470-2045(11)70386-2. [DOI] [PubMed] [Google Scholar]
- 156.Angiolillo AL, Yu AL, Reaman G, Ingle AM, Secola R, Adamson PC. A phase II study of Campath-1H in children with relapsed or refractory acute lymphoblastic leukemia: a Children’s Oncology Group report. Pediatr Blood Cancer. 2009;53:978–83. doi: 10.1002/pbc.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Greaves M. The Evolutionary Legacy. Oxford: Oxford University Press; 2000. Cancer. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.