

Abstract
A series of 2-aminothiazoles was synthesized based on a HTS scaffold from a whole-cell screen against Mycobacterium tuberculosis (Mtb). The SAR shows the central thiazole moiety and the 2-pyridyl moiety at C-4 of the thiazole are intolerant to modification. However, the N-2 position of the aminothiazole exhibits high flexibility and we successfully improved the antitubercular activity of the initial hit by more than 128-fold through introduction of substituted benzoyl groups at this position. N-(3-Chlorobenzoyl)-4-(2-pyridinyl)-1,3-thiazol-2-amine (55) emerged as one of the most promising analogues with a MIC of 0.024 μM or 0.008 μg/mL in 7H9 media and therapeutic index of nearly ~300. However, 55 is rapidly metabolized by human liver microsomes (t1/2 = 28 min) with metabolism occurring at the invariant aminothiazole moiety and Mtb develops spontaneous resistance with a high frequency of ~10−5.
Keywords: Mycobacterium tuberculosis, aminothiazole, high-throughput screening, antibacterial agent
1. Introduction
At the beginning of the 20th century tuberculosis (TB), also referred to as the White Plague and consumption in the middle ages, was a leading cause of mortality.1 Poor public health and sanitation, inadequate diet as well as crowded housing and working conditions contributed to this endemic. The development of modern chemotherapy to treat TB began in 1943 with the momentous discovery of streptomycin, the first antibiotic effective against Mycobacterium tuberculosis (Mtb).2 Dr. Selman Waksman predicted the end of the TB was “now virtually within sight” during his Nobel lecture in 1952 where he described his labs work that led to the isolation of streptomycin from soil microbes.3 Indeed the discovery of other TB drugs followed in rapid succession with para-aminosalicylic acid (1946), pyrazinamide (1952), isoniazid (1952), ethambutol (1961), and rifampin (1965), among many others.4 Introduction of these antibiotics led to a staggering drop in TB incidence in the developing world.
The standard treatment regimen in use today established over several decades through dozens of clinical trials by the British Medical Council (BMC) involves a 2-month intensive phase with isoniazid, rifampin, ethambutol, and pyrazinamide followed by a 4-month continuation phase of isoniazid and rifampin.5 Combination therapy is required since the first clinical trials with streptomycin demonstrated that drug resistance rapidly developed to a single agent.6 The extraordinarily long treatment course is necessary since Mtb is inherently slow growing, drug penetration into TB lessions is poor,7 Mtb periodically switches its metabolism to a dormant state,8 and a subpopulation of the bacteria exhibit reversible phenotypic drug tolerance.9
Unfortunately, the emergence of drug resistant strains is undermining the great advances made in the 20th century to control TB, which is now the second leading cause of infectious disease mortality with over 1.4 million deaths in 2011.10 Multidrug resistant (MDR) TB is defined as resistance to the two most effective antitubercular drugs isoniazid and rifampin. Treatment of MDR-TB is typically accomplished by replacement of isoniazid and rifampin with an injectable aminoglycoside (i.e. amikacin), a fluoroquinolone (i.e. moxifloxacin), and other second-line drugs such as ethionamide, para-aminosalicylic acid, and cycloserine that are more toxic and less effective requiring 18–24 months of chemotherapy. Extensively drug resistant TB (XDR-TB) has been documented in 84 countries, which possesses the MDR phenotype and is additionally resistant to one of the injectable antibiotics and any fluoroquinolone.10 Treatment options for XDR-TB are severely limited. The development of new TB drugs and ultimately of an entirely new treatment regimen for MDR- and XDR-TB is needed. The recent approval of Sirturo by the FDA (also known as bedaquiline, TMC-207, and R207910) as the first new TB drug in over forty years marks the first step toward this goal.11
There are two diametrically opposed strategies to antibacterial drug discovery: biased target-led approaches and un-biased screening using whole-cell assays.12 In target-based strategies, one seeks to identify small molecule inhibitors or modulators of a “validated” target, which is usually an enzyme or receptor employing a number of methods including high-throughput or fragment-based screening as well as the rationale design of multisubstrate, transition-state, or mechanism-based inhibitors. In whole cell assays compounds or natural product extracts are typically screened for frank growth inhibition of bacteria without a priori knowledge of the target. Target-based approaches are intellectually appealing and have been aggressively pursued over the last couple of decades. However, they have several unique challenges including the problems of converting a potent biochemical inhibitor/modulator into compound with whole-cell activity, rapid evolution of resistance to single targets, lack of target vulnerability (the extent to which a target must be inhibited to elicit an effect), and inability to predict if target inhibition by a small molecule will lead to arrest of growth (bacteriostatic) or cell death (bactericidal).12, 8 Consequently, it is increasingly recognized that this strategy is potentially limiting and has not resulted in a single new FDA-approved antibacterial agent. By contrast, all FDA-approved antibiotic scaffolds including all TB drugs have been identified through whole cell screening. As a result of these challenges, attention is being re-focused on traditional whole cell assays for antibacterial drug discovery. All of the promising clinical candidates for TB chemotherapy including the nitroimidazoles PA-82413 and OPC-67683,14 the 1,2-diamine SQ-109,15 and the benzothiazinone BTZ-04316 were obtained by whole cell screening. Additionally, the most recently approved TB drug Sirturo, a diarylquinoline, was discovered by high-throughput screening (HTS) using a Mycobacterium smegmatis whole cell assay.17
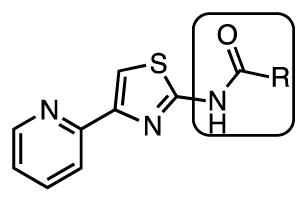
The Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) and the National Institutes of Health MLSCN Roadmap Initiative programs were the first to publically disclose results from a Mtb whole-cell HTS assay (which can be found at PubChem, search AID: 1594, 1626, and 2842 at http://pubchem.ncbi.nlm.nih.gov/).18–20 We were intrigued by an aminothiazole cluster of compounds (see Figure 1) discovered from these screens based on the scaffolds reported potent activity, excellent therapeutic index, clearly defined structure-activity relationships, chemical tractability for analogue synthesis, and prevalence of the aminothiazole scaffold in several approved drugs21–23 (Figure 1). Herein we describe the systematic and comprehensive SAR analysis of this aminothiazole scaffold and in vitro drug metabolism studies that resulted in compounds with improved activity profiles over the parent hit compounds.
Figure 1.

Aminothiazole HTS scaffold and some representative approved drugs containing an aminothiazole moiety.
2. Results and discussion
2.1 Chemistry
The lead structure can be split into three parts: the central aminothiazole, the 2-pyridyl substituent at C-4, and the aryl substituent at N-2 (Figure 1). Based on the SAR provided by Ananthan et al.18 describing the strict requirement of the 2-pyridyl substituent at C-4 for potency and the converse tolerance of N-2 aryl substituents, we elected to initially prepare a series of N-2 aryl analogues. The desired target molecules 1–19 were synthesized via the classic Hantzsch thiazole synthesis starting from 2-bromoacetylpyridine hydrobromide and the respective N-substituted thiourea (Scheme 1).24–26 The substituted thioureas that were not commercially available were synthesized by condensation of the appropriate aniline or 2-aminopyridine derivative with benzoylisothiocyanate followed by saponification to remove the benzoyl group.27–29
Scheme 1.
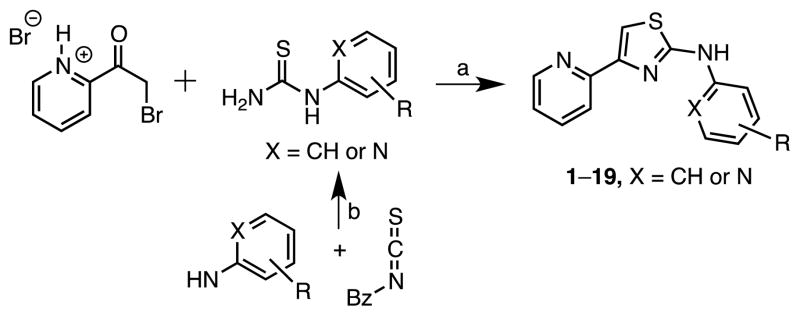
Variation of the 2-amino-residue. Reaction conditions: (a) EtOH, reflux, conc. NH3. (b) (i) EtOH, 40°C, (ii) NaOH, Δ.
We next synthesized a series of N-2 acyl analogues. The key building block 2-amino-4-(pyrid-2-yl)thiazole 20 was prepared by condensation of 2-bromoacetylpyridine hydrobromide with thiourea (Scheme 2). Subsequent coupling of 20 with various aliphatic, aromatic, and heteroaromatic acids provided the desired amides 21–70. Although the coupling of aminothiazole 20 with benzoyl chloride employing stoichiometric DMAP or N-methylimidazole in dichloromethane was high yielding, the method was not broadly applicable, most likely because of solubility issues with many of the substrates. We therefore employed either of the conditions shown in scheme 2 that compromised on some yields, but were more universally applicable.
Scheme 2.
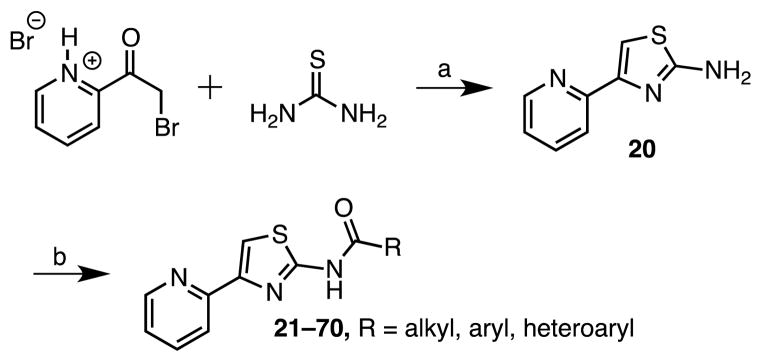
Reaction Conditions: (a) H2O; 7% NH3, (b) EDC, DMAP, DMF, compounds 21–31; or CDI, DBU, DMF, compounds 32–70.
As a result of the increased activity of the N-benzoyl derivatives 35–66, we explored the importance of the amide linkage through N-methylation with compound 71 and inversion of the amide with compound 73 (Scheme 3). Synthesis of analogue 71 was achieved in low yield by direct methylation of 35 employing NaH and iodomethane. The inverted amide 73 was prepared in two steps from 2,4-dibromothiazole by regioselective halogen-metal exchange of the more reactive 2-bromo group30, 31 using isopropylmagnesium chloride and subsequent reaction with phenyl isocyanate to afford 72. Negishi cross-coupling of 72 with 2-pyridylzinc bromide32 furnished the desired target molecule 73.
Scheme 3.

Reaction Conditions: (a) NaH, MeI, DMF, 0°C to rt, (13%); (b) (i) iPrMgCl, THF, −65 °C, 10 min, 0°C, 30 min, (ii) PhNCO, THF, −65 °C to rt, (52%); (c) (i) Pd2(dba)3, XPhos, THF, 65 °C, 15 min, (ii) 2-pyridylzinc bromide, THF, 65 °C, 16 h, (40%).
After exploration of the N-residue and the amide linkage, we examined the importance of the heteroatoms within the thiazole ring. We first attempted to prepare a thiophene analogue through a Gewald multicomponent condensation33, 34 starting from 2-acetylpyridine, ethyl cyanoacetate and sulfur, but were unable to isolate any desired product. Instead we chose a route similar to that employed for the preparation of 73, but starting from 2,4-dibromothiophene, which was first coupled with benzamide to afford 74 using a copper catalyzed amidation procedure developed by Buchwald’s group (Scheme 4).35 Subsequent Negishi cross-coupling with pyridylzinc bromide yielded the desired thiophene analogue 75.32
Scheme 4.
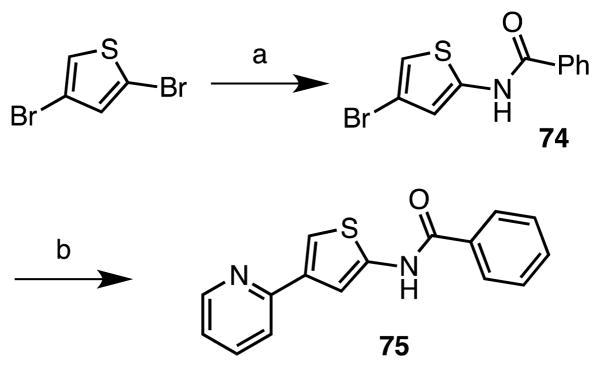
Reaction Conditions: (a) benzamide, CuI, K3PO4, (±)-trans-1,2-diaminocyclohexane, 1,4-dioxane, 110 °C, 12 h, (38%); (b) 2-pyridine zincbromide, Xphos, Pd2(dba)3, THF, reflux, 16 h, (49%).
Synthesis of imidazole 78 was accomplished in analogy to a procedure reported by Fang et al.36 through synthesis of an imidazo[1,2-a]pyrimidine intermediate by condensation of 2-bromoacetylpyridine hydrobromide with 2-aminopyrimidine to afford 76 (Scheme 5). This was then transformed into 2-aminoimidazole 77 through nucleophilic cleavage with hydrazine following the procedure described by Ermolat’ev and coworkers.37 Acylation with benzoyl chloride employing N-methylimidazole yielded the desired amide 78.
Scheme 5.
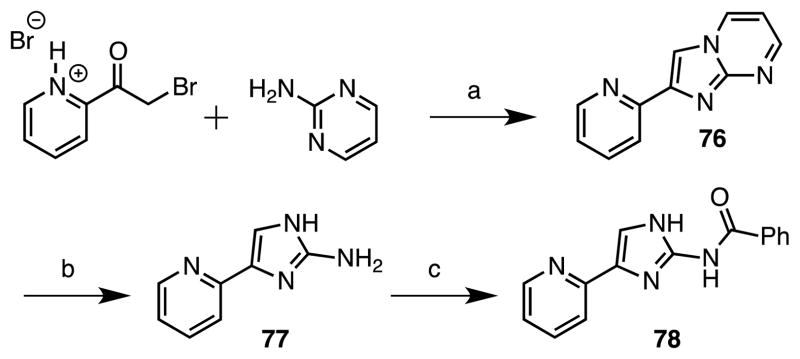
Reaction Conditions: (a) K2CO3, EtOH, reflux, 14 h, (49%); (b) hydrazine monohydrate, EtOH, 130 C, 2.5 h, (49%); (c) benzoyl chloride, 1-methylimidazole, DCM, rt, 18 h, (42%).
The oxazole derivative 82 was prepared as shown in Scheme 6. Our initial attempts to directly synthesize the key oxazole-2-amine intermediate 81 starting from 2-bromoacetylpyridine hydrobromide and urea in analogy to the Hantzsch thiazole synthesis was not successful. Crank and co-workers had described the syntheses of oxazole-2-amines through condensation of α-hydroxyketones with cynamide.38 Therefore we converted 2-bromoacetylpyridine hydrobromide to 2-hydroxyacetylpyridine 80 via nucleophilic displacement with acetate followed by ester hydrolysis. Condensation of 80 with cyanamide successfully provided the key intermediate 81, albeit in a modest overall yield that was benzoylated to furnish oxazole 82.38
Scheme 6.
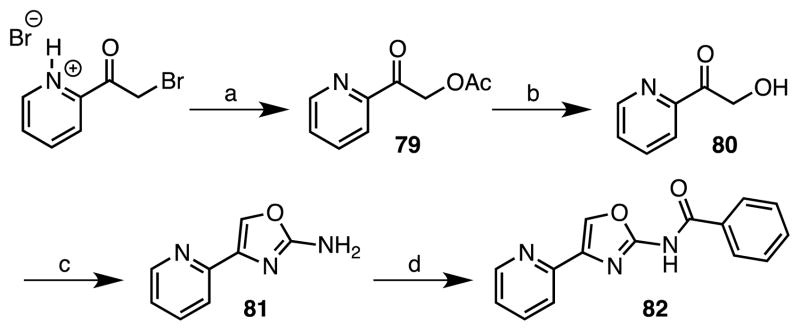
Reaction Conditions: (a) NaOAc, EtOH, reflux, 2 h; (b) 1 M HCl, EtOH, reflux, 1.5 h; (c) NH2CN, EtOH, reflux, 14 h, 10% (3 steps); (d) benzoyl chloride, 1-methylimidazole, DCM, rt, 18 h, 65%.
The oxadiazole 83 and thiadiazole 86 analogues were synthesized following procedures reported by White and coworkers.39 Condensation of picolinohydrazide with cyanogen bromide provided an intermediate 2-aminooxadiazole, which was acylated with 2-fluorobenzoic acid to furnish oxadiazole 83 (Scheme 7). Coupling of picolinoyl chloride with thiosemicarbazide afforded 84 that was cyclized to 85 and acylated to yield the desired thiadiazole 86 (Scheme 7).
Scheme 7.

(a) BrCN, KHCO3, dioxane, reflux, 15 h, 29%; (b) CDI, DBU, DMF, 60 C, 17 h, 72% (X = O), 56% (X = S); (c) THF, rt, 7 d; (d) MeSO3H, toluene, reflux, 92% (two steps).
As the last part of our studies, the aryl ring at C-4 of the aminothiazole was varied. Analogues 90–92 containing a 2-pyrazine, benzene, and thiazole were prepared as shown in Scheme 8 from the corresponding methyl ketone derivatives 87a–c, which were brominated to afford bromomethyl ketones 88a–c. Condensation with thiourea provided the respective 2-aminothiazoles 89a–c that were benzoylated to furnish the desired analogues 90–92.
Scheme 8.
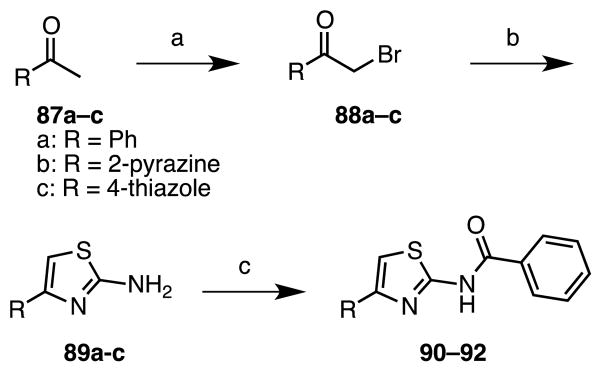
Reaction Conditions: for R = Ph: (a) Br2, CHCl3, rt, 2 h; (b) thiourea, AcOH, reflux, 3.5 h, 27% (2 steps); (c) benzoyl chloride, 1-methylimidazole, rt, 18 h, 28%; for R = 2-pyrazine: (a) HBr (33% in AcOH), Br2, AcOH, rt, 3 h, 71%; (b) thiourea, DIPEA, EtOH, reflux, 3 h, 95%; (c) benzoyl chloride, 1-methylimidazole, DMF, rt, 18 h, 17%; for R = thiazole: (a) pyridinium tribromide, DCM, rt; (b) thiourea, DIPEA, EtOH, reflux, 72 h, 57% (2 steps); (c) benzoyl chloride, 1-methylimidazole, DMF, 70°C, 70 h, 16%.
2.2 Whole cell activity against M. tuberculosis
Compounds were evaluated for whole cell activity against M. tuberculosis H37Rv to determine the minimum inhibitory concentration (MIC) that resulted in complete (>99%) inhibition of growth. In order to eliminate the potential for carbon-dependent activity as observed by Pethe and co-workers40 we employed Middlebrook 7H9 broth with either glucose or butyrate as the sole carbon source and defined GAST media with glycerol and alanine as the carbon sources.
We prepared a systematic set of N-aryl analogues 1–19 containing electron-withdrawing and donating substituents at the o-, m-, and p-positions since the reported SAR showed the N-aryl moiety was tolerant to modification.18 Our initial compound set possesses some overlap with the reported aminothiazoles (i.e. 2, 8, 12, 13, 18, and 19)18 (Table 1). The SAR is relatively flat with MIC values generally ranging from 12.5–25 μM for the simple phenyl derivatives and the potency is much less than reported [i.e. aminothiazoles 2, 8, 12, 13 had reported MIC values ranging from <0.3 μM (for 13) to 2 μM (for 12)]. However, analogues containing a 2-pyridyl substituent (17–19) are much more potent with MIC values of 0.39–0.78 μM in GAST media, which is concordant with the reported HTS results (0.35 μM for 18–19).18 The MICs for the 2-pyridyl analogues 17–19 exhibit a modest dependence on the carbon source with values ranging 2 to 8-fold across the three media conditions with the best activity observed in GAST media. We speculated that the enhanced activity could be due to a potential hydrogen bond formed between the pyridine nitrogen and the putative target(s).
Table 1.
| ||||
|---|---|---|---|---|
| Cmpd | R | MIC, μM (7H9-glucose) | MIC, μM (7H9-butyrate) | MIC, μM (GAST) |
| 1 | Phenyl | 25 | 12.5 | 12.5 |
| 2 | 2-Tolyl | 12.5 | 12.5 | 3.1–6.3 |
| 3 | 3-Tolyl | 25 | 12.5 | 6.3–12.5 |
| 4 | 4-Tolyl | 12.5 | 12.5 | 6.3–12.5 |
| 5 | 2,3-Di-Me-phenyl | 12.5 | 12.5 | 3.1 |
| 6 | 2,4-Di-Me-phenyl | 12.5 | 12.5 | 6.3 |
| 7 | 2,6-Di-Me-phenyl | 12.5 | 12.5 | 3.1 |
| 8 | 2,4-Di-MeO-phenyl | 6.3 | 6.3 | >12.5 |
| 9 | 2,5-Di-MeO-phenyl | >50 | >50 | >12.5 |
| 10 | 2-Fluorophenyl | 50 | 25 | >12.5 |
| 11 | 3-Fluorophenyl | 25 | 25 | >12.5 |
| 12 | 4-Fluorophenyl | 25 | 25 | 12.5 |
| 13 | 2-CF3-phenyl | 12–25 | 6.3 | 6.3 |
| 14 | 3-CF3-phenyl | 25 | 25 | >12.5 |
| 15 | 4-CF3-phenyl | 25 | 12.5 | ≥12.5 |
| 16 | 2-OH-phenyl | 50 | 25 | >12.5 |
| 17 | 2-Pyridyl | 0.78–1.6 | 0.39 | 0.39 |
| 18 | 5-Cl-2-pyridyl | 1.6 | 1.6 | 0.39 |
| 19 | 6-Me-2-pyridyl | 6.3 | 3.1 | 0.78–1.6 |
Based upon this assumption, we decided to introduce an amide linkage in the aminothiazole wherein the carbonyl would provide a hydrogen bond acceptor in place of the 2-pyridyl moiety. A series of N-acyl substituted aminothiazoles was synthesized with a wide variety of N-acyl groups including alkanoyl, cycloalkanoyl, and heteroaroyl, which are shown in Table 2 and discussed below as well as an extensive set of aroyl analogues that are shown in Table 3 and discussed in the following paragraph. The n-alkanoyl 21–24 and cycloalkanoyl 25–29 compounds display modest activity with potency increasing with chain-length from 12–25 μM for a 3-carbon chain to 3.1–6.3 μM for a 6-carbon chain, but the potency plateaus at 6-carbons with no further increase in potency observed up to 10 carbons (Table 2). We also examined a small series of 5-membered heterocyclic analogues 30–34 (Table 2). Among these compounds potency appears to weakly correlate with hydrophobicity with thiophene analogues 32 and 33 (MIC 1.6–3.1 μM) displaying an incremental enhancement in potency relative to the furan analogues 30 and 31 (MIC 3.1–6.3 μM) while the thiazole 34 is inactive (MIC > 50 μM).
Table 2.
| ||||
|---|---|---|---|---|
| Cmpd | R | MIC, μM (7H9-glucose) | MIC, μM (7H9-butyrate) | MIC, μM (GAST) |
| 21 | n-Propyl | 12–25 | 25 | 12.5 |
| 22 | n-Pentyl | 6.3 | 3.1 | 3.1 |
| 23 | n-Heptyl | 6.3 | 6.3 | 0.78–1.6 |
| 24 | n-Nonyl | 6.3 | 3.1 | 1.6 |
| 25 | Cyclopropyl | 12–25 | 12.5 | 6.3–12.5 |
| 26 | Cyclobutyl | 12.5 | 6.3 | 6.3 |
| 27 | Cyclopentyl | 12.5 | 3.1 | 3.1–6.3 |
| 28 | Cyclohexyl | 6.3 | 3.1 | 1.6–3.1 |
| 29 | 2-Methylcyclopropyl | 6.3–12.5 | >50 | 6.3 |
| 30 | 2-Furyl | 3.1–6.3 | 6.3 | 1.6–3.1 |
| 31 | 3-Furyl | 3.1–6.3 | 6.3 | 3.1 |
| 32 | 2-Thiophene | 1.6–3.1 | 1.6 | 1.6 |
| 33 | 3-Thiophene | 1.6–3.1 | 3.1 | 0.78 |
| 34 | 4-Thiazolyl | >50 | >50 | >50 |
Table 3.
| ||||||
|---|---|---|---|---|---|---|
| Cmpd | R | MIC, μM (7H9-glucose) | MIC, μM (7H9-butyrate) | MIC, μM (GAST) | EC50, μM | EC50/MIC |
| 35 | Phenyl | 0.39 | 0.19 | 0.19 | 7.1 | 18–37 |
| 36 | 2-Tolyl | 0.39 | 0.19–0.39 | 0.19 | 6.3 | 16–33 |
| 37 | 3-Tolyl | 0.19–0.39 | 0.049–0.098 | 0.049–0.098 | 4.1 | 11–84 |
| 38 | 4-Tolyl | 0.39 | 0.19 | 0.098 | 10.5 | 27–107 |
| 39 | 2-Benzyloxyphenyl | >50a | >50a | 50a | insol. | n.a.b |
| 40 | 3-Benzyloxyphenyl | 0.39–0.78 | 0.049 | 0.19 | 14.4 | 32–294 |
| 41 | 4-Benzyloxyphenyl | 0.19–0.39 | 0.19 | 0.098 | 2.3 | 6–23 |
| 42 | 2-Methoxyphenyl | 6.3 | 3.1–6.3 | 1.6 | 43.5 | 7–27 |
| 43 | 3-Methoxyphenyl | 0.19–0.39 | 0.19 | 0.098–0.19 | 2.8 | 7–29 |
| 44 | 4-Methoxyphenyl | 0.39 | 0.19–0.39 | 0.098–0.19 | 9.2 | 24–94 |
| 45 | 2-Hydroxyphenyl | 1.6–3.1 | 1.6–3.1 | 0.19–0.39 | n.d. c | n.a. |
| 46 | 2-Acetylphenyl | 12.5 | 12.5 | 3.13 | 86.7 | 7–28 |
| 47 | 3-Acetylphenyl | 0.39 | 0.19 | 0.098–0.19 | 1.5 | 4–15 |
| 48 | 4-Acetylphenyl | 0.39 | 0.19–0.39 | 0.19 | 10.5 | 27–55 |
| 49 | 2-Nitrophenyl | 1.6–3.1 | n.d. | 0.78 | n.d | n.a. |
| 50 | 3-Nitrophenyl | 1.6–3.1 | n.d. | 0.78 | n.d | n.a. |
| 51 | 2-Bromophenyl | 0.39–0.78 | 0.39 | 0.098–0.19 | 3.5 | 4–36 |
| 52 | 3-Bromophenyl | 0.098–0.19 | 0.024 | 0.049 | 16.1 | 115–671 |
| 53 | 4-Bromophenyl | 0.19 | 0.098 | 0.049 | 4.7 | 25–96 |
| 54 | 2-Chlorophenyl | 0.39–0.78 | 0.19–0.39 | 0.19 | 2.0 | 3–1 |
| 55 | 3-Chlorophenyl | 0.19–0.39 | 0.024 | 0.049–0.098 | 7.0 | 30–292 |
| 56 | 4-Chlorophenyl | 1.6 | 0.78 | 0.39–0.78 | n.d. | n.a. |
| 57 | 2-Fluorophenyl | 1.6–3.1 | 0.39 | 1.6–3.1 | n.d. | n.a. |
| 58 | 3-Fluorophenyl | 0.19–0.39 | 0.098 | 0.049 | 1.2 | 3–24 |
| 59 | 4-Fluorophenyl | 0.39 | 0.19 | 0.098–0.19 | 2.5 | 6–26 |
| 60 | 2,3-Difluorophenyl | 0.39 | 0.19 | 0.098–0.19 | 7.2 | 18–73 |
| 61 | 2,4-Difluorophenyl | 0.39–0.78 | 0.39 | 0.19 | 7.3 | 9–38 |
| 62 | 2,5-Difluorophenyl | 0.39 | 0.19 | 0.098–0.19 | 3.3 | 8–34 |
| 63 | 2,6-Difluorophenyl | 0.78 | 0.78 | 0.19 | 3.2 | 4–17 |
| 64 | 3,4-Difluorophenyl | 1.6–3.1 | 3.1 | 0.39 | n.d. | n.a. |
| 65 | 3,5-Difluorophenyl | 0.19 | 0.098 | 0.049 | 1.5 | 8–31 |
| 66 | 2-Fluoro-6-chlorophenyl | 0.78 | 0.39–0.78 | 0.19 | 1.3 | n.a. |
| 67 | 2-Chloropyridin-3-yl | 0.39–0.78 | 0.39 | 0.19 | 1.9 | 2–10 |
| 68 | 2-Pyridyl | 6.3 | 3.1–6.3 | 1.6 | 5.4 | 0.9–3.4 |
| 69 | 3-Pyridyl | 6.3 | 3.1 | >12.5a | n.d. | n.a. |
| 70 | 4-Pyridyl | 1.6–3.1 | 3.1 | 0.78–1.6 | n.d. | n.a. |
Very poor solubility.
Not applicable.
Not determined.
Modification with N-aroyl substituents led to a substantial increase in potency as illustrated with N-benzoyl derivative 35 (Table 3), whose MIC is 0.19–0.39 μM or 64-fold better than the corresponding N-phenyl 1 (MIC = 12.5–25 μM). Due to the remarkable increase in potency, we explored the SAR of the benzoyl substituent through the preparation an extensive and systematic series of compounds (Table 3). We first performed a methyl scan at the o-, m-, and p-positions with 36–38. The MICs are relatively insensitive to substitution, but show a modest preference for the m-position with 37 showing a 2- to 4-fold increase in potency relative to benzoyl 35. To further define the steric requirements for activity, we prepared o-, m-, and p-benzyloxybenzoyl analogues 39–41. While the poor solubility of o-benzyloxybenzoyl 39 precluded determination of its activity, we observed little steric discrimination with this large substituent at the m- and p-positions with both analogues resulting in a modest 2- to 4-fold increase in potency relative to benzoyl 35. To assess the importance of electronic effects on activity we prepared analogues with electron donating and withdrawing substituents on the benzoyl group including methoxy and hydroxy 42–45, acetyl 46–48, nitro 49–50, bromo 51–53, chloro 54–56, fluoro 57–59, difluoro 60–65, and 2-fluoro-6-chloro 66. The SAR from these compounds is relatively flat with MICs similar to benzoyl 35. 2-Acetyl 46 possesses the poorest activity with MICs ranging from 3.1 to 12.5 μM against the three different media while 3-bromo 52 and 3-chloro 55 are the most potent with MICs of 0.024–0.39 μM. We additionally prepared pyridine analogues 67–70. Picolinoyl 68, nicotinoyl 69, and isonicotinoyl 70 are less potent than benzoyl 35 with MICs of 1.6–6.3 μM. However, the activity of nicotinoyl 69 is improved 8- to 16-fold through substitution with a 2-chloro group in 2-chloronicotinyl 67.
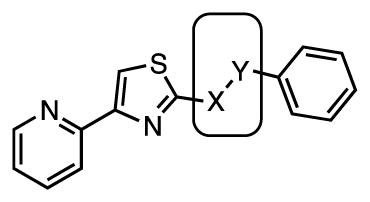
The role of the amide linkage in the benzoyl analogues was next evaluated with N-methyl 71 and the inverted amide 73 (Table 4). Methylation of the amide nitrogen as well as inversion of the amide bond completely abolishes antitubercular activity (MIC > 25 μM). These results suggest the amide moiety likely participates in specific hydrogen-bonding to its’ putative target(s).
Table 4.
| ||||
|---|---|---|---|---|
| Cmpd | X | Y | MIC, μM (7H9-glucose) | MIC, μM (GAST) |
| 71 | NMe | C(O) | >25 | ≥25 |
| 73 | C(O) | NH | >25 | >25 |
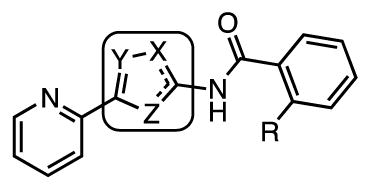
The importance of the central thiazole ring for activity was assessed with a small series of isosteric 5-membered heterocycle derivatives (Table 5). Deletion of the thiazole nitrogen in thiophene 75 is poorly tolerated resulting in a drastic ≥64-fold loss of potency relative to the parent thiazole 35. Similarly, replacement of the thiazole sulfur atom with either a nitrogen or oxygen atom in imidazole 78 and oxazole 82 provides a 16- to 64-fold loss in activity compared to thiazole 35. Introduction of another nitrogen atom into oxazole 82 affords oxadiazole 83, which led to a modest 2–4 regain in potency relative to 82, but this analogue is still considerably less active than 35. Thiadiazole 86 was prepared, but this compound is poorly soluble and a MIC could not be determined. Collectively, these results demonstrate the thiazole is optimal since all modifications either abolish activity or result in a drastic loss in potency.
Table 5.
| ||||||
|---|---|---|---|---|---|---|
| Cmpd | R | X | Y | Z | MIC, μM (7H9-glucose) | MIC, μM (GAST) |
| 75 | H | S | CH | CH | >25 | 12.5–25 |
| 78 | H | NH | CH | N | 25 | 3.13–6.25 |
| 82 | H | O | CH | N | 25 | 25 |
| 83 | F | N | N | O | 12.5 | 6.25 |
| 86 | F | N | N | S | >6.25a | >6.25a |
poor solubility precluded evaluation at higher concentrations.
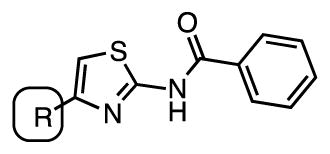
The pyridine ring at C-4 of the aminothiazole was the last part of the original scaffold to be explored. Deletion of the nitrogen atom in phenyl 90 obliterates all activity (Table 6). Surprisingly, even the isosteric pyrazine 91 is inactive, which differs only from the parent 2-pyridyl 35 by an additional nitrogen atom. The loss of the activity of pyrazine 91 is perplexing, but potentially could be due to the lower basicity of the pyrazine nitrogen atom compared to the pyridine nitrogen atom. Thiazole 92 was the last compound in this series examined and is also inactive. Collectively, these results demonstrate a strict requirement of the 2-pyridyl group for antitubercular activity. This SAR is in accord with the HTS results of Ananthan and coworkers.18
Table 6.
| |||
|---|---|---|---|
| Cmpd | R | MIC, μM (7H9-glucose) | MIC, μM (GAST) |
| 90 | phenyl | > 25 | ≥25 |
| 91 | 2-pyrazine | >25 | >25 |
| 92 | 4-thiazole | >25 | ≥25 |
2.3. Cytotoxicity
Compounds were evaluated for their cytotoxicity as measured by the concentration that inhibited 50% viability (EC50) against Vero cells using a MTT assay (see Experimental Section). We observed the cytotoxicity against Vero cells correlated with antitubercular activity with few exceptions resulting in low therapeutic indexes for the majority of compounds (MIC/EC50) (Table 3). The correlation of activity and cytotoxicity might indicate similar mechanism of action in both mammalian and bacterial cells. As an example the 3,5-difluorobenzoyl 65 exhibits potent antitubercular activity with MICs of 0.049–0.19 μM and an EC50 of 1.46 μM to provide a therapeutic index of 8–30. By contrast, 2-acetylbenzoyl 46 possesses relatively weak Mtb whole-cell activity with MICs of 3.13–12.5 μM and an EC50 of 86.7 μM to provide a very similar therapeutic index of 7–28. However, 3-benzyloxybenzoyl 40, 3-bromobenzoyl 52, and 3-chlorobenzoyl 55 have respectable therapeutic indices (as high as 671 for 52). Notably, all of these are meta-substituted benzoyl derivatives.
2.4. Resistance frequency
The frequency of spontaneous resistance development was evaluated using a concentration of 10× the MIC. Mutants to 44 and 52, arose at a frequency of ~10−5 in M. tuberculosis H37Rv with a resistance range of 2–8-fold above the MIC level of the parental strain. These frequencies are similar to isoniazid (~5 × 10−5) and suggest the aminothiazoles may function as prodrugs. The resistant mutants were found not to be cross-resistant to ethionamide suggesting that inactivation of the monooxygenase encoded by etaA did not account for resistance in these mutants.46
2.5. In vitro microsomal stability
Based on their potent activity and favorable therapeutic indices 3-methylbenzoyl 37, 3-methoxybenzoyl 43, 3-bromobenzoyl 52, 4-bromobenzoyl 53 and 3-chlorobenzoyl 55, were evaluated for microsomal stability using mouse liver microsomes (MLM, see Experimental Section). All compounds were rapidly metabolized with half-lives ranging from 6 minutes for 37 to 19 minutes for 55. Compound 55 was additionally evaluated against pooled human liver microsomes (HLM) and exhibited a half-life of 28 minutes (table 6).
In order to determine which regions of the scaffold was metabolized via Phase I metabolism, 55 was chosen as a model compound. Compound 55 was incubated with human liver microsomes and NADPH and the metabolites were analyzed by LC-MS/MS. Two metabolites were identified and their tentative structures are shown in Figure 2. From its MS/MS fragmentation pattern, the M1 metabolite, which corresponds to the parent aminothiazole +16 mass units, can be assigned to modification on the thiazole ring, but the precise modified position was not determined (See Supporting Information for MS/MS spectra). Based on reported thiazole bioactivation, the plausible metabolite is either the the 4,5-epoxide or the 5-hydroxythiazole (via NIH-shift of the epoxide), whose tautomer is a thiazolone as shown in Figure 2.41 The M2 metabolite [M + H]+ mass corresponds to the parent aminothiazole +34 mass units. The MS/MS fragmentation showed this must occur on the 4-(pyrid-2-yl)-1,3-thiazole fragment and we hypothesize this is an acylthiourea due to ring opening of the thiazolone M1, which is precedented for related acylaminothiazoles (Figure 2).41
Figure 2.
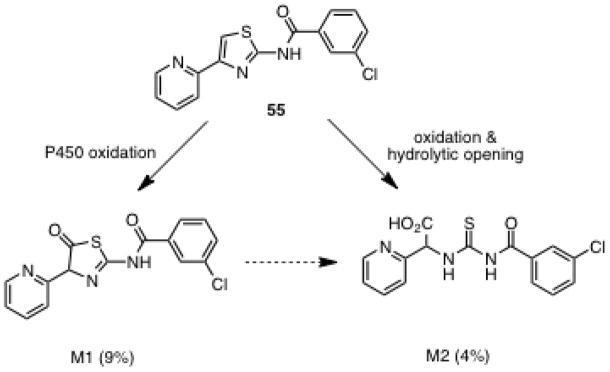
Proposed metabolites of aminothiazole 55.
2.6. Potential for chelation
The 4-(2-pyridyl)thiazole moiety of these inhibitors can coordinate metals in a bidendate fashion using the pyridine and thiazole nitrogens. The stability constants (K1) of the ML species (i.e. one metal and one ligand) have been reported for the parent 4-(2-pyridyl)thiazole ligand and these range from 104 for Fe(II) and Zn(II) to 107 for Cu(II).42 Consequently, 4-(2-pyridyl)thiazole is considered a relatively weak ligand and is comparable to many common biological ligands such as salicylic acid, glycine, and inosine, but far less than EDTA whose K1 for Fe(II) is 1014, and K1 for Cu(II) is 1019.43
3. Conclusion
We synthesized a systematic series of aminothiazole analogues based on a HTS hit from whole-cell screening against M. tuberculosis. The SAR demonstrates that the central aminothiazole and 2-pyridyl substituent at C-4 are required for potent activity. Even conservative modifications to this region completely obliterate activity. By contrast, the SAR shows substantial flexibility at N-2 of the aminothiazole. We observed the N-aryl analogues (from the initial HTS results) are much less active than reported. However, introduction of an N-benzoyl group results in a dramatic increase in potency as illustrated by N-benzoyl derivative 35 whose MIC is 0.19–0.39 μM or 64-fold better than the corresponding N-phenyl 1 (MIC = 12.5–25 μM). Among the more than thirty-five benzoyl derivatives examined, 3-bromobenzoyl 52 and 3-chlorobenzoyl 55 are the most potent analogues identified with MICs of 0.024 μM or 0.008 μg/mL in 7H9-butyrate media. In general, the SAR of the benzoyl series is relatively flat and the cytotoxicity and antitubercular activity are unfortunately strongly correlated implicating a common mechanism of inhibition. However, a few meta-substituted compounds including 3-benzyloxybenzoyl 40 (MIC~0.049–0.78 μM), 3-bromobenzoyl 52 (MIC~0.024–0.19 μM), and 3-chlorobenzoyl 55 (MIC~0.024–0.39 μM) show improved selectivity with therapeutic indices greater than 250, primarily through an increase in antitubercular activity rather than lowering intrinsic cytotoxicity. The frequency of spontaneous resistance development in M. tuberculosis H37Rv to these potent aminothiazoles is very high at 10−5. We also showed that the aminothiazoles are rapidly metabolized by liver microsomes through oxidation of the invariant 4-(pyrid-2-yl)-1,3-thiazole moiety. Further efforts will be required to improve the metabolic stability and decrease the frequency of resistance prior to pre-clinical assessment of this class of compounds.
4. Materials and methods
4.1. General Procedures
All commercial reagents (Sigma-Aldrich, Acros, Oakwood Inc., TCI America) were used as provided unless otherwise indicated. Human liver microsomes were purchased from BD Biosciences (San Jose, CA) and NADPH was supplied by EMD Millipore (Billerica, MA). Water and acetonitrile for the LCMS was Optima LCMS grade from Fisher Scientific (Pittsburgh, PA), while formic acid was from Sigma-Aldrich (St. Louis, MO). An anhydrous solvent dispensing system (J. C. Meyer) using two packed columns of molecular sieves were used to dry DMF, THF and DCM and the solvents were dispensed under argon. Flash chromatography was performed manually or using Combiflash® Companion® equipped with Luknova flash column silica cartridges (www.luknova.com) with the indicated solvent system. All reactions were performed under an inert atmosphere of dry Ar or N2 in oven-dried (150 °C) glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual methanol (3.31 ppm), DMSO (2.50 ppm) or chloroform (7.26 ppm), and carbon chemical shifts are reported using an internal standard of residual methanol (49.1 ppm), DMSO (39.5 ppm) or chloroform (77.0 ppm). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, hep = heptet, m = multiplet), coupling constant, integration. High resolution mass spectra were obtained on an Agilent TOF II TOF/MS instrument equipped with either an ESI or APCI interface. Analytical HPLC were obtained on an Agilent 1100 Series HPLC system with a PDA detector. LC-MS/MS analysis was performed using a Agilent (Santa Clara, CA) 1260 Infinity gradient solvent delivery system, an Agilent 1260 Infinity HiP ALS autosampler, an Agilent 1260 column oven, and an AB Sciex QTRAP 5500 (Framingham, MA) mass spectrometer fitted with an electrospray ionization source.
4.1.1. General procedure A (for synthesis of compounds 1–19)
To 2-bromoacetylpyridine hydrobromide (1.0 equiv) in anhydrous ethanol (5 mL) was added the corresponding thiourea (1.0 equiv, 0.2 g) and the reaction mixture refluxed for 4 h. After cooling to ambient temperature the reaction mixture was poured into water. The pH of the mixture was adjusted to pH 8 with concentrated aqueous NH4OH and the mixture stirred for 2 h. The precipitate was filtered, washed with ethanol and dried to afford the title compound.
4.1.2. General procedure B (for synthesis of compounds 21–31)
To the carboxylic acid substrate (50 mg, 1.0 equiv) in DMF (1.5 mL) was added EDC (1.5 equiv) and DMAP (0.1 equiv). The reaction was stirred for 30 min at 23 °C. Next, 2-amino-4-(2-pyridyl)thiazole 20 (1.5 equiv) was added and the reaction mixture was stirred for 16 h at 23 °C. The reaction was concentrated in vacuo under reduced pressure and the residue was dissolved in CHCl3 (8 mL) and washed successively with saturated aqueous NaHCO3 (5 mL) and H2O (5 mL). The organic layer was separated, dried over MgSO4 and concentrated. The crude product was purified by silica gel flash chromatography (0–30% MeOH–CHCl3) to afford the title compound.
4.1.3. General procedure C
To the carboxylic acid substrate (0.42 mmol, 1.2 equiv) in DMF (1.5 mL) was added CDI (0.50 mmol, 1.1 equiv) and DBU (0.67 mmol, 1.9 equiv). The reaction was stirred for 30 min at 23 °C. Next, 2-amino-4-(2-pyridyl)thiazole 20 (0.35 mmol, 1.0 equiv) was added and the reaction mixture was stirred for 16 h at 23 °C. A small amount of silica gel was added to the reaction mixture, which was concentrated in vacuo under reduced pressure. The crude product impregnated on the silica gel was purified by silica gel flash chromatography (0–10% MeOH–CH2Cl2) to afford the title compound.
4.1.4. N-Phenyl-4-(2-pyridinyl)-1,3-thiazol-2-amine (1)
A representative experimental for General Procedure A. Characterization data of 2–19 is provided in the supporting information.
The title compound was synthesized from phenylthiourea (0.20 g, 1.3 mmol) using general procedure A to afford t (0.27 g, 82%) of a cream colored powder: Rf = 0.53 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 7.05 (t, J = 7.2 Hz, 1H), 7.20 (dd, J = 7.2, 4.8 Hz, 1H), 7.35 (t, J = 8.4 Hz, 2H), 7.41 (s, 1H), 7.43 (d, J = 8.4 Hz, 2H), 7.74 (td, J = 7.5, 1.8 Hz, 1H), 7.99 (d, J = 7.2 Hz, 1H), 8.57 (d, J = 4.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 106.3, 118.2, 120.9, 122.5, 123.0, 129.4, 136.9, 140.3, 149.4, 151.0, 152.5, 164.6; HRMS (ESI+) calcd for C14H12N3S [M + H]+ 254.0746, found 254.0748 (error 0.8 ppm).
4.1.5. 2-Amino-4-(2-pyridyl)-thiazole (20)
A solution of thiourea (1.2 g, 15 mmol) in H2O (10 mL) was added to a solution of 2-bromoacetylpyridine hydrobromide (4.2 g, 15 mmol) in H2O (20 mL). The reaction mixture was stirred for 30 minutes at 23 °C. The crystalline product was collected by filtration. The solid was suspended in H2O (50 mL) and made alkaline by adding 7% aqueous ammonium hydroxide. The product was isolated as a white solid (2.4 g, 90% yield). 1H NMR (600 MHz, CDCl3) δ 5.05 (bs, 2H), 7.18 (ddd, J = 7.3, 4.7, 1.2 Hz, 1H), 7.31 (s, 1H), 7.71 (td, J = 7.6, 1.5 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 8.58 (d, J = 4.4 Hz, 1H); 13C NMR and HRMS matched the reported values.40
4.1.6. N-[4-(2-pyridinyl)-1,3-thiazol-2-yl)butyramide (21)
A representative experimental for General Procedure B. Characterization data of 22–34 is provided in the supporting information.
The title compound was synthesized from butanoic acid (50 mg, 0.57 mmol) using general procedure B to afforded (90 mg, 64%) of a white colored powder: Rf = 0.49 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 0.84 (t, J = 7.5 Hz, 3H), 1.63 (sext, J = 7.2 Hz, 2H), 2.28 (t, J = 7.2 Hz, 2H), 7.16 (t, J = 6.6 Hz, 1H), 7.59 (s, 1H), 7.67 (t, J = 7.5 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 8.54 (d, J = 4.8 Hz, 1H), 11.15 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 13.4, 18.3, 37.7, 111.8, 120.6, 122.6, 137.0, 148.8, 149.1, 152.1, 158.7, 171.5; HRMS (ESI+) calcd for C12H14N3OS [M + H]+ 248.0852, found 248.0850 (error 0.8 ppm).
4.1.7. 2-[N-(Benzoyl)amino]-4-(pyridin-2-yl)thiazole (35)
A representative experimental for General Procedure C. Characterization data of 36–70 is provided in the supporting information.
The title compound was synthesized from benzoic acid (52 mg, 0.43 mmol) using general procedure C to afford (64 mg, 65%) of a slightly yellowish crystalline solid: Rf = 0.84 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 7.19 (dd, J = 7.8, 4.5 Hz, 1H), 7.49 (t, J = 7.8 Hz, 2H), 7.58 (t, J = 7.8 Hz, 1H), 7.71 (dt, J = 7.2, 1.8 Hz, 1H), 7.73 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.92 (d, J = 7.8 Hz, 2H), 8.62 (d, J = 4.7 Hz, 1H), 9.98 (s, 1H, NH); 13C NMR (150 MHz, CDCl3) δ 112.2, 120.4, 122.5, 127.4, 128.7, 131.8, 132.7, 136.6, 149.4, 149.7, 151.9, 159.0, 165.2; HRMS (ESI+) calcd for C15H12N3OS [M + H]+ 282.0696, found 282.0691 (error 1.8 ppm).
4.1.8. 2-[N-Methyl-N-(benzoyl)amino]-4-(pyridin-2-yl)thiazole (71)
A solution of (106 mg, 0.38 mmol, 1.0 equiv) 35 in DMF (2.5 mL) was cooled to 0 °C then NaH (60% suspension in oil, 28 mg, 0.70 mmol, 1.9 equiv) was added and the suspension stirred for 5 min. Next, iodomethane (40 μL, 0.64 mmol, 1.7 equiv) was added and the reaction was stirred for 2 h at 0 °C, slowly warmed to 23 °C over 2 h and stirred an additional 90 h at 23 °C. The reaction was partitioned between H2O (5 mL) and CH2Cl2 (5 mL). The aqueous layer was extracted with CH2Cl2 (3 × 5 mL) and the combined organic layers were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (stepwise gradient of 10–50% EtOAc in hexane ) afforded the title compound (14 mg, 13%) as a colorless solid: Rf = 0.78 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 3.76 (s, 3H), 7.24 (t, J = 5.7 Hz, 1H), 7.50 (t, J = 7.5 Hz, 2H), 7.53 (dd, J = 7.2, 6.6 Hz, 1H), 7.58 (d, J = 7.6 Hz, 2H), 7.79 (t, J = 7.6 Hz, 1H), 7.93 (s, 1H), 8.12 (d, J = 7.6 Hz, 1H), 8.64 (d, J = 4.1 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 38.4, 114.3, 121.0, 122.6, 127.5, 128.6, 130.9, 134.4, 137.6, 148.4, 148.7, 152.2, 160.3, 170.4; HRMS (APCI+) calcd for C16H14N3OS [M + H]+ 296.0852, found 296.0856 (error 1.4 ppm).
4.1.9. N-Phenyl-4-bromothiazole-2-carboxamide (72)
A solution of isopropylmagnesium chloride (2.0 M in THF, 0.43 mL, 0.86 mmol, 1.04 equiv) was slowly added down the side of pre-cooled flask to a solution of 2,4-dibromothiazole (200 mg, 0.82 mmol, 1.0 equiv) in THF (6.0 mL) at −65°C. The reaction was stirred for 10 min at −65 °C, then the reaction was rapidly warmed to 0 °C and stirred for 30 min at 0 °C to afford a light yellow solution. The reaction was re-cooled to −65°C and a solution of phenylisocyanate (0.16 mL, 1.5 mmol, 1.8 equiv) in THF (1 mL) was slowly added to the reaction mixture. The reaction slowly warmed to 23 °C over ~8 h and was stirred for another 14 h at 23 °C. The reaction was quenched by the addition of saturated aqueous ammonium chloride (20 mL). The reaction was extracted with ethyl acetate (3 × 25 mL) and the combined organic layers were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (3:1 hexane–EtOAc) afforded the title compound (120 mg, 52%) as a light yellow solid: Rf = 0.68 (3:1 hexane–EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.19 (t, J = 7.2 Hz, 1H), 7.39 (t, J = 7.6 Hz, 2H), 7.54 (s, 1H), 7.70 (d, J = 8.2 Hz, 2H), 8.93 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 119.9, 123.7, 125.1, 129.0, 129.2, 136.7, 155.9, 164.2; HRMS (APCI+) calcd for C10H8BrN2OS [M + H]+ 282.9535, found 282.9531 (error 1.4 ppm).
4.1.10. N-Phenyl-4-(pyridin-2-yl)thiazole-2-carboxamide (73)
A mixture of Pd2(dba)3 (15 mg, 0.016 mmol, 0.10 equiv), 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (9.0 mg, 0.021 mmol, 0.13 equiv) and 72 in THF (6.0 mL) was heated to 65 °C for 10 min. Next, a solution of pyridylzinc bromide (0.5 M in THF, 0.60 mL, 0.30 mmol, 1.9 equiv) was slowly added and the reaction mixture stirred at 68 °C for 16 h. The reaction was cooled to rt and saturated aqueous NaHCO3 (20 mL) was added. The mixture was extracted with EtOAc (3 × 25 ml) and the combined organic layers were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (linear gradient of hexane to 3:1 hexane–EtOAc) afforded the title compound (18 mg, 40%) as a light yellow solid: Rf = 0.41 (1:1 hexane–EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.19 (t, J = 7.2 Hz, 1H), 7.31 (t, J = 5.4 Hz, 1H), 7.41 (t, J = 7.2 Hz, 2H), 7.76 (d, J = 7.6 Hz, 2H), 7.84 (t, J = 7.2 Hz, 1H), 8.17 (d, J = 7.6 Hz, 1H), 8.36 (s, 1H), 8.67 (d, J = 2.4 Hz, 1H), 9.15 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 119.9, 121.1, 123.1, 123.4, 124.9, 129.2, 137.0, 137.2, 149.6, 151.5, 156.0, 157.1, 163.9; HRMS (APCI+) calcd for C15H12N3OS [M + H]+ 282.0696, found 282.0695 (error 0.4 ppm).
4.1.11. 2-[N-(Benzoyl)amino]-4-bromothiophene (74)
Benzamide (35 mg, 0.29 mmol, 1.4 equiv), copper iodide (13 mg, 0.07 mmol, 33 mol%) and potassium phosphate (95 mg, 0.45 mmol, 2.2 equiv) were placed in a vial. The vial was evacuated and backfilled with Argon twice, then 2,4-dibromothiophene (23.4 μL, 0.21 mmol), racemic trans-1,2-diaminocyclohexane (7.5 mL, 0.06 mmol, 30 mol%) and 1,4-dioxane (0.6 ml) were added. The vial was sealed and heated at 110 °C for 22 h. The resulting suspension was cooled to rt, filtered through a pad of silica gel washing with CH2Cl2 and the filtrate concentrated. Purification by silica gel flash chromatography (3:1 hexane EtOAc) afforded the title compound (22 mg, 38%) as colorless crystals: Rf = 0.74 (1:1 hexane–EtOAc); 1H NMR (600 MHz, DMSO-d6 δ 6.73 (s, 1H), 6.82 (s, 1H), 7.45 (t, J = 7.6 Hz, 2H), 7.54 (t, J = 7.6 Hz, 1H), 7.86 (d, J = 7.6 Hz, 2H), 9.09 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6) δ 106.6, 113.7, 115.0, 127.6, 128.6, 132.2, 132.6, 141.5, 163.8; HRMS (APCI+) calcd for C11H9BrNOS [M + H]+ 281.9583, found 281.9574 (error 3.2 ppm).
4.1.12. 2-[N-(Benzoyl)amino]-4-(pyridin-2-yl)thiophene (75)
Pd2(dba)3 (35 mg, 0.038 mmol, 0.11 equiv), XPhos (20 mg, 0.047 mmol, 0.14 equiv) and 74 (96 mg, 0.34 mmol, 1.0 equiv) in THF (10 mL) were heated at reflux for 15 min. Next, a solution of 2-pyridylzinc bromide (0.5 M in THF, 1.8 mL, 0.90 mmol, 2.6 equiv) was slowly added to this refluxing solution (washing with 1.5 mL THF) and the mixture heated at reflux for 16 h. The reaction was quenched with saturated aqueous NH4Cl (30 mL) and extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (1% MeOH–CH2Cl2) afforded the title compound (47 mg, 49%) as a light orange solid: Rf = 0.75 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CD3OD) δ 7.27 (dd, J = 7.2, 5.4 Hz, 1H), 7.45 (s, 1H), 7.53 (t, J = 7.8 Hz, 2H), 7.54 (s, 1H), 7.59 (d, J = 7.2 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.82 (t, J = 7.8 Hz, 1H), 7.98 (d, J = 7.6 Hz, 2H), 8.52 (d, J = 4.7 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 111.9, 117.2, 121.6, 122.8, 128.2, 129.3, 132.8, 133.7, 138.3, 138.6, 141.4, 149.4, 154.2, 166.0; HRMS (APCI+) calcd for C16H13N2OS [M + H]+ 281.0743, found 281.0742 (error 0.4 ppm).
4.1.13. 2-(Pyridin-2-yl)imidazo[1,2-a]pyrimidine (76)
To a solution of 2-bromoacetylpyridine hydrobromide (0.91 g, 3.2 mmol, 1.0 equiv) and 2-aminopyrimidine (0.31 g, 3.2 mmol, 1.0 equiv) in EtOH (15 mL) was added potassium carbonate (0.45 g, 3.3 mmol, 1.02 equiv) and the reaction heated at reflux for 20 h. The reaction was cooled to rt and concentrated, then the residue was partitioned between CH2Cl2 (50 mL) and H2O (50 mL). The aqueous layer was extracted with CH2Cl2 (3 × 30 mL) and the combined organic layers were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (linear gradient CH2Cl2 to 4% MeOH–CH2Cl2) afforded the title compound (0.31 g, 49%) as a brown solid: Rf = 0.24 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CD3OD) δ 7.06 (dd, J = 6.2, 4.4 Hz, 1H), 7.37 (t, J = 6.6 Hz, 1H), 7.92 (t, J = 7.6 Hz, 1H), 8.16 (d, J = 7.6 Hz, 1H), 8.34 (s, 1H), 8.57–8.59 (m, 2H), 8.90 (d, J = 6.5 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 111.0, 111.3, 122.5, 124.9, 136.8, 139.0, 146.6, 150.0, 150.6, 153.0, 153.2; HRMS (APCI+) calcd for C11H9N4 [M + H]+ 197.0822, found 197.0818 (error 2.0 ppm).
4.1.14. 2-Amino-4-(pyridin-2-yl)imidazole (77)
To a suspension of 76 (290 mg, 1.48 mmol, 1.0 equiv) in ethanol 95% (11 mL) was added hydrazine monohydrate (2.4 mL, 49.5 mmol, 33 equiv). The pressure tube was sealed and the mixture heated at 130 °C for 2.5 h. The reaction was cooled to rt and concentrated in vacuo. Purification by silica gel flash chromatography (linear gradient 5–10% MeOH–CH2Cl2) afforded the title compound (116 mg, 49%) as a yellow foam: Rf = 0.09 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CD3OD) δ 7.08 (t, J = 6.2 Hz, 1H), 7.20 (s, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 8.37 (d, J = 4.1 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 117.7, 119.8, 122.1, 133.6, 138.4, 149.8, 152.6, 152.9; HRMS (APCI+) calcd for C8H9N4 [M + H]+ 161.0822, found 161.0824 (error 1.2 ppm).
4.1.15. 2-[N-(Benzoyl)amino]-4-(pyridin-2-yl)imidazole (78)
A solution of 77 (115 mg, 0.72 mmol), benzoyl chloride (124 μL, 1.08 mmol, 1.5 equiv) and 1-methylimidazole (143 μL, 1.79 mmol, 2.5 equiv) in CH2Cl2 (15 mL) was stirred at 23 °C for 18 h. The reaction was concentrated in vacuo and the residue purified by silica gel flash chromatography (linear gradient CH2Cl2 to 2% MeOH–CH2Cl2) to afford the title compound (79 mg, 42 %) as a colorless solid: Rf = 0.13 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, DMSO-d6) δ 7.19 (ddd, J = 6.9, 4.7, 1.8 Hz, 1H), 7.48 (s, 1H), 7.53 (t, J = 7.5 Hz, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.78 (td, J = 7.8, 1.8 Hz, 1H), 7.81 (d, J = 7.2 Hz, 1H); 8.09 (d, J = 7.0 Hz, 2H), 8.51 (d, J = 4.7 Hz, 1H), 11.76 (s, 1H, NH), 12.03 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6) δ 109.9, 114.6, 118.6, 121.7, 128.2, 128.8, 132.6, 133.2, 137.2, 142.2, 149.4, 152.3, 166.2; HRMS (APCI+) calcd for C15H13N4O [M + H]+ 265.1084, found 265.1088 (error 1.5 ppm).
4.1.16. 2-(2-Acetoxy)acetylpyridine (79)
A mixture of 2-bromoacetylpyridine hydrobromide (0.20 g, 0.72 mmol, 1.0 equiv) and sodium acetate (0.17 g, 2.0 mmol, 2.8 equiv) in 95% EtOH (6.0 mL) were heated at reflux for 2 h. The reaction mixture was cooled to rt and the suspension was filtered over Celite washing with CH2Cl2. The filtrate was concentrated to afford a brown solid (150 mg, quant.) which was used without further purification. Silica gel flash chromatography (stepwise gradient from hexanes to 50% EtOAc in hexanes) provided an analytically pure sample for characterization: Rf = 0.50 (1:1 hexane–EtOAc); 1H NMR (600 MHz, CDCl3) δ 2.24 (s, 3H), 5.62 (s, 2H), 7.51 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 7.86 (td, J = 7.8, 1.5 Hz, 1H), 8.03 (d, J = 7.6 Hz, 1H), 8.66 (d, J = 4.7 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 20.6, 67.9, 122.8, 129.5, 138.7, 150.4, 152.5, 172.3, 194.9; HRMS (APCI+) calcd for C9H10NO3 [M + H]+ 180.0655, found 180.0657 (error 1.1 ppm).
4.1.17. 2-Amino-4-(2-pyridyl)oxazole (81)
Crude 79 (~0.72 mmol) prepared above was dissolved in 6 M aqueous HCl (5.0 mL) and stirred at 50 °C for 1.5 h. The mixture was cooled to rt and neutralized with saturated aqueous NaHCO3. The aqueous phase was extracted with EtOAc (3 × 20 ml) and the combined organic extracts were dried over MgSO4 and concentrated to a brown oil. The oil was redissolved in 95% EtOH (5.0 mL) then cyanamide (46 mg, 1.1 mmol, 1.5 equiv) was added and the solution was heated at reflux for 14 h. After cooling to rt, the pH was adjusted to 10 using 1 M aqueous NaOH. The aqueous phase was extracted with EtOAc (3 × 20 ml) and the combined extracts were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (5% MeOH–CH2Cl2) afforded the title compound (11 mg, 0.068 mmol, 10% over three steps from 2-bromoacetylpyridine hydrobromide) as ayellow solid: Rf = 0.09 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 7.19 (t, J = 6.0 Hz, 1H), 7.34 (s, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.78 (t, J = 7.9 Hz, 1H), 8.43 (d, J = 4.7 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 119.2, 123.0, 126.9, 139.0, 145.0, 148.8, 150.2, 164.2; HRMS (ESI+) calcd for C8H8N3O [M + H]+ 162.0662, found 162.0658 (error 2.5 ppm).
4.1.18. 2-[N-(Benzoyl)amino]-4-(pyridin-2-yl)oxazole (82)
Benzoyl chloride (30 μL, 0.26 mmol, 1.5 equiv) was added to a solution of 81 (28 mg, 0.17 mmol, 1.0 equiv) and 1-methylimidazole (36 μL, 0.45 mmol, 2.6 equiv) in CH2Cl2 (3.0 mL) and the reaction mixture was stirred at 23 °C for 18 h. The reaction was partitioned between saturated aqueous ammonium chloride (15 mL) and CH2Cl2 (20 mL). The aqueous layer was extracted with CH2Cl2 (2 × 20 mL) and the combined organic layers were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (gradient from CH2Cl2 to 3% MeOH in DCM) afforded the title compound (30 mg, 65%) as a colorless solid: Rf = 0.63 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 7.18–7.22 (m, 1H), 7.44 (t, J = 6.3 Hz, 2H), 7.50–7.55 (m, 2H), 7.64 (d, J = 7.6 Hz, 1H), 7.74 (t, J = 6.6 Hz, 1H), 7.96 (d, J = 7.0 Hz, 2H), 8.47 (d, J = 3.0 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 118.6, 122.7, 125.1, 128.2, 128.4, 129.3, 132.4, 137.1, 146.5, 149.5, 154.1, 165.1, 167.6; HRMS (APCI+) calcd for C15H12N3O2 [M + H]+ 266.0924, found 266.0917 (error 2.6 ppm).
4.1.19. 2-[N-(2-Fluorobenzoyl)amino]-5-(pyridin-2-yl)-1,3,4-oxadiazole (83)
Picolinyl hydrazide (5.0 g, 36.5 mmol, 1.0 equiv), cyanogen bromide (4.2 g, 40.1 mmol, 1.1 equiv), and potassium bicarbonate (4.0 g, 40.1 mmol, 1.1 equiv) in 1:1 dioxane–H2O (72 mL) were heated at reflux for 15 h. The reaction was cooled to rt and the suspension was filtered to collect the solid product, which was washed with 1:1 dioxane–H2O to afford crude 2-amino-4-(pyridin-2-yl)oxadiazole (3.0 g, 51%) that was used in the next step without purification.
A mixture of 2-fluorobenzoic acid (64 mg, 0.46 mmol, 1.3 equiv), CDI (85 mg, 0.52 mmol, 1.5 equiv) and DBU (0.10 mL, 0.67 mmol, 1.9 equiv) in DMF (1.5 mL) was stirred at 60 °C for 1 h. Next, crude 2-amino-4-(pyridin-2-yl)oxadiazole prepared above (57 mg, 0.35 mmol, 1.0 equiv) was added and the reaction mixture was stirred at 60° C for 17 h. The reaction was concentrated in vacuo onto silica gel. Purification by silica gel flash chromatography (linear gradient CH2Cl2 to 5% MeOH–CH2Cl2) afforded the title compound (72 mg, 72%) as a colorless solid: Rf = 0.55 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 7.24 (dd, J = 12.3, 8.8 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.46 (dd, J = 7.3, 5.0 Hz, 1H), 7.60–7.64 (m, 1H), 7.89 (t, J = 7.9 Hz, 1H), 8.21–8.24 (m, 2H), 8.77 (d, J = 4.7 Hz, 1H), 9.47 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 116.5 (d, 2JC-F = 25 Hz), 118.9, 122.8, 125.5, 125.8, 132.8, 135.5 (d, 3JC-F = 9.2 Hz), 137.2, 143.1, 150.3, 157.5, 159.5, 160.7 (d, 1JC-F = 247 Hz), 160.8; HRMS (ESI+) calcd for C14H10FN4O2 [M + H]+ 285.0782, found 285.0775 (error 2.5 ppm).
4.1.20. 2-Amino-5-(pyridin-2-yl)-1,3,4-thiadiazole (85)
A suspension of picolinoyl chloride hydrochloride (5.0 g, 28.1 mmol, 1.0 equiv) and hydrazinecarbothioamide (5.1 g, 56.2 mmol, 2 equiv) in THF (170 mL) was stirred at 23 °C for 7 d. The solvent was evaporated to afford crude 2-picolinoylhydrazinecarbothioamide 84 in quantitative yield.
Methanesulfonic acid (1.5 mL, 22.9 mmol, 1.3 equiv) was added to crude picolinoylhydrazinecarbothioamide 84 prepared above (3.0 g, 15.3 mmol, 1.0 equiv) in toluene (104 mL) at 23 °C, which led to precipitation. The reaction mixture was heated at reflux for 16 h. After cooling to rt, the toluene was decanted and the remaining solid (i.e. crude product) was dissolved in H2O. The aqueous solution was treated with concentrated ammonium hydroxide until basic, which led to the formation of a precipitate. The precipitate was collected and dried to afford the title compound (2.5 g, 92%) as a yellow solid: Rf = 0.48 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CD3OD) δ 7.39 (ddd, J = 7.2, 4.8, 1.2 Hz, 1H), 7.88 (td, J = 7.9, 1.8 Hz, 1H), 8.09 (d, J = 7.6 Hz, 1H), 8.54 (d, J = 4.7 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 120.8, 125.8, 138.6, 150.7, 151.0, 161.4, 172.9; HRMS (APCI+) calcd for C7H7N4S [M + H]+ 179.0386, found 179.0382 (error 2.2 ppm).
4.1.21. 2-[N-(2-Fluorobenzoyl)amino]-5-(pyridin-2-yl)-1,3,4-thiadiazole (86)
A mixture of 2-fluorobenzoic acid (63 mg, 0.45 mmol, 1.3 equiv), CDI (85 mg, 0.52 mmol, 1.5 equiv) and DBU (0.10 mL, 0.67 mmol, 1.9 equiv) in DMF (1.5 mL) was stirred at 60 °C for 1 h. Next, 85 (62 mg, 0.35 mmol) was added and the reaction was stirred at 60 °C for 17 h. The reaction mixture was concentrated in vacuo onto silica gel. Purification by silica gel flash chromatography (linear gradient CH2Cl2 to 1% MeOH–CH2Cl2) afforded the title compound (59 mg, 56%) as a colorless solid: Rf = 0.75 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, DMSO-d6) δ 7.30 (dd, J = 10.2, 9.0 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.46 (dd, J = 6.7, 5.0 Hz, 1H), 7.61 (ddd, J = 13.8, 7.2, 1.8 Hz, 1H), 7.77 (td, J = 7.2, 1.2 Hz, 1H), 7.93 (td, J = 7.8, 1.5 Hz, 1H), 8.21 (d, J = 8.2 Hz, 1H), 8.64 (d, J = 4.7 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 116.5 (d, 2JC-F = 22 Hz), 120.1, 121.8 (d, 2JC-F = 13 Hz), 124.7 (d, 3JC-F = 3.5 Hz), 125.3, 130.6, 134.2 (d, 3JC-F = 9.2 Hz), 137.6, 149.4, 150.0, 159.9 (d, 1JC-F = 252 Hz), 160.4, 163.0, 164.5; HRMS (ESI+) calcd for C14H10FN4OS [M + H]+ 301.0554, found 301.0551 (error 1.0 ppm).
4.1.22. 2-Amino-4-(phenyl)thiazole (89a)
To a solution of acetophenone (1.2 mL, 10.3 mmol, 1.0 equiv) in CHCl3 (5.0 mL) was slowly added a solution of bromine (0.60 mL, 11.6 mmol, 1.1 equiv) in CHCl3 (5.0 mL). The reaction was stirred for 2 h at 23 °C, then another aliquot of bromine (0.60 mL, 11.6 mmol, 1.1 equiv) in CHCl3 (5.0 mL) was added and the reaction was stirred a further 2 h at 23 °C. The reaction mixture was concentrated and cooled to afford crystals of phenacyl bromide 88a that were collected and used in the next step without further purification.
Thiourea (0.77 g, 10 mmol, 1 equiv) was added to a solution of phenacyl bromide 88a prepared above in glacial acetic acid (15 mL) and the reaction was heated at reflux for 3.5 h. The mixture was cooled to rt, and the solid product was collected and dissolved in H2O (10 mL). The pH was adjusted to 4 by the addition of sat. aqueous NaHCO3 and the solid precipitate was collected. Purification by flash chromatography (gradiant of 1–8% MeOH in DCM) afforded the title compound (0.47 g, 27%) as a yellow solid: Rf = 0.50 (9:1 CH2Cl2–MeOH); Analytical data matched previously reported values.41
4.1.23. 2-[N-(Benzoyl)amino]-4-(phenyl)thiazole (90)
A mixture of 2-amino-4-(phenyl)thiazole 89a (0.10 g, 0.57 mmol), benzoyl chloride (0.10 mL, 0.87 mmol, 1.5 equiv) and N-methylimidazole (0.13 mL, 1.6 mmol, 2.9 equiv) in CH2Cl2 (15 mL) was stirred at 23 °C for 18 h. Saturated aqueous NH4Cl (25 mL) was added and the mixture extracted with CH2Cl2 (3 × 20 mL). The combined extracts were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (CH2Cl2) afforded the title compound (45 mg, 28%) as light yellow crystals: Rf = 0.48 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 7.11 (s, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.25 (t, J = 7.6 Hz, 2H), 7.29 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.66 (d, J = 7.6 Hz, 2H), 7.75 (d, J = 7.6 Hz, 2H), 10.59 (s, 1H, NH); 13C NMR (150 MHz, CDCl3) δ 108.0, 126.0, 127.4, 128.0, 128.6, 128.8, 131.8, 132.7, 134.1, 150.2, 158.7, 165.0; HRMS (APCI+) calcd for C16H13N2OS [M + H]+ 281.0743, found 281.0736 (error 2.5 ppm).
4.1.24. 2-Amino-4-(pyrazin-2-yl)thiazole (89b)
Hydrobromic acid (33 wt% in AcOH, 3.1 mL, 17.1 mmol, 1.05 equiv) and bromine (0.85 mL, 16.5 mmol, 1.0 equiv) were sequentially, slowly added to a solution of 2-acetylpyrazine (2.0 g, 16.4 mmol, 1.0 equiv) in glacial acetic acid (18 mL) and the reaction was stirred for 4 h at 23 °C. The yellow solid was collected, washed with glacial acetic acid (7 mL) and dried to afford crude 2-(2-bromo)acetylpyrazine hydrobromide 88b (3.3 g, 71% crude) as a colorless solid, which was used without further purification.
To a solution of 2-(2-bromo)acetylpyrazine hydrobromide 88b prepared above (1.0 g, 3.55 mmol, 1.0 equiv) in 95% EtOH was added thiourea (0.33 g, 4.3 mmol, 1.2 equiv) and DIPEA (1.8 mL, 10.3 mmol, 2.9 equiv) and the brown suspension was heated at reflux for 3 h. After cooling to rt the solvent was evaporated and the residue washed with CH2Cl2. The solid was dried under reduced pressure to afford the title compound (0.60 g, 95%) as a yellow solid: Rf = 0.27 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CD3OD) δ 7.32 (s, 1H), 8.40 (d, J = 2.3 Hz, 1H), 8.51 (s, 1H), 9.01 (d, J = 1.2 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 109.5, 142.5, 143.0, 144.6, 147.5, 148.6, 170.4; HRMS (APCI+) calcd for C7H7N4S [M + H]+ 179.0386, found 179.0382 (error 2.2 ppm).
4.1.25. 2-[N-(Benzoyl)amino]-4-(pyrazin-2-yl)thiazole (91)
To a solution of 2-amino-4-(pyrazin-2-yl)thiazole 89b (63 mg, 0.35 mmol, 1.0 equiv) in DMF (1.5 mL) was added benzoyl chloride (50 μL, 0.43 mmol, 1.2 equiv) and 1-methylimidazole (0.10 mL, 1.3 mmol, 3.5 equiv) and the reaction mixture stirred at 23 °C for 18 h. The reaction was concentrated in vacuo onto silica gel. Purification by silica gel flash chromatography afforded the title compound (17 mg, 17%) as a yellow crystalline solid: Rf = 0.72 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CDCl3) δ 7.57 (t, J = 7.8 Hz, 2H), 7.66 (t, J = 7.8 Hz, 1H), 8.05 (s, 1H), 8.15 (d, J = 7.0 Hz, 2H), 8.61 (d, J = 2.3 Hz, 1H), 8.69 (s, 1H), 9.23 (d, J = 1.2 Hz, 1H), 12.91 (s, 1H, NH); 13C NMR (150 MHz, CDCl3) δ 114.3, 128.2, 128.6, 131.8, 132.7, 141.5, 143.7, 144.5, 146.7, 147.4, 159.4, 165.4; HRMS (APCI+) calcd for C14H11N4OS [M + H]+ 283.0648, found 283.0640 (error 2.8 ppm).
4.1.26. 2-Amino-4-(thiazol-2-yl)thiazole (89c)
Pyridinium tribromide (1.26 g, 3.94 mmol, 1.0 equiv) was added to a solution of 2-acetylthiazole (0.46 mL, 4.63 mmol, 1.2 equiv) in CH2Cl2 (10 mL) and the reaction was stirred at 23 °C for 2 d. The reaction mixture was concentrated to approximately half the volume and the solid product was collected washing with 1:1 EtOAc–hexane (50 mL) to afford crude 2-(2-bromo)acetylthiazole hydrobromide 88c as a light yellow solid, which was used directly in the next step.
To a solution of crude 2-(2-bromo)acetylthiazole hydrobromide 88c prepared above in 95% EtOH (10 mL) was added thiourea (0.35 g, 4.64 mmol, 1.2 equiv) and DIPEA (1.6 mL, 9.19 mmol, 2.3 equiv) and the reaction was heated at reflux for 72 h. The reaction was cooled to 23 °C, concentrated in vacuo, and the residue partitioned between CH2Cl2 (40 mL) and saturated aqueous NaHCO3 (40 mL). The aqueous layer was extracted with CH2Cl2 (3 × 30 mL), the combined organic layers dried over MgSO4 and concentrated. The brown solid was recrystallized from EtOAc to afford the title compound (0.41 g, 57% over two steps) as a yellow solid: Rf = 0.35 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CD3OD) δ 7.19 (s, 1H), 7.52 (d, J = 2.4 Hz, 1H), 7.78 (d, J = 2.4 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 106.5, 120.7, 144.2, 145.5, 165.4, 171.5; HRMS (APCI+) calcd for C6H6N3S2 [M + H]+ 183.9998, found 183.9997 (error 0.5 ppm).
4.1.27. 2-[N-(Benzoyl)amino]-4-(thiazol-2-yl)thiazole (92)
To a solution of 2-amino-4-(thiazol-2-yl)thiazole 89c (65 mg, 0.34 mmol, 1.0 equiv) in DMF (4 mL) were added benzoyl chloride (0.11 mL, 0.95 mmol, 2.7 equiv) and 1-methylimidazole (0.11 mL, 1.38 mmol, 3.9 equiv) and the solution was heated at 70 °C for 70 h. The reaction mixture was cooled to rt, concentrated in vacuo and the residue was partitioned between H2O (20 mL) and CH2Cl2 (20 mL). The aqueous layer was extracted with CH2Cl2 (3 × 15 mL) and the combined organic layers were dried over MgSO4 and concentrated. Purification by silica gel flash chromatography (stepwise gradient of hexanes to 20% EtOAc in hexanes) afforded the title compound (16 mg, 16%) as colorless crystals: Rf = 0.78 (9:1 CH2Cl2–MeOH); 1H NMR (600 MHz, CD3OD) δ 7.46 (d, J = 2.4 Hz, 1H), 7.52 (t, J = 7.5 Hz, 2H), 7.60 (t, J = 7.2 Hz, 1H), 7.67 (s, 1H), 7.80 (d, J = 2.4 Hz, 1H), 8.02 (d, J = 7.6 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 111.4, 119.2, 127.4, 129.0, 131.6, 133.1, 143.7, 144.0, 158.6, 162.6, 164.7; HRMS (APCI+) calcd for C13H10N3OS2 [M + H]+ 288.0260, found 288.0258 (error 0.7 ppm).
4.2. M. tuberculosis H37Rv MIC Assays
All compounds’ MICs were experimentally determined as previously described.44 Minimum inhibitory concentrations (MICs) were determined in quadruplicate in standard 7H9/glucose/glycerol/Tween, 7H9/glucose media, 7H9/butyrate media and BSA-free GAST/Fe medium according to the broth microdilution method using compounds from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. 7H9/glucose medium consisted of 7H9 Middebrook broth supplemented per liter with 0.8g NaCl, 5g bovine serum albumin fraction V, 4g glucose and 0.05% Tyloxapol. 7H9/butyrate medium consisted of 7H9 Middebrook broth supplemented per liter with 0.8g NaCl, 5g bovine serum albumin fraction V, 2.5mM butyrate and 0.05% Tyloxapol. Isoniazid was used as positive control while DMSO was employed as a negative control. All measurements reported herein used an initial cell density of 104–105 cells/assay, plates were incubated at 37 °C (100 μL/well) and growth monitored at both 7 and 14 days. Growth was recorded using an inverted enlarging mirror and the MIC was recorded as the concentration of compound that completely inhibits all visible growth.
4.3. Vero Cell Cytotoxicity Assay
Cytotoxicity of each compound was determined with a standard tetrazolium assay using Vero green monkey kidney cells (ATCC CCL-81).45 All tissue culture reagents were purchased from Gibco, Invitrogen (Carlsbad, California). Cells were grown in MEM media supplemented with 10% fetal bovine serum (FBS), 1% Penn-Strep and 1% Glutamax. Cells were seeded at 3.0 × 104 cells per well in a 96-well microtiter plate (Corning) and allowed to adhere overnight. Medium was carefully aspirated and replaced with 195 μL fresh medium, and compound solutions in DMSO (5 μL) were added to give a final concentration ranging from 100–0.16 μM. All concentrations were tested in triplicate. Plates were incubated for 72 h at 37 °C and 4.5% CO2 in a humidified chamber. The solutions were carefully removed and RPMI without phenol red containing 1.0 mg/mL 3-(4,5-dimethyl-thiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) was added (200 μL) and incubated for 3 h, after which the MTT medium was carefully removed. Isopropyl alcohol (200 μL) was added to dissolve the precipitated purple formazan crystals and the plates were read at 570 nm using a plate reader (Molecular Devices Spectramax M5e); plate background (690 nm) was subtracted and cell viability was estimated as the percentage absorbance relative to the DMSO control. Dose response curves were generated using GraphPad Prism 5 software and used to determine the EC50 concentrations (minimal concentration that inhibits 50% of growth).
4.4. Resistance Frequency
M. tuberculosis H37Rv was grown to an OD650nm of 0.5 in standard 7H9/glucose/glycerol/Tween medium. Cells and dilutions thereof were plated onto Middlebrook 7H11 agar medium plates supplemented per with 0.06g oleic acid, 2g glucose, 4g glycerol, 5g bovine serum albumin fraction V and 0.8g NaCl containing compound 44 or 52 at 0, 1-, 5- and 10-fold MIC levels. Colonies were counted after 4–5 weeks of incubation at 37 °C and resistance frequency calculated by scoring colonies relative to the drug-free control plates. Colonies (5 from each compound) from the plates containing compound at 10-fold MIC levels were picked and grown up to an OD650nm of 0.5 in standard 7H9/glucose/glycerol/Tween medium after which the MIC to compounds 44 or 52 as well as ethionamide and isoniazid as controls was determined as described above.
4.5. Microsomal stability testing
4.5.1. Mouse liver microsomal assay
A mixture of 20 μL compound solution (2 mM in DMSO), 575 μL 0.1 M sodium phosphate buffer (pH 7.4), 340 μL water and 50 μL NADPH solution (20 mM in water) was preincubated at 37°C in a water bath for 5 min. The mixture was added to 25 μL mouse liver microsomes (CD-1 mice, 20 mg/mL) and incubated in the water bath at 37 °C. After 0, 1, 2, 5, 10, 15, 30, 45 and 60 min aliquots of 100 μL were removed and added to 100 μL ice-cold acetonitrile to stop the reaction. After the last time point the aliquots were centrifuged (13,000 g, 5 min, 4°C), the supernatant transferred to spin filters, centrifuged for 30 s (13,000 g, 4°C) and kept at −80°C until analyzed. Reactions were done in duplicate. As a control NADPH was replaced by water and time points taken at 0 and 60 min. Quantitative analysis was accomplished on a Agilent 1100 series HPLC system on an analytical column (Agilent Zobrax Eclipse XDB-C8, 3.5 μm, 150 × 3.0 mm, operated at 0.5 mL/min, detection at 220 nm) using an isocratic method with 30% MeCN in 0.05% aqueous formic acid as solvent system. Retention times: 37 5.89min, 44 4.54 min, 52 3.31 min, 53 3.57min, 55 4.04min,
4.5.2. Human liver Microsomal assay
Pooled human liver microsomes (0.5 mg/mL) were incubated in 100 mM potassium phosphate buffer, pH 7.4 with 10 mM MgCl2 and 1.0 μM 54 in a volume of 900 μL. This solution was preincubated for 5 min at 4 °C followed by 2 min at 37 °C. NADPH (100 μL of a 10 mM solution in phosphate buffer) was added and this solution was incubated for 60 min at 37 °C. At various times, 100 μL from the reaction was added to 300 μL acetonitrile; the resulting suspension was vortexed, centrifuged (14,000g, 5 min) to remove precipitated protein, and the supernatant was analyzed directly via LC-MS/MS. Aminothizole 55 was analyzed using the same chromatographic conditions as described below for Metabolite Identification.’ For mass spec detection, an MRM was developed using electrospray in the positive mode to detect the transition from 139.0 ([M+H]+ precursor ion) to 111.0 (product ion). The following compound and source/gas parameters were used: declustering potential = 115 V; entrance potential = 10 V; colission energy = 63 V; collision cell exit potential = 18 V; curtain gas = 20 psi; collision gas = medium; ionspray voltage = 1500 V; temperature = 600 °C; ion source gas 1 = 40 psi; ion source gas 2 = 40 psi. Nitrogen was used for the nebuliser and collision gas. Analyst software (version 1.5.2) was used to detect analytes and MultiQuant software (version 2.0.2) was used to quantitate peak areas. % remaining values were calculated by dividing the peak area at the particular time by the peak area of time zero. The ln (% remaining) versus time was fit by linear regression analysis using GraphPad Prism (version 6.0) to provide the fractional rate of elimination (ke). The half-life was calculated by dividing the ln(2) by ke.
4.6. Metabolite identification
4.6.1. Human Liver Microsome Reaction for Metabolite Identification
Pooled human liver microsomes (0.5 mg/mL) were incubated in 100 mM potassium phosphate buffer, pH 7.4 with 10 mM MgCl2 and 1.0 μM 55 in a volume of 450 μL. This solution was preincubated for 5 min at 4 °C followed by 2 min at 37 °C. Water (50 μL, control reaction) or 10 mM NADPH (50 μL, microsomal reaction) were added and the two solutions were incubated for 60 min at 37 °C. After 60 min, 100 μL from each reaction was added to 200 μL acetonitrile; the resulting suspension was vortexed, centrifuged (14,000g, 5 min) to remove precipitated protein, and the supernatent was analyzed via LC-MS/MS.
4.6.2. Chromatography
HPLC analysis was performed using gradient HPLC on a Kinetix C18 column (50 × 2.1 mm, 2.6 μm particle size; Phenomenex, Torrance, CA). Mobile phase A was 0.1% aqueous formic acid while mobile phase B was 0.1% formic acid in acetonitrile. Initial conditions were 20% B from 0 to 0.5 min, after which the %B was increased to 95% from 2 to 8 min. The column was washed in 95% B for 0.5 min, returned to 20% over 0.5 min, and allowed to reequilibrate for 4 min in 20% B to provide a total run time of 13 min. The flow rate was 0.5 mL/min and the column oven was maintained at 40 °C. The injection volume was 10 μL.
4.6.3. Mass spectrometry
Multiple Reaction Monitoring (MRM) and Enhanced Product Ion (EPI) were created and run using Analyst Software (version 1.5.2, AB SCIEX). The following mass spectrometry settings were used: declustering potential = 115 V; entrance potential = 10 V; collision energy = 63 V; collision cell exit potential = 18 V; curtain gas = 25 psi; collision gas = high; ionspray voltage = 5500 V; temperature = 625 °C; ion source gas 1 = 45 psi; ion source gas 2 = 45 psi. Nitrogen was used for the nebuliser and collision gas. LightSight software (AB SCIEX) was used to predict MRM transitions for various Phase I metabolites. LightSight was also used to compare control reactions (water only, no metabolism) with metabolic reactions (NADPH included) to identify metabolites from data acquired in Analyst software.
Supplementary Material
Table 6.
| |||
|---|---|---|---|
| Cmpd | R | t1/2 (MLM) [min] | t1/2 (HLM) [min] |
| 37 | 3-methyl | 6.7 | n.d. |
| 44 | 4-methoxy | 16 | n.d. |
| 52 | 3-bromo | 11 | n.d. |
| 53 | 4-bromo | 6.8 | n.d. |
| 55 | 3-chloro | 19 | 28 |
Acknowledgments
This research was supported by a National Institutes of Health Grant AI070219 (to C.C.A) and the Intramural Research Program of the NIAID, NIH.
Footnotes
Supplementary data associated with this article can be found in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.xx.xx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Corbett L, Raviglione M. In: Tuberculosis and the Tubercle Bacillus. Cole ST, Eisenach KS, McMurray DN, Jacobs WR Jr, editors. ASM Press; Washington, DC: 2005. pp. 3–12. [Google Scholar]
- 2.Schatz A, Bugie E, Waksman SA. Proc Soc Exp Biol Med. 1944;55:66. [Google Scholar]
- 3.Waksman SA. Nobel Lectures, Physiology or Medicine 1942–1962. Elsevier Publishing Company; Amsterdam: 1964. [Google Scholar]
- 4.Aldrich CC, Boshoff HI, Remmel RP. Antitubercular Agents. In: Rotella D, Abraham DJ, editors. Burgers Medicinal Chemistry. 7. Wiley; New York: 2010. pp. 713–812. [Google Scholar]
- 5.Mitchison D, Davies G. Int J Tuberc Lung Dis. 2012;16:724. doi: 10.5588/ijtld.12.0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfuetze KH, Pyle MM, Hinshaw HC, Feldman WH. Am Rev Tuberc. 1955;71:752. doi: 10.1164/artpd.1955.71.5.752. [DOI] [PubMed] [Google Scholar]
- 7.Kjellsson MC, Via LE, Goh A, Weiner D, Low KM, Kern S, Pillai G, Barry CE, 3rd, Dartois V. Antimicrob Agents Chemother. 2012;56:446. doi: 10.1128/AAC.05208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. Nat Rev Microbiol. 2009;7:845. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wakamoto Y, Dhar N, Chait R, Schneider K, Signorino-Gelo F, Leibler S, McKinney JD. Science. 2013;339:91. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 10.World Health Organization. [Accessed March 2013];Fact sheet on tuberculosis. http://www.who.int/mediacentre/factsheets/fs104/en/index.html.
- 11.Cohen J. Science. 2013;339:130. doi: 10.1126/science.339.6116.130. [DOI] [PubMed] [Google Scholar]
- 12.Silver LL. Clin Microbiol Rev. 2011;24:71. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. Nature. 2000;405:962. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, Sasaki H, Shimokawa Y, Komatsu M. PLoS Med. 2006;3:e466. doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee RE, Protopopova M, Crooks E, Slayden RA, Terrot M, Barry CE., 3rd J Comb Chem. 2003;5:172. doi: 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- 16.Makarov V, Manina G, Mikusova K, Mollmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A, De Rossi E, Belanova M, Bobovska A, Dianiskova P, Kordulakova J, Sala C, Fullam E, Schneider P, McKinney JD, Brodin P, Christophe T, Waddell S, Butcher P, Albrethsen J, Rosenkrands I, Brosch R, Nandi V, Bharath S, Gaonkar S, Shandil RK, Balasubramanian V, Balganesh T, Tyagi S, Grosset J, Riccardi G, Cole ST. Science. 2009;324:801. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. Science. 2005;307:223. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 18.Ananthan S, Faaleolea ER, Goldman RC, Hobrath JV, Kwong CD, Laughon BE, Maddry JA, Mehta A, Rasmussen L, Reynolds RC, Secrist JA, 3rd, Shindo N, Showe DN, Sosa MI, Suling WJ, White EL. Tuberculosis. 2009;89:334. doi: 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddry JA, Ananthan S, Goldman RC, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Reynolds RC, Secrist JA, 3rd, Sosa MI, White EL, Zhang W. Tuberculosis. 2009;89:354. doi: 10.1016/j.tube.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reynolds RC, Ananthan S, Faaleolea E, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Sosa MI, Thammasuvimol E, White EL, Zhang W, Secrist JA., 3rd Tuberculosis (Edinb) 2012;92:72. doi: 10.1016/j.tube.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brogden RN, Heel RC. Drugs. 1986;31:96. doi: 10.2165/00003495-198631020-00002. [DOI] [PubMed] [Google Scholar]
- 22.Noble S, Balfour JA. Drugs. 1996;51:424. doi: 10.2165/00003495-199651030-00007. [DOI] [PubMed] [Google Scholar]
- 23.Bryson HM, Fulton B, Benfield P. Drugs. 1996;52:549. doi: 10.2165/00003495-199652040-00010. [DOI] [PubMed] [Google Scholar]
- 24.Hantzsch A. Ber Dtsch Bot Ges. 1927;60:2537. [Google Scholar]
- 25.Egan RS, Tadanier J, Garmaise DL, Gaunce AP. J Org Chem. 1983;33:4422. [Google Scholar]
- 26.Taurins A, Blaga A. J Heterocyclic Chem. 1970;7:1137. [Google Scholar]
- 27.Narayana B, Vijaya Raj KK, Ashalatha BV, Suchetha Kumari K, Sarojini BK. Eur J Med Chem. 2004;39:867. doi: 10.1016/j.ejmech.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Hay MP, Turcotte S, Flanagan JU, Bonnet M, Chan DA, Sutphin PD, Nguyen P, Giaccia AJ, Denny WA. J Med Chem. 2010;53:787. doi: 10.1021/jm901457w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Press NJ, Taylor RJ, Fullerton JD, Tranter P, McCarthy C, Keller TH, Brown L, Cheung R, Christie J, Haberthuer S, Hatto JD, Keenan M, Mercer MK, Press NE, Sahri H, Tuffnell AR, Tweed M, Fozard JR. Bioorg Med Chem Lett. 2005;15:3081. doi: 10.1016/j.bmcl.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 30.Schröter S, Stock C, Bach T. Tetrahedron. 2005;61:2245. [Google Scholar]
- 31.Delgado O, Heckmann G, Müller HM, Bach T. J Org Chem. 2006;71:4599. doi: 10.1021/jo060462g. [DOI] [PubMed] [Google Scholar]
- 32.Luzung MR, Patel JS, Yin J. J Org Chem. 2010;75:8330. doi: 10.1021/jo1018798. [DOI] [PubMed] [Google Scholar]
- 33.Tormyshev VM, Trukhin DV, Rogozhnikova OY, Mikhalina TV, Troitskaya TI, Flinn A. Synlett. 2006:2559. [Google Scholar]
- 34.Gewald K, Schinke E, Böttcher H. Chem Ber. 1966;99:94. [Google Scholar]
- 35.Klapars A, Antilla JC, Huang X, Buchwald SL. J Am Chem Soc. 2001;123:7727. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- 36.Fang QK, Wu FX, Grover PT, Hopkins SC, Campbel U, Chytil M, Spear KL. WO 2009/143156 (A2)
- 37.Ermolat’ev DS, Svidritsky EP, Babaev EV, Van der Eycken E. Tetrahedron Lett. 2009;50:5218. [Google Scholar]
- 38.Crank G, Khan HR. Aust J Chem. 1985;38:447. [Google Scholar]
- 39.White AD, Creswell MW, Chucholowski AW, Blankley CJ, Wilson MW, Bousley RF, Essenburg AD, Hamelehle KL, Krause BR, Stanfield RL, Dominick MA, Neub M. J Med Chem. 1996;39:4382. doi: 10.1021/jm960404v. [DOI] [PubMed] [Google Scholar]
- 40.Pethe K, Sequeira PC, Agarwalla S, Rhee K, Kuhen K, Phong WY, Patel V, Beer D, Walker JR, Duraiswamy J, Jiricek J, Keller TH, Chatterjee A, Tan MP, Ujjini M, Rao SP, Camacho L, Bifani P, Mak PA, Ma I, Barnes SW, Chen Z, Plouffe D, Thayalan P, Ng SH, Au M, Lee BH, Tan BH, Ravindran S, Nanjundappa M, Lin X, Goh A, Lakshminarayana SB, Shoen C, Cynamon M, Kreiswirth B, Dartois V, Peters EC, Glynne R, Brenner S, Dick T. Nat Commun. 2010;1:57. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Chem Res Toxicol. 2002;15:269. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 42.Eilbeck WJ, Holmes F, Thomas TW, Williams G. J Chem Soc (A) 1968:2348. [Google Scholar]
- 43.Motekaitis RJ. NIST standard reference database 46. U.S. Dept. of Commerce, National Institute of Standards and Technology, Standard Reference Data Program; Gaithersburg, Md: 2003. 1 CD. [Google Scholar]
- 44.Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. J Med Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 45.Mosmann T. J Immunol Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 46.DeBarber AE, Mduli K, Bosman M, Bekker LG, Barry CE., 3rd Proc Natl Acad Sci USA. 2000;97:9677. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.