

Abstract
The SR family of proteins plays important regulatory roles in the control of alternative splicing in a wide range of organisms. These factors affect splicing through both positive and negative controls of splice site recognition by pre-spliceosomal factors. Recent studies indicate that the Drosophila SR factor Transformer 2 (Tra2) activates and represses splicing through distinct and separable effector regions of the protein. While the interactions of its Arg-Ser-rich activator region have been well studied, cofactors involved in splicing repression have yet to be found. Here we use a luciferase-based splicing reporter assay to screen for novel proteins necessary for Tra2-dependent repression of splicing. This approach identified Half pint, also known as Puf68, as a co-repressor required for Tra2-mediated autoregulation of the M1 intron. In vivo, Half pint is required for Tra2-dependent repression of M1 splicing but is not necessary for Tra2-dependent activation of doublesex splicing. Further experiments indicate that the effect of Hfp is sequence-specific and that it associates with these target transcripts in cells. Importantly, known M1 splicing regulatory elements are sufficient to sensitize a heterologous intron to Hfp regulation. Two alternative proteins deriving from Hfp transcripts, Hfp68, and Hfp58, were found to be expressed in vivo but differed dramatically in their effect on M1 splicing. Comparison of the cellular localization of these forms in S2 cells revealed that Hfp68 is predominantly localized to the nucleus while Hfp58 is distributed across both the nucleus and cytoplasm. This accords with their observed effects on splicing and suggests that differential compartmentalization may contribute to the specificity of these isoforms. Together, these studies reveal a function for Half pint in splicing repression and demonstrate it to be specifically required for Tra2-dependent intron inclusion.
Keywords: RNA splicing, Transformer2, Half pint, splicing repression, Drosophila
Introduction
Alternative splicing of pre-mRNA plays a vital role in producing a flexible and complex diversity of proteins from a more fixed genome. Transcriptome analyses indicate that a majority of genes in metazoans produce alternative transcripts.1,2 Although a large number of factors affecting alternative splicing have been found, the mechanisms by which these factors interact with pre-mRNA to alter splicing in developmentally specific patterns are still under investigation. Moreover, the mechanisms by which these factors control splice site selection are not completely understood.
The serine/arginine (SR)-rich proteins and SR-related factors play important roles in the regulation of alternative pre-mRNA splicing. Numerous studies have now documented the ability of these RNA binding proteins to activate alternative splice sites through interactions with nearby exonic splicing enhancers (ESEs) (for review, see ref. 3). SR-ESE complexes activate splicing through the recruitment and stabilization of pre-spliceosomal complexes at the alternative splice site, committing the pre-mRNA to selection of the activated site.4 SR proteins bind to ESEs through their RNA recognition motifs (RRMs) and facilitate pre-spliceosome formation through interactions of their Arg-Ser-rich effector domains.5-11
While activation of splicing through binding to ESEs has been widely observed, it is not the only function of SR proteins in the regulation of splicing. In some transcripts, SR factors have been found to negatively regulate splice site recognition.12-15 The Transformer 2 (Tra2) protein, a component of the Drosophila sex determination pathway and one of the founding members of the SR protein family, provides a case in point. When bound to ESE elements within an exon of the doublesex (dsx) pre-mRNA, Tra2 robustly activates a nearby sex-specific splice site by forming an enhancer complex with Transformer (Tra) and SR factors.16-18 This complex recruits U2AF to the splice site and facilitates pre-spliceosome formation. In contrast, Tra2 also binds directly to regulatory elements within the M1 intron of its own pre-mRNA where it inhibits pre-spliceosome assembly and causes repression of splicing through intron retention.13,14 Similar inhibitory functions of SR factors have also been observed in mammalian cells.12,19,20
Recent findings indicate that the effect of splicing factors on splice site recognition is highly dependent on the position where the protein binds relative to the affected splice site.21-24 In the studies of Tra2, it was observed that Tra2 bound at intronic positions elicits splicing repression, but binding at exonic positions causes splicing activation.25,26 Moreover, tethering experiments show that while activation of splicing is mediated by effector functions of the Arg-Ser-rich region of the protein, repression depends on an effector function intrinsic to the RRM-linker region.26,27 This suggests that these opposing functions are performed through distinct molecular interactions with the splicing machinery.24 However, the nature of the latter interactions and the specific cofactors involved have not yet been determined.
To better understand how Tra2 represses splicing of its M1 intron, we performed an RNAi-based screen in search of co-repressors. Among the factors identified using this approach was Half pint (Hfp), a conserved protein that has previously been shown to play roles in the regulation of alternative splicing as well as in the direct transcriptional regulation of the myc oncogene in both flies and mammals.28-30 We find that Hfp is required for repression of M1 splicing by Tra2 both in cultured cells and within the male germline in vivo, but is not required for Tra2-dependent activation of dsx splicing. Moreover, we report the expression of alternative forms of the Hfp protein that differ in their ability to repress M1 splicing uncovering an additional layer of regulation of Tra2-dependent splicing repression. Collectively, these results illustrate that differing Tra2 activities are mediated through unique interactions with distinct cofactors.
Results
Half-pint is required for Tra2-dependent repression of M1 splicing
To identify factors that contribute to Tra2-dependent splicing repression, we designed a reporter plasmid (pM-Luc) in which the expression of firefly luciferase depends on splicing of the M1 intron (Fig. 1A). This reporter plasmid contains the intact M1 intron as well as exons 3 and 4 positioned upstream of a firefly luciferase open reading frame. The intron includes the intronic splicing silencer (ISS) sequences previously shown to be sufficient for both binding of Tra2 and repression of splicing in nuclear extracts.14 A naturally occurring translation initiation codon is split by the M1 intron and is positioned as the only initiator that is in-frame with luciferase coding sequences. As a control, we created an analogous reporter using the ftz exons 1 and 2, which has been shown previously to be recalcitrant to Tra2 regulation.13 Transfection of either reporter into S2 cells resulted in efficient and detectable luciferase expression (Fig. 1B). However, cotransfection of these reporters into Drosophila S2 cells along with a plasmid (pT2) that express Tra2 resulted in a specific reduction in luciferase (Fig. 1B) from the M1 intron containing reporter in a dose-dependent fashion. No repression was observed for the control pftz-luc reporter (Fig. 1C). Semi-quantitative RT-PCR analysis of RNA isolated from transfected cells revealed a correlative increase in the levels of unspliced pM-Luc-derived mRNA, while no change in splicing was observed for the control pftz-luc mRNA. These results show that the pM-Luc reporter is a specific indicator of Tra2-dependent splicing repression.
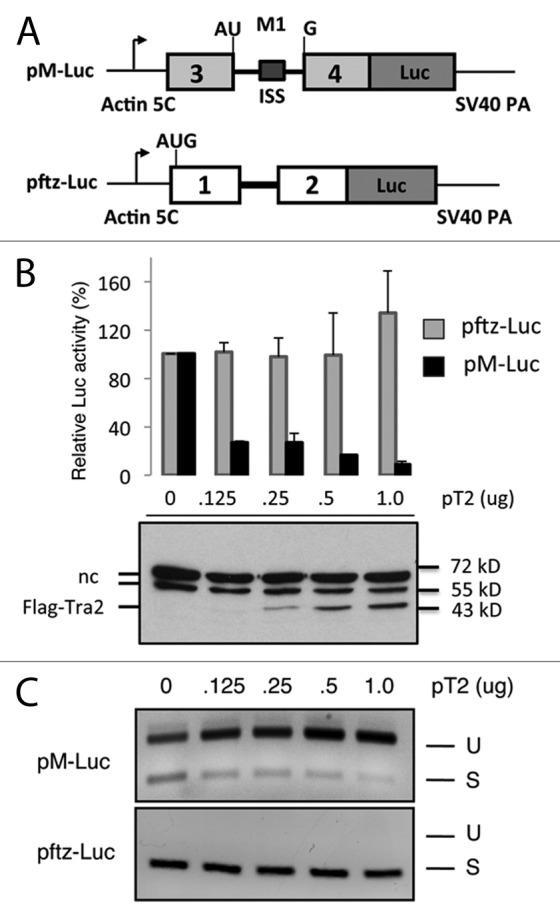
Figure 1. A luciferase reporter detects specific repression of M1 splicing. (A) The organization of reporter plasmids used to screen for Tra2 cofactors in Drosophila S2 cells is shown. Splicing of the M1 intron from pM-Luc transcripts leads to expression of firefly luciferase (Luc). The light gray exons and M1 intron derive from the endogenous Drosophila Tra2 gene. The initiation codon shown is naturally split by the intron and is in frame with luciferase coding sequences. The ISS element is indicated within M1 intron. Transcript from pftz-Luc contains exons and a constitutive intron from the Drosophila ftz gene. The same promoter and polyA signals are used in both constructs. (B) Luciferase activity from pM-Luc but not pftz-Luc is repressed in the presence of pT2 a plasmid expressing the Flag-tagged Tra2-PC isoform of Tra2 in cotransfection experiments. Results from luciferase assays (graph) and immunoblots probed with anti-Flag antibodies are shown. The position of Flag-Tra2 and two non-specific bands (nc) typically observed in such assays as well as molecular weight markers are indicated. (C) The effects of Tra2 on splicing of transcripts from both reporters, as detected by RT-PCR, is also shown. As expected, the ratio of amplification products from unspliced (U) to those of spliced (S) transcripts deriving from pM-Luc increases with higher levels of Tra2.
Given these proof-of-principle results, we next tested the effect that individual dsRNA knockdowns of 247 factors with known or potential roles in RNA metabolism31 had on pM-Luc splicing in the presence of Tra2. Luciferase assays on these knockdowns identified a group of 10 candidate factors that exhibited larger reductions in pM-Luc activity than did parallel positive controls in which Tra2 protein levels were reduced with dsRNA (Fig. S1). Among these candidates, the Half-pint (Hfp) protein, also known as Puf68, was selected for further analysis as it has previously been implicated in the regulation of alternative splicing.30,32 To address any potential off-target effects of the RNAi, we first tested whether a second dsRNA targeted at Hfp also produced significant effects on pM-Luc splicing in the presence of Tra2. Western blot analysis confirmed that, like the original dsRNA tested (Fig. 2C), this dsRNA was effective in depleting Hfp protein levels and derepressed luciferase activity from the reporter (Fig. 2A). This demonstrates that Tra2-mediated repression indeed requires Hfp expression. As expected, RT-PCR experiments showed that the higher luciferase activity was indicative of an increase in M1 splicing from the reporter (Fig. 2B) confirming that knockdown of Hfp impairs M1 repression. We also examined whether the effect on M1 splicing might result from an indirect effect on the amount of Tra2; however, we found that the level of plasmid-derived protein was unchanged by Hfp knockdown (Fig. 2C). Taken together, the above results indicate that Hfp participates in the repression of M1 splicing in this system.
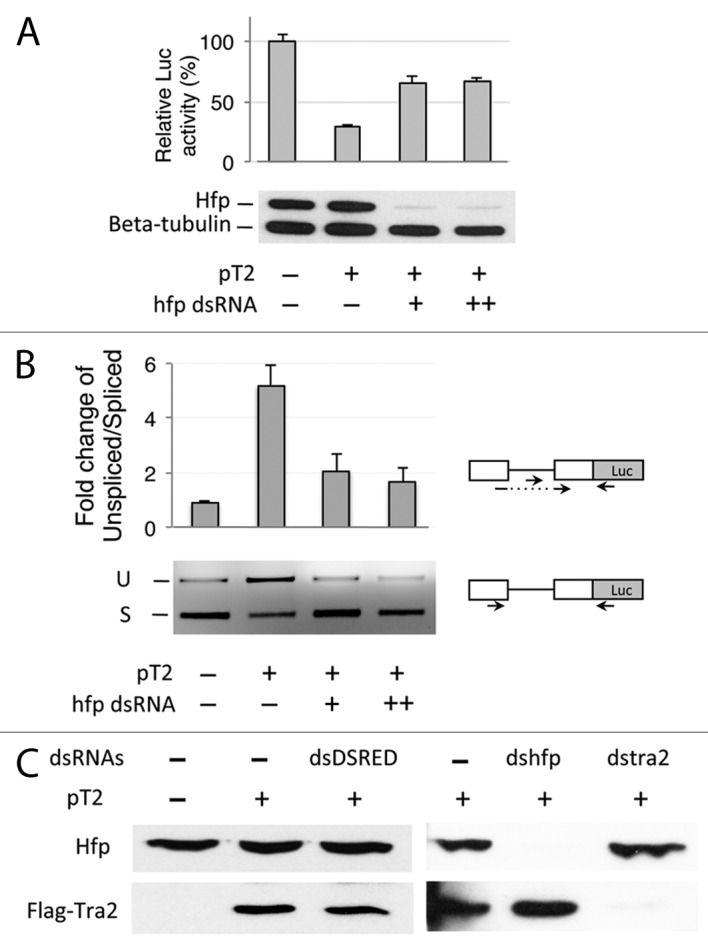
Figure 2. Half pint is required for repression of M1 splicing in pM-Luc transcripts. (A) The effect of 2 µg and 6 µg of Hfp dsRNA on luciferase activity from Drosophila S2 cells cotransfected with pT2 and pM-Luc is shown. The graph shows percent luciferase activity in relation to cells transfected with an empty expression vector. Hfp68 and tubulin protein levels from the same cells are analyzed in the western blot below the graph. All samples are transfected with the same amount of pM-Luc. Error bars represent SD values (n = 3). (B) The ratio of unspliced to spliced pM-Luc transcripts as determined by qRT-PCR of cells treated with Hfp dsRNA is shown in the graph. Error bars represent SE values (n = 2). Primers positions are indicated by arrows in the diagram. A gel showing unspliced (U) and spliced products (S) from a parallel semi-quantitative PCR analysis with different primers is shown below the graph. (C) western blots detecting expression of Flag Tra2 from cells transfected with pT2 and endogenous Hfp after treatment with Hfp dsRNA and control dsRNAs are shown. In all experiments, a monoclonal anti-Hfp antibody was used to detect level of Hfp. The M2 monoclonal anti-Flag antibody was used to detect transfected Flag-Tra2.
Half-pint associates with Tra2 transcripts
To determine if Hfp physically associates with Tra2 transcripts, we next performed RNA immunoprecipation experiments with anti-Hfp antibody from S2 cell lysates cotransfected with three separate reporters, pM-Luc, pftz-Luc, and pAct-GFP (Fig. 3A). The precipitated material was then subjected to RT-PCR with primers spanning the intron and flanking exons to detect associated RNA sequences. As shown in Figure 3A and Figure S2, specific association of Hfp was observed with both the spliced and unspliced transcripts of pM-Luc, but was not found to associate with pftz-Luc or pAct-GFP transcripts. Furthermore, Hfp was found to associate with endogenous Tra2 in S2 cells. Again, association was observed in transcripts both with and without the M1 intron (Fig. S3). Notably in both pM-Luc and endogenous transcripts containing M1 were preferentially precipitated over those lacking the intron. Taken together, these results indicate that the association of Hfp depends on sequences located both within and outside of the M1 intron with the stronger associations specified by intron sequences.
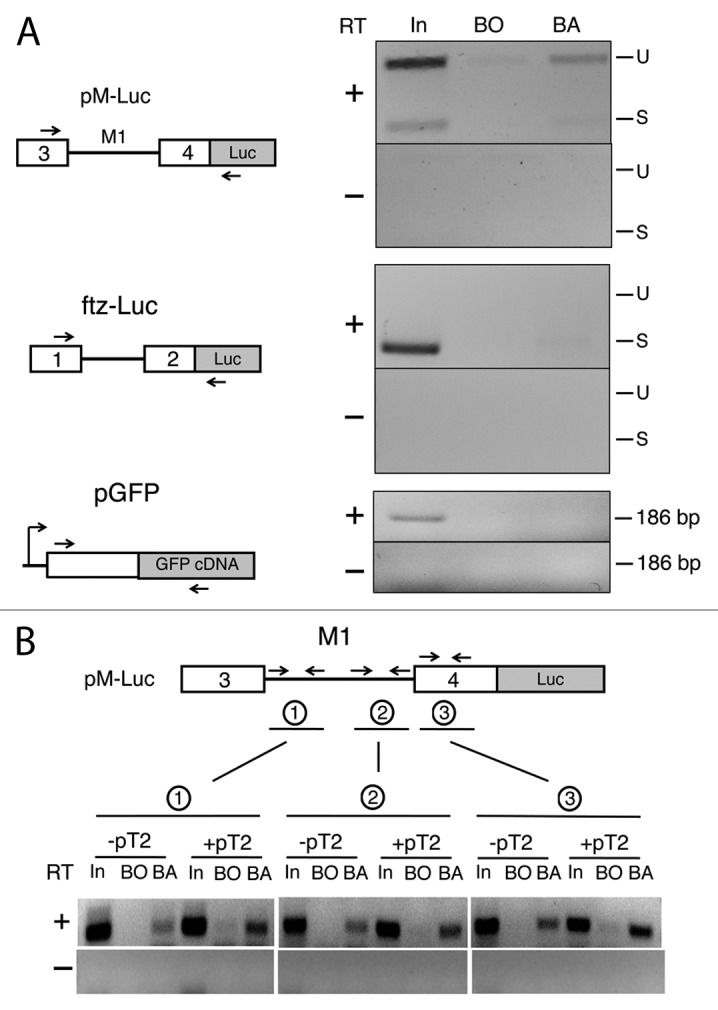
Figure 3. Half pint is associated with Tra2 pre-mRNA. (A) RNA-protein complexes from S2 cells cotransfected with equimolar amounts of pM-Luc and two control reporter constructs were immunopreciptiated with anti-Hfp antibodies and analyzed by RT-PCR. Primer positions for each reporter are diagrammed to the left and products are shown to the right. Lanes are loaded with products amplified from 20% of input lysate (In), eluate from beads only (BO) and eluate from beads coated with anti-Hfp antibody (BA). The three reporter constructs contain the same promoter and polyA signal sequences. (B) Similar RNA immunoprecipitation assays were performed with anti-Hfp antibody on various regions near or within the M1 intron of pM-Luc after cotransfection with and without pT2. The numbered primers correspond to the individual immunoprecipitations as indicated. Products from amplification of 50% input (In), eluate from beads only (BO) and eluate from beads with antibodies (BA) are shown.
As Tra2 is known to repress splicing by binding to the ISS sequences within the M1 intron, we also tested whether the interaction of Hfp with pM-Luc transcript was affected when levels of Tra2 were increased (see +pT2 in Fig. 3B). These experiments revealed a small but consistent increase in Hfp binding in lysates from pT2-transfected cells for all three amplicons spanning unique regions with the pM-Luc reporter. The small increase in binding signals observed could be due to the increased M1 retention that increases the amount of the proteins preferred target.
Previous crosslinking studies on Tra2 indicated that it binds ISS sequences with moderate affinity and high specificity in the presence of Drosophila nuclear extracts, but fails to do so in the absence of complementing extracts.13,14 We therefore next examined whether Hfp alone can bind directly to an RNA containing the M1 intron and it can facilitate Tra2 binding in the absence of extract. Gel shift assays (Fig. S4 lane 3 and Fig. S5) performed with low micromolar concentrations of recombinant Hfp protein produced an shift in the mobility of RNA containing the entire M1 intron suggesting that the protein can bind RNA directly with at least low affinity. Addition of Tra2 and Hfp proteins together (Fig. S4, lane 5) led to the appearance of a new complex with lower mobility than obtained with either protein alone. Together, these results suggest that Hfp and Tra2 are capable of co-occupying the M1 intron and that associations observed in vivo are likely to be further facilitated by additional unknown factors.
The effect of Hfp on splicing depends on the M1 ISS and a suboptimal 3′ splice site
We next tested whether sequences previously found to support binding and repression by Tra2 are also sufficient for the observed effects of Hfp.14,33 As illustrated in Figure 4A, the native M1 intron contains two conserved sequences known to be important in M1 splicing. Based on assays in S2 nuclear extracts, the 80 nt ISS alone was found to be sufficient to confer Tra2-dependent repression of splicing on an intron that is otherwise recalcitrant to regulation by Tra2.14,26 Furthermore, experiments performed in transgenic flies determined that the intron’s weak 3′ splice site context is also required for repression by Tra2.33 Notably, a weak 3′ splice site from the myosin heavy chain gene (mhc) is sufficient to replace the native weak M1 sequences, suggesting this requirement is general and not specific to the native Tra2 3′ sequences. The strong 3′ splice site from the ftz gene was also found not compatible with splicing repression in vivo.33
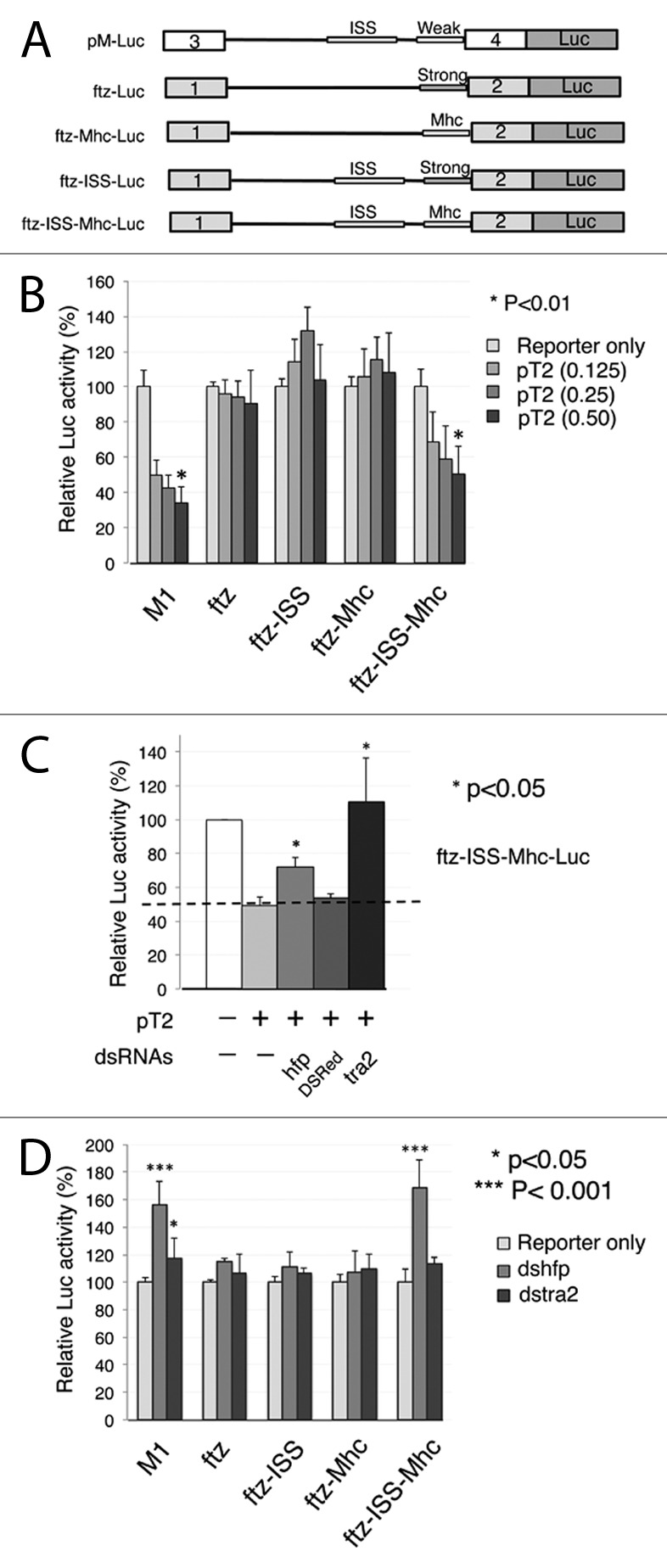
Figure 4. Repression of splicing by Hfp and Tra2 depend on both the ISS and a suboptimal 3′ splice site. (A) The diagram shows various constructs used to test the specificity of Hfp for the ISS and a weak 3′ splice site. The organization of the Tra2 M1 intron (top) is shown only for reference. All experiments were performed in constructs based on the ftz intron (thin line) and its flanking exons (gray boxes). Thickened lines indicate the positions where the ISS and the polypyrimidine tract/3′ splice site. The ftz intron naturally has a strong 3′ splice site and no ISS. In ftz-mhc, a weak 3′ splice site of mhc intron 5 is substituted into this intron, in ftz ISS the Tra2 ISS is substituted, and in ftz-ISS-mhc both are present in combination. (B) Luciferase assays performed after cotransfection with different amounts of pT2. Significant repression is only observed with the native M1 intron (n = 7) and in ftz-ISS-mhc (n = 9), but not in ftz (n = 7), ftz-ISS (n = 6), and ftz-mhc (n = 10), compared with reporter only in each group. Error bars represent SD values. (C) Knockdown of Hfp in cells transfected with pT2 derepresses luciferase expression from ftz-ISS-mhc (n = 3). The graph shows relative luciferase levels from treatment of cells various dsRNAs. Significant derepression (*) was observed only with dsRNA directed at Tra2 and Hfp, compared with pT2 transfected only. Error bars represent SD values. (D) RNA interference with dsRNA directed at endogenous Hfp and Tra2 in S2 cells. Knockdown of Hfp significantly increases luciferase from the M1 and ftz-ISS-mhc reporter but not from other reporters, compared with reporter only group (n = 7–10). Error bars represent SD values.
To determine if the ISS and a weak 3′ splice site are sufficient for repression by Hfp in a manner similar to Tra2, a series of splicing reporters based on pftz-Luc was constructed in which expression depends on splicing of the ftz intron (Fig. 4A). Each of these splicing reporters was then cotransfected with increasing amounts of Tra2 and relative luciferase activity was then measured. We observed that when a combination of the ISS and the mhc weak 3′ splice site (ftz-ISS-mhc-Luc) are substituted into this intron they are sufficient to allow dose-dependent repression of reporter activity by Tra2 (Fig. 4B). In contrast, constructs missing either of these individual elements failed to undergo significant repression in response to elevated Tra2. These results confirm that the ISS and weak 3′ splice site are both required for Tra2-dependent splicing repression in S2 cell transfections as inferred from other systems.
To determine if the pftz-ISS-Mhc luciferase reporter requires Hfp expression for Tra2-mediated splicing repression, we repeated the transfections under conditions that deplete Hfp. Notably, the knockdown of Hfp led to a significant reversal of the Tra2-induced repression when the ftz intron contained both the ISS and the weak Mhc 3′ splice site (Fig. 4C). These results are consistent with the data shown in Figure 2A and also strengthen the conclusion that Hfp is a cofactor for Tra2 as a heterologous reporter sensitive to Tra2 also requires Hfp expression for full splicing repression. Moreover, knockdown of Hfp also led to a similar reversal of both pM-Luc and ftz-ISS-Mhc-Luc (Fig. 4D) when tested in the presence of only endogenous Tra2. Together, these results indicate that Hfp affects splicing through interactions with the ISS in cooperation with Tra2.
Hfp is required in vivo for repression of M1 splicing but not for activation of an alternative splice site in dsx pre-mRNA
In Drosophila development, repression of M1 splicing is most prominently observed in the male germline where about 50% of Tra2 mRNA retains the M1 intron.34 To determine if Hfp plays a significant role in splicing repression in vivo we dissected testes from wild-type and hfp mutant adults. Because strong loss of function results in lethality at earlier stages, this analysis was performed with hypomorphic hfp alleles that allow adult survival.30 As shown in Figure 5A, endogenous M1 splicing efficiency was found to be significantly increased when Hfp function was reduced. For comparison, a parallel RT-PCR assay with RNA from tra2 mutant testes is shown in which M1 splicing is also increased. These results demonstrate that Hfp is required for repression of M1 splicing in the male germline.
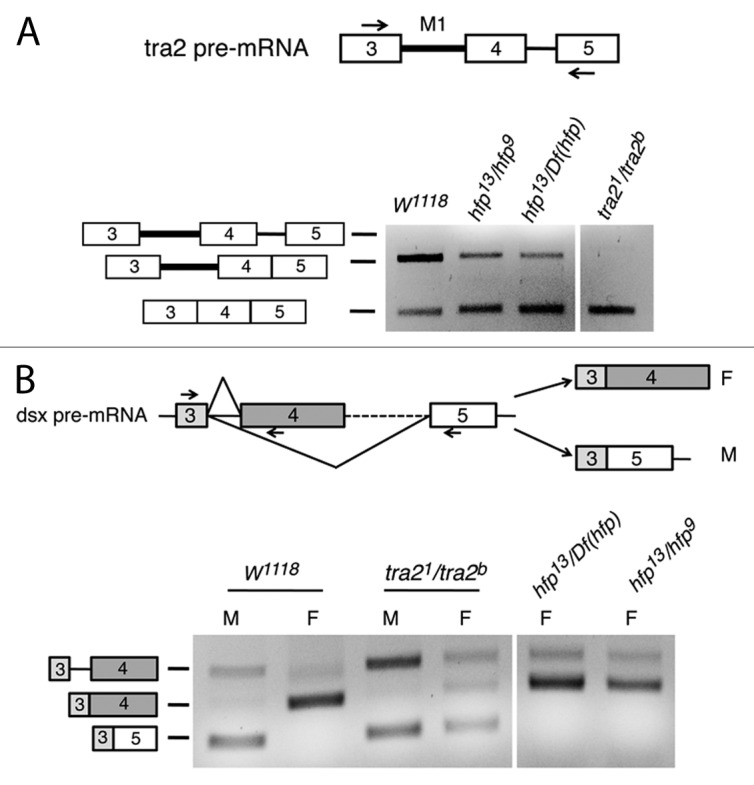
Figure 5. Half pint loss of function affects M1 splicing but not dsx splicing in vivo. (A) RT-PCR analysis of M1 splicing in adult testis mRNA from various genotypes is shown. Primer locations are indicated by arrow the diagram. In w1118 flies that are wild type for both tra2 and hfp, the M1 intron is retained in a majority of transcripts. Loss of function for either tra2 or hfp result in reduced M1 retention. tra21 and tra2b are point mutations that do not alter the total amount of Tra2 mRNA. Df(hfp) corresponds to Df(3L)AR148 is a large deletion that spans the entire hfp gene. (B) Similar RT-PCR analysis was performed on dsx RNA from whole adult males (M) and females (F) of various genotypes. Three primers, as indicated by arrows in the diagram, were used together to amplify both male and female-specific splicing products of dsx. As expected mature male mRNA was detected in RNA from a loss-of-function tra2 mutant genotype, but was not found in females from either of two strong loss-of-function hfp mutant genotypes tested.
We next tested whether the female-specific splicing of the dsx pre-mRNA, which is activated by Tra2, also depends on Hfp function. Splicing was analyzed in wild-type, tra2, and hfp mutant adult flies by RT-PCR performed on total RNA with primers that detect the male and female-specific dsx transcripts as distinct products diagrammed in Figure 5B. The results of this experiment, shown in the lower part of Figure 5B, indicate that Hfp loss of function did not reduce the activation of the Tra2-dependent female-specific dsx 3′ splice site. Thus, Hfp is not generally required for all Tra2-dependent alternative splicing events but rather is a specific cofactor required in the regulation of M1 splicing.
Alternative forms of Half pint differ in their effects on M1 splicing
Analysis of the Drosophila transcriptome indicate that it contains at least nine alternatively spliced mRNAs from the hfp gene.35 These mRNAs are predicted to encode either of two protein forms that differ by the presence of a 92 amino acid N-terminal sequence, which contains four arginine-serine dipeptides (Fig. 6A; Fig. S6). Type A transcripts encode a 68 kDa protein (Puf68) predicted to initiate translation in the first exon and consistent with the major Hfp product previously reported.30 All other Hfp transcripts contain in-frame stop codons a short distance downstream of this initiation site. However, initiation at a downstream start codon found in these transcripts could potentially produce a 58 kDa protein (Fig. 6A; Fig. S6). Consistent with this prediction, immunoblots performed on Drosophila lysates with Hfp monoclonal antibody directed to the protein's C terminus detect both 68 and 58 kDa proteins (Fig. 6B and C; Fig. S7). Importantly, both bands were reduced in level in flies with a hemizygous hfp mutant genotype (Fig. 6B). In S2 cells, Hfp68 is the naturally expressed form with much higher level than Hfp58 (Fig. 7A). The longer 5′UTR of Hfp58 transcripts, compared with that of Hfp68, might be responsible for its low expression level in cells. However, expression of either Hfp58 (Fig. 6C) or both isoforms (Fig. 7A) was found to be increased when the corresponding hfp cDNAs were transfected into Drosophila S2 cells, as expected if they are products of hfp mRNA. Notably, these products also showed sex and tissue specificity with the 68 kDa product being preferentially expressed in males and 58 kDa in females (Fig. 6C). The gender bias of expression also suggests that protein degradation is not the source of the lower product.

Figure 6. Hfp has two isoforms expressed in vivo. (A) The exon-intron patterns giving rise to various Hfp (Puf68) encoding mRNAs. The diagram was created based on the information in Gbrowse. Boxes correspond to exons, lines to introns. Gray shaded regions are predicted to be noncoding, orange box is predicted protein coding. Note that only the RA transcript has the potential to encode the Hfp68 isoform, all other transcripts are predicted to encode Hfp58. The initiation codons and stop condons are indicated. (B) A comparison of Hfp68 expression in lysates from a mixture of Drosophila male and female adult flies with various genotypes is shown. Both forms are observed in w1118 adults (carrying two wild type alleles of hfp), but are both reduced in hfp13/Df(hfp). Both hfp13 and hfp9 are partial loss-of-function mutations. (C) Expression of Hfp68 and Hfp58 in w1118 male (M) and female (F) adult flies is shown as detected by immunoblotting. For comparison, lysates from S2 cells (-) and S2 cells transfected with Flag-Hfp58 (F-Hfp58) were loaded on the same gel.

Figure 7. The alternative N-terminal region of Half pint is required for repression of M1 intron splicing. (A) Transfection experiments performed with pM-Luc splicing reporter and constructs expressing Flag-Hfp58 or Flag-Hfp68. Partial loss of endogenous Hfp using a dsRNA targeted at the 3′ UTR (hfp 3′UTR dsRNA) derepresses Luc expression. The ability of each Hfp isoform to restore repression was tested by transfection of the cells with expression constructs containing 3′ UTR sequence from SV40 that are not affected by the dsRNA. Levels of Hfp58 and Hfp68 as detected by immunoblotting with Hfp is shown below the chart. The 92 kD band shown is a non-specific cross-reacting protein from the same gel that is used to represent loading. (B) Quantitative RT-PCR analysis of total cellular RNA from cells treated as in (A) (n = 3). The ratio of spliced/unspliced pM-Luc transcripts was determined using the same primers as were used in Figure 2B. For all panels, results that affected significantly from controls treated with hfp-3′UTR dsRNA only are indicated (*). Error bars represent SE values.
To test if these isoforms differ in their ability to support repression of M1 splicing, we knocked down endogenous Hfp protein using a dsRNA targeted to its 3′UTR (hfp-3′UTR dsRNA). These cells were then cotransfected with plasmids expressing either of the Hfp isoforms from mRNA with a distinct 3′UTR sequence as well as the pT2 and pM1-Luc plasmids. As shown in the immunoblots in Figure 7A, endogenous Hfp was effectively reduced by treatment with a dsRNA targeting the Hfp mRNA 3′ UTR (Fig. 7A, lane 1 and 2) and the levels of luciferase activity increased due to reduced M1 intron retention. As expected, expression of Hfp68 restored luciferase activity to nearly its original levels (Fig. 7A, lanes 5 and 6), but unexpectedly, expression of Hfp58 did not reverse this effect (Fig. 7A, lane 3, 4). These results suggest that Hfp68, but not Hfp58, is capable of mediating repression of M1 splicing. This conclusion is supported by quantitative RT-PCR analysis of mRNA isolated from S2 cells transfected analogously (Fig. 7B). The spliced/unspliced ratio of pM-Luc transcripts was found to significantly increase upon Hfp knockdown and to be restored after expression of Hfp68 but not Hfp58.
Hfp68 and Hfp58 differ in their nuclear/cytoplasmic localization
The Hfp58 isoform lacks sequences at the N terminus of Hfp68 that are of unknown function. Immunofluorescence staining of endogenous Hfp in S2 cells (which primarily express Hfp68) indicated that most Hfp protein in these cells is localized to the nucleus (Fig. 8A, Endo-Hfp). To determine if Hfp58 and Hfp68 are likely to differ in this regard, we used an anti-Flag antibody to similarly stain S2 cells transfected with either Flag-Hfp58 or Flag-Hfp68 (Fig. 8A). This revealed that while Flag-Hfp68 is predominantly nuclear (Fig. 8B), Flag-Hfp58 is distributed almost equally between the nucleus and cytoplasm (Fig. 8B). These results suggest that the reduced ability of Hfp58 to repress M1 splicing is in part due to reduced localization to the nucleus.
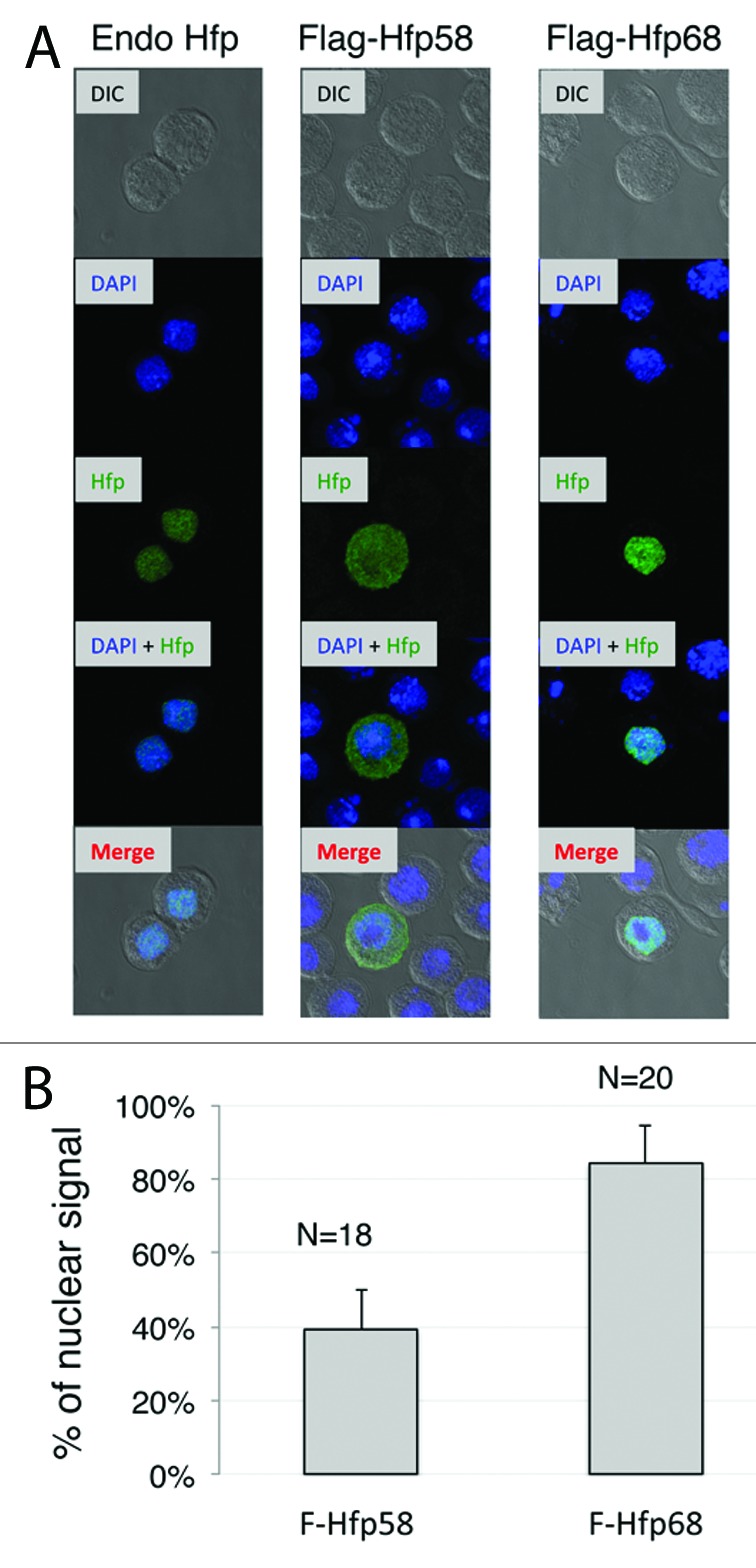
Figure 8. Hfp isoforms differ in nuclear/cytoplasmic localization. (A) Endogenous Hfp localization (Endo Hfp) detected with a monoclonal antibody directed at the protein's C-terminal region is shown in comparison to the localization of Flag-Hfp58 and Flag-Hfp68 in cells transfected with constructs expressing each form. Anti-Flag antibody was used to stain transfected Hfp protein. Both Hfp (green) and DAPI immunfluoescent staining (blue) are overlaid on DIC images of the same cells in the bottom row. (B) The percentage of signal detected within the nucleus as determined by Imaris, is shown for both Flag-Hfp58 and Flag-Hfp68. The number of analyzed cells is indicated as N. Error bars represent SD values.
Discussion
The results reported here indicate the Hfp/Puf68 protein of Drosophila is required for the negative regulation of splicing by Tra2. This finding was initially based on an RNAi screen performed in Drosophila cultured cells with a splicing reporter construct and was extended to living flies by examining how loss of function mutations in Hfp affects repression of M1 splicing in a developmental context where it normally occurs. We observed that Hfp is required for detection of intron retaining transcripts in mRNA derived from male testes where Tra2 repression normally blocks splicing of M1 in about half of Tra2 mRNA molecules in response to a negative feedback mechanism.34 Splicing repression in this tissue is thus dependent on both Tra2 and Hfp.
Previous studies show that the dependence of M1 repression on the Tra2 protein in the male germline is complete. In its absence, the intron is spliced from all detectable germline mRNA.36 Thus, the endogenous Hfp present in the male germline is unable to cause M1 repression in the absence of Tra2. Whether Tra2 can cause repression independently of Hfp is presently unclear because only partial loss-of-function mutations in Hfp are available and they display low residual M1 repression.
The results presented here depend on the use of S2 cells to study an alternative splicing event that normally occurs primarily in the male germline. Although it is possible that germline-specific splicing factors are needed in vivo for M1 splicing repression, extensive in vitro analysis of splicing has demonstrated that S2 nuclear extracts are competent to support sequence-specific repression of M1 splicing by Tra2.13,14,26 Consistent with this, previous studies on transgenic fly strains clearly showed that Tra2 has the potential to repress M1 splicing in somatic cells in vivo when it is expressed at high levels comparable to those normally found in the germline.25 This observation is important as Tra2 transcripts are generated from a distinct promoter in the male germline that is significantly more active than the somatic promoter. The appearance of M1 retaining RNA through negative feedback on splicing in the male germline is most likely to arise because Tra2 is transcribed at its highest levels there. Thus, the factors responsible for tissue specificity of this splice choice are thought to act at the level of transcription. Using the promoter from the Drosophila actin5C gene here, we have provided high levels of Tra2 in S2 cells sufficient to cause significant levels of M1 repression.
It is worth noting here however that using a splicing reporter in S2 cells we also observed basal M1 retention that occurred without overexpression of Tra2. Notably, this basal repression did not respond dramatically to knockdown of endogenous Tra2 protein but was nonetheless sensitive to knockdown of endogenous Hfp and its response to Hfp required the presence of both the M1 ISS and a weak 3′ splice site. These observations suggests that while Hfp is required for repression of M1 induced by high levels of Tra2, it also has the ability to elicit a low level of sequence-specific repression of splicing that is independent of Tra2 in S2 cells.
Although Hfp plays a required role in Tra2-dependent splicing repression, it does not appear to be needed for the well-characterized function of Tra2 in activation of splicing through exonic splicing enhancers as illustrated by the absence of any visible effect of Hfp mutations on female sexual differentiation or on the sex-specific splicing of dsx mRNA. The failure to affect dsx splicing is unlikely to be the result of tissue-limited expression of Hfp, as this protein is known to be widely expressed and functional within a variety of somatic tissues such as the imaginal discs in which regulation of dsx splicing normally occurs.28,37 Therefore, it is likely to be available in such tissues but simply does not participate in dsx splicing. Thus, Hfp behaves as a specific co-repressor of Tra2 but lacks a discernible co-activator function.
Hfp has previously been reported to play a role in the regulation of alternative splicing in the Drosophila female germline, but the results presented here are the first suggesting that Hfp can contribute specifically to repression of splice site recognition. Indeed, Hfp was previously observed to be required to promote inclusion of an alternative exon in transcripts from the ovarian tumor gene expressed in the adult female germline.30 Moreover, studies on PUF60/FIR, the human ortholog of Hfp, showed that it binds to and activates 3′ splice sites that have suboptimal polypyrimidine tracts.38 PUF60 function in this regard is intertwined with that of U2AF65, which generally promotes 3′ splice site recognition.39 In vitro studies show that PUF60 can synergize with U2AF65 to promote splicing and in some situations is able to functionally substitutes for this factor.38 Thus, both Hfp and its human ortholog, PUF60, have previously been regarded as factors that promote rather than repress splice site recognition. Our finding that Hfp performs a negative function and collaborates with Tra2 thus broadens the range of effects on regulated splicing that this factor has been associated with.
A connection between Hfp and Tra2 was also found in studies on ATR dependent alternative splicing of Taf1 transcripts in Drosophila S2 cells.32,40,41 These two factors were both among a small group of RNA binding proteins shown to be required to regulate splicing of mRNA encoding the Taf1–3 and Taf1–4 isoforms from the Drosophila Taf1 gene. The similar dependence of Taf1 on these two factors suggests that perhaps regulation occurs through a similar mechanism. However, it is not yet known whether Tra2 and Hfp alter Taf1 pre-mRNA splicing primarily through positive or negative effects on the alternative splice sites.
The identification of a specific protein co-repressor for Tra2 is of particular significance in light of recent studies indicating that the known activation and repression functions of Tra2 are encoded by distinct and separable effector regions of the protein. The first evidence for this came from the observation that certain mutations in Tra2 disrupt repression but have little if any effect on splicing activation.42 More recent experiments in which various Tra2 protein sequences were tethered to target RNAs through fusion to the MS2 coat protein and its cognate binding sites showed that activation depends on effector functions of the protein's Arg-Ser rich regions, while repression is mediated through effector functions within the RRM-linker region.26,43 This separation of functions supports the idea that distinct cofactors are likely to be involved in activation and repression.
Known Tra2 co-activators include a number of proteins that bind cooperatively with Tra2 to form the dsx splicing enhancer complex.18,44,45 Identified among these are several non-sex-specific SR proteins and the female-specific Transformer (Tra) protein which both a required and limiting factor in vivo. Tra2, Tra, and these SR factors are believed to activate splicing through the combined action of their Arg-Ser-rich domains, which directly facilitate the assembly of pre-spliceosomal complexes at the dsx 3′ splice site. The results we have reported here indicate that Hfp has properties expected of a co-repressor that could instead take part in functions specific to repressive effector domain of Tra2. Consistent with this idea, the effects of Hfp on splicing were found to depend on the same intronic sequence elements in the M1 intron that mediate RNA binding and repression of splicing by Tra2. This suggests a close collaboration between these factors and the possibility that Hfp is recruited to the transcript upon Tra2 binding. Our immunoprecipitation data also indicates that Hfp affects M1 splicing through either a direct or indirect physical interaction with the target transcript in the presence of Tra2. Aside from Tra2 and Hfp the only other cofactor known to affect M1 splicing is Rbp1, a non-sex-specific SR protein that is also a component of the dsx enhancer complex.14,46,47 The dual role of Rbp1 is not well understood, but curiously it does not cooperate with Tra2 in binding to the ISS.14 In summary, our results suggest that Hfp directly facilitates splicing repression by Tra2 in a manner unique among known Tra2 cofactors.
A key question that remains is mechanistically how Tra2, in cooperation with Hfp, and perhaps other factors inhibit early steps in splicing. Insight to this question comes from the recent observation that the direction of Tra2-dependent regulation of splicing depends on whether this protein and its associated factors are bound at an intronic or exonic position.26,43 When Tra2 is tethered to RNA at different intronic positions, or is bound to native ISS elements placed at different locations in the intron, the protein acts as a splicing repressor. But when the ISS or tethering sites are placed at exonic positions then Tra2 promotes activation of splicing in the same system. Repression of splice sites by intronically positioned Tra2 and activation of splice sites by exonically positioned Tra2 both result in the same outcome—the exonic identity of the bound sequences. In the case of the M1 intron repression of its splice sites by Tra2 and Hfp blocks pre-spliceosome assembly26 and results in retention of the intron within a larger exon. As the intron's 3′ splice site is a weak match to the Drosophila consensus and the recognition of weak 3′ splice sites is a characteristic feature of Hfp's mammalian ortholog, one possibility is that Hfp assists Tra2 in identifying such sites for repressive interactions.
In addition to the above mechanistic insights, we also report here for the first time the detection of two distinct forms of the Hfp protein that differ by the presence of an alternative N-terminal region. These two forms differ functionally in both their nuclear/cytoplasmic distribution and their ability to affect splicing with the larger Hfp68 being most localized to the nucleus and displaying the stronger effects. These isoforms appear to arise through alternative splicing of Hfp pre-mRNA itself. Thus, regulation of splicing may occur at multiple levels to affect expression of targets of Hfp and Tra2. Notably, we have observed that the intracellular distribution of Hfp differs between various cell types, and that males and females differ in their relative accumulation of these two isoforms. The existence of multiple protein forms is particularly interesting in light of the observation that, aside from its role in splicing, Hfp has also been reported to play a conserved and direct role in the transcriptional regulation of the c-myc oncogene.28,29
Materials and Methods
Plasmids and primers
The plasmid pM-Luc was constructed based on pBluescript SK+ vector (pSK+) by inserting amplified fragment containing the actin 5C promoter, intact Tra2 sequences from M1 intron and its two flanking exons, and firefly luciferase protein coding sequences. An SV40 polyadenylation signal derived from pFastback-Flag-Tra2 was inserted downstream of the luciferase termination codon. Primers are listed in the Table S1. The plasmid pT2, is designed to express Tra2-PC, an isoform that was genetically defined as sufficient for both repression and activation of splicing.36 It was generated by amplifying sequences from pFastBac 3XFlag-Tra2-PC and then inserting these into pSK+ contaning Actin5C promoter and SV40 poly A signal (pSK-AS).
pFastBac 3XFlag-Tra2-PC was made by inserting a synthetic DNA fragment (Integrated DNA Technologies) encoding a 3XFlag tag at the N terminus of Tra2 PC coding sequence in pFastBac 6XHis Tra2 PC.14
The plasmid pftz-Luc was constructed as described for pM-Luc, except that sequences from the ftz gene including its intron were inserted instead of those from Tra2. The plasmids pftz-ISS-Luc, pftz-mhc-Luc, and pftz-ISS-mhc-Luc are all modifications of the pftz-Luc reporter. The sequences used in ftz-ISS were amplified from pftz12014 and ligated into pftz-Luc by replacing an ApaI and XhoI fragment. The plasmid pftz-mhc-Luc was made using through three PCR amplifications with the mhc 3′ splice site sequence integrated into two different primers:
ftz RNA: Sense-ATGGACTACTTGGACGTCTACTCG
mhc 3′ 1: Antisense- CTTGTTTGCAAGGGGATAAGTTCAATGGGTTAGCTAATGAGTTTT;
mhc 3′ 2: Antisense- GTCTGACGGGTGCGTTTCGAGTCTTTGCAATCTTGTTTGCAAGGGGATAAG;
mhc 3′ 3: Antisense- CTCGAGCTCCAGGGTCTGGTAGCGGGTGTACGTCTGACGGGTGCGTTTCGA.
The plasmid pftz-ISS-mhc-Luc was derived from sequences of pftz-mhc-Luc in a similar way. Only first step primer is different:
ISS-mhc 3′ 1: 3- CTTGTTTGCAAGGGGATAAGTTCAAAAATAAGATTATCTTGCGGTTCG.
The plasmid pFlag-Hfp68 was generated using the same vector as pT2, which has the actin 5C promoter and SV40 PA signal sequence. Flag tagged Hfp68 sequences were amplified from a cDNA (AT08368) purchased from the Drosophila Genome Resource Center using primers with integrated Flag sequences. pFlag-Hfp58 was generated by a similar strategy just with a different start position.
Cell culture and transfection
Dmel/S2 cells were cultured in SFII-900 medium at 28 degrees and split at regular intervals. For transfections, 2.5 × 105 cells were seeded in 1.9 cm2 wells and a total of 1.5 μg of DNA was mixed with 3 μL cellfectin in 100 μL of the same medium. A plasmid with no insert that contained both the Actin5C promoter and SV40 polyA signal was used as a control and to equalize DNA amounts in all transfections. For the RNAi screen, 1.5 × 106 cells were seeded in 9.5 cm2 wells and transfected with 5 μg DNA and 10 μL cellfectin. After 5–6 h, the cells were resuspended and counted. They were then reseeded into 96-well plates with 3 × 104 cells per well.
Synthesis of dsRNA
Double-stranded RNAs were synthesized following the approach of Park and Graveley.48 Briefly, cDNA fragments amplified and inserted into PCR vectors (Invitrogen). M13 reverse and M13 forward primers were used to amplify linear DNA template. These fragments were transcribed using the Megascript kit (Ambion) with SP6, T3, and T7 RNA polymerases to produce single strand RNAs. Single strand RNAs were mixed in annealing buffer (100 mM NaCl, 20 mM TRIS-HCl, pH 8.0, 1 mM EDTA), incubated at 80 degrees for 10 min and then cooled at room temperature for 30 min, followed by storage on ice. Primer sequences used to amplify fragments for dsRNA synthesis are provided in Table S1.
RNA interference
In all secondary RNA interference experiments dsRNAs was added directly to cells growing in SFII-900 media in the amounts of 4 μg per well in 24-well plates, 20 μg per well in 6-well pate. To maximize the extent the knockdown on target expression, the RNAi treatment was repeated at both 24 and 48 h after the initial treatment.
RNA immunoprecipitation
For RNA immunopreciptation (RIP) experiments, 1.0 × 107cells were seeded in a 78 cm2 dish and transfected with a total of 30 μg plasmid DNA. After 48 h, the cells were washed two times with 1X PBS buffer. They were dounced repeatedly in RIP buffer (150 mM NaCl, 50 mM Tris pH 7.5, 5 mM EDTA, and 0.05% NP-40). Cell lysates were pre-absorbed with Sepharose Gammabind beads (Amersham) for 1 h and then incubated with activated beads with or without conjugated anti-Hfp antibody at 4 degrees for 4 h. After washing beads three times, the whole precipitates were treated with Trizol (Invitrogen). RNA isolated from precipitates was analyzed by RT-PCR.
Splicing reporter luciferase assays
Luciferase assay were performed following the protocol described in the manual of Dual-Luciferase reporter assay system kit (Promega). Briefly, 100 μL passive lysis buffer (Promega) was added to cells in each well of a 24-well plates. Then 25 μL of the lysate was used to measure luciferase signals. Luciferase signals were measured on a Perkin Elmer VICTOR™ X5 Multilabel Plate Reader.
Immunoblotting
Cultured cells were lysed in passive lysis buffer (Promega). A monoclonal antibody against C terminus of Hfp30 or the M2 anti-Flag epitope (Sigma) were detected using appropriate secondary antibodies and the ECL system (General Electric). After incubating with antibodies for one hour at room temperature, blots were washed 5 min with TBST (50 mM TRIS-HCl pH 7.4, 150mM NaCl, 2.5mM KCl, 0.05% Tween 20) for three times then exposed to film. Fly samples were dounced in lysis buffer (20 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100), spin at 10,000 rpm for 1 min at 4 degrees. The supernatant was heated in SDS loading buffer for 10 min.
Real-time PCR
For quantitative PCR experiments cDNA was synthesized from 1 μg of total cellular RNA using the Superscript first strand RNA kit (Invitrogen). The cDNA was mixed with a 2X Syber Green PCR mix (ABI) and DNA primers at a final concentration of 0.625 ng/μL. Q-PCR was performed on 7900 FAST PCR machine (Applied Biosystems) with SDS 2.3 software (Applied Biosystem). Fragments were amplified by first incubating at 50 °C for 2 min and 95 °C for 10 min, and then for 40 cycles at 95 °C for 15 sec each and 60 °C for 1 min. Relative quantification analysis was performed by using the delta delta CT method.49
S2 cell immunostaining and image analysis
Localization of proteins was determined after S2 cells were seeded into 12-well plate with 5 × 105 cells/well. Coated coverslips (BD Biocoat) were placed into the wells and cells were allowed to settle on them overnight. Plasmids were transfected the next day. After 36–48 h incubation at 28 °C, cell medium was removed. Cells were washed once with 1XPBS, then fixed with 4% paraformaldehyde in PBS at 37 °C for 30 min. After washing with PBS, the cells were incubated in PBX (0.2% triton X-100 in PBS) at room temperature for 10–15 min. Blocking was performed with 1% BSA at room temperature for 1 h, cells were then incubated with primary antibody (1:200 in PBS with 1% BSA) at 4 °C overnight. They were then washed with PBS four times and incubated in secondary antibody buffer (1:500 in PBS with 1% BSA) at room temperature for 1 h. Coverslips were removed and samples were mounted with CellMask (Invitrogen). Images were obtained using a Nikon Eclipse Ti Confocal microscope. Images were analyzed for nucleus/cytoplasm distribution using Imaris 7.3 software (Bitplane) after deconvolution by AutoQuant X3 (MediaCybernetics). The total protein signal across the entire three dimensional nucleus was divided by that across the whole cell.
Supplementary Material
Acknowledgments
We thank Shihuang Su for the assistance in preparing media and supplies, Rong Dong for support with Drosophila cultures and media. We also thank Dr Clifford Stephen in the William S. Dunn Chemical Genomics Screening Center for his help arraying dsRNA used in the RNAi screen. We are grateful to Trudi Schupbach for providing hfp mutant flies, Hank Adams for assistance with confocal microscopy, Dr Marco Marcelli and Huiying Sun for providing facilities of luciferase measurement and quantitative PCR. This work was supported by an NIH grant R01GM070892 and a supplement from the American Recovery and Reinvestment Act (WM) and NIH grant R00GM080447 and R01 CA167752 (EJW). DNA sequencing was performed at the U.T. MD Anderson DNA Analysis Facility and supported by a grant (no. CA16672, DAF) from the National Cancer Institute.
Submitted
04/15/2013
Revised
06/21/2013
Accepted
07/05/2013
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/25645
References
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–5. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 3.Bourgeois CF, Lejeune F, Stévenin J. Broad specificity of SR (serine/arginine) proteins in the regulation of alternative splicing of pre-messenger RNA. Prog Nucleic Acid Res Mol Biol. 2004;78:37–88. doi: 10.1016/S0079-6603(04)78002-2. [DOI] [PubMed] [Google Scholar]
- 4.Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–211. doi: 10.1017/S1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graveley BR, Maniatis T. Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell. 1998;1:765–71. doi: 10.1016/S1097-2765(00)80076-3. [DOI] [PubMed] [Google Scholar]
- 6.Shen H, Kan JL, Green MR. Arginine-serine-rich domains bound at splicing enhancers contact the branchpoint to promote prespliceosome assembly. Mol Cell. 2004;13:367–76. doi: 10.1016/S1097-2765(04)00025-5. [DOI] [PubMed] [Google Scholar]
- 7.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 8.Kohtz JD, Jamison SF, Will CL, Zuo P, Lührmann R, Garcia-Blanco MA, et al. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–24. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 9.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–70. doi: 10.1016/0092-8674(93)90316-I. [DOI] [PubMed] [Google Scholar]
- 10.Tacke R, Manley JL. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 1995;14:3540–51. doi: 10.1002/j.1460-2075.1995.tb07360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tacke R, Manley JL. Determinants of SR protein specificity. Curr Opin Cell Biol. 1999;11:358–62. doi: 10.1016/S0955-0674(99)80050-7. [DOI] [PubMed] [Google Scholar]
- 12.Simard MJ, Chabot B. SRp30c is a repressor of 3′ splice site utilization. Mol Cell Biol. 2002;22:4001–10. doi: 10.1128/MCB.22.12.4001-4010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandler DS, Qi J, Mattox W. Direct repression of splicing by transformer-2. Mol Cell Biol. 2003;23:5174–85. doi: 10.1128/MCB.23.15.5174-5185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi J, Su S, Mattox W. The doublesex splicing enhancer components Tra2 and Rbp1 also repress splicing through an intronic silencer. Mol Cell Biol. 2007;27:699–708. doi: 10.1128/MCB.01572-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibrahim EC, Schaal TD, Hertel KJ, Reed R, Maniatis T. Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc Natl Acad Sci USA. 2005;102:5002–7. doi: 10.1073/pnas.0500543102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue K, Hoshijima K, Higuchi I, Sakamoto H, Shimura Y. Binding of the Drosophila transformer and transformer-2 proteins to the regulatory elements of doublesex primary transcript for sex-specific RNA processing. Proc Natl Acad Sci USA. 1992;89:8092–6. doi: 10.1073/pnas.89.17.8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian M, Maniatis T. A splicing enhancer complex controls alternative splicing of doublesex pre-mRNA. Cell. 1993;74:105–14. doi: 10.1016/0092-8674(93)90298-5. [DOI] [PubMed] [Google Scholar]
- 18.Lynch KW, Maniatis T. Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev. 1996;10:2089–101. doi: 10.1101/gad.10.16.2089. [DOI] [PubMed] [Google Scholar]
- 19.Kanopka A, Mühlemann O, Akusjärvi G. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature. 1996;381:535–8. doi: 10.1038/381535a0. [DOI] [PubMed] [Google Scholar]
- 20.Buratti E, Stuani C, De Prato G, Baralle FE. SR protein-mediated inhibition of CFTR exon 9 inclusion: molecular characterization of the intronic splicing silencer. Nucleic Acids Res. 2007;35:4359–68. doi: 10.1093/nar/gkm444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–54. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erkelenz S, Mueller WF, Evans MS, Busch A, Schöneweis K, Hertel KJ, et al. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA. 2013;19:96–102. doi: 10.1261/rna.037044.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun S, Zhang Z, Fregoso O, Krainer AR. Mechanisms of activation and repression by the alternative splicing factors RBFOX1/2. RNA. 2012;18:274–83. doi: 10.1261/rna.030486.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi J, Su S, McGuffin ME, Mattox W. Concentration dependent selection of targets by an SR splicing regulator results in tissue-specific RNA processing. Nucleic Acids Res. 2006;34:6256–63. doi: 10.1093/nar/gkl755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen M, Mattox W. Activation and repression functions of an SR splicing regulator depend on exonic versus intronic-binding position. Nucleic Acids Res. 2012;40:428–37. doi: 10.1093/nar/gkr713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sciabica KS, Hertel KJ. The splicing regulators Tra and Tra2 are unusually potent activators of pre-mRNA splicing. Nucleic Acids Res. 2006;34:6612–20. doi: 10.1093/nar/gkl984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinn LM, Dickins RA, Coombe M, Hime GR, Bowtell DD, Richardson H. Drosophila Hfp negatively regulates dmyc and stg to inhibit cell proliferation. Development. 2004;131:1411–23. doi: 10.1242/dev.01019. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell NC, Johanson TM, Cranna NJ, Er AL, Richardson HE, Hannan RD, et al. Hfp inhibits Drosophila myc transcription and cell growth in a TFIIH/Hay-dependent manner. Development. 2010;137:2875–84. doi: 10.1242/dev.049585. [DOI] [PubMed] [Google Scholar]
- 30.Van Buskirk C, Schüpbach T. Half pint regulates alternative splice site selection in Drosophila. Dev Cell. 2002;2:343–53. doi: 10.1016/S1534-5807(02)00128-4. [DOI] [PubMed] [Google Scholar]
- 31.Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA. 2004;101:15974–9. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katzenberger RJ, Marengo MS, Wassarman DA. Control of alternative splicing by signal-dependent degradation of splicing-regulatory proteins. J Biol Chem. 2009;284:10737–46. doi: 10.1074/jbc.M809506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandler DS, McGuffin ME, Mattox W. Functionally antagonistic sequences are required for normal autoregulation of Drosophila tra-2 pre-mRNA splicing. Nucleic Acids Res. 2001;29:3012–9. doi: 10.1093/nar/29.14.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattox W, Baker BS. Autoregulation of the splicing of transcripts from the transformer-2 gene of Drosophila. Genes Dev. 1991;5:786–96. doi: 10.1101/gad.5.5.786. [DOI] [PubMed] [Google Scholar]
- 35.McQuilton P, St Pierre SE, Thurmond J, FlyBase Consortium FlyBase 101--the basics of navigating FlyBase. Nucleic Acids Res. 2012;40(Database issue):D706–14. doi: 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattox W, McGuffin ME, Baker BS. A negative feedback mechanism revealed by functional analysis of the alternative isoforms of the Drosophila splicing regulator transformer-2. Genetics. 1996;143:303–14. doi: 10.1093/genetics/143.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinett CC, Vaughan AG, Knapp JM, Baker BS. Sex and the single cell. II. There is a time and place for sex. PLoS Biol. 2010;8:e1000365. doi: 10.1371/journal.pbio.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hastings ML, Allemand E, Duelli DM, Myers MP, Krainer AR. Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF(65) PLoS One. 2007;2:e538. doi: 10.1371/journal.pone.0000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Page-McCaw PS, Amonlirdviman K, Sharp PA. PUF60: a novel U2AF65-related splicing activity. RNA. 1999;5:1548–60. doi: 10.1017/S1355838299991938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katzenberger RJ, Marengo MS, Wassarman DA. ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. Mol Cell Biol. 2006;26:9256–67. doi: 10.1128/MCB.01125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marengo MS, Wassarman DA. A DNA damage signal activates and derepresses exon inclusion in Drosophila TAF1 alternative splicing. RNA. 2008;14:1681–95. doi: 10.1261/rna.1048808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dauwalder B, Mattox W. Analysis of the functional specificity of RS domains in vivo. EMBO J. 1998;17:6049–60. doi: 10.1093/emboj/17.20.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graveley BR, Hertel KJ, Maniatis T. A systematic analysis of the factors that determine the strength of pre-mRNA splicing enhancers. EMBO J. 1998;17:6747–56. doi: 10.1093/emboj/17.22.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burtis KC, Baker BS. Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoding related sex-specific polypeptides. Cell. 1989;56:997–1010. doi: 10.1016/0092-8674(89)90633-8. [DOI] [PubMed] [Google Scholar]
- 45.Ryner LC, Baker BS. Regulation of doublesex pre-mRNA processing occurs by 3′-splice site activation. Genes Dev. 1991;5:2071–85. doi: 10.1101/gad.5.11.2071. [DOI] [PubMed] [Google Scholar]
- 46.Heinrichs V, Ryner LC, Baker BS. Regulation of sex-specific selection of fruitless 5′ splice sites by transformer and transformer-2. Mol Cell Biol. 1998;18:450–8. doi: 10.1128/mcb.18.1.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S, Lopez AJ. Negative feedback regulation among SR splicing factors encoded by Rbp1 and Rbp1-like in Drosophila. EMBO J. 2005;24:2646–55. doi: 10.1038/sj.emboj.7600723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park JW, Graveley BR. Use of RNA interference to dissect the roles of trans-acting factors in alternative pre-mRNA splicing. Methods. 2005;37:341–4. doi: 10.1016/j.ymeth.2005.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.