

Abstract
In this paper we consider a problem from hematopoietic cell transplant (HCT) studies where there is interest on assessing the effect of haplotype match for donor and patient on the cumulative incidence function for a right censored competing risks data. For the HCT study, donor’s and patient’s genotype are fully observed and matched but their haplotypes are missing. In this paper we describe how to deal with missing covariates of each individual for competing risks data. We suggest a procedure for estimating the cumulative incidence functions for a flexible class of regression models when there are missing data, and establish the large sample properties. Small sample properties are investigated using simulations in a setting that mimics the motivating haplotype matching problem. The proposed approach is then applied to the HCT study.
Keywords: Binomial modeling, Bone marrow transplant, Competing risks, Haplotype effects, Haplotype match, Missing covariates, Inverse-censoring probability weighting, Nonparametric effects, Non-proportionality, Regression effects
1 Introduction
For the medical studies involving competing risks, one often wishes to estimate and model the cumulative incidence probability, the marginal probability of failure for a specific cause. The cumulative incidence curves and cause specific hazard functions for all causes contain the same information but are represented in a different ways and thus leads to different interpretations. Both quantities are generally of interest. Let λ1(t; z) be the cause-specific hazard of a cause one event and λ2(t; z) be the cause-specific hazard of an event of other causes than one, where both hazards are conditionally given by a set of covariates z. Assuming that cause one is the primary cause of interest, the cumulative incidence curve for cause one, that is the probability of experiencing cause one before time t, given covariates z which is given by
where ε indicates the type cause of failure. Recently, several new methods have been developed to directly model the cumulative incidence probability of a specific cause of failure (Fine and Gray 1999; Scheike et al. 2008; Scheike and Zhang 2008).
The aim of this paper is to consider the situation where there are missing covariates for all individuals. We consider data from a hematopoietic cell transplantation (HCT) study. HCT is a life saving procedure for many cancer patients. With shrinking family sizes, the lack of human leukocyte antigen (HLA) matched sibling is common, and this has increased the use of alternative donor graft sources. One of the alternatives is to use a donor who is not from the patient’s family. Unfortunately, in addition to its curative effect, HCT also has potentially lethal complications, especially for unrelated transplants. Severe graft-versus-host disease (GVHD) is one of the major causes of treatment related death. To reduce GVHD and to increase engraftment, a fully HLA-matched graft needs to be selected. For this purpose, the genotypes of the patient and potential donors are determined. Scientists found that many of the more than 400 genes have immune-related functions, and haplotypes that share the same HLA alleles may also share discrete blocks of highly conserved sequences in linkage disequilibrium with those HLA alleles. HLA haplotype analysis has be considered in a HCT study (Petersdorf et al. 2007).
For the HCT data, the donor and patient HLA-genotypes are fully observed and matched, but haplotypes are unknown in current practice, since standard genotyping techniques cannot distinguish the two homologous chromosomes of an individual, thus they cannot determine the haplotype pairs, that is the specific sequence of nucleotides on the chromosomes. In a fully HLA-matched unrelated transplant, the donor and patient have identical genotypes, but may not necessarily have matched haplotype pairs. For example, consider HLA loci A and B. Suppose the donor and patient have identical genotypes of G = { A = (1, 3), B = (7, 8)}. There are two potential haplo-types that are consistent with this genotype, namely (h1, h2) = ((A1, B7), (A3, B8)) and ((A1, B8), (A3, B7)). When the donor and the patient have identical haplotypes, then they are haplotype matched in addition to being genotype matched. Otherwise, they are haplotype mismatched. With more HLA loci considered the number of possible haplotypes corresponding to a single genotype is much larger; Petersdorf et al. (2007) give a detailed example with three loci A, B and DRB1. Laboratory techniques have been used to determine haplotypes, but these methods required for HLA haplotyping are technically complex and not easily adaptable for routine diagnostic use. The method previously described by Petersdorf et al. (2007) requires the manual purification of intact, long strand (2 Mbp-long) DNA, construction of probe-based DNA arrays, and hybridization of long strand DNA to solid phase probes. These are very complex techniques that are not easily adaptable to routine diagnostic testing and would be challenging even for advanced research laboratories, and often these methods are cost-prohibitive (Fallin and Schork 2000). The existence of a haplotype matching effect on transplant survival outcomes among fully HLA-genotype matched unrelated transplants has not been fully investigated.
In HCT studies, aGVHD (acute GVHD) and death without aGVHD are two competing risks, and cancer recurrence (relapse) and treatment-related-mortality (TRM, defined as death without relapse) are another pair of commonly studied competing risks. It has not been fully investigated whether there is a haplotype matching effect on cumulative incidence function of aGVHD, TRM and relapse among those HLA-genotype fully matched unrelated transplants. If a positive beneficial haplotype matching effect can be identified then patient and physician should search for a haplotype matched donor among those available to improve the transplant result. It has been shown that the probability of a HLA-haplotype match is around 80 % among the HLA-genotype matched patients and donors in the HCT setting (Petersdorf et al. 2007).
The Petersdorf et al. (2007) analysis was based on observing the haplotype directly and was carried out as a simple logistic regression analysis or a simple Cox regression analysis. They showed that haplotype mismatched HCT had a higher aGVHD rate and a lower relapse rate, and had no effect on TRM and overall mortality. Their study was based on a relative small sample size with 246 cases. This is the only available study for the haplotype matching effect. Their results have not been confirmed by others. Recently, Scheike et al. (2008) proposed using inverse weighting technique to directly model the cumulative incidence function and proposed a class of flexi-ble regression models. In this paper we study the haplotype matching effect for the competing risks data only based on HLA-genotype data, this makes it possible to directly use the vast amounts of available data for studying the possible effects of haplotype match. We also stress that there are many other related blood and marrow transplant treatments that have similar issues, and where the methods we develop here can be used. We here develop our techniques for the HCT study, and consider a similar cohort of patients with HLA-genotype identical unrelated HCT for leukemia. The data was selected from the center for international blood and marrow transplant research (CIBMTR). More generally our techniques can make use of already available data and lead to new knowledge in the field. Our analysis indicates that haplotype matching has same direction of effects as in the Petersdorf, but are non significant for all events. To further verify our conclusion, we consider a simulation study, which shows that proposed method works well.
We have in earlier work assessed the effect of haplotype match on the overall survival in HCT studies using missing data techniques for hazard estimation (Scheike et al. 2010, 2011). Also Flanders et al. (2005) considered testing of haplotype association on the hazard scale based on genotype data. These techniques can not be directly applied in a competing risks setting because the competing risk may also depend on the unobserved haplotye match thus leading to dependent censoring if the cause specific hazard is estimated. We also stress that we here consider the cumulative incidence regression models that models the absolute risk of experiencing the different causes of death in the competing risks setting and that this quantity depends on the underlying cause specific hazards for all causes.
The paper is structured as follows. In Sect. 2 we develop a model and show how one can estimate the parameters of the model and derive the asymptotic results that can be used for inference. Simulation results to examine the small sample performs are presented in Sect. 3. Section 4 contains a worked example based on HCT data. Finally, Sect. 5 contains some discussion.
2 Model specification
Let T̃i and Ci be the event time and right censoring time of the i th individual and let ε̃i ε {1, 2} denote the failure type. Let Ni (t) =
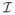 (T̃i ≤t, ε̃i = 1) be the underlying counting process associated with cause 1 and define the indicator Δi =
(T̃i ≤ Ci) that is one when the observation is uncensored. Note that Ni (t) are not fully observed for censored individuals. However, the observed counting process, {Δi Ni (t)} are computable for all t. In addition to the risk covariates (Xi,
Zi), we also observe the common genotype of the patient and donor, that we denote Gi. We assume that we observe n independent identically distributed (i.i.d.) replications of {(Ti, εi, Xi, Zi, Gi), i = 1,…, n}, where Ti = min(T̃i,
Ci), εi = ε̃i Δi, Xi = (1, Xi,1,…, Xi,p)⊤, and Zi = (Zi,1,…, Zi,q)⊤. We assume that (T̃i, ε̃i) are independent of Ci given covariates of (Xi,
Zi,
Gi).
(T̃i ≤t, ε̃i = 1) be the underlying counting process associated with cause 1 and define the indicator Δi =
(T̃i ≤ Ci) that is one when the observation is uncensored. Note that Ni (t) are not fully observed for censored individuals. However, the observed counting process, {Δi Ni (t)} are computable for all t. In addition to the risk covariates (Xi,
Zi), we also observe the common genotype of the patient and donor, that we denote Gi. We assume that we observe n independent identically distributed (i.i.d.) replications of {(Ti, εi, Xi, Zi, Gi), i = 1,…, n}, where Ti = min(T̃i,
Ci), εi = ε̃i Δi, Xi = (1, Xi,1,…, Xi,p)⊤, and Zi = (Zi,1,…, Zi,q)⊤. We assume that (T̃i, ε̃i) are independent of Ci given covariates of (Xi,
Zi,
Gi).
Let Hd = (H d1, Hd2) and H p = (H p1, H p2) be the underlying unobserved haplotype pairs for the donor and patient, respectively. To assess the effect of haplo-type matching, we consider various regression models for the cumulative incidence function on the form
| (1) |
for a known link function g and a known regression function g1.
These flexible models allow some covariates, X (H d, H p), to have time-varying effects and other covariates, Z (H d, H p), to have constant effects. This distinction becomes very important for the latter application to the HCT study data. Model (1) contains the Fine and Gray (1999) proportional subdistribution hazards model as a special case and Scheike and Zhang (2008) used this model to provide goodness-of-fit procedures for model identification and to check whether a specific covariate has a time-varying effect. The proposed models rely on the unobserved haplotype pairs for the patient and donor. The specific covariate designs we have in mind are to model the effect of matching haplotype nonparametrically, by letting x (H d, H p) = {1, x, I (H d = H p)}, or parametrically by letting z(H d, H p) = {z, I (H d = H p)}.
Commonly used “cloglog” and “log” link functions can be considered here, which lead to a semiparametric multiplicative model
| (2) |
and a semiparametric additive model
| (3) |
respectively.
When the haplotype model that links the observed genotype to the haplotype frequencies can be identified, then we can estimate these parameters consistently and derive the large sample properties.
To model the haplotype matching effect based on observed genotype data, we also need a model that relates the observed genotypes to the underlying haplotypes. With the haplotype pair H = (h1, h2), we assume Hardy-Weinberg equilibrium such that
where πi, i = 1,.., K gives the frequencies of the considered haplotypes. Based on this we can infer the conditional distribution of the underlying haplotypes given the observed genotype, P(H = h|G = g). Specifically, P{H = (hi,hj)|G = g} = P{H = (hi, h j: hi * h j = g)}/P(G = g), where hi * h j = g means that the haplotypes hi and h j are consistent with observed genotype g, and P (G = g) = Σhi,hj:hi*hj = g P{H = (hi, h j)}. In the HCT data we have a very large haplotype space (with many haplotypes) and limited observed data. We therefore make further simplifying assumptions about the haplotype frequencies letting several rare haplo-types share common frequencies in our model specifications concerning the haplotype frequencies that follows in the next section.
For the fully observed covariate V = (X, Z, G), the cumulative incidence function has the form:
where h p * hd ε G if h p1 * h p2 = hd1 * hd2 = g ε G. The last equality follows by the assumption that the risk covariates do not affect the haplotype distribution given the genotype, and the patient and donor have independent haplotypes given G. The last assumption is consistent with the fact that the donor and patient are unrelated, but for related donor and patient this part of the expression must be changes appropriately.
To estimate the parameters of the underlying model (1) we consider an inverse probability censoring weighting method as used in Scheike et al. (2008). This is based on the fact that
where SC (t |X i, Z i, Gi) is the survival distribution for the censoring time. We will for simplicity in the presentation assume that the censoring distribution does not depend on any of the observed covariates, denoted as SC (t).
These calculations show how the basic principle for modelling the missing data relies on computing the conditional mean of the missing covariate, (Hd, Hp), given the observed data (X, Z, G). It is evident how our approach more generally deal with missing covariates in the context of cumulative incidence estimation, but we here focus on the haplotype match problem that has been motivating our developments.
2.1 Estimation
We suggest a simple and robust two-stage procedure for estimation, where we first estimate the haplotype-probabilities and then use these estimated frequencies for estimating the regression parameters given in model (1) for the cumulative incidence functions. This will lead to some loss of efficiency but keeps things simple, and in our experience this loss of efficiency will typically be minor.
To estimate the haplotype parameters we consider the log-likelihood of the genotype data Πi P (Gi = gi). We consider the logistic regression model for the haplotype frequencies,
where (α1,…,αK–1)⊤ = Xθ, and X is a design matrix of size (K –1) × m, with m ≤ K–1 and θ = (θ1,…,θm)⊤
Let Ũθ(θ) = Σi Ũθ,i (θ), where Ũθi (θ)is the score of the log-likelihood for the i th subject. θ̂ is estimated by solving Ũθ(θ̂) = 0. By standard asymptotic theory of the MLE, is asymptotically normal and asymptotically equivalent to
where Uθ,i (θ) =
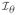 (θ)}−1
Ũθ,i(θ), and
(θ) = −∂Ũθ(θ)/∂θ.
(θ)}−1
Ũθ,i(θ), and
(θ) = −∂Ũθ(θ)/∂θ.
Now, given θ̂ we can solve score equations for η(t) and γ simultaneously using inverse-censoring weighted technique. Let be the n × 1 vector of P1(t; xi, zi, gi) for i = 1,…, n, let R(t) be the n × 1 vector of adjusted responses Δi Ni (t)/ŜC (Ti), and let Dη (t, η(t),γ,θ) and Dγ (t,η (t),γ, θ) be matrices with with the i th rows equal to Dη,i (t,η (t),γ, θ) = ∂P1(t; xi, zi, gi)/∂η (t) and Dγ,i (t, η(t),γ) = ∂P1(t; xi, zi, gi)/∂γ, respectively. Define similarly Dθ (t, η(t),γ, θ) as the matrix with i th row equal to ∂P1(t; xi, zi, gi)/∂ θ.
The regression functions η(t) and regression parameters γ can be estimated based on the following estimation equations for fixed θ:
| (4) |
| (5) |
where τ is the last event time point. Note that the estimates of η(t) will be piecewise constant functions that change their value only after events of type one, so we only need to consider the score equations for η(t) in the jump times.
The large sample properties in the case of known haplotype parameters, θ, follows the developments of Scheike et al. (2008) and Scheike and Zhang (2008). We show that how the asymptotics are changed due to the additional uncertainty that comes from the estimates of the haplotype frequencies.
For known θ, we have shown that the distributions of and are asymptotically equivalent to the following i.i.d. decomposition of
respectively, where
and detailed formula of (W1i (t), W2i) and its consistent estimates (Ŵ1i (t), Ŵ2i) are given in Scheike et al. (2008) and Scheike and Zhang (2008).
For unknown θ, based on a two-stage approach using the MLE θ̂ and under regularity conditions, it therefore can be shown that and are jointly asymptotically Gaussian, both zero mean, and with distributions that are asymptotically equivalent to
respectively, where
Let
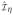 (t), Ŵη,i (t),
(t), Ŵη,i (t),
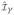 and Ŵγi be the estimators of their population counter parts by plugging estimates of all needed quantities. The asymptotic distributions of
and
are asymptotically equivalent to the following conditional multiplier version of the i.i.d. decompositions
and
, respectively, where E1,.., En are i.i.d. standard normals. It follows that the asymptotic variance of
and
can be estimated consistently by
and Ŵγi be the estimators of their population counter parts by plugging estimates of all needed quantities. The asymptotic distributions of
and
are asymptotically equivalent to the following conditional multiplier version of the i.i.d. decompositions
and
, respectively, where E1,.., En are i.i.d. standard normals. It follows that the asymptotic variance of
and
can be estimated consistently by
respectively, where a⊗ 2 = aa⊤.
A (1 - α) × 100 % asymptotic confidence band for η̂(t) over a fixed time interval can be constructed using resampling technique. These resampling results can be used to construct confidence band for the predicted cumulative incidence function as in Scheike et al. (2008).
3 Simulation study
To investigate the fixed sample properties we did a simulation study that mimics the data example analyzed in Sect. 4. We considered 3,712 patients with genotypes equivalent to the observed ones. Based on the haplotype frequencies described earlier we then sampled haplotype pairs consistent with the observed genotypes for both patient and donor, and then simulated a cumulative incidence function of additive form with approximately 815 events of interest for different levels of the effect of haplotype match. We generate data from a semiparametric multiplicative model (2),
with γ = {−0.5, −0.3, −0.1, 0, 0.1, 0.3, 0.5}. Observed coverage probabilities are reported in Table 1.
Table 1.
Mean of estimates (mean est.), the standard deviation of estimates (sd. est.), the mean of estimated standard errors (mean SE), and observed coverage of 95 % confidence intervals (coverage %) based on 1,000 realizations for different effect sizes (β)
| γ | Mean est. | Sd. est. | Mean SE | Coverage (%) |
|---|---|---|---|---|
| −0.5 | −0.470 | 0.097 | 0.097 | 94 |
| −0.3 | −0.277 | 0.063 | 0.065 | 93 |
| −0.1 | −0.089 | 0.049 | 0.051 | 96 |
| 0.0 | 0.000 | 0.041 | 0.041 | 95 |
| 0.1 | 0.093 | 0.050 | 0.050 | 94 |
| 0.3 | 0.285 | 0.065 | 0.063 | 95 |
| 0.5 | 0.479 | 0.087 | 0.088 | 94 |
The results of the simulations are given in Table 1. We see that the estimator is almost unbiased for all considered levels of the effect size. The variance is well estimated and the coverage is close the nominal level. All in all we conclude that the finite sample performance is quite good in a sample size similar to the one in the considered data.
4 HCT patients
Petersdorf et al. (2007) studied the effect of MHC haplotype match based on 246 leukemia patients who received a HLA-genotype fully matched unrelated HCT from 1986 to 2003. Their analysis was based on observing the haplotypes directly. This is a very costly and time-consuming procedure (Fallin and Schork 2000). They identi-fied 191(78 %) and 55(22 %) transplants were haplotype matched and miss-matched, respectively. They demonstrated that haplotype matched transplant had a lower incidence rate of grade III–IV aGVHD (odds-ratio=0.22, p < 0.0001) and a higher cancer relapse rate (hazard-ratio=2.22, p = 0.03), but had no impact on the TRM and overall mortality. In this study, we show how modeling of the missing haplotype data can also be used to address these issues. For the illustration purpose, a similar transplant patient cohort was selected from the statistical center of the center for international blood and marrow transplant research (CIBMTR). The analysis has not been reviewed or approved by the Advisory or Scientific Committee of the CIBMTR. The CIBMTR is comprised of clinical and basic scientists who confidentially share data on their blood and bone marrow transplant patients with CIBMTR Data Collection Center located at the Medical College of Wisconsin. The CIBMTR is a repository of information about results of transplants at more than 450 transplant centers worldwide.
The example data consists of 3,712 leukemia patients (1,822 for acute myeloid leukemia (AML), 982 for acute lymphoblastic leukemia (ALL) and 908 for chronic myelogenous leukemia (CML)). All patients in the study were HLA-A, B, DRB1 allele matched in high resolution with their donors and transplanted between 1995 and 2007. 1,651; 1,047 and 1,014 patients were transplanted in low, intermediate or high risk of disease statuses, respectively. 647 Males patients received graft from a female donor. 81 % of patients received myeloablative conditioning regimen, 53 % of patients were treated with methotrexale (MTX) + cyclosporin (CsA) ± other or CsA± other for GVHD prophylaxis, and 56 and 44 % of patients received bone-marrow (BM) or peripheral-blood (PB) transplant, respectively.
The genotype data is based on high resolution classification of alleles at three-loci HLA-A, HLA-B and HLA-DRB1. We consider two-stage procedure for this study. First, we need to estimate the haplotype frequencies, πi or θ = (θ1,…,θm)⊤ (see Sect. 2). Two estimation approaches can be considered. One is using MLE method based on current study data. Potentially, we will have a very rich haplotype space. To reduce the total number of possible haplotypes, some additional structure is needed. We here suggest to group the rare frequencies into groups with a common haplotype frequency. Alternatively, we can also use other available data for estimation of the hap-lotype frequencies (Excoffier and Slatkin 1995; Hawley and Kidd 1995; Long et al. 1995). Clearly using this additional data leads to a more accurate and stable estimation for the haplotype frequencies.
Both methods were used for this HCT study and gave similar results. In this paper, we report the results based on the additional data for the estimation of the haplotype frequencies. The National Marrow Donor Program (NMDP)’s existing potential donor pool as the background population cohort. The NMDP is a nonprofit organization dedicated to creating an opportunity for all patients to receive the bone marrow or umbilical cord blood transplant they need, when they need it. Currently, there are about eight million volunteer adult donors are registered in the NMDP’s potential donor pool. It is known that the haplotype frequency is determined by racial category. The donor’s and patient’s racial categories are fully observed for our study cohort and utilized in our haplotype frequency calculation. Among 3,712 HLA-identical unrelated HCT, the estimated average probability of haplotype matching is 80.7 % which is similar to Petersdorf et al. (2007)’s report.
To excess the haplotype matching effect on aGVHD, TRM, relapse and treatment failure which is defined as TRM or relapse, we fit the multiplicative model
| (6) |
where the first element of X is constant one, and adjusting remaining covariates X of disease type (AML versus ALL versus CML), and covariates Z of patient age (>45 versus 19–45 versus ≤18 years old), disease status at pre-transplant (advanced disease versus low or intermediate disease), donor–patient gender match (female to male versus other), conditioning regimen (NST/RIC versus myeloablative), GVHD prophylaxis (MTX+CsA ± Other or CsA±Other versus others), graft type (PB versus BM) and year of transplant (2002–2007 versus 1995–2001). In our study sample, 815(22 %) patients developed grade III–IV aGVHD, 1,488 patients died without aG-VHD and 1,409 patients were censored at end of study. 2,268 Patients were considered as treatment failure in which 1,255 patients died in compete remission (TRM) and 1,013 patients relapsed. Our analysis showed that haplotype match has no effect on aGVHD, relapse, TRM and treatment failure (Table 2).
Table 2.
Adjusted effect of HLA-A, -B, -CRB1 haplotype matching of risks of grades III–IV aGVHD, relapse, TRM and treatment-failure after HCT from HLA-identical unrelated donors
| Outcome | exp(γ̂ 0) | 95 % CI | P-value | Nonparametric test P-value |
|---|---|---|---|---|
| aGVHD III–IV | 0.98 | 0.69–1.38 | 0.98 | 0.46 |
| Relapse | 1.03 | 0.76–1.41 | 0.83 | 0.94 |
| TRM | 0.93 | 0.69–1.24 | 0.62 | 0.27 |
| Treatment-failure | 0.99 | 0.77–1.25 | 0.91 | 0.15 |
We also considered the model with non-parametric haplotype match effect where
| (7) |
where we subsequently performed a resampling test for the constant effect of H0: η0(t) ≡ η0 using a supremum Kolmogoroff–Smirnov test. If this test is significant it also suggests that the haplotype match effect is significant with time-varying effect. We stress that the test differs from the test for non-significant effects H0: η0(t) ≡ 0, that are not significant for all outcome events. We note that haplotype match has non-significant effect on the probability of aGVHD, relapse, TRM and treatment failure (see Table 2). Figure 1 shows the effect of haplotype match versus haplotype mismatch for TRM, a negative (beneficial) effect for the haplotype matched HCT within first 90 months of transplant and a positive late effect, however, it is not significant since 95 % confidence band contains some straight lines. This is further confirmed by non-significance of the constant effect test (H0: η0(t) ≡ η0, for a constant η0). Constant haplotype matching effects have been observed in all other outcomes. Thus, multiplicative model (6) with constant haplotype matching effect can be considered for all outcomes, which validates the the parametric test from Table 2. This model validation is a critical an important part of such an analysis, and easily done by the developed methodology.
Fig. 1.

Effect of haplo-match for CIF of TRM with corresponding 95 % confidence bands (dashed lines)
Based on fitted models, we can compute the predicted cumulative incidence functions (CIF) of aGVHD, relapse, TRM, and leukemia-free survival probability for a given set values of the covariates with 95 % confidence intervals and confidence bands. For the illustration purpose, we compute the predicted CIF of TRM by haplotype matched versus miss-matched for a patient with AML disease, transplanted in early or intermediate disease stage, donor–patient gender match of male to male (MM) or (MF) or (FF), received myeloablative conditioning regimen, CsA±Other for GVHD prophylaxis, and bone marrow graft source, and transplanted between 2003 and 2007. Resampling method based on 1,000 realizations was used to construct confidence band (See Fig. 2). Figure 2a shows the predicted CIF based on constant multiplicative model (6). Figure 2b shows the predicted CIF based on the alternative non-parametric haplotype match effect model (7), which is a more flexible model allowing haplotype matching effect change over time. Figure 2b shows that haplotype matched transplant has a lower cumulative incidence of TRM initially and a higher incidence rate of TRM later although this change is not significant.
Fig. 2.

Thick solid and dashed lines are the CIF of TRM for haplo-matched and haplo-mismatched HCT, respectively. Light solid and dashed lines represent corresponding 95 % confidence bands. a based on parametric model, and b non-parametric effect of haplo-match
5 Discussion
We have demonstrated how to assess the haplotype matching effect on the competing risks for hematopoietic cell transplant studies based on modeling of the missing data. This opens up for using the huge amounts of available data for studying detailed aspects of the HLA haplotypes on the outcome for blood and marrow transplants studies without cost-prohibitive laboratory typing for the haplotypes for patient and donor.
Another situation where the effect of haplotype matching is of interest is transplantations using umbilical cord blood (CB) which has recently been accepted as an alternative graft source to bone marrow (BM) for HCT (Eapen et al. 2007). Most CB transplants are mismatched at one or two HLA loci. For HLA-genotype mismatched unrelated transplants, the haplotype pairs of the donor and the patient can be either matched on a single haplotype or mismatched on both haplotypes. For example, with patient HLA-genotype G p = { A = (1, 3), B = (7, 8)} and donor genotype Gd = { A = (1, 3), B = (7, 13)} which are mismatched at the B locus, it is possible that they share a single haplotype (A1, B7). As far as we know, the question of whether there is an effect of a single haplotype matching versus both haplotypes being mismatched for HLA-mismatched unrelated transplants has not yet been investigated. The methods developed here can be used to study such questions.
We found no significant haplotype matching effect on grade III–IV aGVHD, relapse and TRM when fitting a model with constant effects. A more careful model examination using the non-parametric models validated these conclusions.
We have implemented the methods in the R-package HaploSurvival that is available from R-forge.
An issue for further research is a further study of the robustness to incorrect modeling of the haplotype distribution. Here it could be of interest to develop robust estimating equation along the lines of Allen and Satten (2005) and Allen et al. (2005), but extending these methods to our setting is not obvious.
Acknowledgments
The research was supported by National Cancer Institute Grant 2 R01 CA54706-10. Thomas Scheike was also supported by a grant from the Danish Research Council on “Point process modeling and statistical inference” and the excellence program at the University of Copenhagen.
Contributor Information
Thomas H. Scheike, Email: ts@biostat.ku.dk, Department of Biostatistics, University of Copenhagen, Copenhagen, Denmark
Martin J. Maiers, National Marrow Donor Program, Minneapolis, MA, USA
Vanderson Rocha, Hematology Bone Marrow Transplant Department, Hospital Saint-Louis, Paris, France.
Mei-Jie Zhang, Division of Biostatistics, Medical College of Wisconsin, Milwaukee, WI, USA.
References
- Allen AS, Satten GA. Robust testing of haplotype/disease association. BMC Genet. 2005;6(Suppl 1):S69. doi: 10.1186/1471-2156-6-S1-S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AS, Satten GA, Tsiatis AA. Locally-efficient robust estimation of haplotype-disease association in family-based studies. Biometrika. 2005;92:559–571. [Google Scholar]
- Eapen M, Rubinstein P, Zhang M-J, Stevens C, Kurtzberg J, Scaradavaou A, Loberiza FRECR, Klein JP, Horowitz MM, Wagner JE. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet. 2007;369:1947–1954. doi: 10.1016/S0140-6736(07)60915-5. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M. Maximum-likelihood estimation of polecular haplotype frequenceis in a deiploid population. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- Fallin D, Schork NJ. Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet. 2000;67:947–959. doi: 10.1086/303069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- Flanders WD, Khoury MJ, Yang QH, Austin H. Test of trait—haplotype association when linkage phase is ambiguous, appropriate for matched case–control and cohort studies with competing risks. Stat Med. 2005;24:2299–2316. doi: 10.1002/sim.2156. [DOI] [PubMed] [Google Scholar]
- Hawley M, Kidd K. Haplo: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered. 1995;86:409–411. doi: 10.1093/oxfordjournals.jhered.a111613. [DOI] [PubMed] [Google Scholar]
- Long J, Williams R, Urbanek M. An EM algorithm and testing strategy for multi-locus haplotypes. Am J Hum Genet. 1995;56:799–810. [PMC free article] [PubMed] [Google Scholar]
- Petersdorf E, Malkki M, Gooley T, Martin P, Guo Z. MHC haplotype matcing for unrelated hema-topoietic cell transplantation. PLOS Med. 2007;4:59–68. doi: 10.1371/journal.pmed.0040008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheike T, Martinussen T, Silver J. Estimating haplotype effects for survival data. Biometrics. 2010;66:705–715. doi: 10.1111/j.1541-0420.2009.01329.x. [DOI] [PubMed] [Google Scholar]
- Scheike T, Martinussen T, Zhang M. The additive risk model for estimation of haplotype effects. Scand J Stat. 2011;38:409–423. doi: 10.1111/j.1467-9469.2010.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheike TH, Zhang M-J. Flexible competing risks regression modelling and goodness-of-fit. Lifetime Data Anal. 2008;14:464–483. doi: 10.1007/s10985-008-9094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheike TH, Zhang M-J, Gerds T. Predicting cumulative incidence probability by direct binomial regression. Biometrika. 2008;95:205–220. [Google Scholar]