

The net removal of two adjacent (vicinal) hydrogen atoms from an alkane to produce an alkene is a fundamental process of long-standing interest1,2 that is critical to, for example, lipid and terpene biosynthesis. Desaturases (and acetylenases) are adept at achieving this essential, often ubiquitous, oxidative functionalization [e.g., biosynthesis of unsaturated fatty acids3(oleates, linoleates, etc.), eicosanoids (leukotrienes, arachadonic acid, etc.), gibberellins4, and carotenoids5]. More broadly, alkane to alkene conversion always involves one or more chemical intermediates in a multistep reaction pathway. These may be either isolable species (such as alcohols or alkyl halides) or reactive intermediates (such as carbocations, alkyl radicals, or σ-alkyl-metal species). Here we report a desaturation reaction of simple, unactivated alkanes that is mechanistically unique. We show that benzynes are capable of simultaneous (i.e., concerted), bimolecular removal of two vicinal hydrogen atoms from a hydrocarbon. The discovery of this exothermic, net redox process (benzyne reduction and alkane oxidation) was enabled by the simple thermal generation of reactive benzyne intermediates through the hexadehydro-Diels–Alder cycloisomerization reaction of triyne substrates6. We are not aware of any single-step, bimolecular reaction in which two hydrogen atoms are simultaneously transferred from a saturated alkane (Fig. 1a). We present here results that are best described by this process. Computational studies indicate a preferred geometry having eclipsed vicinal C–H bonds in the alkane donor. A variety of structurally diverse benzynes (including o-benzyne itself!) effect this reaction.
Figure 1. Introduction and background.
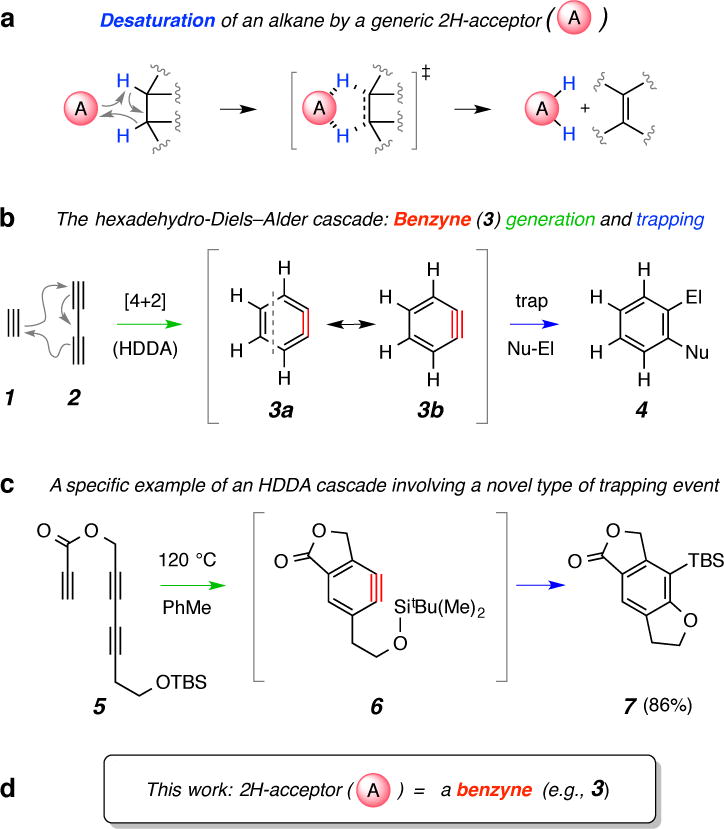
a. A = a potential double hydrogen atom (2H) acceptor, in which the accepting moiety could be either monoatomic (e.g., metal, metal-oxo, carbene, or nitrene) or polyatomic (e.g., a π-bonded species) in nature. b, The prototypical HDDA cascade. c, Intramolecular HDDA cycloisomerization followed by silyl ether trapping.6 d, Here we show that benzynes (like 3 or 6) will extract two hydrogen atoms from adjacent carbon atoms of suitable 2H-donor substrates.
Arynes7,8,9 engage in myriad trapping reactions that functionalize adjacent sp-hybridized carbons in the o-aryne ring. We recently reported a general strategy for the formation and subsequent in situ trapping of benzynes via the hexadehydro-Diels–Alder (HDDA) reaction6,10,11. The simplest imaginable variant (Fig. 1b) is the reaction of 1,3-butadiyne (2) with ethyne (1, the diynophile) to produce o-benzyne (3). The free energy change for this process is computed to be exothermic by ca. 50(!) kcal•mol−1 6,12. Trapping of 3 permits the synthesis of many useful benzene derivatives (4). In practice (Fig. 1c), the HDDA cycloisomerization is effected intramolecularly simply by heating a tethered triyne substrate like 5 to produce a fused bicyclic benzyne intermediate like 6. Trapping leads to a highly substituted benzenoid product like 7. In addition to the preparative value of this de novo generation of benzynes, the HDDA reaction provides the opportunity to uncover previously unprecedented aryne trapping modes13,14 (e.g., the insertion of the strained benzyne into the silyl ether bond as 6 proceeds to 7). This is largely because HDDA cyclizations produce reactive benzyne intermediates in the absence of added reagents, byproducts, or catalysts.
We now report a double hydrogen atom (2H) transfer reaction in which a HDDA-generated benzyne simultaneously accepts two vicinal hydrogen atoms from a suitable alkane 2H-donor (H–Csp3Csp3–H). This gives the corresponding (oxidized) alkene and (reduced) benzenoid products. For example, when we heated triyne 8 in cyclooctane to 85 °C, the only isolated product (89%) was the reduced fluorenone derivative 10-h2 (Fig. 2a). Using 1H NMR spectroscopy, we observed that a comparable amount of cyclooctene had been formed via desaturation15 (see Fig. 3b). The only well-characterized example of benzyne reduction via the net addition of two hydrogen atoms is the work of Sterenberg and coworkers in which a benzyne intermediate derived from a bis-diyne-bridged, dinuclear metal complex was reduced to the arene16. They demonstrated that the solvent (THF) was the source of the hydrogen (and, in the case of THF-d8, deuterium17) atoms that appeared in the reduced benzenoid product. When we heated substrate 8 in THF-h8, 10-h2 was the only product isolated (75%, Fig. 2a). Similarly, when 8 was heated in THF-d8, the dideuterated analog 10-d2 (MS and 1H NMR) was the only isolated product.
Figure 2. Both hydrogen atoms come from the same donor molecule.
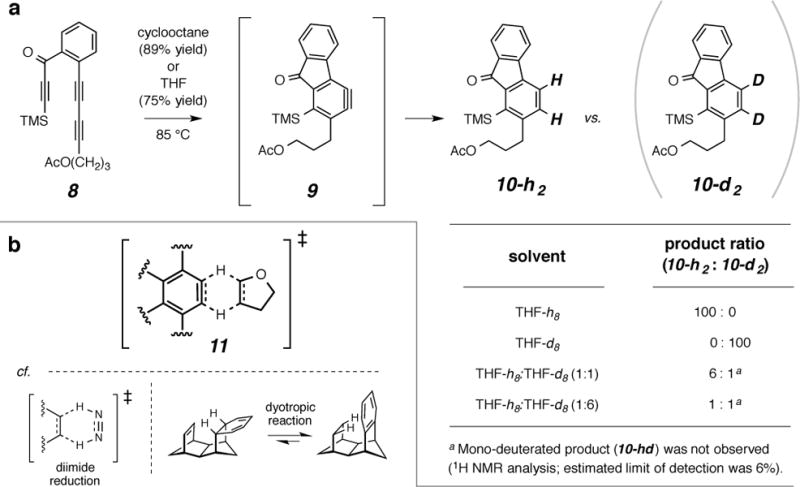
a. Dihydrogen transfer reactions between the HDDA-generated benzyne 9 and the 2H-donor solvents cyclooctane and tetrahydrofuran (THF) give the benzenoid 10; isotope profiling using THF-h8, THF-d8, and mixtures thereof (Table, lower right) shows that both hydrogen atoms in the product originate from a single molecule of 2H-donor. b, A representation (11) of simultaneous double hydrogen transfer between an aryne and a THF molecule. Analogous six-atom arrays are involved in the TSs of 2H-transfer by diimide to an alkene acceptor18,19 and in the class of intramolecular reorganizations known as dyotropic reactions.20,21
Figure 3. Dihydrogen transfer between arynes and cyclic hydrocarbons.
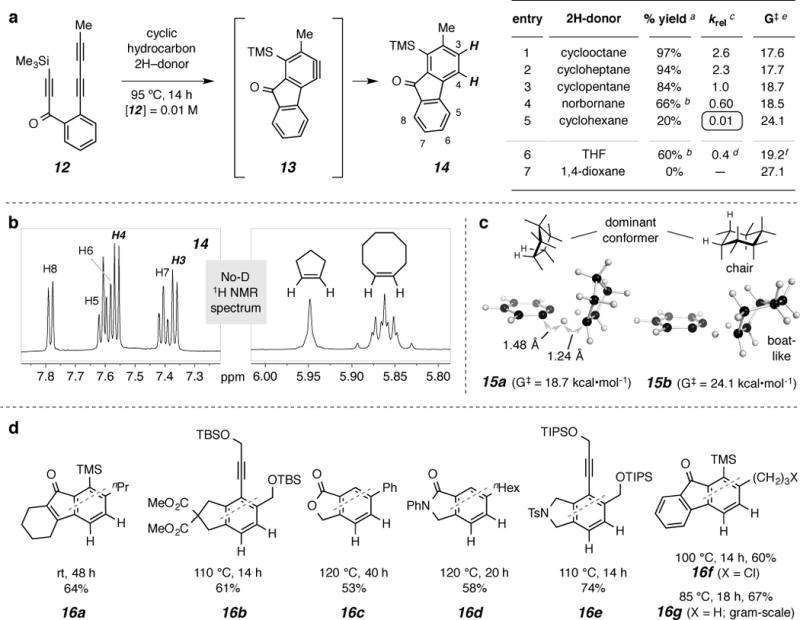
a. Relative efficiency (% yield and krel vs. cyclopentane) of various hydrocarbon (and cyclic ether) 2H-donors for the reduction of aryne13 to arene14. For tabular inset notes a-f see Supplementary Information. b, A representative No-D 1H NMR22 spectrum [this of the reaction solution arising from heating 12 (at 10 mM) in a 1.5:1 molar ratio of cyclopentane:cyclooctane at 95 °C]; thisshows the overall efficiency of the reaction and validates the krel value (1:2.6) obtained as described above. c, Computed TS geometries and ΔG‡ for transfer of two hydrogen atoms to benzyne (3) from cyclopentane (15a) and cyclohexane (15b). d, Reduced benzenoid products 16a-g generated by heating the triyne precursor in cyclooctane under the indicated conditions (starting substrate concentration of 10mM).
To further probe the mechanism of this process and, in particular, to distinguish between pathways involving sequential hydrogen atom abstractions from two solvent molecules vs. a transfer of two hydrogen atoms from a single molecule, we repeated the generation and trapping of benzyne 9, this time in the presence of an equimolar mixture of THF-h8 and THF-d8. Intriguingly, only the diprotio-and dideuterio-benzenoid products 10-h2 and 10-d2 were produced; none of the mono-H/mono-D analog (10-hd) was detected. The observed 10-h2:10-d2 product ratio was 6:1, indicating a significant H/D kinetic isotope effect for the 2H-transfer. In a complementary experiment, we used a 1:6 molar ratio of THF-h8:THF-d8, which gave a nearly 1:1 ratio of products 10-h2:10-d2. The lack of an observable level of monodeuterated product in any of these experiments is consistent with the concerted transfer to the benzyne of two hydrogen atoms from a single THF molecule as represented in the transition structure (TS) depiction 11 (Fig. 2b). Although such a description might seem unusual, it can be noted that the generally accepted mechanism for (i) the reduction of alkenes by diimide (HN=NH)18,19 and (ii) dyotropic reactions in which two hydrogen atoms are shuffled intramolecularly20,21 (Fig. 2b) involves a similar simultaneous transfer.
We next screened a series of cyclic hydrocarbons to explore their relative ability to engage an aryne in a similar hydrogen transfer reaction (Fig. 3). We were surprised to observe that cyclohexane was significantly less efficient than the other cycloalkanes in its reduction of the benzyne 13 (entries 1–5, Fig. 3a). This was initially seen from simple comparison of the chemical yields following purification of reduced (benzenoid) product 14. It is relevant that when the HDDA cyclization is carried out in the absence of a suitably good trapping agent, we routinely observe the formation of intractable, dark-colored mixtures of oligomeric substances. We hypothesize that this is because the reactive benzyne (e.g., 13) engages the conjugated diyne unit in another molecule of substrate triyne (e.g., 12) competitively with its abstraction of two hydrogen atoms from a solvent molecule. Thus, for these processes the yield of 14 is a meaningful reflection of the 2H-transfer rate from each donor solvent. Use of the acyclic hydrocarbon n-heptane as solvent also resulted in the production of 14, now in 30% (isolated) yield—i.e., heptane is intermediate in reactivity between cyclopentane and cyclohexane. Notably, when the starting concentration of triyne 12 was reduced by an order of magnitude (i.e., 10 vs. 1 mM), the isolated yield of 14 more than doubled for the reaction in cyclohexane (20% to 53%) or n-heptane (30% to 73%). This is consistent with the hypothesis that the rate of the primary decomposition process is dependent on the triyne concentration.
We then turned to the use of No-D NMR spectroscopy22 to both positively identify the alkene byproduct and to demonstrate that it was formed in nearly equimolar amount vis-à-vis the reduced benzenoid product 14. A typical No-D spectrum, this one from heating 12 in an equivolume mixture of cyclooctane and cyclopentane ([12] = 0.01 M), is shown in Fig. 3b. The ratio of the alkene resonances of cyclooctene to cyclopentene (adjusted for the molar ratio of solvents) provides the krel (2.6 for this example). The relative intensity of alkene to arene resonances (from 14) as well as the absence of resonances indicative of aromatic byproducts shows the overall cleanliness of the reaction.
As stated earlier, cyclohexane (entry 5, tabular inset in Fig. 3a) is a considerably poorer 2H-donor compared to the other cyclic hydrocarbons (entries 1–4). We hypothesized that this implies a preference for an eclipsed geometry for the relevant HCsp3Csp3H subunit within the 2H-donor molecule. Accordingly, cyclohexane, dominated by the chair conformation, is least disposed toward transfer of two of its hydrogen atoms, whereas the other hydrocarbons all have low-lying conformers with HCsp3Csp3H dihedral angles much smaller than 60°. That is, those cyclic hydrocarbons populated to a significant extent by conformers having less highly staggered vicinal C–H bonds are the more reactive 2H-donors. To further test this thinking, we examined 1,4-dioxaneas a potential 2H-donor. Not surprisingly, use of this chair-like compound gave none of the reduced product 14 (entry 7, Fig. 3a). On the other hand, norbornane, having a boat-like cyclohexane embedded in its framework (and an associated HCsp3Csp3H moiety with a 0° dihedral angle) is a kinetically competent donor (entry 4), even though the product norbornene comprises a strained alkene.
Next, using DFT methods (see Supplementary Information) we computed the TS geometry and the free energy of activation (ΔG‡) for the double hydrogen atom transfer21 between o-benzyne (3) and each of the seven cyclic donors shown in entries 1–7 (Fig. 3a). For all 2H-donors the calculations indicate a relatively early TS (cf. the two distances shown in 15a). This is consistent with the highly exothermic nature of the 2H-transfer step [e.g., we computed the free energy of reaction to be −65.6 kcal mol−1 for o-benzyne (3) + cyclopentane going to benzene + cyclopentene]. The computed geometries for the cyclopentane (15a) and cyclohexane (15b) TSs are shown in Fig. 3c. It is not accidental that the energy difference between the chair and boat conformers of cyclohexane (ca. 6 kcal•mol−1) is similar to the computed difference in ΔG‡ between 15a and (the boat-like) 15b. The ΔG‡s computed for all seven donors are given in the tabular inset in Fig. 3a. There is a remarkably good correlation between the computed ΔG‡s and the observed krel values. These observations are most consistent with the idea of substantial dihedral angle dependence for the process, which can only be true if the double hydrogen atom transfer event is concerted.
Products 16a-g (Fig. 2d) arose from incubating the corresponding triyne precursor [inferred from the dashed line in each structure (see Supplementary Information for details)] in cyclooctane under the indicated conditions. Notable features include: (i) a variety of functional groups, present in both the triyne precursor and benzenoid product, are readily tolerant of these benign reducing conditions; (ii) benzynes representing a breadth of electronic activation and/or perturbation engage in the reaction; (iii)most of the products 16 have a 1,2,3,4-tetrasubstituted motif, a substitution pattern that can be challenging to access by classical aromatic synthesis strategies; (iv)the double hydrogen atom transfer process occurs readily even at ambient temperature (cf. 16a); and(v) the reaction is not limited by scale (cf. 16g).
Finally, an ancillary but important practical consideration has emerged. The most common method for generating simple benzyne derivatives, including the parent 3, is that of Kobayashi23 in which 2-trimethylsilylphenyl triflate (o-TMSPhOTf) is exposed to a fluoride ion source (commonly CsF) in, most often, THF as the solvent. We speculated that some known trapping reactions of benzynes generated in THF are compromised in their efficiency due to competitive reduction by that solvent. Indeed, when we exposed o-TMSPhOTf to CsF in THF-d8 in the absence of any other trapping agent, we observed the production of benzene (C6H4D2, by 1H NMR analysis). Similarly, benzene (and cyclopentene) was seen when CsF and o-TMSPhOTf were reacted in CD3CN that contained cyclopentane (ca. 25 equiv). We suggest that all traditional benzyne generation methods performed in the presence of a potential 2H-donor (most typically, THF) are at risk to the unwanted, benzyne-depleting, 2H-transfer process, especially when the benzyne trapping event is inherently slow. Indeed, we can infer that this has already been encountered. Recent reports show THF to be an inferior medium(vs. 1,4-dioxane24 or diethyl ether25) for some benzyne trapping reactions. This is consistent with (i) the results we reported above for the relative efficiency of THF vs. 1,4-dioxane as a2H-donor (entries 6 vs. 7, tabular inset in Fig. 3a) and (ii) our arguments for angle-dependency during the 2H-transfer.
In summary, we have described the essential mechanistic features of a double hydrogen atom transfer process. Both (vicinal) hydrogen atoms come from the same donor molecule. There is substantial dihedral angle dependence–donors having a greater degree of eclipsing among their low-energy conformers are more reactive. This is reinforced by the nearly planar geometry of the six reacting atoms in the computed transition structures. Our observations support a pathway in which both hydrogen atoms are transferred simultaneously from the saturated alkane to the benzyne carbon atoms—a process that could be viewed as a metal-free, double C–H activation event26.
Methods Summary
A typical double hydrogen atom transfer reaction comprised heating a solution of HDDA triyne precursor (substrate) in cyclooctane (ca. 0.01 M) in a closed glass reaction vessel (e.g., a screw-capped vial or culture tube). After the specified time, the reaction mixture was loaded directly onto a bed of silica gel and eluted first with hexanes to remove the excess cyclooctane and then with ethyl acetate to capture the reduced benzenoid products. These were further purified by chromatography on silica gel. Relative rate data (Fig. 3a and 3b) were collected by 1H NMR spectroscopy at 500 MHz using No-D22 and qNMR27 techniques. Details are given in the Supplementary Information (SI). Details for the preparation of all new compounds, their full spectroscopic characterization data, and the computational methods used are also provided in the SI.
Supplementary Material
Acknowledgments
D.N. and P.H.W. thank the University of Minnesota Graduate School Doctoral Dissertation Fellowship and National Science Foundation Graduate Research Fellowship program, respectively. We thank Professor Christopher J. Cramer for helpful discussions about the computational studies. Financial support from the National Institute of General Medical Sciences (GM65597) and the National Cancer Institute (CA76497) of the U.S. Department of Health and Human Services is acknowledged. Portions of this work were performed using hardware and software resources available through the University of Minnesota Supercomputing Institute (MSI).
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions D.N. made the initial key observations and performed the majority of the scope studies. P.H.W. performed the majority of the mechanistic studies. B.B. and B.P.W. also performed aspects of the experimental work. All authors interpreted the data and wrote the manuscript.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
References
- 1.Linstead RP, Braude EA, Mitchell PWD, Wooldridge KRH, Jackman LM. Transfer of hydrogen in organic systems. Nature. 1952;169:100–103. [Google Scholar]
- 2.Bloomfield DK, Bloch K. The formation of Δ9-unsaturated fatty acids. J Biol Chem. 1960;235:337–345. [PubMed] [Google Scholar]
- 3.Buist PH. Fatty acid desaturases: selecting the dehydrogenation channel. Nat Prod Rep. 2004;21:249–262. doi: 10.1039/b302094k. [DOI] [PubMed] [Google Scholar]
- 4.Bhattacharya A, Kourmpetli S, Ward DA, Thomas SG, Gong F, Powers SJ, Carrera E, Taylor B, Gonzalez FN de C, Tudzynski B, Phillips AL, Davey MR, Hedden P. Characterization of the fungal gibberellin desaturase as a 2-oxoglutarate-dependent dioxygenase and its utilization for enhancing plant growth. Plant Physiol. 2012;160:837–845. doi: 10.1104/pp.112.201756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moran NA, Jarvik T. Lateral transfer of genes from fungi underlies carotenoid production in aphids. Science. 2010;328:624–627. doi: 10.1126/science.1187113. [DOI] [PubMed] [Google Scholar]
- 6.Hoye TR, Baire B, Niu D, Willoughby PH, Woods BP. The hexadehydro-Diels–Alder reaction. Nature. 2012;490:208–212. doi: 10.1038/nature11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann RW. Dehydrobenzene and Cycloalkynes. Vol. 11. Academic; 1967. (Organic Chemistry, A Series of Monographs). [Google Scholar]
- 8.Tadross PM, Stoltz BM. A comprehensive history of arynes in natural product total synthesis. Chem Rev. 2012;112:3550–3577. doi: 10.1021/cr200478h. references therein. [DOI] [PubMed] [Google Scholar]
- 9.Baire B, Niu D, Willoughby PH, Woods BP, Hoye TR. Synthesis of complex benzenoids via the intermediate generation of o-benzynesthrough the hexadehydro-Diels–Alder reaction. Nature Protocols. 2013;8:501–508. doi: 10.1038/nprot.2013.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyawaki K, Suzuki R, Kawano T, Ueda I. Cycloaromatization of a non-conjugated polyenyne system: Synthesis of 5H-benzo[d]fluoreno[3,2-b]pyrans via diradicals generated from 1-[2-{4-(2-alkoxymethylphenyl)butan-1,3-diynyl}]phenylpentan-2,4-diyn-l-ols and trapping evidence for the 1,2-didehydrobenzene diradical. Tetrahedron Lett. 1997;38:3943–3946. [Google Scholar]
- 11.Bradley AZ, Johnson RP. Thermolysis of 1,3,8-nonatriyne: Evidence for intramolecular [2+4] cycloaromatization to a benzyne intermediate. J Am Chem Soc. 1997;119:9917–9918. [Google Scholar]
- 12.Ajaz A, Bradley AZ, Burrell RC, Li WHH, Daoust KJ, Bovee LB, DiRico KJ, Johnson RP. Concerted vs. stepwise mechanisms in dehydro-Diels−Alder reactions. J Org Chem. 2011;76:9320–9328. doi: 10.1021/jo201567d. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann RW, Suzuki KA. “hot, energized” benzyne. Angew Chem Int Ed. 2013;52:2–4. doi: 10.1002/anie.201209041. [DOI] [PubMed] [Google Scholar]
- 14.Yun SY, Wang K-P, Lee N-K, Mamidipalli P, Lee D. Alkane C–H insertion by aryne intermediates with a silver catalyst. J Am Chem Soc. 2013;135:4668–4671. doi: 10.1021/ja400477r. [DOI] [PubMed] [Google Scholar]
- 15.Voica AF, Mendoza A, Gutekunst WR, Fraga JO, Baran PS. Guided desaturation of unactivated aliphatics. Nat Chem. 2012;4:629–635. doi: 10.1038/nchem.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsui JA, Sterenberg BT. A metal-templated 4 + 2 cycloaddition reaction of an alkyne and a diyne to form a 1,2-aryne. Organometallics. 2009;28:4906–4908. [Google Scholar]
- 17.de Almeida G, Townsend LC, Bertozzi CR. Synthesis and reactivity of dibenzoselenacycloheptynes. Org Lett. 2013;15:3038–3041. doi: 10.1021/ol401225n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hünig S, Müller H, Thier W. Reduktionen mit diimid. Tetrahedron Lett. 1961;2:353–357. [Google Scholar]
- 19.Corey EJ, Pasto DJ, Mock WL. Chemistry of diimide. II. Stereochemistry of hydrogen transfer to carbon-carbon multiple bonds. J Am Chem Soc. 1961;83:2957–2958. [Google Scholar]
- 20.Fernández I, Cossío FP, Sierra MA. Dyotropic reactions: Mechanisms and synthetic applications. Chem Rev. 2009;109:6687–6711. doi: 10.1021/cr900209c. [DOI] [PubMed] [Google Scholar]
- 21.Fernández I, Sierra MA, Cossío FP. In-plane aromaticity in double-group transfer reactions. J Org Chem. 2007;72:1488–1491. doi: 10.1021/jo062310r. [DOI] [PubMed] [Google Scholar]
- 22.Hoye TR, Eklov BM, Ryba TD, Voloshin M, Yao LJ. No-D NMR (no deuterium proton NMR) spectroscopy: A simple yet powerful method for analyzing reaction and reagent solutions. Org Lett. 2004;6:953–956. doi: 10.1021/ol049979+. [DOI] [PubMed] [Google Scholar]
- 23.Himeshima Y, Sonoda T, Kobayashi H. Fluoride-induced 1,2-elimination of o-trimethylsilylphenyl triflate to benzyne under mild conditions. Chem Lett. 1983;12:1211–1214. [Google Scholar]
- 24.Ma Z-X, Feltenberger JB, Hsung RP. Total syntheses of chelidonine and norchelidonine via an enamide–benzyne–[2+2] cycloaddition cascade. Org Lett. 2012;14:2742–2745. doi: 10.1021/ol300967a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sumida Y, Kato T, Hosoya T. Generation of arynes via ate complexes of arylboronic esters with an ortho-leaving group. Org Lett. 2013;15:2806–2809. doi: 10.1021/ol401140d. [DOI] [PubMed] [Google Scholar]
- 26.Davies HML, Du Bois J, Yu J-Q. C–H Functionalization in organic synthesis. Chem Soc Rev. 2011;40:1855–1856. doi: 10.1039/c1cs90010b. [DOI] [PubMed] [Google Scholar]
- 27.Paul GF, Jak B, Lankin DA. Routine experimental protocol for qHNMR illustrated with taxol. J Nat Prod. 2007;70:589–595. doi: 10.1021/np060535r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.