

Summary
We have constructed derivatives of Streptomyces coelicolor M145 as hosts for the heterologous expression of secondary metabolite gene clusters. To remove potentially competitive sinks of carbon and nitrogen, and to provide a host devoid of antibiotic activity, we deleted four endogenous secondary metabolite gene clusters from S. coelicolor M145 – those for actinorhodin, prodiginine, CPK and CDA biosynthesis. We then introduced point mutations into rpoB and rpsL to pleiotropically increase the level of secondary metabolite production. Introduction of the native actinorhodin gene cluster and of gene clusters for the heterologous production of chloramphenicol and congocidine revealed dramatic increases in antibiotic production compared with the parental strain. In addition to lacking antibacterial activity, the engineered strains possess relatively simple extracellular metabolite profiles. When combined with liquid chromatography and mass spectrometry, we believe that these genetically engineered strains will markedly facilitate the discovery of new compounds by heterologous expression of cloned gene clusters, particularly the numerous cryptic secondary metabolic gene clusters that are prevalent within actinomycete genome sequences.
Introduction
The increasing incidence of antibiotic resistance in a wide range of bacterial pathogens is a major global health concern (Boucher et al., 2009). At the same time, the discovery of new antimicrobials, and particularly of new classes of antibiotics, has declined markedly (Baltz, 2008). Consequently, there is an urgent need to develop new strategies for antibiotic discovery.
One approach to the discovery of new bioactive compounds is genome mining of actinomycetes, Gram‐positive mycelial bacteria with DNA of high GC content that produce about two‐thirds of all known antibiotics of microbial origin (Baltz, 2008). Genome sequencing of actinomycetes has revealed the presence of numerous cryptic secondary metabolite gene clusters of unknown function that could provide future clinically useful antibiotics (Zerikly and Challis, 2009). Strategies to access these compounds fall into two classes: induction of expression of cryptic gene clusters in the natural host and heterologous expression in a surrogate strain. The first approach is largely empirical, relying heavily on the use of different growth media and/or, if possible, the ability to genetically manipulate individual and often quite disparate actinomycetes. The second approach involves heterologous expression of gene clusters in a surrogate host. Recent advances in DNA sequencing provide relatively facile and inexpensive access to large numbers of biosynthetic gene clusters by genome scanning. In addition to targeting readily cultivated bacteria, this approach can also be applied to difficult‐to‐culture microorganisms, including many rare terrestrial and marine actinomycetes (Fenical and Jensen, 2006; Baltz, 2008) and myxobacteria (Wenzel and Müller, 2009) that may not have been screened by conventional means. Growing many of these organisms at large scale for production purposes is unlikely to be practical and their biosynthetic gene clusters are particularly attractive targets for heterologous expression in a genetically amenable host.
The genus Streptomyces produces about half of all known antibiotics of microbial origin, as well as metabolites with other medicinal and biotechnological applications. Streptomyces coelicolor A3(2) is the model species for the study of actinomycete genetics and biology, and a large array of genetic tools, including its genome sequence, is available to understand and manipulate the organism (Kieser et al., 2000; Bentley et al., 2002). Streptomyces coelicolor produces at least four compounds with antibiotic activity: the Type II polyketide actinorhodin, the NPRS/PKS‐derived prodiginines, the NRPS‐derived calcium‐dependent antibiotic (CDA) and the Type I polyketide CPK (Bentley et al., 2002; Pawlik et al., 2006).
We set out to develop S. coelicolor as a host for the expression of heterologous gene clusters, and focused initially on deleting the four endogenous antibiotic gene clusters. This was predicted to remove competing sinks of carbon and nitrogen, thus increasing precursor availability and the productivity of heterologous cloned gene clusters. These deletions were also predicted to simplify the identification and characterization of the products of cloned gene clusters by activity screening and metabolite profiling. In addition, and importantly, we introduced mutations that markedly enhance, in a non‐specific manner, the level of secondary metabolite production. We believe that these strains can make a major contribution to the discovery of novel natural products and may contribute significantly to addressing the urgent need for new antibiotics.
Results
Deletion of endogenous gene clusters
We deleted the four antibiotic gene clusters from S. coelicolor M145 [a derivative of the wild‐type strain A3(2) lacking plasmids SCP1 and SCP2 (Kieser et al., 2000)] by homologous recombination by cloning 2 kb PCR‐amplified fragments from both ends of each cluster into a suicide vector (Tables S1 and S2). The extent of three of the gene clusters (act, red and cda) was taken from Challis and Hopwood (2003) while that of the cpk gene cluster was taken from Pawlik and colleagues (2006). The strategy used to make the deletions and the constructs used (Table S2 and Experimental procedures in Supporting information) are summarized in Fig. 1A.
Figure 1.
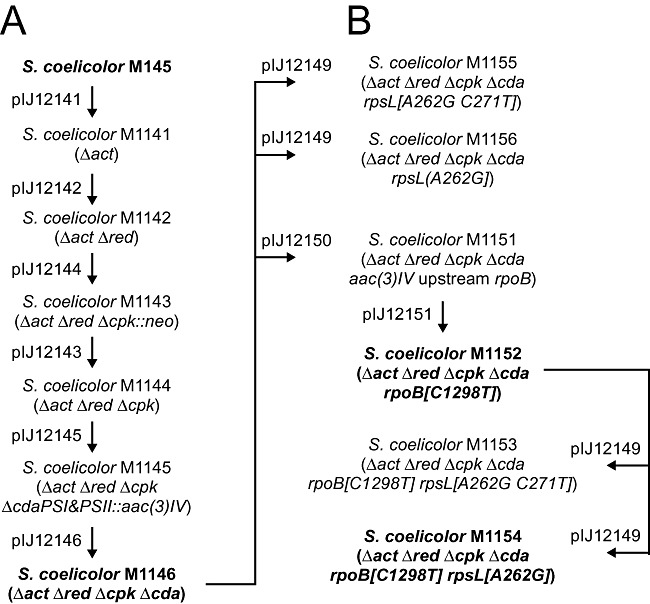
Pedigree of mutant strains. Steps involved in deleting the four endogenous secondary metabolite gene clusters (A) and in the introduction of point mutations in rpsL and rpoB (B). Constructs used in each step are indicated.
We first deleted most of the actinorhodin gene cluster [16 985 bp deletion from 5 515 934 bp to 5 532 918 bp of the genome sequence (Bentley et al., 2002)], followed by most of the prodiginine gene cluster (27 262 bp deletion from 6 434 879 bp to 6 462 140 bp); double cross‐over deletion mutants were obtained by screening for loss of blue and red pigment production, respectively. We then deleted the cpk gene cluster in two steps: we replaced most of the cluster with the kanamycin resistance gene neo, and then removed the neo marker using the same PCR‐amplified flanking DNA fragments (50 193 bp deletion from 6 896 195 bp to 6 946 388 bp). Finally, we deleted the cda gene cluster, again in two steps. First, we replaced cdaPSI and most of cdaPSII (31 355 bp deletion from 3 543 337 bp to 3 574 691 bp) by PCR‐targeting of cosmid StE63 with the apramycin resistance gene (aac(3)IV) of pIJ773 amplified with primers shown in Table S1. We then removed the rest of the cda cluster (from 3 521 521 bp to 3 600 278 bp, deleting a total of 78 758 bp) and aac(3)IV by homologous recombination with 2 kb PCR‐amplified fragments from each end of the cda cluster. Removal of the four secondary metabolite gene clusters resulted in the deletion of 173 kb of genome sequence and yielded the quadruple deletion strain M1146.
Introduction of mutations enhancing secondary metabolite production
The production of secondary metabolites in streptomycetes is regulated at both the transcriptional and translational levels (Bibb, 2005). We next introduced point mutations in rpoB and rpsL, genes that encode the RNA polymerase β‐subunit and ribosomal protein S12 respectively. Each mutation had been shown to enhance levels of antibiotic production in Streptomyces without growth impairment (Shima et al., 1996; Okamoto‐Hosoya et al., 2000; Hu et al., 2002). We introduced rpoB[S433L] (conferring resistance to rifampicin), and in addition either rpsL[K88E] or rpsL[K88E] and rpsL[P91S] (conferring streptomycin and paromomycin resistance, respectively). Site‐directed mutagenesis was performed with a subcloned fragment of rpoB or rpsL and oligo‐nucleotide pairs given in Table S1. The mutant alleles were incorporated into a suicide plasmid and substituted for the corresponding wild‐type genes by homologous recombination (Table S2 and Experimental procedures in Supporting information). For mutation of rpoB, we first introduced aac(3)IV upstream of rpoB to allow facile identification of subsequent double cross‐overs, and hence allele replacement, by loss of apramycin resistance. For mutation of rpsL, we identified double cross‐overs, and hence allele replacement, by selecting for exconjugants capable of growing on 5 µg ml−1 of streptomycin, thus simultaneously introducing both rpsL[K88E] and rpsL[P91S]. We also isolated spontaneous streptomycin‐resistant clones that contained rpsL[K88E]. Construction of the desired mutants was confirmed by PCR‐amplification and sequencing of the rpoB and rpsL mutant alleles. The genotypes of the constructed strains are given in Fig. 1B and Table S3.
Verification of strains
The construction of each strain was verified using three different criteria. Sequencing: Each 2 kb flanking fragment used to delete the four gene clusters by homologous recombination was sequenced to confirm that the desired fragment had been correctly PCR‐amplified. The mutagenized fragments used for gene replacement were also sequenced to ensure that only the desired point mutations had been introduced. Once double cross‐over recombinants were obtained, total DNA was isolated from selected clones, the targeted regions amplified by PCR and sequenced to confirm the presence of the required rpoB and/or rpsL mutations. Southern blotting and hybridization: The occurrence of the required recombination event during each deletion or allele replacement was confirmed by Southern analysis using the flanking DNA fragments used for homologous recombination as probes. Genomotyping: Lack of additional, unwanted deletions and amplifications was confirmed by comparing the genomes of S. coelicolor M1146 and S. coelicolor M1154 with that of the parental strain S. coelicolor M145 by genomotyping using Affymetrix microarrays (Fig. S1A and B; see Experimental procedures in Supporting information for a detailed explanation).
Growth and sporulation
Deletion mutants M1141–M1146 grew and sporulated as well as the parental strain M145 (Figs 2, 3 and S2A). Introduction of rpoB[S433L] (to yield M1152) delayed sporulation on agar media and spore pigmentation failed to reach that of M145 and M1146 (Fig. S2A). Addition of rpsL[K88E] (M1154) delayed sporulation further, and introduction of rpsL[P91S] to yield the triple point mutant M1153 impaired both growth and differentiation even more (Fig. 2). M1152 and M1154 grew in a less dispersed manner than M145 and M1146 in liquid culture, but both strains reached biomass yields similar to those of the parental strains when inoculated from seed cultures (Fig. 3C) but not when germinated spores were used as inoculum (Fig. 3A). Antimicrobial activity detected in supernatants from M145 grown in GYM liquid medium and presumed to be actinorhodin was not detected in M1154 (Fig. S2B).
Figure 2.

Actinorhodin production by strains containing a reintroduced act gene cluster. Plates are shown from on top after 3 days (scanned, A) and 7 days (with trans‐illumination, B). Results specifically referred to in the text are highlighted in coloured boxes. ‘1A’ and ‘LabM’ indicates the use of agar from two different suppliers (Kieser et al., 2000).
Figure 3.
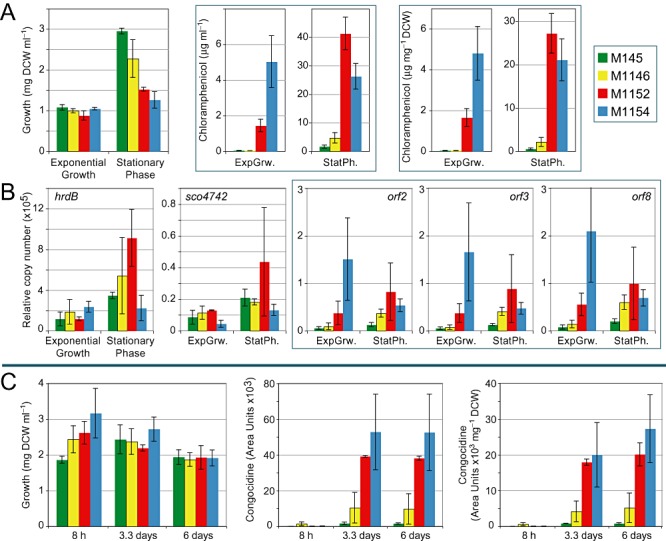
Heterologous expression of gene clusters. Production of chloramphenicol (A), qRT‐PCR analysis of chloramphenicol gene expression (B) and congocidine production (C). Strains were grown in liquid GYM medium inoculated with pre‐germinated spores (chloramphenicol gene cluster) or with mycelium from a seed culture (congocidine gene cluster). Average values and standard deviations are given in Table S4. Bars indicate average values of three biological replicates and error bars represent standard deviation (see Experimental procedures for inocula used and a discussion of growth differences).
Strain validation: increased actinorhodin production
To assess their potential utility, we first analysed actinorhodin production after integration of pAH88, a BAC derivative of SCBAC28G1 containing the actinorhodin gene cluster, at the φBT1 attachment site of each strain. Actinorhodin production was assessed using several agar media (Fig. 2). M1146 derivatives with the rpoB mutation (M1152) or with, additionally, one or both of the rpsL mutations (M1154 and M1153, respectively) gave higher levels of actinorhodin production than M145 and M1146 exconjugants on several agar media (Fig. 2), most notably those on which the M145 and M1141–M1146 derivatives produced little of the blue‐pigmented antibiotic (e.g. SFM, DNA, SMS). Similar results were obtained with two liquid media, where actinorhodin production by M1152 and M1154 containing the reintroduced act gene cluster was 1.5 to 9 times higher than that of M145.
Expression of heterologous gene clusters
To assess the performance of the strains when used for heterologous expression, we introduced two gene clusters representing different classes of antibiotics at the φC31 att site of strains M145, M1146, M1152 and M1154: the chloramphenicol gene cluster from Streptomyces venezuelae ATCC 10712, and the congocidine gene cluster from Streptomyces ambofaciens ATCC23877 (Juguet et al., 2009). Strains were grown in liquid GYM medium inoculated with germinated spores (chloramphenicol gene cluster) or with mycelium from a seed culture (congocidine gene cluster) (see Experimental procedures for detailed methods and a discussion of growth differences). GYM medium was also used by Ochi and coworkers during their original studies of rpoB and rpsL mutants of S. coelicolor and Streptomyces lividans (Shima et al., 1996; Okamoto‐Hosoya et al., 2000; Hu et al., 2002). In all cases, the highest levels of antibiotic production were obtained with M1152 and M1154, which produced four to ten times more antibiotic than M1146, which in turn produced three to five times more than M145. Overall, depending on the heterologous gene cluster, M1152 and M1154 produced 20–40 times more antibiotic than the parental strain M145 (Fig. 3). Interestingly, whereas detection of chloramphenicol production in M1152 and M1154 proved relatively facile, and could be accomplished with simple bioassays, production by S. venezuelae was possible only by using a more sensitive HPLC protocol, and even then on an inconsistent basis (A. Hesketh, pers. comm.).
Higher transcript levels are responsible for increased antibiotic production
Levels of transcription of three of the chloramphenicol biosynthetic genes (open reading frames 2, 3 and 8 of He et al., 2001) were determined in each of the four strains by quantitative RT‐PCR. Samples taken for RNA extraction were also used to estimate growth and antibiotic production. Two genes were used as internal controls: hrdB, encoding the major vegetative sigma factor of S. coelicolor, and SCO4742, both of which are expressed at relatively constant levels throughout growth (Hesketh et al., 2007). All three chloramphenicol biosynthetic genes showed similar temporal patterns of expression (Fig. 3B) that correlated well with levels of chloramphenicol production. M1154 gave the highest level of production and mRNA abundance during exponential growth, while in stationary phase M1152 gave slightly higher chloramphenicol production and also higher mRNA abundance. Thus the enhanced level of chloramphenicol reflects, at least in part, increased levels of biosynthetic gene transcripts.
A simplified HPLC chromatogram for easier compound identification
The HPLC chromatogram of a culture supernatant from S. coelicolor M145 contains a large number of peaks and a high baseline. Removal of the four endogenous gene clusters (to yield M1146) resulted in a much simpler chromatogram with fewer peaks, and a lower and more stable baseline (Fig. 4). Consequently, when combined with liquid chromatography and mass spectrometry, these engineered strains are likely to markedly facilitate the discovery of new compounds from heterologously expressed gene clusters. Indeed, their utility was demonstrated by the relatively facile identification of peaks corresponding to the congocidine gene cluster. This would have been much more difficult with M145. The high level of congocidine production obtained with M1154 also allowed facile fractionation and MS/MS analysis to confirm the identity of the heterologously produced compound (Figs 4 and S3).
Figure 4.
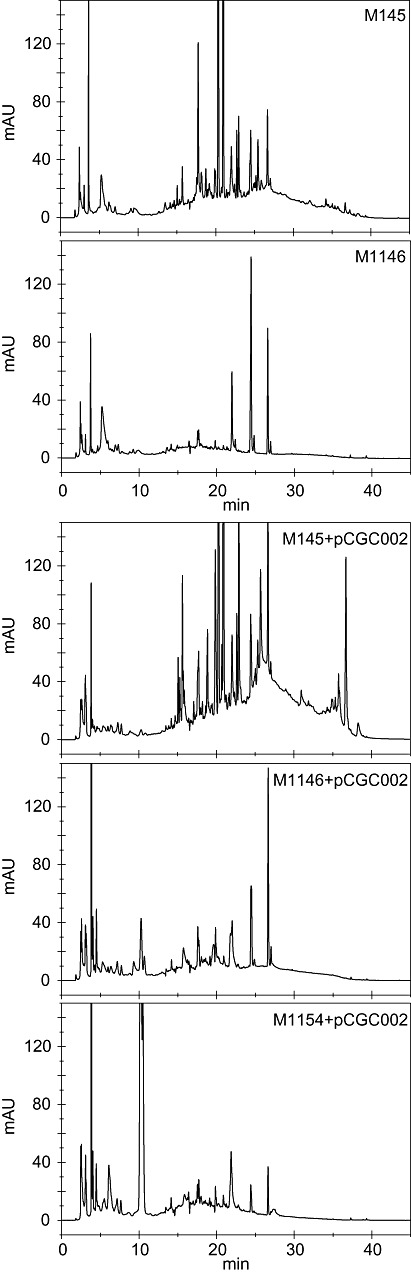
Comparative metabolic profiling. HPLC chromatograms of supernatants of M145 and the quadruply deleted mutant M1146, and of the same strains containing the congocidine gene cluster (indicated as ‘+pCGC002’) after growth in liquid GYM medium. Note the simplified chromatogram of M1146 compared with M145, and the higher levels of congocidine production in the mutant strains, especially M1154. Fractions that eluted at around 10 min were collected from the M1154 + pCGC002 sample and analysed by MS‐MS to verify the production of congocidine (Fig. S3).
Discussion
We have described the construction of S. coelicolor strains that have been specifically engineered for enhanced levels of secondary metabolite production. We have demonstrated their utility by heterologously expressing two actinomycete gene clusters for structurally distinct antibiotics, but they may also prove useful for gene clusters from other high G + C bacteria. Removal of four of the endogenous secondary metabolite gene clusters from S. coelicolor M145 provided consistently higher levels of secondary metabolite production, demonstrated by the increased productivity of M1146 versus M145 when expressing the chloramphenicol and congocidine gene clusters (Fig. 3). This presumably reflects increased availability of precursors for biosynthesis. While similar deletions in Streptomyces avermitilis also resulted in increased levels of production from heterologous gene clusters, no attempt was made to further improve the strain as an expression host (Komatsu et al., 2010).
Introduction of point mutations in rpoB and rpsL into the quadruply deleted mutant increased productivity of chloramphenicol and congocidine 40‐ and 30‐fold, respectively, compared with M145 (Fig. 3). Such a dramatic effect on production could make a crucial difference when attempting to detect a potentially novel compound or activity by heterologous gene expression. Although the underlying molecular mechanism is not understood, these mutations have a pleiotropic effect on levels of secondary metabolite production that is achieved, at least in part, by elevated levels of transcript abundance (Fig. 3). Furthermore, the increase in actinorhodin production conferred by these mutations was observed using a variety of different growth media (Fig. 2) and there is no reason to believe that this will not be true for other secondary metabolites. This may potentially reduce the amount of medium optimization required to yield sufficient quantities of material for analysis of biological activity and/or chemical characterization. Moreover, increases in chloramphenicol and congocidine production were obtained using two different types of inoculum, germinated spores and mycelium.
In contrast to previous studies (e.g. Wang et al., 2008), we introduced the rpoB mutation before introducing mutations into rpsL, and our results suggest that much of the increase in antibiotic production previously attributed to the accumulative effect of both sets of mutations, particularly those in rpsL, can be obtained by introducing the rpoB mutation alone. Depending on the growth medium, further addition of an rpsL mutation can incrementally increase production (e.g. chloramphenicol in exponential growth phase, Fig. 3A; actinorhodin in MM(labM) medium, Fig. 2). M1155 and M1153, containing rpsL[K88E, P91S], grew and sporulated poorly and gave lower levels of actinorhodin production than M1152 and M1154, and are not considered suitable hosts for heterologous gene expression. While M1154 (rpoB[S433L], rpsL[K88E]) often gave the highest levels of antibiotic production (Fig. 3), M1152 (rpoB[S433L]) sporulates better (at least in our laboratory), thus facilitating strain preservation and manipulation, while yielding only slightly lower production levels. Consequently, we recommend that both M1152 and M1154 be considered for use when attempting the heterologous expression of cloned gene clusters.
The production of novel compounds by heterologous expression of secondary metabolic genes clusters of diverse origin by genome mining or library screening is likely to play a significant role in future drug discovery. However, expression of foreign gene clusters is often associated with low levels of production. The application of these specifically engineered strains, with their simplified metabolic profiles, is likely to play an important role not only in the de novo discovery of novel compounds, but also in the production of new chemical structures by combinatorial biosynthesis, which is also often characterized by low levels of production.
Experimental procedures
Strains and growth conditions
The strains used and generated during this study are listed in Table S3. Escherichia coli strains were grown and manipulated following standard methods (Sambrook et al., 1989; Gust et al., 2003; 2004). Streptomyces coelicolor strains were grown in SFM, DNA, R5, R2, MM, SMS (Kieser et al., 2000), GYM (Shima et al., 1996) and MYM (Stuttard, 1982). Liquid cultures were performed in 50 ml of medium in 250 ml flasks. Streptomyces cultures were grown at 30°C in siliconized flasks containing stainless steel springs with shaking at 250 r.p.m.
Nucleic acid manipulations
These were performed using standard methods (Sambrook et al., 1989) or instructions provided by the manufacturers of restriction enzymes and kits. Southern blotting and hybridization were performed with a DIG Filter Hybridization Kit (Roche, Penzberg, Germany). RNA was isolated with a RNA Easy kit (Qiagen, Crawley, UK) from mycelium treated with RNA Protect Bacteria Reagent (Qiagen) following published procedures (Hesketh et al., 2007). Quantitative RT‐PCR was performed with Invitrogen SYBR GreenER 2‐step qRT‐PCR Universal Kit using a Chromo4 thermocycler (Bio‐Rad, Hercules, CA, USA) and Opticon 2 Monitor software (MJ Research, Waltham, MA, USA); serial dilutions of genomic DNA from S. coelicolor M145 and from S. venezuelae were used as controls and to derive a standard curve for copy number determinations.
Strain construction and confirmation
See Experimental procedures in Supporting information.
Assay of antimicrobial activity
Micrococcus luteus ATCC4698 was grown overnight in Luria‐Bertani (LB) medium and 1.5 ml of the culture were added to 150 ml of LB‐agar medium at about 45°C and poured into 24 × 24 cm plates; 100 µl of culture supernatant was added to wells made in the solidified agar; plates were incubated at 4°C for 2 h and then at 30°C for 24–48 h, after which the diameters of inhibitory halos were measured.
Actinorhodin production
BAC pAH88 (A. Hesketh, pers. comm.), which contains the entire actinorhodin gene cluster from S. coelicolor A3(2), was conjugated into S. coelicolor M145, M1146, M1152 and M1154 following the standard protocol (Gust et al., 2003; 2004). Spore stocks were prepared from three different exconjugants for each strain. For the 25‐well plate assays (Fig. 1) 3 ml of the stated medium was pipetted into each well; 20 µl of a spore stock dilution containing 106 spores was pipetted onto the agar and allowed to dry; plates were incubated at 30°C for the indicated time; the assay was repeated twice with two different exconjugants for each strain.
Chloramphenicol production
Cosmid pAH91 (A. Hesketh, pers. comm.), which contains the entire chloramphenicol gene cluster from S. venezuelae, was conjugated into S. coelicolor M145, M1146, M1152 and M1154 following the standard protocol (Gust et al., 2003; 2004). Spore stocks were prepared from three different exconjugants for each strain. 108 spores of each clone (12 in total, three from each strain) were germinated in 10 ml of 2xYT for 7 h at 30°C, harvested by centrifugation, re‐suspended in approximately 10 ml of GYM medium, dispersed by sonication in a water bath for 1 min, and inoculated into 250 ml flasks containing a final volume of 50 ml of GYM medium (Kieser et al., 2000). Because the germination and growth of M1152 and M1154 were delayed compared with M145 and M1146, exponential phase samples were harvested at 16.5 h for M145 and M1146, and at 41 h for M1152 and M1154; stationary phase samples were harvested at 4 days for all of the strains. Samples were taken and processed as follows: 10 ml of culture was harvested in 15 ml tubes by centrifugation; the supernatant was divided into 2 ml aliquots and frozen at −80°C; the mycelial pellet was re‐suspended in water and filtered through pre‐weighed 0.45 µm cellulose acetate filters for dry cell weight estimation. Two millilitre of culture was harvested for RNA extraction. Chloramphenicol production was estimated by HPLC: culture supernatants were filtered through VectaSpin Micro Polysulphone 0.2 µm columns (Whatman, Maidstone, UK), injected onto a Spherisorb 5 µm ODS2 4.6 × 250 mm C18 column (Waters, Milford, MA, USA) fitted to an Agilent 1100 HPLC system with Diode Array detector and analysed using a method modified from He and colleagues (2001) (A. Hesketh, pers. comm.): gradient water : methanol; min 0, 0% methanol; min 2, 25% methanol; min 12, 50% methanol, min 14, 100% methanol; min 20, 100% methanol; min 22, 0% methanol. Chloramphenicol eluted at about 15.8 min, and was detected at 273 nm. Chloramphenicol (Sigma, catalogue number C0378) was used as a standard.
Congocidine production
Cosmid pCGC002, which contains the entire congocidine gene cluster from S. ambofaciens ATCC23877 (Juguet et al., 2009), was conjugated into S. coelicolor M145, M1146, M1152 and M1154 following the standard protocol (Gust et al., 2003; 2004). One exconjugant from each strain was selected. Because M1152 and M1154 and their derivatives sporulated less well than M1146 and its derivatives, we used mycelial inocula to determine congocidine production. This also allowed us to assess the performance of the strains using two different types of inoculum. Homogenized frozen mycelial stocks were prepared from exponentially growing TSB : SuperYEME 50:50 liquid cultures and stored in 20% glycerol. Seed cultures for each strain were inoculated into TSB : SuperYEME 50:50 and grown for 21 h. Growth was estimated by determining the OD at 600 nm, and the inoculum required to obtain 50 ml of culture medium at 0.25 OD at 600 nm was calculated and the corresponding mycelium harvested by centrifugation, re‐suspended in fresh GYM medium and added to 250 ml flasks containing 50 ml of GYM. Because both M1152 and M1154 grow in a less dispersed fashion than their parental strains, monitoring the growth of their seed cultures by absorbance may have resulted in an underestimate of biomass, and might account for the more abundant growth of M1152 and M1154 compared with M145 and M1146 after 8 h of culture (Fig. 3C). Samples were harvested and processed as above for chloramphenicol. Congocidine production was estimated by HPLC using the method of Juguet and colleagues (2009): 0.1% formic acid in water(A):0.1% formic acid in acetonitrile(B); 0 min, 5% B; 7 min, 5% B; 30 min, 60% B; 35 min 100% B; 45 min, 100% B; 50 min, 5% B. Congocidine eluted at about 10.5 min and was detected at 297 nm. Confirmation of the identity of congocidine was achieved by mass spectrometry and by comparison with data reported in Juguet and colleagues (2009). Fractions eluting at 10.5 min were collected and analysed using a Surveyor HPLC equipped with a DecaXPplus ion trap mass spectrometer (Thermo, Waltham, MA, USA) by loop injection without chromatography, using a flow of 80 µl min−1 of 50% methanol in water. MS data were collected by electrospray ionization, in positive mode, with 50 units sheath gas, 5 units aux gas, 350°C capillary temperature and 5.2 kV spray voltage. Full MS and data‐dependent MS2 and MS3 data were collected at 35% collision energy and an isolation width of 3.0 amu.
Acknowledgments
We thank Andy Hesketh for providing the actinorhodin and chloramphenicol gene clusters and details thereof, and for advice on genomotyping and qRT‐PCR, Maureen Bibb for advice on genomotyping, Lionel Hill for mass spectrometric analysis of congocidine, and Jean‐Luc Pernodet and Silvie Lautru for providing the congocidine gene cluster and the HPLC protocol for congocidine. This work was supported by grants from the EU (Integrated Project ActinoGen contract No. 005224) and UK BBSRC to MB.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Genomotyping analysis. (A) Scatter plot of normalized data from the quadruple deletion mutant (QDM) M1146 (Y‐axis) versus M145 (X‐axis). (B) Scatter plot of normalized data from M1154 (Y‐axis) versus M145 (X‐axis). In green, positive controls (fragments used for homologous hybridization); in red, negative controls (deleted genes); in grey, rest of the genes; diagonal lines from top to bottom, two‐fold increase in intensity, 1:1 ratio and two‐fold decrease in intensity. (C) Southern blot of digested DNA from M145 and M1146 hybridized with a probe derived from some of the genes deleted from the cda gene cluster that gave higher than expected intensities upon genomotyping. The expected fragments are detected only in M145 and not in M1146, while several cross‐hybridizing fragments are observed in both strains.
Fig. S2. Growth and differentiation. (A) Growth and sporulation on SFM agar medium. Plates were scanned from above (top row) and below (bottom row; the image was inverted horizontally for easier interpretation) at the indicated times. Note the delayed growth and reduction in spore pigmentation in M1152 and M1154. (B) Antimicrobial activity detected in M145 but not in M1154 GYM culture supernatants.
Fig. S3. Identification of congocidine. MS/MS analysis of a fraction of the large peak eluting at 10.5 min in the chromatogram from M1154 containing the congocidine gene cluster (Fig. 4). 431 m z−1 corresponds to congocidine + 1 hydrogen ion ; 453 m z−1 corresponds to the sodium adduct; 216 m z−1 corresponds to doubly charged congocidine + 2 hydrogen ions; See Juguet and colleagues (2009) for further explanation of the MS/MS data and fractionation spectra.
Table S1. Oligonucleotides used in this study.
Table S2. Vectors and constructs used in this study.
Table S3. Strains used in this study.
Table S4. Data for Fig. 3 for chloramphenicol (CP) (Fig. 3A) and congocidine (CGC) (Fig. 3B) production. Strains were grown in liquid GYM medium inoculated with pre‐germinated spores (chloramphenicol gene cluster) or with mycelium from a seed culture (congocidine gene cluster).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Baltz R.H. Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol. 2008;8:557–563. doi: 10.1016/j.coph.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Bentley S.D., Chater K.F., Cerdeño‐Tárraga A.‐M., Challis G.L., Thomson N.R., James K.D. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. et al. [DOI] [PubMed] [Google Scholar]
- Bibb M.J. Regulation of secondary metabolism in streptomycetes. Curr Opin Microbiol. 2005;8:208–215. doi: 10.1016/j.mib.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Boucher H.W., Talbot G.H., Bradley J.S., Edwards J.E., Gilbert D., Rice L.B. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. et al. [DOI] [PubMed] [Google Scholar]
- Challis G.L., Hopwood D.A. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci USA. 2003;100:14555–14561. doi: 10.1073/pnas.1934677100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenical W., Jensen P.R. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat Chem Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- Gust B., Challis G.L., Fowler K., Kieser T., Chater K.F. PCR‐targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci USA. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust B., Chandra G., Jakimowicz D., Yuqing T., Bruton C.J., Chater K.F. Lambda red‐mediated genetic manipulation of antibiotic‐producing Streptomyces. Adv Appl Microbiol. 2004;54:107–128. doi: 10.1016/S0065-2164(04)54004-2. [DOI] [PubMed] [Google Scholar]
- He J., Magarvey N., Piraee M., Vining L.C. The gene cluster for chloramphenicol biosynthesis in Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular non‐ribosomal peptide synthetase gene. Microbiology. 2001;147:2817–2829. doi: 10.1099/00221287-147-10-2817. [DOI] [PubMed] [Google Scholar]
- Hesketh A., Chen W.J., Ryding J., Chang S., Bibb M. The global role of ppGpp synthesis in morphological differentiation and antibiotic production in Streptomyces coelicolor A3(2) Genome Biol. 2007;8:R161. doi: 10.1186/gb-2007-8-8-r161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H., Zhang Q., Ochi K. Activation of antibiotic biosynthesis by specified mutations in the rpoB gene (encoding the RNA polymerase beta subunit) of Streptomyces lividans. J Bacteriol. 2002;184:3984–3991. doi: 10.1128/JB.184.14.3984-3991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juguet M., Lautru S., Francou F.‐X., Nezbedová S., Leblond P., Gondry M., Pernodet J.‐L. An iterative nonribosomal peptide synthetase assembles the pyrrole‐amide antibiotic congocidine in Streptomyces ambofaciens. Chem Biol. 2009;16:421–431. doi: 10.1016/j.chembiol.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Kieser T., Bibb M.J., Buttner M.J., Chater K.F., Hopwood D.A. John Innes Foundation; 2000. [Google Scholar]
- Komatsu M., Uchiyama T., Omura S., Cane D.E., Ikeda H. Genome‐minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad Sci USA. 2010;107:2646–2651. doi: 10.1073/pnas.0914833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto‐Hosoya Y., Sato T.A., Ochi K. Resistance to paromomycin is conferred by rpsL mutations, accompanied by an enhanced antibiotic production in Streptomyces coelicolor A3(2) J Antibiot. 2000;53:1424–1427. doi: 10.7164/antibiotics.53.1424. [DOI] [PubMed] [Google Scholar]
- Pawlik K., Kotowska M., Chater K., Kuczek K., Takano E. A cryptic type I polyketide synthase (cpk) gene cluster in Streptomyces coelicolor A3(2) Arch Microbiol. 2006;187:87–99. doi: 10.1007/s00203-006-0176-7. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shima J., Hesketh A., Okamoto S., Kawamoto S., Ochi K. Induction of actinorhodin production by rpsL (encoding ribosomal protein S12) mutations that confer streptomycin resistance in Streptomyces lividans and Streptomyces coelicolor A3(2) J Bacteriol. 1996;178:7276–7284. doi: 10.1128/jb.178.24.7276-7284.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuttard C. Temperate phages of Streptomyces venezuelae: lysogeny and host specificity shown by phages SV1 and SV2. J Gen Microbiol. 1982;128:115–121. [Google Scholar]
- Wang G., Hosaka T., Ochi K. Dramatic activation of antibiotic production in Streptomyces coelicolor by cumulative drug resistance mutations. Appl Environ Microbiol. 2008;74:2834–2840. doi: 10.1128/AEM.02800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel S.C., Müller R. The impact of genomics on the exploitation of the myxobacterial secondary metabolome. Nat Prod Rep. 2009;26:1385–1407. doi: 10.1039/b817073h. [DOI] [PubMed] [Google Scholar]
- Zerikly M., Challis G.L. Strategies for the discovery of new natural products by genome mining. Chembiochem. 2009;10:625–633. doi: 10.1002/cbic.200800389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Genomotyping analysis. (A) Scatter plot of normalized data from the quadruple deletion mutant (QDM) M1146 (Y‐axis) versus M145 (X‐axis). (B) Scatter plot of normalized data from M1154 (Y‐axis) versus M145 (X‐axis). In green, positive controls (fragments used for homologous hybridization); in red, negative controls (deleted genes); in grey, rest of the genes; diagonal lines from top to bottom, two‐fold increase in intensity, 1:1 ratio and two‐fold decrease in intensity. (C) Southern blot of digested DNA from M145 and M1146 hybridized with a probe derived from some of the genes deleted from the cda gene cluster that gave higher than expected intensities upon genomotyping. The expected fragments are detected only in M145 and not in M1146, while several cross‐hybridizing fragments are observed in both strains.
Fig. S2. Growth and differentiation. (A) Growth and sporulation on SFM agar medium. Plates were scanned from above (top row) and below (bottom row; the image was inverted horizontally for easier interpretation) at the indicated times. Note the delayed growth and reduction in spore pigmentation in M1152 and M1154. (B) Antimicrobial activity detected in M145 but not in M1154 GYM culture supernatants.
Fig. S3. Identification of congocidine. MS/MS analysis of a fraction of the large peak eluting at 10.5 min in the chromatogram from M1154 containing the congocidine gene cluster (Fig. 4). 431 m z−1 corresponds to congocidine + 1 hydrogen ion ; 453 m z−1 corresponds to the sodium adduct; 216 m z−1 corresponds to doubly charged congocidine + 2 hydrogen ions; See Juguet and colleagues (2009) for further explanation of the MS/MS data and fractionation spectra.
Table S1. Oligonucleotides used in this study.
Table S2. Vectors and constructs used in this study.
Table S3. Strains used in this study.
Table S4. Data for Fig. 3 for chloramphenicol (CP) (Fig. 3A) and congocidine (CGC) (Fig. 3B) production. Strains were grown in liquid GYM medium inoculated with pre‐germinated spores (chloramphenicol gene cluster) or with mycelium from a seed culture (congocidine gene cluster).