

Summary
Bacteriophage endolysins are peptidoglycan hydrolases employed by the virus to lyse the host at the end of its multiplication phase. They have found many uses in biotechnology; not only as antimicrobials, but also for the development of novel diagnostic tools for rapid detection of pathogenic bacteria. These enzymes generally show a modular organization, consisting of N‐terminal enzymatically active domains (EADs) and C‐terminal cell wall‐binding domains (CBDs) which specifically target the enzymes to their substrate in the bacterial cell envelope. In this work, we used individual functional modules of Listeria phage endolysins to create fusion proteins with novel and optimized properties for labelling and lysis of Listeria cells. Chimaeras consisting of individual EAD and CBD modules from PlyPSA and Ply118 endolysins with different binding specificity and catalytic activity showed swapped properties. EAD118–CBDPSA fusion proteins exhibited up to threefold higher lytic activity than the parental endolysins. Recombineering different CBD domains targeting various Listeria cell surfaces into novel heterologous tandem proteins provided them with extended recognition and binding properties, as demonstrated by fluorescent GFP‐tagged CBD fusions. It was also possible to combine the binding specificities of different single CBDs in heterologous tandem CBD constructs such as CBD500‐P35 and CBDP35‐500, which were then able to recognize the majority of Listeria strains. Duplication of CBD500 increased the equilibrium cell wall binding affinity by approximately 50‐fold, and the enzyme featuring tandem CBD modules showed increased activity at higher ionic strength. Our results demonstrate that modular engineering of endolysins is a powerful approach for the rational design and optimization of desired functional properties of these proteins.
Introduction
In recent years, peptidoglycan‐degrading enzymes and bacteriophage endolysins in particular have received increasing attention as antimicrobial agents. In view of emerging and spreading resistance of pathogenic bacteria against classical antibiotics, there is an increasing demand for alternative antimicrobials. Especially in case of Gram‐positive bacteria, the application of phage endolysins is a promising approach (Loessner et al., 1995a; Nelson et al., 2001; Fischetti, 2005; Loessner, 2005; Loessner and Rees, 2005; Borysowski et al., 2006; Hermoso et al., 2007; Fischetti, 2010). Due to the absence of an outer membrane in Gram‐positives, these enzymes also work when added from without, i.e. they can cause rapid lysis of susceptible cells. Numerous potential applications of lysins in food safety, agriculture, and medicine have been reported. Aiming towards the control of the opportunistic food‐borne pathogen Listeria monocytogenes, genes encoding Listeria‐specific phage endolysins were introduced into starter organisms used in cheese production; and secretion of the active enzymes from the bacteria provided them with lytic activity against Listeria (Gaeng et al., 2000; Turner et al., 2006). Other reports include the detection and killing of Bacillus anthracis (Schuch et al., 2002), Staphylococcus aureus in milk (Obeso et al., 2008; Garcia et al., 2010) and MRSA in vitro (Becker et al., 2008), and the control of Streptococcus pneumoniae in different models (Loeffler et al., 2001; Nelson et al., 2001; Jado et al., 2003; Loeffler and Fischetti, 2003; Cheng et al., 2005).
Like all endolysins from a Gram‐positive background, Listeria phage endolysins show a modular organization in which catalytic activity and substrate recognition are separated: they consist of two functional domains, an N‐terminal enzymatically active domain (EAD) and a C‐terminal cell wall‐binding domain (CBD) (Loessner et al., 1995a; Loessner et al., 2002). The EAD determines the enzymatic mechanism of the endolysin. The CBD of an endolysin binds to a certain ligand within the bacterial cell wall, thereby bringing the EAD in proximity to its substrate. In general, cell wall lytic enzymes can assume at least four different enzymatic activities (Young and Blasi, 1995; Loessner, 2005). Among the Listeria phage endolysins, two classes exist: N‐acetylmuramoyl‐l‐alanine amidases cleave between the sugar and the peptide moieties, and the unique l‐alanoyl‐d‐glutamate peptidases target the bond between l‐alanine and d‐glutamate in the peptide chain (Loessner et al., 1995a).
The CBD confers high specificity and binding affinity to the endolysin. Listeria phage CBDs bind independent of species boundaries, but recognize certain carbohydrate components associated with the murein which also determine the serovar groups within the genus of Listeria; they show very little or no cross‐reactions with other bacteria (Loessner et al., 2002; Schmelcher et al., 2010). These attractive properties have been harnessed for development of specific diagnostics tools for Listeria (Loessner et al., 2002); even multiplex differentiation of strains by fluorescent reporter–CBD fusion proteins is possible (Schmelcher et al., 2010). In addition, Listeria phage CBDs have been used for affinity‐based magnetic separation (CBD‐MS) of target cells from food matrices, and this method was demonstrated to be superior to standard detection protocols (Kretzer et al., 2007; Schmelcher et al., 2010; Walcher et al., 2010).
Until now, the PlyPSA amidase from L. monocytogenes phage PSA (Zimmer et al., 2003) and the Ply500 peptidase domain from phage A500 (Loessner et al., 1995a) are the only Listeria endolysins for which structural data exist (Korndoerfer et al., 2006; 2008). The PlyPSA crystal structure revealed two distinct domains connected by a short, but highly flexible linker region (Korndoerfer et al., 2006). Sequence alignments showed the presence of similar linkers in other Listeria phage endolysins, and revealed strong sequence homologies among the enzymes (Loessner et al., 2002; Schmelcher et al., 2010). This may include entire domains, such as in PlyPSA, whose CBD is highly similar to that of Ply500, whereas the EADs are unrelated. The EAD of Ply500, however, is nearly identical to that of Ply006 (Schmelcher et al., 2010). This suggests frequent exchange of these functional modules by horizontal gene transfer, enabling a highly variable mix‐and‐match of domain combinations within the phage population. It was therefore reasonable to assume that this variability could be further extended by artificial recombination of functional domains, thereby creating chimeric proteins with desired properties. In this study, we have created chimeric proteins by defined fusions of EADs and CBDs from various Listeria phage endolysins, using GFP as a fluorescent reporter. We were able to modulate lytic properties of endolysins by exchange of EADs and CBDs, to extend the binding range of reporter–CBD fusion proteins by combination of CBDs with different specificities, and created proteins with duplicated CBDs, which represents a novel and unique strategy to significantly increase binding affinity to bacterial cell surfaces.
Results
Combination of EADs and CBDs of different origin generates proteins with swapped specificity and enhanced lytic activity
The N‐acetylmuramoyl‐l‐alanine amidase PlyPSA features a CBD specifically targeting cell walls of Listeria strains belonging to serovars 4, 5 or 6 (Korndoerfer et al., 2006). In contrast, the CBD of the l‐alanoyl‐d‐glutamate peptidase Ply118 (Loessner et al., 1995a) only recognizes strains of serovars 1/2, 3 and ‘7’ (Loessner et al., 2002; see Discussion below). Based on the crystal structure of PlyPSA (Korndoerfer et al., 2006) and the in silico identified putative linker region between EAD and CBD of Ply118 (Schmelcher et al., 2010), a series of fusion proteins was created featuring CBDPSA including its linker fused to the C‐termini of putative EAD118 fragments in three different length variants. In a complementary approach, the EAD of PlyPSA including the linker was fused to the N‐termini of six different CBD118 fragments (Fig. 1A). The four longer variants (CBD118 D, C, B, A1), which include the putative linker region of Ply118 and portions of different length of the C‐terminal end of the EAD118, had been shown to be sufficient for directing the green fluorescent protein in GFP–CBD fusion proteins to the cell surface of susceptible strains (Loessner et al., 2002). The two shorter variants, A2 and A3, only comprise the CBD and parts of the putative linker.
Figure 1.
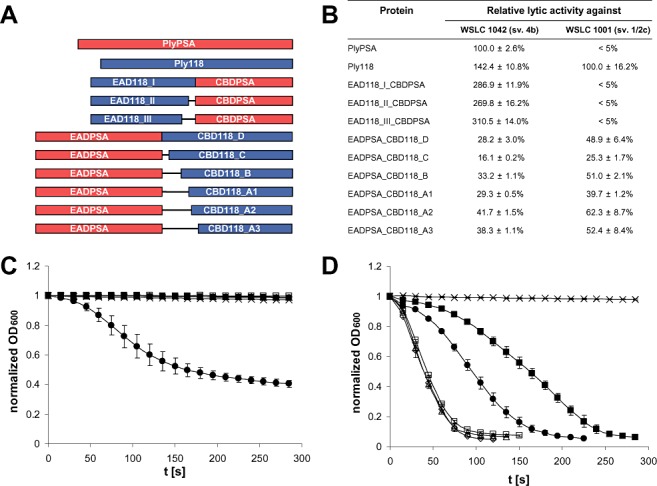
Lytic activities of native endolysins and engineered fusion proteins against Listeria cells. Schematic representations of the different constructs tested here are shown in (A). The table in (B) lists relative lytic activities (mean values and standard deviations) determined from lysis curves as shown for serovar 1/2c strain WSLC 1001 (C) and serovar 4b strain WSLC 1042 (D). PlyPSA and Ply118 served as references. The changes in optical density over a period of 300 s after addition of EAD118_I_CBDPSA (□), EAD118_II_CBDPSA (◊), EAD118_III_CBDPSA (Δ), PlyPSA ( ) and Ply118 (●) to cell suspensions are shown. Buffer was added as control (x). Curves were normalized and recalculated to an initial OD at 600 nm of 1.0.
) and Ply118 (●) to cell suspensions are shown. Buffer was added as control (x). Curves were normalized and recalculated to an initial OD at 600 nm of 1.0.
Following overexpression and purification, lytic activities of all chimeric enzymes were determined by photometric lysis assays using L. monocytogenes WSLC 1001 (serovar 1/2c) and WSLC 1042 (serovar 4b) cells in suspension. The parental endolysins Ply118 (Loessner et al., 1996) and PlyPSA (Korndoerfer et al., 2006) (Fig. 1B) were used as controls. As expected with respect to the specificity of CBDPSA for WSLC 1042, the full‐length endolysin PlyPSA exhibited strong lytic activity against this strain, whereas it was not able to lyse WSLC 1001. Surprisingly, Ply118 did not only work against WSLC 1001 which its CBD recognizes, but was also able to very efficiently lyse cells of WSLC 1042 (142.4% activity compared with PlyPSA; please see Discussion).
The lysis of cells of both serovar groups by the chimeras containing EAD118 and CBDPSA as well as those of the native endolysins are shown in Fig. 1C and D. None of the EAD118_CBDPSA fusions was able to lyse strain WSLC 1001, while all of them displayed strong activity against WSLC 1042. For all three length variants, lytic activity against the serovar 4b strain was considerably higher than that of both parental endolysins (up to threefold compared with the reference protein PlyPSA) (Fig. 1B). These results demonstrate that functional chimeric proteins with swapped lytic specificities can be generated by exchange of the CBD of a Listeria phage endolysin. By replacing the native CBD in Ply118 by that of PlyPSA, the lytic activity of this enzyme against the serovar 1/2 strain was abolished, whereas that against the serovar 4 strain was enhanced.
All EADPSA_CBD118 fusion proteins constructed in this work exhibited lytic activity against both WSLC 1001 and WSLC 1042, which was, however, generally lower than the activities of the parental endolysins (Fig. 1B). The exchange of the native CBD of PlyPSA, which is not able to lyse WSLC 1001, by that of Ply118 rendered the enzyme active against the serovar 1/2 strain. Activity against the serovar 4b strain was not completely abolished, but considerably reduced by this substitution. The effect of different length variants of single modules on the lytic activity of similar fusion proteins is apparent, especially among the different EADPSA_CBD118 constructs. The EADPSA_CBD118_A2 enzyme displayed the highest lytic activity, with 41.7% compared with PlyPSA against WSLC 1042 and 62.3% compared with Ply118 against WSLC 1001.
Combination of CBDs of different specificity extends binding range
Fusion proteins consisting of an N‐terminal GFP domain and a C‐terminal CBD from any Listeria phage endolysin can be used to fluorescently decorate bacterial cell walls, which enables rapid qualitative determination of CBD association to various bacteria (Loessner et al., 2002; Schmelcher et al., 2010). Based on the complementary specificities of CBD118 and CBD500 (Loessner et al., 2002), we aimed to create fusion proteins with an extended binding range by combination of both CBDs in one heterodimer. CBD500 exhibits binding properties highly similar to those of CBDPSA; it evenly decorates the entire surface of Listeria cells of serovars 4, 5 and 6, but features stronger affinity and higher binding density than the CBDPSA (Schmelcher et al., 2010). CBD118 is specific for serovar 1/2 and 3 cells (Loessner et al., 2002), where it primarily targets polar and septal regions of the rod‐shaped murein sacculus.
Schematic representations of double CBD constructs created in this work are shown in Fig. 2. The results of binding assays using a large set of Listeria strains comprising the six major species and all serovars with all constructs described here and single CBDs as reference are summarized in Table 1. The GFP_CBD500‐118 and GFP_CBD118‐500 proteins, in which both CBDs are directly fused to each other in both orientations and are tagged by an N‐terminal GFP, revealed binding to serovar 4, 5 and 6 cells, albeit with a somewhat weaker intensity. This demonstrated proper function of CBD500 in both N‐terminal and C‐terminal position. However, because this corresponded to the binding pattern of CBD500 only, it also indicated that CBD118 is non‐functional in these conformations.
Figure 2.

Schematic representations of all GFP and CBD fusions created and tested in this work, as well as the reference constructs GFP_CBD500 and GFP_CBD118 (Loessner et al., 2002). GFP = green fluorescent protein; CBD500, CBD118, CBDP35 = cell wall‐binding domains of Listeria phage endolysins Ply500, Ply118 and PlyP35 respectively; L = linker region of PlyPSA.
Table 1.
Binding of various GFP‐tagged CBD constructs from Listeria phage endolysins to Listeria cells from different species and serovars.
| Species | Strain (WSLC) | Source | SV | Binding of GFP_CBD_ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 500a | 118a | P35b | 500‐118 | 118‐500 | 500L118 | 118L500 | 500‐GFP‐118 | 118‐GFP‐500 | 500‐P35 | P35‐500 | ||||
| L. monocytogenes | EGDe | J. Kreft | 1/2a | − | ++ | ++ | − | − | + | (+) | (+) | − | ++ | ++ |
| L. monocytogenes | 10403S | D. Portnoy | 1/2a | − | ++ | ++ | − | − | − | − | (+) | − | ++ | + |
| L. monocytogenes | 1442c | Food | 1/2a | − | ++ | − | − | − | + | + | (+) | + | − | − |
| L. monocytogenes | 1066 | SLCC 8800 | 1/2b | − | ++ | ++ | − | − | − | − | (+) | − | ++ | ++ |
| L. monocytogenes | 1001 | ATCC 19112 | 1/2c | − | ++ | ++ | − | − | − | − | (+) | − | ++ | ++ |
| L. seeligeri | 4007 | ATCC 35967 | 1/2b | − | ++ | ++ | − | − | + | + | (+) | − | ++ | ++ |
| L. welshimeri | 50149 | SLCC 5877 | 1/2b | − | ++ | + | − | − | + | + | (+) | − | (+) | (+) |
| L. monocytogenes | 1485 | Soft cheese | 3a | − | + | + | − | − | − | − | − | − | ++ | ++ |
| L. monocytogenes | 1031 | SLCC1694 | 3b | − | + | ++ | − | − | − | − | − | − | ++ | ++ |
| L. monocytogenes | 1032 | SLCC 2479 | 3c | − | + | ++ | − | − | − | − | − | − | ++ | ++ |
| L. seeligeri | 40127 | SLCC 8604 | 3b | − | + | ++ | − | − | − | − | − | − | ++ | ++ |
| L. monocytogenes | 1034c | SLCC 2482 | ‘7’ | − | + | − | − | − | − | − | − | − | − | − |
| L. monocytogenes | 1020 | ATCC 19114 | 4a | ++ | − | ++ | + | + | + | ++ | + | ++ | ++ | ++ |
| L. monocytogenes | 1042 | ATCC 23074 | 4b | ++ | − | − | + | + | + | ++ | + | ++ | ++ | ++ |
| L. monocytogenes | ScottA | J. Jay | 4b | ++ | − | − | + | + | + | ++ | + | ++ | ++ | ++ |
| L. monocytogenes | 1019 | ATCC 19116 | 4c | ++ | − | ++ | + | + | + | ++ | + | ++ | ++ | ++ |
| L. monocytogenes | 1033 | ATCC 19117 | 4d | ++ | − | + | + | + | + | ++ | + | ++ | ++ | ++ |
| L. monocytogenes | 1018 | ATCC 19118 | 4e | ++ | − | (+) | + | + | + | ++ | + | ++ | ++ | ++ |
| L. ivanovii | 3009 | SLCC 4769 | 5 | ++ | − | ++ | + | + | + | ++ | + | ++ | ++ | ++ |
| L. ivanovii (ssp. ivanovii) | 3010 | ATCC 19119 | 5 | ++ | − | ++ | + | + | + | ++ | + | ++ | ++ | ++ |
| L. ivanovii (ssp. londoniensis) | 3060 | SLCC 3765 | 5 | ++ | − | − | + | + | + | + | (+) | + | + | + |
| L. innocua | 2011 | ATCC 33090 | 6a | ++ | − | − | + | + | + | + | (+) | + | ++ | ++ |
| L. innocua | 2012 | ATCC 33091 | 6b | ++ | − | ++ | + | + | + | ++ | + | ++ | ++ | ++ |
| L. welshimeri | 50146 | SLCC 7622 | 6a | ++ | − | + | + | + | + | ++ | + | ++ | ++ | ++ |
| L. grayi (ssp. grayi) | 6036 | ATCC 19120 | − | (+) | (+) | ++ | − | − | − | − | − | − | + | ++ |
| L. grayi (ssp. murrayi) | 6037 | ATCC 25401 | − | (+) | (+) | ++ | − | − | − | − | − | − | ++ | ++ |
Strains 1442 and 1034 are mutants and lack N‐acetyl glucosamine in their wall teichoic acids.
++, strong; +, weak; (+), very weak; −, no binding.
Assuming that enhanced flexibility of both binding domains in a fusion construct might render CBD118 functional, GFP_CBD500L118 and GFP_CBD118L500 were created, which employ the linker peptide of PlyPSA (Korndoerfer et al., 2006) for separating the CBDs. Again, both constructs nicely decorated serovar 4, 5 and 6 cells, similar to single GFP_CBD500 (Fig. 3D–F). Moreover, it also labelled four out of seven of the serovar 1/2 strains tested. The latter were predominantly marked at the poles and septa (Fig. 3B and C), as observed for single GFP_CBD118 (Fig. 3A) (Loessner et al., 2002). Obviously, these heterologous dual CBD fusions combined the recognition and binding properties from both CBDs, even though binding among the serovars 1/2 and 3 strains seems to be more selective than that of GFP_CBD118 alone. The dimeric CBD fusion GFP_CBD118L500 displayed binding to serovar 4, 5 and 6 strains almost indistinguishable from monomeric GFP_CBD500. In conclusion, introduction of the native linker peptide from PlyPSA not only enabled CBD118 to better access its ligands, but it also permitted full functionality of CBD500 in the C‐terminal location.
Figure 3.
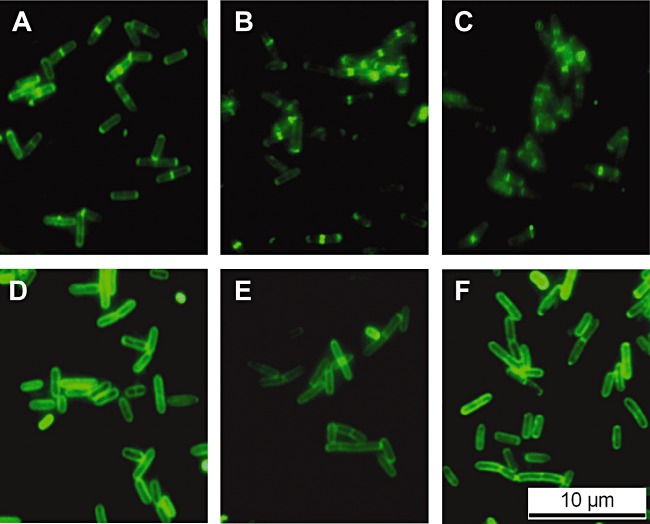
Decoration of Listeria monocytogenes cells by fluorescently tagged phage endolysin CBDs. Listeria cells of strains WSLC 1442 (A–C) and WSLC 1042 (D–F) were labelled using GFP_CBD118 (A) and GFP_CBD500 (D) (controls), and dual CBD fusions GFP_CBD500L118 (B, E) and GFP_CBD118L500 (C, F). Images were taken using epifluorescent microscopy.
We also created fusions featuring GFP in a central position acting as a spacer, with CBD500 and CBD118 located at the N‐ or C‐termini. In CBD500_GFP_CBD118, where CBD118 is located similarly to the situation in the native enzyme or in monomeric GFP_CBD118, it recognized all serovar 1/2 strains, even though the decoration was weak. In CBD118_GFP_CBD500, the orientation of the CBDs is reversed, and only weak labelling of one serovar 1/2 strain was observed. As a conclusion, CBD118 function is highly dependent on domain location, while CBD500 functionality seemed to be less influenced by these constraints.
To further optimize recognition of and binding to Listeria serovar 1/2 cells by recombineered CBDs, CBD118 was replaced by CBDP35 in another series of heterodimeric dual CBD proteins. The CBDP35 polypeptide had recently been shown to strongly bind to strains of serovars 1/2, 3, and even some strains of serovars 4, 5 and 6, in a CBD500‐like fashion, i.e. over the entire cell surface (Schmelcher et al., 2010) (see Table 1). In fact, labelling of cells by recombinant GFP_CBD500‐P35 and GFP_CBDP35‐500 nicely combined the properties of the single CBD500 and CBDP35. Both constructs showed strong binding to all strains which are recognized by CBD500 or by CBDP35 or both. This was regardless of the spatial location of the single CBDs within the fusions, indicating that both CBDs are fully functional within the chimeric proteins.
Tandem duplication of CBD500 strongly increases binding affinity
We questioned whether the duplication of a CBD would result in higher binding affinity to the cell wall, and constructed GFP_CBD500‐500, which features tandem CBD500 domains fused to an N‐terminal GFP (Fig. 4A). Kinetic data of the interaction of both GFP_CBD500 and GFP_CBD500‐500 with the cell wall of L. monocytogenes WSLC 1042 cells were obtained by surface plasmon resonance (SPR) analysis. Real‐time kinetic data were measured by applying GFP_CBD500 or GFP_CBD500‐500 onto immobilized bacterial cells. The equilibrium association constants obtained for three different concentrations of each protein reflect the affinities to the cell wall (Table 2). GFP_CBD500‐500 was shown to bind to the Listeria cells with approximately 50‐fold higher affinity compared with GFP_CBD500, with equilibrium constants in the pico‐molar range. When overlaying sensorgrams of both the single and the double CBD protein (Fig. 4B), it became obvious that the proteins mainly differ in the dissociation phase. In other words: when bound to the cell surface, GFP_CBD500 detaches easier than GFP_CBD500‐500, resulting in the higher overall affinity of the double CBD construct.
Figure 4.
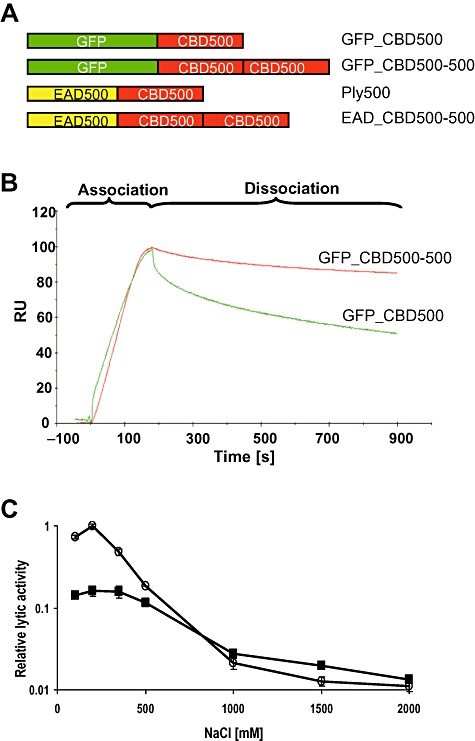
Effect of CBD500 duplication on binding properties and lytic activity. A. Schematic representation of tandem CBD500 fusion proteins and the single CBD500 reference proteins. GFP = green fluorescent protein; EAD500 = enzymatically active domain (l‐alanoyl‐d‐glutamate peptidase) of Listeria phage endolysin Ply500; CBD500 = cell wall‐binding domain of Ply500. B. Overlay of SPR sensorgrams showing association and dissociation phases of GFP_CBD500 and GFP_CBD500‐500, respectively, to the cell walls of L. monocytogenes WSLC 1042, measured at a protein concentration of 50 nM. RU = relative response units. C. Relative lytic activity of Ply500 (○) and EAD_CBD500‐500 ( ) on WSLC 1042 cells at different NaCl concentrations. All assays were carried out in triplicate.
) on WSLC 1042 cells at different NaCl concentrations. All assays were carried out in triplicate.
Table 2.
Equilibrium affinity constants (KA) of GFP_CBD500 and GFP_CBD500‐500 binding to the cell wall of Listeria monocytogenes serovar 4b strain WSLC 1042.
| GFP_CBD500 | GFP_CBD500‐500 | ||
|---|---|---|---|
| Concentration (nM) | KA (M−1) | Concentration (nM) | KA (M−1) |
| 200 | 5.61 × 108 | 50 | 1.00 × 1010 |
| 100 | 6.50 × 108 | 25 | 5.61 × 1010 |
| 50 | 5.96 × 108 | 12.5 | 2.19 × 1010 |
| Mean | 6.02 × 108 | Mean | 2.93 × 1010 |
Final values are arithmetic means calculated from three different analyte concentrations tested.
Increased affinity improves endolysin activity at high ionic strength
It was suggested that ionic interaction by way of charge plays a major role as the molecular basis for association of CBDs to their cell wall ligands. Monomeric CBD500 shows optimum binding at 100–150 mM NaCl, which decreases with increasing salt concentration, i.e. ionic strength (Loessner et al., 2002). Therefore, we reasoned that an endolysin featuring two identical binding domains such as a duplicated CBD500 should exhibit stronger activity than native Ply500 at high salt concentrations, due to higher affinity of the double CBD domain. In order to test this, EAD_CBD500‐500 (see Fig. 4A) was made and its lytic activity determined against L. monocytogenes WSLC 1042 cells at different salt concentrations. As shown in Fig. 4C, lytic activity of the tandem CBD construct was approximately sixfold lower than that of Ply500 at the standard NaCl concentration. However, with increasing ionic strength, the differences in activity between both enzymes became smaller and both curves intersected between 500 and 1000 mM. At even higher NaCl concentrations, EAD_CBD500‐500 exhibited higher lytic activity than Ply500 (approximately twofold at 1500 M), demonstrating that the enhanced affinity also supports binding and thereby enzyme activity at higher ionic strength.
Discussion
The available data suggest that many proteins encoded by bacteriophage genomes such as murein hydrolases share a common ancestry with related enzymes from the bacterial hosts, and have co‐evolved by recombination‐driven interchange of functional domains (Hendrix, 2002; Hermoso et al., 2007). This characteristic modular organization, in which the specific functions of substrate recognition and catalysis are physically separated and independent, appears beneficial for enzymes degrading polymeric insoluble substrates such as peptidoglycan (Wren, 1991; Croux et al., 1993a). These domains represent stable globular polypeptides that can fold autonomously (Rashin, 1981), and can be recombined to create novel functions (Khosla and Harbury, 2001). Strong evidence for a modular mix‐and‐match in Listeria phages endolysins was provided by a recent comprehensive study on these endolysins (Schmelcher et al., 2010).
We here demonstrate that the natural variability found within the Listeria endolysins can be further extended by artificial design and recombination of functional modules, generating novel proteins with optimized properties. Fusions of EADs and CBDs from endolysins PlyPSA and Ply118 provided proof of concept for interchangeability of domains within these enzymes. Switching the CBDs of two endolysins resulted in a swap of their binding and lysis specificity. Similar observations have been reported for autolysins from Streptococcus and Clostridium, and pneumococcal and lactococcal phage endolysins (Diaz et al., 1990; Croux et al., 1993a,b; Sheehan et al., 1996). Recently, Becker and co‐workers reported that streptococcal lysins became active against S. aureus cells following exchange of their CBDs against Staphylococcus‐specific binding modules (Becker et al., 2009).
With respect to cell lysis in suspension, Ply118 was found to efficiently lyse cells of WLSC 1042, whereas its isolated CBD118 alone does not recognize and bind to these cells (Loessner et al., 2002). This suggest that in suspension of cells and lytic enzyme, where the enzyme is added at a large excess, the CBD is not required for lytic activity and presence of a high enough concentration of EAD may be sufficient to cause lysis from without. In its natural context, i.e. when the endolysin is produced inside the bacterial host cell at the end of the phage multiplication cycle, enzyme concentration is much lower, and high‐affinity targeting might be necessary to achieve timely and efficient cell lysis. Moreover, after disruption of the host cell it is important that the liberated endolysins remain attached to cell wall debris of the host, thereby preventing collateral damage to neighbouring host cells which have not yet produced phage (Loessner et al., 2002). Cell lysis by an ‘isolated’ catalytic domain has also been reported for LytC, an autolysin of S. pneumoniae (Garcia et al., 1999), as well as PlyPSA, whose EAD could still lyse Listeria cells, albeit activity was reduced to only 15% (Korndoerfer et al., 2006). The extent of such non‐specific lysis presumably depends on the susceptibility of the cells and the nature of the EAD. In this context, it was interesting to note that EAD118_CBDPSA, which combines lytic activity of EAD118 (serovar 1/2 background) with specificity of CBDPSA (serovar 4 background), was found to have higher activity on serovar 4 strains than any of the parental endolysins. However, in contrast to the serovar 4 strain WSLC 1042, the serovar 1/2 strain WSLC 1001 was only lysed by enzymes containing a corresponding CBD118 module, which indicates that this cell wall type requires a certain binding affinity of the enzyme.
CBD500 retained its functionality and binding properties regardless of its N‐ or C‐terminal location within a heterologous construct. The majority of bacteriophage endolysins and bacterial peptidoglycan hydrolases from Gram‐positive organisms feature at least one catalytic domain at the N‐terminus, and a CBD at the C‐terminus. However, inverse orientations have also been reported (Garcia et al., 1999). On the other hand, the various constructs made during the course of this work also disclose some limitations of ‘artificial’ domain engineering. CBD118 never exhibited full functionality as in native Ply118 or GFP_CBD118 in any of the chimaeras; modifications resulted in either a narrower binding range, weaker binding or complete loss of function. The most likely explanation is improper overall conformation of the recombinant constructs, whose folding and stability depend on domain interactions and the nature of linker regions which influences the orientation of the proteins relative to their ligands in the cell wall. Accessibility of the ligands recognized by CBD118 might play a role, and this could be limited by sterical considerations. This possibility is supported by our findings that inclusion of a suitable linker to provide more flexibility between the domains also enhanced the recognition function of CBD118. We previously showed that CBDs from Listeria phage endolysins fused to fluorescent proteins can serve as tools for specific detection of Listeria cells, making it possible to even simultaneously detect and distinguish different strains (Loessner et al., 2002; Schmelcher et al., 2010), and can be used for efficient immobilization and capture of bacteria when coated onto paramagnetic beads (Kretzer et al., 2007). The findings presented here demonstrate that modular engineering of CBDs can provide a tailor‐made toolbox ideally suited for such purposes requiring high specificity and high‐affinity proteins. Both CBD500 and CBDP35 exhibit affinity constants in the nanomolar range and show little cross‐reactions with other bacteria than Listeria (Loessner et al., 2002; Schmelcher et al., 2010). Since the GFP_CBD500‐P35 and GFP_CBDP35‐500 fusions combine the binding ranges of both CBDs, they represent ideal reagents for CBD‐based magnetic separation assays (CBD‐MS) (Kretzer et al., 2007). They recognize the vast majority of all Listeria strains, except for those few strains (WSLC 1442, WSLC 1034) lacking the N‐acetylglucosamine substitution at the C4 position of their poly‐ribitol phosphate teichoic acids, which are specifically recognized and bound by CPBP35. However, these strains are extremely rare, and actually feature mutations in the serovar 1/2‐type teichoic acid glycosylation pathway (M. Eugster and M.J. Loessner, submitted).
Several murein hydrolases feature specific motifs or repeats in their cell wall‐recognizing domains (Garcia et al., 1990; 1999; Joris et al., 1992), and it was suggested that the number of such motifs and repeats may determine the binding affinity (Yother and White, 1994). Our data regarding the tandem repeat in CBD500‐500 demonstrate that this is also applicable to the domain level, as the duplication of CBD500 resulted in a drastic increase in equilibrium affinity, brought about by lowering the dissociation rate (kd), whereas the association (ka) was similar to that of the monomeric polypeptide (Loessner et al., 2002) (Fig. 4B). An elegant explanation is that the detachment of the single CBD in GFP_CBD500 would always result in displacement of the protein. In contrast, if one of the two CBDs of GFP_CBD500‐500 detaches, the protein remains bound with the second CBD, providing it with ample time to re‐attach to its ligand.
Presence of a CBD is required for maximum lytic activity of a murein hydrolase (Sanz et al., 1992; Korndoerfer et al., 2006). By attaching to a ligand in the cell wall, the local enzyme concentration is elevated and the EAD is positioned in proximity to its substrate. Other authors reported that absence or presence of a putative CBD may not influence lytic activity (Cheng and Fischetti, 2006). We found that duplication of CBD in Ply500, which increases binding affinity of the protein, reduces lytic activity against intact cells in suspension, suggesting that stronger binding does not necessarily correlate with increasing cell lysis. Apparently, a lytic enzyme should retain a certain degree of surface mobility for optimal lytic activity, as suggested for Cpl‐1 and modular bacterial cellulases (Jervis et al., 1997; Hermoso et al., 2003). However, under specific environmental conditions, stronger binding may also support lysis, as shown by the fact that at elevated ionic strength, EAD_CBD500‐500 works better than the non‐modified enzyme. Thus, engineered enzymes could be useful for lysis of target bacteria in specific environments such as foods with elevated salt content, e.g. cheese surfaces, cured fish and meat products.
Our work shows that modular engineering of bacteriophage endolysins is helpful to provide specifically tailored, optimized tools for the detection and lysis of bacterial pathogens. A recent report of an endolysin fused to a membrane‐disrupting peptide demonstrated that Gram‐negative organisms can also be targeted by these enzymes (Briers et al., 2010). Considering that the Caudovirales represent the most abundant self‐replicating units on earth, the toolbox of enzymatically active and cell wall‐binding modules seems virtually unlimited.
Experimental procedures
Bacteria, culture conditions, plasmids and phages
Escherichia coli XL1‐Blue MRF' (Stratagene) served for cloning and overexpression of 6xHis‐tagged phage endolysins and the various constructs made throughout this work, and it was cultured in LB (Luria–Bertani) medium at 30°C with 100 µg ml−1 ampicillin and 30 µg ml−1 tetracycline for plasmid selection. Photometric lysis assays of cells in suspension were performed with L. monocytogenes WSLC 1001 (serovar 1/2c) and WSLC 1042 (serovar 4b). Twenty‐six Listeria strains comprising the six major species and all serovars (Table 1) were used for the CBD binding assays. Listeria were cultured in TB (tryptose broth) medium at 30°C.
The plasmids pHPLPSA (Korndoerfer et al., 2006), pHPL118 and pHPL500 (Loessner et al., 1996), and genomic DNA of phage P35 (Dorscht et al., 2009) were used as templates for amplification of EAD and/or CBD coding regions of endolysins PlyPSA, Ply118, Ply500 and PlyP35 respectively. The plasmid pHGFP (Loessner et al., 2002) served as template for the amplification of the gfp‐coding sequence. Plasmid pQE‐30 (Qiagen) and its derivatives pHGFP, pHGFP_CBD118, pHGFP_CBD500 (Loessner et al., 2002), and pHEADPSA (Korndoerfer et al., 2006), were used as backbones for the construction of vectors encoding N‐terminally 6xHis‐tagged fusion proteins (see Table S2).
DNA techniques and cloning procedures
Standard DNA manipulation and cloning procedures (Sambrook et al., 1989) were used for construction of plasmids encoding endolysin‐based fusion proteins. Enzymes were purchased from New England Biolabs, MBI Fermentas, Qiagen and Roche, and used according to manufacturer instructions. DNA fragments coding for functional domains of Listeria phage endolysins, as well as the GFP‐coding fragment were amplified from templates and with primers listed in Table S1 (Supporting information), using Proof Start DNA Polymerase (Qiagen) or High Fidelity PCR Enzyme Mix (MBI Fermentas). Restriction sites needed for insertion of the fragments into the plasmids were introduced via primer design. Concentrations of DNA fragments and plasmids were determined spectrophotometrically (NanoDrop ND‐1000).
All plasmids used and created in this work are listed in Table S2 (Supporting information). Plasmid pHCBDPSA was constructed by inserting a DNA fragment coding for the CBD of PlyPSA, including the flexible linker region between EAD and CBD, into SacI/SalI sites of pQE‐30. The position of the linker was derived from the resolved crystal structure of PlyPSA (Korndoerfer et al., 2006). In order to create a series of EAD118–CBDPSA chimeric proteins, three EAD118‐coding fragments of different length were cloned into BamHI/SacI sites of pHCBDPSA, upstream of the CBDPSA fragment, creating genetic fusions. In analogy, five different CBD118 fragments were inserted into SacI/SalI sites of pHEADPSA (Korndoerfer et al., 2006), yielding a series of EADPSA‐CBD118 constructs. Similar strategies were employed for creation of GFP‐double CBD constructs. CBD118 and CBD500‐coding fragments were joined via compatible EcoRI/MunI sites in both orientations, and then inserted into SacI/SalI sites of pHGFP, yielding pHGFP_CBD118‐500 and pHGFP_CBD500‐118. The plasmids pHGFP_CBDP35‐500 and pHGFP_CBD500‐P35 were created the same way. In case of pHGFP_CBD118L500 and pHGFP_CBD500L118, the fragment coding for the PlyPSA linker was introduced between the two CBDs by PCR‐based Gene Splicing by Overlap Extension PCR (SOE PCR) (Horton et al., 1990) before insertion into pHGFP. As for pHCBD500_GFP_118 and pHCBD118_GFP_500, the 5′ CBD and the GFP fragments were first joined via KpnI sites or by SOE PCR, and then ligated into BamHI/SacI sites of pHGFP_CBD118 and pHGFP_CBD500, respectively, replacing the gfp sequence in these plasmids. For construction of pHGFP_CBD500‐500, the CBD500 fragment was cloned into the SacI site of pHGFP_CBD500, resulting in a duplication of CBD500. pHEAD_CBD500‐500 was created by inserting the complete ply500 gene into BamHI/SacI sites of pHGFP_CBD500, replacing gfp.
The translational stop TAA was generally introduced as stop codon at the 3′ ends of the artificial coding sequences. All constructs were verified by nucleotide sequencing.
Synthesis and purification of His‐tagged protein constructs
Overproduction of His‐tagged fusion proteins in E. coli XL1‐Blue MRF' was carried out as described before (Loessner et al., 2002). The respective strains were grown in modified LB medium (15 g l−1 tryptose, 8 g l−1 yeast extract, 5 g l−1 NaCl) containing 100 µg ml−1 ampicillin and 30 µg ml−1 tetracycline for plasmid selection at 30°C, with 1 mM IPTG added as inducer once an OD600 of 0.4 was reached. After further incubation at 30°C for 4 h, cultures producing proteins that contain a GFP domain were further stored overnight at 4°C before harvesting and resuspension in 5 ml of buffer A (500 mM NaCl, 50 mM Na2HPO4, 5 mM imidazole, 0.1% Tween 20, pH 8.0) per 250 ml of culture. If no GFP was present, cultures were centrifuged 4 h after induction. Bacterial cells were disrupted by two passages through a French Press cell (SLM Aminco) at 100 MPa, cell debris removed by centrifugation (10 000 g, 10 min) and the supernatants filter‐sterilized (0.2 µM PES membrane, Millipore).
Then, His‐tagged proteins in the extracts were purified by immobilized metal affinity chromatography (IMAC) with Ni‐NTA Superflow resin (Qiagen) using Micro Biospin columns (Bio‐Rad). Buffer B (500 mM NaCl, 50 mM Na2HPO4, 250 mM imidazole, 0.1% Tween 20, pH 8.0) served as elution buffer. Eluates were dialysed against two changes of Dialysis Buffer (100 mM NaCl, 50 mM NaH2PO4, 0.005–0.1% Tween 20, pH 8.0), again filtered (0.2 µM PES membrane, Millipore), and stored at −20°C after addition of 50% (v/v) of glycerol. For each protein, intracellular synthesis and purification were monitored by SDS‐PAGE, and protein concentrations were determined spectrophotometrically (NanoDrop ND‐1000).
Photometric lysis assays
The lytic activity of native His‐tagged endolysins and chimeric lytic proteins was determined by a photometric lysis assay as described before (Loessner et al., 1995b; 1996). Substrate cells of L. monocytogenes strains WSLC 1001 and WSLC 1042 were prepared by growing the bacteria in TB medium until late log phase, harvesting by centrifugation, resuspension in PBS (50 mM NaH2PO4, 120 mM NaCl, pH 8.0) at 50‐fold concentration and freezing at −80°C. Lysis assays were performed in a total volume of 1 ml, with cells diluted in PBS to an initial OD600 of approximately 1.0. All tested endolysins and chimeric enzymes were added to the cells in equimolar amounts (ranging from 30 to 152 pmol ml−1), and OD600 was measured at intervals of 15 s for total of 10 min. As negative control, PBS buffer was added to the cells. All assays were carried out in triplicate.
To examine the effect of NaCl concentration on lytic activity of Ply500 and EAD_CBD500‐500, assays were performed as described above, with final NaCl concentrations ranging from 100 mM to 2 M.
Data processing was carried out as described before (Korndoerfer et al., 2006). Lysis curves were normalized and corrected by subtraction of the controls [corrected value = value + (1 − control value)]. The resulting curves were fitted using the sigmoid function f = y0 + a/{1 + exp[−(x − x0)/b]}∧c, employing SigmaPlot 9.0 (Systat Software). The steepest slope of the function was used to calculate the relative lytic activity.
Cell wall binding assays and fluorescence microscopy
The binding properties of GFP–CBD fusion proteins were examined by binding assays as described before (Loessner et al., 2002; Schmelcher et al., 2010). In brief, late‐log‐phase cells in PBST buffer (50 mM NaH2PO4, 120 mM NaCl, pH 8.0, 0.01% Tween 20) were incubated with 10–20 ng of the GFP–CBD proteins for 5 min at room temperature. After washing twice with buffer, the cells were prepared for fluorescence microscopy, using an Axioplan microscope and an appropriate filter set (Carl Zeiss AG). Images of the fluorescently labelled cells were obtained by using a Leica DFC320 camera, and processed using appropriate software (Adobe Photoshop).
Determination of affinities by SPR analysis
Affinities of GFP_CBD500 and GFP_CBD500‐500 to the cell wall of L. monocytogenes WSLC 1042 were determined by SPR analysis, using a BIAcore X instrument and the flat surface C1 sensor chips (BIAcore, Uppsala, Sweden). Surface coating and monitoring of real‐time binding kinetics and interaction analysis were performed essentially as described (Schmelcher et al., 2010). In brief, the chip surface was activated with the amine coupling method and coated with GFP–CBD500 molecules in both flow cells (70 µl of 0.5 mg ml−1 protein in 10 mM sodium acetate buffer, pH 5, at a flow rate of 5 µl min−1). Heat‐inactivated WSLC 1042 cells in HBS buffer (10 mM Hepes, 150 mM NaCl, 3.4 mM EDTA, 0.005% Tween 20, pH 7.8) were then bound to the immobilized CBDs in flow cell Fc2 (3.0 × 1010 cells per ml; 15 µl at a flow rate of 3 µl min−1). Finally, interactions between the immobilized cells and three different concentrations of both GFP_CBD500 (50 nM, 100 nM, 200 nM) and GFP_CBD500‐500 (12.5 nM, 25 nM, 50 nM) in HBS buffer were measured (30 µl at 10 µl min−1). Fc1 served as reference cell. The association phase was measured for 3 min, the dissociation phase for 12 min. All steps were carried out at 25°C. Evaluation of kinetic data was performed with the BIAevaluation software, version 4.1 (BIAcore), employing a ‘1:1 binding with mass transfer’ model.
Acknowledgments
We thank Marcel Eugster and Julia Dorscht for sharing unpublished information, and are very grateful to Monique Herensperger and the late Ursula Schuler for their excellent technical assistance. This work was supported by the WWTF, Vienna, Austria.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Primers for PCR amplification and sequencing used in this study.
Table S2. Plasmids used in this study.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Becker S.C., Foster‐Frey J., Donovan D.M. The phage K lytic enzyme LysK and lysostaphin act synergistically to kill MRSA. FEMS Microbiol Lett. 2008;287:185–191. doi: 10.1111/j.1574-6968.2008.01308.x. [DOI] [PubMed] [Google Scholar]
- Becker S.C., Foster‐Frey J., Stodola A.J., Anacker D., Donovan D.M. Differentially conserved staphylococcal SH3b_5 cell wall binding domains confer increased staphylolytic and streptolytic activity to a streptococcal prophage endolysin domain. Gene. 2009;443:32–41. doi: 10.1016/j.gene.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Borysowski J., Weber‐Dabrowska B., Gorski A. Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med (Maywood) 2006;231:366–377. doi: 10.1177/153537020623100402. [DOI] [PubMed] [Google Scholar]
- Briers Y., Lavigne R., Volckaert G. 2010.
- Cheng Q., Fischetti V.A. Mutagenesis of a bacteriophage lytic enzyme PlyGBS significantly increases its antibacterial activity against group B streptococci. Appl Microbiol Biotechnol. 2006;74:1284–1291. doi: 10.1007/s00253-006-0771-1. [DOI] [PubMed] [Google Scholar]
- Cheng Q., Nelson D., Zhu S., Fischetti V.A. Removal of group B streptococci colonizing the vagina and oropharynx of mice with a bacteriophage lytic enzyme. Antimicrob Agents Chemother. 2005;49:111–117. doi: 10.1128/AAC.49.1.111-117.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croux C., Ronda C., Lopez R., Garcia J.L. Interchange of functional domains switches enzyme specificity: construction of a chimeric pneumococcal‐clostridial cell wall lytic enzyme. Mol Microbiol. 1993a;9:1019–1025. doi: 10.1111/j.1365-2958.1993.tb01231.x. [DOI] [PubMed] [Google Scholar]
- Croux C., Ronda C., Lopez R., Garcia J.L. Role of the C‐terminal domain of the lysozyme of Clostridium acetobutylicum ATCC 824 in a chimeric pneumococcal‐clostridial cell wall lytic enzyme. FEBS Lett. 1993b;336:111–114. doi: 10.1016/0014-5793(93)81621-6. [DOI] [PubMed] [Google Scholar]
- Diaz E., Lopez R., Garcia J.L. Chimeric phage‐bacterial enzymes: a clue to the modular evolution of genes. Proc Natl Acad Sci USA. 1990;87:8125–8129. doi: 10.1073/pnas.87.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorscht J., Klumpp J., Bielmann R., Schmelcher M., Born Y., Zimmer M. Comparative genome analysis of Listeria bacteriophages reveals extensive mosaicism, programmed translational frameshifting, and a novel prophage insertion site. J Bacteriol. 2009;191:7206–7215. doi: 10.1128/JB.01041-09. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischetti V.A. Bacteriophage lytic enzymes: novel anti‐infectives. Trends Microbiol. 2005;13:491–496. doi: 10.1016/j.tim.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Fischetti V.A. Bacteriophage endolysins: a novel anti‐infective to control Gram‐positive pathogens. Int J Med Microbiol. 2010;300:357–362. doi: 10.1016/j.ijmm.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeng S., Scherer S., Neve H., Loessner M.J. Gene cloning and expression and secretion of Listeria monocytogenes bacteriophage‐lytic enzymes in Lactococcus lactis. Appl Environ Microbiol. 2000;66:2951–2958. doi: 10.1128/aem.66.7.2951-2958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia P., Garcia J.L., Garcia E., Sanchez‐Puelles J.M., Lopez R. Modular organization of the lytic enzymes of Streptococcus pneumoniae and its bacteriophages. Gene. 1990;86:81–88. doi: 10.1016/0378-1119(90)90116-9. [DOI] [PubMed] [Google Scholar]
- Garcia P., Paz Gonzalez M., Garcia E., Garcia J.L., Lopez R. The molecular characterization of the first autolytic lysozyme of Streptococcus pneumoniae reveals evolutionary mobile domains. Mol Microbiol. 1999;33:128–138. doi: 10.1046/j.1365-2958.1999.01455.x. [DOI] [PubMed] [Google Scholar]
- Garcia P., Martinez B., Rodriguez L., Rodriguez A. Synergy between the phage endolysin LysH5 and nisin to kill Staphylococcus aureus in pasteurized milk. Int J Food Microbiol. 2010;141:151–155. doi: 10.1016/j.ijfoodmicro.2010.04.029. [DOI] [PubMed] [Google Scholar]
- Hendrix R.W. Bacteriophages: evolution of the majority. Theor Popul Biol. 2002;61:471–480. doi: 10.1006/tpbi.2002.1590. [DOI] [PubMed] [Google Scholar]
- Hermoso J.A., Monterroso B., Albert A., Galan B., Ahrazem O., Garcia P. Structural basis for selective recognition of pneumococcal cell wall by modular endolysin from phage Cp‐1. Structure. 2003;11:1239–1249. doi: 10.1016/j.str.2003.09.005. et al. [DOI] [PubMed] [Google Scholar]
- Hermoso J.A., Garcia J.L., Garcia P. Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Curr Opin Microbiol. 2007;10:461–472. doi: 10.1016/j.mib.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Horton R.M., Cai Z.L., Ho S.N., Pease L.R. Gene splicing by overlap extension: tailor‐made genes using the polymerase chain reaction. Biotechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- Jado I., Lopez R., Garcia E., Fenoll A., Casal J., Garcia P. Phage lytic enzymes as therapy for antibiotic‐resistant Streptococcus pneumoniae infection in a murine sepsis model. J Antimicrob Chemother. 2003;52:967–973. doi: 10.1093/jac/dkg485. [DOI] [PubMed] [Google Scholar]
- Jervis E.J., Haynes C.A., Kilburn D.G. Surface diffusion of cellulases and their isolated binding domains on cellulose. J Biol Chem. 1997;272:24016–24023. doi: 10.1074/jbc.272.38.24016. [DOI] [PubMed] [Google Scholar]
- Joris B., Englebert S., Chu C.P., Kariyama R., Daneo‐Moore L., Shockman G.D., Ghuysen J.M. Modular design of the Enterococcus hirae muramidase‐2 and Streptococcus faecalis autolysin. FEMS Microbiol Lett. 1992;70:257–264. doi: 10.1016/0378-1097(92)90707-u. [DOI] [PubMed] [Google Scholar]
- Khosla C., Harbury P.B. Modular enzymes. Nature. 2001;409:247–252. doi: 10.1038/35051723. [DOI] [PubMed] [Google Scholar]
- Korndoerfer I.P., Danzer J., Schmelcher M., Zimmer M., Skerra A., Loessner M.J. The crystal structure of the bacteriophage PSA endolysin reveals a unique fold responsible for specific recognition of Listeria cell walls. J Mol Biol. 2006;364:678–689. doi: 10.1016/j.jmb.2006.08.069. [DOI] [PubMed] [Google Scholar]
- Korndoerfer I.P., Kanitz A., Danzer J., Zimmer M., Loessner M.J., Skerra A. Structural analysis of the l‐alanoyl‐d‐glutamate endopeptidase domain of Listeria bacteriophage endolysin Ply500 reveals a new member of the LAS peptidase family. Acta Crystallogr D Biol Crystallogr. 2008;64:644–650. doi: 10.1107/S0907444908007890. [DOI] [PubMed] [Google Scholar]
- Kretzer J.W., Lehmann R., Schmelcher M., Banz M., Kim K.P., Korn C., Loessner M.J. High affinity cell wall‐binding domains of bacteriophage endolysins for immobilization and separation of bacterial cells. Appl Environ Microbiol. 2007;73:1992–2000. doi: 10.1128/AEM.02402-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler J.M., Fischetti V.A. Synergistic lethal effect of a combination of phage lytic enzymes with different activities on penicillin‐sensitive and ‐resistant Streptococcus pneumoniae strains. Antimicrob Agents Chemother. 2003;47:375–377. doi: 10.1128/AAC.47.1.375-377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler J.M., Nelson D., Fischetti V.A. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science. 2001;294:2170–2172. doi: 10.1126/science.1066869. [DOI] [PubMed] [Google Scholar]
- Loessner M.J. Bacteriophage endolysins – current state of research and applications. Curr Opin Microbiol. 2005;8:480–487. doi: 10.1016/j.mib.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Loessner M.J., Rees C.E.D. Listeria phages: basics and applications. In: Waldor M.K., Friedman D.I., Adhya S.L., editors. ASM Press; 2005. pp. 362–379. [Google Scholar]
- Loessner M.J., Wendlinger G., Scherer S. Heterogeneous endolysins in Listeria monocytogenes bacteriophages: a new class of enzymes and evidence for conserved holin genes within the siphoviral lysis cassettes. Mol Microbiol. 1995a;16:1231–1241. doi: 10.1111/j.1365-2958.1995.tb02345.x. [DOI] [PubMed] [Google Scholar]
- Loessner M.J., Schneider A., Scherer S. A new procedure for efficient recovery of DNA, RNA, and proteins from Listeria cells by rapid lysis with a recombinant bacteriophage endolysin. Appl Environ Microbiol. 1995b;61:1150–1152. doi: 10.1128/aem.61.3.1150-1152.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner M.J., Schneider A., Scherer S. Modified Listeria bacteriophage lysin genes (ply) allow efficient overexpression and one‐step purification of biochemically active fusion proteins. Appl Environ Microbiol. 1996;62:3057–3060. doi: 10.1128/aem.62.8.3057-3060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner M.J., Kramer K., Ebel F., Scherer S. C‐terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high‐affinity binding to bacterial cell wall carbohydrates. Mol Microbiol. 2002;44:335–349. doi: 10.1046/j.1365-2958.2002.02889.x. [DOI] [PubMed] [Google Scholar]
- Nelson D., Loomis L., Fischetti V.A. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc Natl Acad Sci USA. 2001;98:4107–4112. doi: 10.1073/pnas.061038398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso J.M., Martinez B., Rodriguez A., Garcia P. Lytic activity of the recombinant staphylococcal bacteriophage PhiH5 endolysin active against Staphylococcus aureus in milk. Int J Food Microbiol. 2008;128:212–218. doi: 10.1016/j.ijfoodmicro.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Rashin A.A. Location of domains in globular proteins. Nature. 1981;291:85–87. doi: 10.1038/291085a0. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Maniatis J., Fritsch E.F. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sanz J.M., Diaz E., Garcia J.L. Studies on the structure and function of the N‐terminal domain of the pneumococcal murein hydrolases. Mol Microbiol. 1992;6:921–931. doi: 10.1111/j.1365-2958.1992.tb01542.x. [DOI] [PubMed] [Google Scholar]
- Schmelcher M., Shabarova T., Eugster M.R., Eichenseher F., Tchang V.S., Banz M., Loessner M.J. Rapid multiplex detection and differentiation of Listeria cells by use of fluorescent phage endolysin cell wall binding domains. Appl Environ Microbiol. 2010;76:5745–5756. doi: 10.1128/AEM.00801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch R., Nelson D., Fischetti V.A. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature. 2002;418:884–889. doi: 10.1038/nature01026. [DOI] [PubMed] [Google Scholar]
- Sheehan M.M., Garcia J.L., Lopez R., Garcia P. Analysis of the catalytic domain of the lysin of the lactococcal bacteriophage Tuc2009 by chimeric gene assembling. FEMS Microbiol Lett. 1996;140:23–28. doi: 10.1111/j.1574-6968.1996.tb08309.x. [DOI] [PubMed] [Google Scholar]
- Turner M.S., Waldherr F., Loessner M.J., Giffard P.M. Antimicrobial activity of lysostaphin and a Listeria monocytogenes bacteriophage endolysin produced and secreted by lactic acid bacteria. Syst Appl Microbiol. 2006;30:58–67. doi: 10.1016/j.syapm.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Walcher G., Stessl B., Wagner M., Eichenseher F., Loessner M.J., Hein I. Evaluation of paramagnetic beads coated with recombinant Listeria phage endolysin‐derived cell‐wall‐binding domain proteins for separation of Listeria monocytogenes from raw milk in combination with culture‐based and real‐time polymerase chain reaction‐based quantification. Foodborne Pathog Dis. 2010;7:1019–1024. doi: 10.1089/fpd.2009.0475. [DOI] [PubMed] [Google Scholar]
- Wren B.W. A family of clostridial and streptococcal ligand‐binding proteins with conserved C‐terminal repeat sequences. Mol Microbiol. 1991;5:797–803. doi: 10.1111/j.1365-2958.1991.tb00752.x. [DOI] [PubMed] [Google Scholar]
- Yother J., White J.M. Novel surface attachment mechanism of the Streptococcus pneumoniae protein PspA. J Bacteriol. 1994;176:2976–2985. doi: 10.1128/jb.176.10.2976-2985.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R., Blasi U. Holins: form and function in bacteriophage lysis. FEMS Microbiol Rev. 1995;17:191–205. doi: 10.1111/j.1574-6976.1995.tb00202.x. [DOI] [PubMed] [Google Scholar]
- Zimmer M., Sattelberger E., Inman R.B., Calendar R., Loessner M.J. Genome and proteome of Listeria monocytogenes phage PSA: an unusual case for programmed +1 translational frameshifting in structural protein synthesis. Mol Microbiol. 2003;50:303–317. doi: 10.1046/j.1365-2958.2003.03684.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers for PCR amplification and sequencing used in this study.
Table S2. Plasmids used in this study.