

Abstract
Fully functional T cells are necessary for the maintenance of protective immunity during chronic infections. However, activated T cells often undergo apoptosis or exhaustion upon chronic stimulation mediated by antigen or inflammation. T cell attrition can be compensated by the production of thymus-derived T cells, although the new naive T cells must undergo T cell priming and differentiation under conditions different from those encountered during acute infection. We have used a murine model of Mycobacterium tuberculosis (Mtb) infection to address how the activation and differentiation of new thymic emigrants is affected by chronic inflammation, and whether the newly developed effector T cells help to maintain peripheral T cell responses. Although new thymic emigrants contributed to the peripheral T cell response early during acute Mtb infection, the relative contribution of new effector T cells to the peripheral CD4 and CD8 T cell pools declined during chronic infection. The decline in new T cell recruitment was a consequence of quantitative and/or qualitative changes in antigen presentation, as during chronic infection both the priming and expansion of naïve T cells was inefficient. Thus, although thymic tolerance is not a major factor that limits protective T cell responses, the chronic environment does not efficiently support naive T cell priming and accumulation during Mtb infection. These studies support our previous findings that long-term protective T cell response can be maintained indefinitely in the periphery, but also suggest that the perturbation of homeostasis during chronic inflammatory responses may elicit immune pathology mediated by new T cells.
Introduction
Infection with a pathogen results in the orchestrated priming, expansion, and differentiation of a T cell response. T cells first encounter antigen in secondary lymphoid tissues, enter the circulation after a period of proliferation, and are subsequently recruited to sites of infection, where they exert their effector functions. For Mycobacterium tuberculosis (Mtb), studies in the mouse have shown that antigen-specific naïve T cells are primed in the lung draining lymph node, approximately 10 days after aerosol infection (1). Primed antigen-specific T cells, undergo extensive proliferation, emigrate from the lymph nodes, and are recruited to the lungs and other sites of inflammation (2, 3). The activity of these T cells, i.e. production of IFNγ, is essential for the control of bacterial replication and survival of the host (4, 5).
Although much is known regarding the early adaptive immune response generated against Mtb, less is known about how T cells responses are maintained during chronic infection. Chronic T cell responses are affected by the inflammatory environment, which for Mtb, includes the granuloma, a hallmark of tuberculosis infection (6). The granuloma is highly ordered and is continually evolving at the structural and cellular level throughout tuberculosis infection (7, 8). How these changes in the granuloma, i.e. the chronic environment, in turn impact T cell immunity is unclear. The granuloma may limit T cell migration and/or access to antigen, as it has been shown that effector T cell functions are diminished in part due to limiting antigen (9, 10). This apparent change in antigen availability could, in part, allow for bacterial persistence.
Persistent antigen stimulation during other chronic infection often leads to T cell exhaustion. Pathogens such as Mtb establish lifelong chronic infection; so the host must maintain a stable population of functioning effector T cells. Many mechanisms may contribute to the maintenance of T cells during chronic infection, and these likely are pathogen-specific. We have proposed that T cell responses are maintained during Mtb infection by a yet uncharacterized self-renewing effector cell population, as T cells do not appear to undergo clonal exhaustion (13, 14) and highly proliferative CD4 T cells undergo rapid turnover in the lung throughout chronic infection (14, 15). Indeed, peripheral T cell responses are maintained for up to four months post-infection in thymectomized mice (15). Nevertheless, these data do not exclude the possibility that newly generated T cells (i.e., recent thymic emigrants; RTEs) also contribute to the maintenance of peripheral T cell responses. Some chronic viruses, including polyoma virus and murine cytomegalovirus, utilize RTEs to maintain protective T cell responses (16, 17), the relative contribution of RTEs during Mtb infection has not been investigated. Moreover, studies of Mycobacterium avium-infected mice demonstrated that infection of the thymus resulted in T cell tolerance and prevented the generation of effector T cell responses to pathogen-specific antigens (18). The findings from the M. avium studies suggested that RTEs do not contribute to peripheral immunity during chronic tuberculosis infections.
In the present study, we investigated whether the infected environment within the mouse was capable of supporting the activation of RTEs, thereby allowing recently generated effector T cells to contribute to the peripheral T cell response during Mtb infection. We demonstrate that new thymus-derived antigen-specific T cells contribute to peripheral immunity, revealing that central tolerance is not a major factor limiting peripheral T cell responses during Mtb infection. However, the contribution of RTEs diminishes during chronic infection, likely a consequence of the diminished capacity of the chronic environment to support the activation and accumulation of new effector T cells. These studies extend our understanding of how T cell responses are maintained in a chronic inflammatory environment during persistent bacterial infections.
Materials and Methods
Animals
C57BL/6J and B6.SJL-Ptprca Pep3b/BoyJ (CD45.1) mice were purchased from The Jackson Laboratory. C57BL/6 were crossed with B6.SJL-Ptprca Pep3b/BoyJ mice to generate B6.SJL-Ptprca/b Pep3b/c, and are indicated herein as (CD45.1/CD45.2). The early secreted antigenic target 6 (ESAT6) transgenic mice, which are specific for ESAT61-20 presented by I-Ab, were described previously (1). Thy1.1+ SM1 transgenic mice, which contain CD4 T cells specific for Salmonella flagellin427-441, have been described (19). T cell transfers were described previously, and utilized 1×106 or 2×103 naïve transgenic T cells, which were transferred by i.v. injection into recipient mice (1). All mice were maintained in specific pathogen-free facilities at the Trudeau Institute. Experimental mice were age and sex-matched, and were infected between 8 and 12 weeks of age. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Mice were handled according to all applicable institutional, state, and federal animal care guidelines, under animal care protocols approved by the Trudeau Institute (Saranac Lake NY). Veterinary technicians or laboratory staff assessed animal health daily. Moribund mice were humanely euthanized.
Bacterial infections
The H37Rv strain of Mtb was grown in Proskauer and Beck medium containing 0.05% Tween 80 to mid-log phase, and was preserved in 1 ml aliquots at −70°C. For aerosol infections, animals were infected with a low dose of bacteria (~75 CFUs), using a Glas-Col airborne infection system, as described previously (20). Bacteria in the lungs were measured by counting viable CFUs in homogenized tissue as described (20).
Lymphocyte Isolation and Flow Cytometry
Lung tissue was prepared for analysis by injecting lungs with a 0.5 mg/ml solution of collagenase (Roche) and DNase (Sigma Aldrich); the tissue was then coarsely chopped and incubated in the same solution for 30–45 minutes at 37°C. Single cell suspensions were prepared from lung tissue, lymph nodes, or spleens by dispersing the tissues through a 70-μm nylon tissue strainer (BD Falcon). The cell suspensions were treated with buffered ammonium chloride solution to lyse erythrocytes. Lymphocytes were enriched from lung tissue by differential centrifugation, using a gradient of 40/80% Percoll (GE Healthcare). Cells were stained with an ESAT64-17/Ab- PE tetramer reagent (14, 21), a Tb10.44-11/Kb-APC tetramer reagent (22), and fluorochrome-labeled antibodies specific for anti-CD4 (clone RM4-5), anti-CD8 (5H10), anti-CD45.2 (104), anti-CD69 (H1.2F3), anti-BrdU, and anti-IFNγ (XMG1.2; all from BD Biosciences); anti-KLRG1 (2F1) (Southern Biotech); anti-CD45.1 (A20), anti-PD-1 (RPM1), and anti-TNFα (MP6-XT22)(BioLegend); anti-CD44 (IM7) (eBioscience), and anti-CD62L (Mel-14; Invitrogen). The samples were analyzed on a LSRII flow cytometer (BD Biosciences), and the data were analyzed using FlowJosoftware (Tree Star, Inc.; Ashland OR). Cells were gated on lymphocyte size and granularity, with doublets being excluded.
In vitro Th1 Effector Cell Generation
Effector T cell populations were generated by stimulating naive ESAT6 transgenic T cells with 2 μM ESAT61-20 peptide presented by mitomycin C-treated B cell blasts. Cells were cultured and expanded in Th1 polarizing conditions: IL-2 (11 ng/mL), IL-12 (10 μg/mL), and anti-IL4 (10 μg/mL). The cells were used for experiments 6 to 8 days post-activation.
Intracellular Cytokine Detection Assays
Lymphocytes were isolated and incubated in a 96-well plate, at a concentration of 3×106 cells per well, in the presence of ESAT-61-20 or Sendai HN421-436 peptides (5 ug/ml), for two hours at 37°C; Brefeldin A (50 ug/ml) was then added, and the incubation was continued for an additional 4h. Surface staining for CD4, CD8, was performed, after which the cells were fixed and permeabilized using a Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences). For detection of intracellular IFNγ and TNFα, the cells were incubated for 30 minutes in Perm/Wash Buffer (BD Biosciences) with anti-IFNγ and anti-TNFα; the cells were analyzed by flow cytometry.
Generation of Bone Marrow Chimeras
Myeloablative drug treatment and bone marrow reconstitution was preformed, as previously described (16), by treatment of naïve or Mtb-infected CD45.1/CD45.2 mice, or allotype congenic mice, with busulfan i.p. (0.6 mg/animal; generously provided by Otsuka America Pharmaceutical, Rockville, MD). One-day later, mice were administered, via i.v. injection, 1.5×107 C57BL/6 bone marrow cells that had been depleted of CD3-, CD4- and CD8-positive T cells.
Statistic Analysis
Statistical significance between groups was preformed with Prism5 GraphPad software using a Two-Way Anova test. Differences with a p-value < 0.05 were considered significant and are indicated in the figures as follows: *p < 0.05; **p < 0.01; and***p < 0.001.
Results
The thymus is a site of tuberculosis infection
To assess whether the chronic infection allows for the development of RTEs, and whether infection of the thymus impacts thymic tolerance during Mtb infection, we first examined the kinetics of bacterial dissemination to the thymus after aerosol infection. Similar to observations made by Nobrega et al. (23), we observed that Mtb colonizes the thymus, but infection in that tissue reached a maximum only after day 60 post-infection, much later than in the lung and secondary lymphoid tissues (Fig. 1 A). We investigated whether the change in the log linear growth of Mtb within the thymus, was correlated with the presence of an effector T cell response. Indeed, the apparent delay in the control of bacterial replication within the thymus was correlated with a delay in the accumulation of ESAT64-17/Ab tetramer-positive CD4 T cells, and Tb10.44-11/Kb tetramer-positive CD8 T cells (Fig. 1 B and C). The thymic effector T cells were functional, as they produced IFNγ and TNFα following antigen encounter in vitro (Fig. 1 D). These results demonstrate that Mtb infects the thymus, and results in the recruitment of effector T cells to the site of infection, albeit later in infection relative to other tissues.
Figure 1. The thymus is a target of Mtb infection.

Mice were infected via the low-dose aerosol route with Mtb (strain H37Rv), and bacterial titers and effector T cell responses were measured. (A) Colonization of the thymus, lung, MLN, spleen, and liver after aerosol Mtb infection. The data shown were accumulated from two experiments. (B) The flow cytometric gating strategy used to analyze ESAT64-17/Ab and Tb10.44-11/Kb antigen- specific T cells in the thymus, on day 11, 33, 68, 98 post-infection, is shown. (C) The data indicate the number of ESAT64-17/Ab and Tb10.44-11/Kb antigen-specific T cells detected in the thymus during Mtb infection. (D) Representative dot plots indicate the frequency of IFNγ̃ and TNFα-producing CD4 and CD8 single-positive thymic T cells detected on day 31, 67, and 100 post-infection. The data are representative of three experiments of similar design, wherein 5 mice were used per group.
RTEs contribute to the peripheral T cell response during acute infection
Mtb infection of the thymus raised the possibility that developing thymocytes could become tolerized against bacterial antigens. Therefore, to determine whether new effector T cells generated from RTEs were tolerized, or could contribute to the maintenance of T cell response during Mtb infection, we transferred 1.5 × 107 C57BL/6 T cell-depleted bone marrow cells into congenic recipient mice that had been infected with Mtb (on either day 30 or 90 post-infection). Recipient animals were treated one day prior to bone marrow transfer with a minimally myeloablative drug (i.e., busulfan) (Fig. 2 A). The loss of bone marrow cells caused by busulfan treatment promotes the engraftment of the donor bone marrow cells that subsequently give rise to thymus-derived naïve T cells (16); donor cells can be distinguished from host T cells based on allelic differences. Drug treatment did not impact bacterial burdens in mice that had been treated with busulfan, and did not detectably affect the host T cell response, as the frequency and number of ESAT64-17/Ab tetramer-positive host CD4 T cells remained normal (Fig. S1; Fig. 2B).
Figure 2. RTEs contribute to the T cell response during Mtb infection.

(A) A schematic illustrating the experimental protocol is shown. CD45.1/CD45.2 mice either remained uninfected, or were infected, via the low-dose aerosol route, with Mtb (strain H37Rv); 30 or 90 days later, the mice were administered busulfan (0.6 mg/mouse). One day later, 1.5×107 T cell-depleted bone marrow cells from C57BL/6 mice were injected, and ESAT64-17/Ab -specific CD44hi thymus-derived donor and host CD4 T cells were quantitated in the lungs 63 days later. (B) Analyses of donor and host T cells are shown. The numbers in the dot plots indicate the frequencies of ESAT64-17/Ab antigen-specific donor or host CD4 T cells after bone marrow reconstitution of uninfected, day 30, or day 90 Mtb-infected animals. Data are representative of two experiments, where five mice were analyzed in each experimental group.
Engraftment of donor bone marrow was observed in all mice, and thymus-derived CD4-positive CD45.2-positive donor T cells were detected in the lungs and secondary lymphoid organs as early as five weeks post-donor bone marrow transfer. Sixty-three days after donor bone marrow transfer, we examined whether the donor cells were present within the peripheral T cell response. A population of donor-derived ESAT64-17/Ab tetramer-positive CD4 T cells was detected in mice that had been reconstituted on day 30 post-aerosol infection, indicating that new thymus-derived T cells contributed to the antigen-specific T cell response. In contrast, few if any donor-derived antigen-specific T cells were detected within the lungs of mice that had received donor bone marrow on day 90 post-infection (Fig. 2 B).
Priming and expansion of naïve CD4 T cell declines during chronic infection
While tolerance of developing lymphocytes may explain the failure to detect thymus-derived effector T cells after busulfan treatment on day 90 post-infection, an alternative explanation was that the chronic environment was insufficient to drive efficient naïve T cell priming and/or expansion. Inefficient T cell activation could have been caused by sequestration of antigen, changes in tissue organization, exclusion of T cells within the granuloma or insufficient antigen. Support for this was demonstrated in two recent reports, in which the ability of Antigen-85b-specific effector cells to respond to antigen in vivo was low during chronic infection (9, 10). To address whether and when antigen was available to prime naive T cells, we transferred 106 naïve ESAT-61-20/Ab-specific transgenic CD4 T cells into infected CD45- and Thy1-congenic recipient mice; 12 hours later, primed T cells were identified by their expression of the early activation marker, CD69 (Fig. 3 A; (1, 24)). This approach allowed us to determine if antigen was present that was capable of stimulating naïve T cells. Transferred antigen-specific CD4 T cells were activated within the mediastinal lymph node (MLN), beginning on 10 post-infection (Fig. 3 B; (1)). T cell activation was specific, because flagellin427-441/Ab-specific SM1 transgenic T cell transferred into infected mice were not activated (Fig. 3 A, Fig. 3 B). T cell priming in the MLN was highest during acute infection (i.e., days 18–25), and declined markedly thereafter. T cell priming was less efficient after day 30, because only about 10% of the donor T cells were activated within 12 hours; nevertheless, antigen was available at this time, because the frequency of primed T cells in the MLN continued to increased 36 hours post-transfer (Fig. S2A). Slightly delayed kinetics of naïve T cell priming were observed in the spleen, although higher frequencies of T cells were primed, relative to the MLN, after day 30 post-infection (Fig. 3 B). While T cell priming predominantly occurred in the spleen and MLN, priming was also observed in the cervical lymph node, mesenteric lymph node, and lung, albeit at reduced efficiencies (Fig. S2 B). These results indicate that antigen is available during chronic infection, although it is less efficient at priming T cells.
Figure 3. Naïve T cell priming during Mtb infection.

Naïve ESAT6 and SM1 transgenic T cells were isolated from mice by negative magnetic bead selection, and 1×106 cells were injected, at a 1:1 ratio, into infected CD45- and Thy1-congenic recipient mice. Mice were analyzed 12 hours after cell transfer on each of the days indicated. (A) Representative flow cytometric analysis of CD69expression on ESAT6 or SM1 transgenic T cells from the spleen on day 25 post-infection. (B) Expression of CD69 on donor ESAT6 transgenic T cells in the MLN and spleen during infection. The datum represents individual mice; the dashed line indicates the mean CD69 expression of SM1 transgenic T cells in mice analyzed throughout the experiment. The data shown were accumulated from two experiments, and are representative of three independent experiments.
Although T cell priming declined during chronic infection, it was not possible in the above studies to determine whether this decrease was due to limiting antigen, or whether antigen presenting cells lacked the capacity to efficiently stimulate naïve T cells. To resolve this issue, we utilized in vitro differentiated ESAT-61-20/Ab-specific Th1 effector cells; such activated T cells are highly sensitive to antigen, and can detect antigen presented on many different APCs in the absence of costimulation. We transferred 106 in vitro differentiated ESAT-61-20/Ab-specific Th1 effector cells into CD45-congenic recipient mice at several times post-infection; 12 hours later, effector T cells that had responded to antigen were identified by their expression of CD69. Transferred effector T cells readily detected antigen in the spleen, MLN, and lungs at all time points examined (Fig. 4). In contrast to naive T cells, effector T cells detected similar levels of antigen within the spleen and lungs, at time points as long as 325 days post-infection. Additionally, effector T cells readily detected antigen in the lung (Fig. 4, Fig. S2 B), a finding that supports our previous studies, which concluded that the lung is not an efficient environment for naïve T cell priming (1). Effector T cells also detected antigen in the MLN, although, like naïve T cells, the effector T cells were stimulated less efficiently in this tissue during chronic infection; suggesting that antigen presentation progressively declined with the MLN during chronic infection. Taken together, these results indicate that the loss of naïve T cells priming observed after day 30 post-infection was not completely a result of declining antigen availability within infected tissues.
Figure 4. Effector T cells detect similar levels of antigen during Mtb infection.
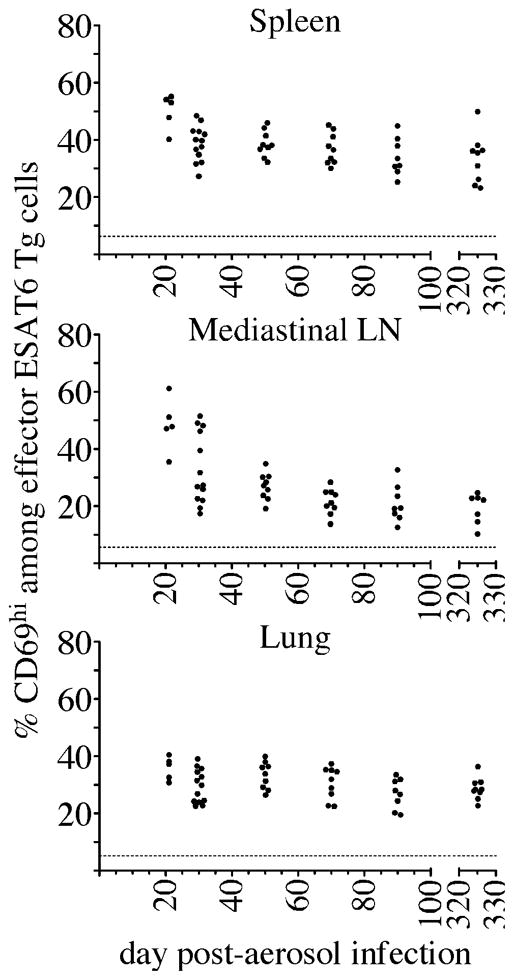
In vitro differentiated ESAT6 Th1 effectors (1×106) were injected into congenic recipient mice, and mice were analyzed 12 hours after cell transfer on each of the days indicated. The percent of cells that expressed of CD69 on donor ESAT6 Th1 effector T cells in the spleen, MLN, and lung is shown. The datum represents individual mice; the dashed line indicates the mean CD69 expression detected on effector Th1 cells that were transferred into uninfected mice. The data shown were accumulated from three experiments.
Since the capacity of the immune system to prime naïve T cell responses declined over time following Mtb infection, we next asked how this decline affected the accumulation and/or expansion of RTEs. For these studies, we transferred 2 X 103 ESAT-61-20/Ab-specific T cells into mice that had been infected with Mtb for 11, 30, 60, or 90 days; donor T cells were then enumerated 2, 7, and 10 days later. When transfer was performed on day 11 or 30 post-infection, donor T cells accumulated rapidly in the spleen, MLN, and lung (Fig. 5). In contrast, donor T cells underwent limited expansion and/or accumulation when transferred on day 60 or 90 post-infection. No further increase in donor T cells expansion was detected after day 10 post-transfer, regardless of the time when T cells were transferred post-infection. The decline in T cell expansion observed during chronic infection was not due to a failure of the donor cells to differentiate into effector cells, as effector cell phenotype, proliferation, and cytokine production all occurred normally in those cells (Fig. S3).
Figure 5. Expansion of naïve transgenic T cells during acute and chronic Mtb infection.

Naïve ESAT6 transgenic T cells (2 × 103) were injected intravenously into CD45-congenic mice that had been aerosol-infected 11, 30, 60, or 90 days prior. The number of transgenic donor cells in the spleen, MLN, and lung, were determined on days 2, 7, and 10 post-transfer. The arrows in each plot indicate the day the transgenic T cells were transferred. The data are representative of two experiments of similar design, wherein 5 mice were used per group.
RTEs accumulate during chronic infection
Our results demonstrate that, although antigen is presented throughout Mtb infection, and is capable of initiating T cell priming, expansion, and differentiation, T cell priming is less efficient during chronic infection. Thus, the failure to detect the accumulation of effector T cells generated from RTEs after day 90 post-infection, is likely a consequence of poor T cell priming, and a slower accumulation of these cells. To address this hypothesis, we examined RTEs at later times following hematopoietic reconstitution. Mice were treated with busulfan, reconstituted on day 30, 60 or 90 post-infection, and antigen-specific effector RTEs were quantitated in lungs 49, 63, 84, and 140 days post-donor bone marrow transfer. Mice that were reconstituted on day 30 post-infection contained a sizable population of antigen-specific effector RTEs as early as 49 days post-donor bone marrow transfer (Fig. 6 A). As the duration following reconstitution increased, so did the accumulation of antigen-specific effector RTEs in the lung. In contrast, few antigen-specific effector RTEs were detected when busulfan was administered 60 days post-infection and analysis was performed 49 days post-donor bone marrow transfer. However, when these mice were analyzed 63 days post-bone marrow transfer, significant numbers of antigen-specific donor T cells were detected in most mice. The accumulation of antigen-specific effector RTEs was delayed further in mice that had been treated with busulfan on day 90 post-infection, because antigen-specific effector RTEs were detected only after 84 days post-donor bone marrow transfer; however, by day 140 post-reconstitution, thymus-derived T cells had accumulated to similar numbers as mice that were treated with busulfan on either day 30 or day 60 post-infection.
Figure 6. RTEs contribute to the peripheral CD4 T cell response during chronic infection.

Infected CD45-congenic mice were administered 0.6 mg of busulfan on day 30, 60, or 90 post-infection. One day later, 1.5×107 T cell-depleted bone marrow cells from C57BL/6 mice were injected, and ESAT64-17/Ab -specific CD4 T cells were quantitated in the lungs 49, 63, 84, and 140 days after reconstitution. (A) The number of ESAT64-17/Ab-specific host and donor CD4 T cells is shown. (B) Representative dot plots are shown of IFNγ̃ and TNFα-producing donor or host CD4 T cells and the frequencies of ESAT64-17/Ab antigen-specific CD4 T cells from busulfan-treated mice that were analyzed 140 days post-reconstitution. Intracellular and tetramer staining were preformed on two independent samples from the same animals. (C) The frequencies of ESAT64-17 / I-Ab tetramer-positive cells within the donor- and host-expressing populations were plotted against the frequency of ESAT61-20 / I-Ab cells that specifically produced IFNγ for each busulfan-treated mice within each group; the mice received busulfan on either day 30 or 90 post-infection, and were analyzed 140 days post-reconstitution. The best-fit line was generated using least squares regression analysis. No statistical difference was found upon comparison of the two slopes (p=0.83 day 30, p=0.87 day 90). The data are representative of two experiments of similar design, wherein five mice were used per group.
Although effector RTEs were detected in chronically infected mice, it was nevertheless possible that the cells were functionally impaired (i.e., anergic). This was not the case, however, as T cells from mice that had been treated with busulfan on day 30 or 90 post-infection, produced IFNγ and TNFα in response to specific peptide antigen, when assayed 140 days post-reconstitution (Fig. 6 B). The lower frequency of effector RTEs producing cytokine compared to host cells was a result of a difference in the frequencies of antigen-specific T cells within the two populations and not due to the inability of donor cells to produce cytokines (Fig. 6B). This was evident from comparison of the frequencies of host and donor ESAT64-17/Ab tetramer-specific CD4 T cells with the abilities of these cells to produce IFNγ in response to ESAT61-20 peptide (Fig. 6C). These results indicate that the antigen-specific effector RTEs were functional and could contribute to the control of bacterial replication during Mtb infection. Our findings indicate that the failure to detect CD4 effector RTEs in the periphery of chronically-infected busulfan-treated mice was likely a consequence of their delayed priming and slow accumulation.
These results demonstrated that RTEs contribute to the maintenance of antigen-specific CD4 T cells during chronic Mtb infection. However, CD4 and CD8 T cells could be maintained by different mechanisms during acute and chronic infections (25). To determine if RTEs could contribute towards the maintenance of antigen-specific CD8 T cells, we examined whether Tb10.44-11/Kb antigen-specific CD8 T cells were present in mice 63 days after donor bone marrow reconstitution day 30 or 90 post-Mtb infection (Fig. 7 A). Similar to our studies of CD4 T cells, mice that were reconstituted on day 30 post-infection contained appreciable numbers of effector RTEs which had expanded and become part of the ongoing antigen-specific T cell response. However, few, if any, detectable Tb10.44-11/Kb antigen-specific CD8 T cells were present in mice that were reconstituted on day 90 post-infection. Moreover, the ability of CD8 RTEs to enter into the response decreased during chronic infection (Fig. 7B). Therefore, during Mtb infection RTEs can contributes towards the maintenance of both the peripheral CD4 and CD8 antigen-specific T cell response.
Figure 7. RTEs contribute to the peripheral CD8 T cell response during chronic infection.

Infected mice were administered 0.6 mg of busulfan on the indicated days post-infection. One day later, 1.5×107 T cell- depleted bone marrow cells from C57BL/6 mice were injected, and Tb10.44-11/Kb antigen-specific CD8 T cells were quantitated in the lungs. (A) Analyses of donor and host T cells are shown. The numbers in the dot plots indicate the frequencies of Tb10.44-11/Kb antigen-specific CD8 T cells after donor-bone marrow transfer in mice treated with busulfan on day 30, day 90 post-infection. (B) The number of Tb10.44-11/Kb -specific host and donor CD8 T cells after donor-bone marrow reconstitution is shown.
Discussion
Our data demonstrates that thymus-derived T cells contribute to the maintenance of T cell responses during acute and chronic Mtb infection, and that thymic tolerance is not a major mechanism impacting the maintenance of protective immunity during chronic infection. The slow accumulation of effector RTEs during infection does not appear to be due to thymic tolerance or anergy, but likely was a consequence of inefficient antigen presentation in the periphery during chronic infection, which limits the accumulation of antigen-specific effector T cells.
The thymus, once thought of as an immune privileged site, can also be infected by viruses (e.g. HIV, lymphocytic choriomeningitis virus, and measles), parasites (T. cruzi), and fungi (Paracoccidioides brasiliensis) (26). The most common consequence of chronic infection is thymic atrophy, but it is not known whether infection of the thymus by such diverse pathogens leads to central tolerance. Our studies indicate that thymic tolerance is not an inevitable outcome of thymic infection, even though a persistent infection is present in the thymus during murine Mtb infection. One explanation for the apparent lack of central tolerance during Mtb infection is that bacterial antigens are not expressed by cells in the thymus that are capable of inducing tolerance in developing T cells. Alternatively, the control of infection by day 60 post-aerosol may limit the expression of bacterial antigens within the thymus thereby limiting the induction of thymic tolerance. Although our studies have utilized a mouse model, Mtb has been detected in the thymi of infected humans (27, 28), although whether this is a common occurrence in actively Mtb-infected patients is not yet known. Our results suggest that, even if Mtb colonizes the thymus in chronically-infected humans, developing T cells are nevertheless able to circumvent central tolerance and contribute to the peripheral immune response.
Our data differ from those in a report that proposed thymic tolerance as a major mechanism of immune evasion during M. avium infection (18). In that study, thymi were transplanted from M. avium-infected TCRα −/− mice into nude recipient mice prior to infection; under these conditions, the authors failed to detect effector T cell responses ex vivo to M. avium antigens. Opposing findings from the studies likely include differences in the species of mycobacterium used, routes of infection, the sensitivity of the techniques used to identify RTEs, and the experimental approaches (i.e., transplantation of infected thymi, versus the use of busulfan). Another possibility is that the numbers of bacteria, or the expression of bacterial antigens, was higher within the transferred thymi used in the study by Nobrega et al., relative to Mtb-infected thymi, where effector T cell response were observed. We cannot exclude the possibility that some developing T cells were tolerized, as we could not directly examine if antigen-specific T cells were deleted from the thymus of chronically infected mice that were treated with busulfan. However, our studies provide definitive evidence that new antigen-specific T cell responses can develop during chronic Mtb infection in the mouse.
A requirement for RTEs in other chronic infections has been documented (16, 17). However, if recruitment of RTEs were the sole mechanism responsible for maintaining the antigen-specific T cell response during chronic infections then eventually all antigen-specific cells would be replaced by donor derived cells. Our data demonstrates that effector RTEs could contribute to the peripheral T cell response, yet they are not solely required for maintenance, as the donor derived cells never fully replaced host cells. These data support our previous findings that the thymus was not required for the maintenance of antigen-specific T cell responses, for at least as long as 125 days post-infection (15). Thus RTE provide only one mechanism by which peripheral T cell responses are maintained during Mtb infection.
The capacity of T cell to be primed during chronic infection is diminished, and is likely due to changes in the infected environment, a consequence of host control of bacterial replication. The temporal changes in T cell responses could be due to reduced antigen availability, changes in the APC populations and/or their expression of costimulation molecules, the cytokine milieu, the ability of a naïve RTEs to interact with APC populations and the ability of naïve RTEs to sustain prolonged contact with APCs. We observed differences in the capacity of lymphoid tissues to present Mtb antigens, as both the priming and expansion of naïve T cells was decreased in the MLN during chronic infection, relative to the spleen. These differences are unlikely a consequence of tissue bacterial loads, as these were similar between the two lymphoid organs after day 30 post-infection (Fig. 1a; (14). Indeed, effector T cells responded similarly to antigen during both acute and chronic infection. One possibility is that chronic tuberculosis infection changes the architecture of the spleen and MLN and that this disruption may alter the capacity of APCs to prime naïve T cells. Differences among the types or strength of costimulatory molecules, the cytokines being produced, and/or the quantity and quality of available antigen in the different lymphoid tissues could influence the ability of APCs to prime and expand naïve T cells (29).
Our finding that naïve T cells accumulate to a lesser extent during chronic Mtb infection has potential clinical implications for Mtb-infected individuals with active disease. Our results would suggest that it maybe advantageous to employ additional strategies that invoke an efficient naïve T cell activation, in addition to current chemotherapeutic treatments, as such a strategy could lead to decreased periods of chemotherapy treatment. The ability to effectively stimulate new naïve T cell responses during chronic Mtb infection may be a mechanism employed by the therapeutic vaccine RUTI, which has been shown to increase effector T cell responses in Mtb infected animals that are undergoing chemotherapy (30). Thus, thymic selection and peripheral T cell priming occurs throughout chronic Mtb infection in the mouse, and contribute to the maintenance of protective T cell responses. Although infection of the thymus occurs in mice and humans, our data also argue that thymic tolerance is not a major mechanism that limits T cell-mediated immunity to Mtb infection.
Supplementary Material
Acknowledgments
We thank Drs. A. Cooper, E. Torrado, M. Freeman and L. Connor for critical review of the manuscript. We would like to thank Larry Johnson for assistance with statistical analysis. We thank Otsuka America Pharmaceutical for the generous gift of the drug busulfan. We also thank Pamela Scott Adams, the Trudeau Institute Molecular Biology Core Facility and Dr. M. Jenkins for the production of the ESAT64-17/Ab tetramer. The authors also acknowledge the contributions of Dr. Karen Chave of the Northeast Biodefense Center Expression Core for MHC tetramer production.
This work was supported by Public Health Service Grants R01AI073564 (to D.L.W. and G.M.W.), R21AI077531 (to G.M.W. and D.L.W.), P01AG02160 (to L.H.) and U54-AI057158 (to I.L.).
Abbreviations used in this article
- Mtb
Mycobacterium tuberculosis
- RTEs
recent thymic emigrants
- MLN
mediastinal lymph node
- ESAT6
early secreted antigenic target 6
References
- 1.Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, Fountain JJ, Martino CA, Roberts AD, Cooper AM, Winslow GM, Woodland DL. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci USA. 2008;105:10961–10966. doi: 10.1073/pnas.0801496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper AM. T cells in mycobacterial infection and disease. Current opinion in immunology. 2009;21:378–384. doi: 10.1016/j.coi.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winslow GM, Cooper A, Reiley W, Chatterjee M, Woodland DL. Early T-cell responses in tuberculosis immunity. Immunol Rev. 2008;225:284–299. doi: 10.1111/j.1600-065X.2008.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.North RJ, Jung YJ. Immunity to tuberculosis. Annu Rev Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 5.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams DO. The granulomatous inflammatory response. A review. Am J Pathol. 1976;84:164–192. [PMC free article] [PubMed] [Google Scholar]
- 7.Rhoades ER, Frank AA, Orme IM. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:57–66. doi: 10.1016/s0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- 8.Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. 2012;12:352–366. doi: 10.1038/nri3211. [DOI] [PubMed] [Google Scholar]
- 9.Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. Intravital Imaging Reveals Limited Antigen Presentation and T Cell Effector Function in Mycobacterial Granulomas. Immunity. 2011;34:807–819. doi: 10.1016/j.immuni.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bold TD, Banaei N, Wolf AJ, Ernst JD. Suboptimal Activation of Antigen-Specific CD4 Effector Cells Enables Persistence of M. tuberculosis In Vivo. PLoS Pathog. 2011;7:e1002063. doi: 10.1371/journal.ppat.1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 12.Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Current opinion in immunology. 2007;19:408–415. doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T Cells Promote Rather than Control Tuberculosis in the Absence of PD-1-Mediated Inhibition. J Immunol. 2011;186:1598–1607. doi: 10.4049/jimmunol.1003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard G, Moon JJ, Jenkins MK, Urdahl KB, Winslow GM, Woodland DL. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 2010;107:19408–19413. doi: 10.1073/pnas.1006298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winslow GM, Roberts AD, Blackman MA, Woodland DL. Persistence and turnover of antigen-specific CD4 T cells during chronic tuberculosis infection in the mouse. J Immunol. 2003;170:2046–2052. doi: 10.4049/jimmunol.170.4.2046. [DOI] [PubMed] [Google Scholar]
- 16.Vezys V, Masopust D, Kemball CC, Barber DL, O’Mara LA, Larsen CP, Pearson TC, Ahmed R, Lukacher AE. Continuous recruitment of naive T cells contributes to heterogeneity of antiviral CD8 T cells during persistent infection. J Exp Med. 2006;203:2263–2269. doi: 10.1084/jem.20060995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29:650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nobrega C, Roque S, Nunes-Alves C, Coelho A, Medeiros I, Castro AG, Appelberg R, Correia-Neves M. Dissemination of mycobacteria to the thymus renders newly generated T cells tolerant to the invading pathogen. J Immunol. 2010;184:351–358. doi: 10.4049/jimmunol.0902152. [DOI] [PubMed] [Google Scholar]
- 19.McSorley SJ, Cookson BT, Jenkins MK. Characterization of CD4+ T cell responses during natural infection with Salmonella typhimurium. J Immunol. 2000;164:986–993. doi: 10.4049/jimmunol.164.2.986. [DOI] [PubMed] [Google Scholar]
- 20.Roberts AD, Cooper AM, Belisle JT, Turner J, Gonzalez-Juarerro M, Orme IM. Murine model of tuberculosis. In: Stefan K, Dieter Kabelitz HE, editors. Methods in Microbiology. Vol. 32. Academic Press; 2002. pp. 433–462. [Google Scholar]
- 21.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woodworth JS, Wu Y, Behar SM. Mycobacterium tuberculosis-specific CD8+ T cells require perforin to kill target cells and provide protection in vivo. J Immunol. 2008;181:8595–8603. doi: 10.4049/jimmunol.181.12.8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nobrega C, Cardona P-J, Roque S, Pinto do O P, Appelberg R, Correia-Neves M. The thymus as a target for mycobacterial infections. Microbes Infect. 2007;9:1521–1529. doi: 10.1016/j.micinf.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Testi R, Phillips JH, Lanier LL. Leu 23 induction as an early marker of functional CD3/T cell antigen receptor triggering. Requirement for receptor cross-linking, prolonged elevation of intracellular [Ca++] and stimulation of protein kinase C. J Immunol. 1989;142:1854–1860. [PubMed] [Google Scholar]
- 25.Stockinger B, Bourgeois C, Kassiotis G. CD4+ memory T cells: functional differentiation and homeostasis. Immunol Rev. 2006;211:39–48. doi: 10.1111/j.0105-2896.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- 26.Savino W. The thymus is a common target organ in infectious diseases. PLoS Pathog. 2006;2:e62. doi: 10.1371/journal.ppat.0020062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simmers TA, Jie C, Sie MCTB. Thymic tuberculosis: A case report. Neth J Med. 1997;51:87–90. doi: 10.1016/s0300-2977(97)00036-3. [DOI] [PubMed] [Google Scholar]
- 28.Stephen T, Thankachen R, Parihar B, Nair S, Shukla V. Multilocular tuberculous cyst of thymus gland. J Thorac Cardiovasc Surg. 2003;126:2093–2094. doi: 10.1016/j.jtcvs.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–266. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 30.Cardona PJ. RUTI: a new chance to shorten the treatment of latent tuberculosis infection. Tuberculosis (Edinb) 2006;86:273–289. doi: 10.1016/j.tube.2006.01.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.