

Abstract
A series of novel (E)-N-aryl-2-arylethenesulfonamides (6) were synthesized and evaluated for their anticancer activity. Some of the compounds in this series showed potent cytotoxicity against a wide spectrum of cancer cell-lines (IC50 values ranging from 5 to 10 nM) including all drug resistant cell-lines. Nude mice xenograft assays with compound (E)-N-(3-Amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6t) showed dramatic reduction in tumor size indicating their in vivo potential as anticancer agents. A preliminary drug development study with compound 6t is predicted to have increased blood-brain barrier permeability relative to many clinically used anti-mitotic agents. Mechanistic studies indicate that 6t and some other analogs disrupted microtubule formation, formation of mitotic spindles and arrest of cells in mitotic phase. Compound 6t inhibited purified tubulin polymerization in vitro and in vivo and circumvented drug resistance mediated by P-glycoprotein. Compound 6t specifically competed with colchicine binding to tubulin and with similar avidity as podophylltoxin indicating its binding site on tubulin.
INTRODUCTION
Microtubules, formed by α- and β-tubulin heterodimers, are essential constituents of the cytoskeleton in eukaryotic cells and are involved in a number of important structural and regulatory functions, including the maintenance of cell shape, intracellular transport machinery, as well as cell growth and mitosis.1 Their highly ordered structures, rigidity, and their ability to grow and shrink via polymerization/depolymerization mechanisms are critical to their function in several cellular processes. Perhaps the most important role of microtubules is during mitosis, where they serve to organize and segregate chromosomes. Tubulin is the major structural component of the microtubules and a well verified target for a variety of highly successful anticancer drugs.2 Thus, vinca alkaloids 1 (vincristine and vinblastine)3 have been successfully used for the therapy of hematological disorders for the past three decades and the seminal discovery of paclitaxel (Taxol) by Wani and Wall4 in 1971 had a profound impact on the treatment of breast and ovarian cancers. The success of these agents has also led to the identification of several new tubulin interactive agents that have found application in cancer chemotherapy.5 Based on the mechanism of action of alternation of microtubule dynamics, drugs can be classified into two categories. Amongst the compounds (Chart 1) that inhibit tubulin polymerization and destabilize microtubules are the combretastatins (2), colchicine (3), Plinabulin (NPI-2358) (4), podophyllotoxin (5), curcumin and the vinca (1) alkaloids, and those that promote the polymerization of tubulins and stabilize the microtubules in their polymerized state include discodermolide, eleutherobins, the epothilones, laulimalide, the sarcodictyins, and the taxanes.6 Both microtubule stabilizers and destabilizers alter the tubulin-microtubule equilibrium causing mitotic arrest at G2/M phase7 and ultimately apoptotic cell death. Because of the clinical success of microtubule-affecting compounds such as paclitaxel, the vinca alkaloids, and epothilone derivatives in the treatment of a wide variety of cancers, it has been argued that microtubules represent the single most important protein target for anticancer therapy.8
Chart 1.

Structures of microtubule depolymerizing agents
These antimitotic drugs, however, are not without limitations. Many, including paclitaxel and the vinca alkaloids, are large (MW>700Da) natural products that display ADME-Tox shortcomings (including poor water solubility, bioavailability, and significant dose-limiting toxicity). In addition, a common problem observed with this class of compounds is that these large natural products are substrates for efflux pumps of the ABC transporter family, such as P-glycoprotein (P-gp) and multidrug resistance (MDR) proteins that can alter their pharmacokinetic characteristics.9 Furthermore, drugs such as taxanes are typically poor chemotherapeutics for the treatment of many brain cancers, as high levels of P-gp in the blood-brain barrier (BBB) and the chemical properties of the molecules themselves prevent significant accumulation of drug in the brain. Because of these factors, there has been an intense search for more effective antimitotics.10
A variety of synthetic small molecules have also been reported11 as inhibitors of polymerization, which compete with the colchicine-binding site of tubulin.12 While no colchicine-site binders are currently approved for cancer chemotherapy, compounds like combretastatin A-4P (CA4P) (2b),13 AVE-8062 (2c),14 ZD6126 (6),15 ABT-751(7),16 T-138067 (8),17 N-(3-Hydroxy-4-methoxyphenyl)-3,4,5-trimethoxybenzenesulfonamide (9) 18 and 1-Methyl-1H-indole-5-sulfonic acid (3,4,5-trimethoxyphenyl)amide (10) 19 are now under clinical investigation as potential new chemotherapeutic agents (Chart 1).20 However, a report of the activity and SAR information for these compounds, especially in vivo efficacy, is limited.
Hence herein we report the synthesis, in vitro evaluations, cell cycle progression and structure activity relationship (SAR) of (E)-N-aryl-2-arylethenesulfonamides, which cause cell death through destabilization of microtubules. In this study, we also report the caspase activation and tubulin depolymerization study along with blood-brain barrier (BBB) permeability of the active compound (E)-N-(3-Amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6t).
CHEMISTRY
The syntheses of (E)-N-aryl-2-arylethenesulfonamides (6) were achieved by multiple synthetic routes as illustrated in Schemes 1, 5 & 6. The initial method involved for the synthesis of 6 by the condensation of chlorosulfonylacetic acid ethyl ester (2) with various anilines 1 in the presence of triethylamine in DCM to obtain the arylsulfamoylacetic acid ethyl esters (3) in high yields. Hydrolysis of 3 with 10% NaOH in water afforded the corresponding arylsulfamoylacetic acids (4) in good yields. Knovenagel condensation of 4 with various aromatic aldehydes 5 either in benzylamine/acetic acid21 or piperidine/benzoic acid22 in toluene afforded 6 in good yields (Scheme 1). All 3-nitro and 3,5-dinitro substituted arylethenesulfonamides were reduced to their corresponding amino analogs either with sodium hydrosulfite in acetone/water or with iron powder in methanol/acetic acid as shown in the Scheme 2.23 The substituted anilines 1g and 1j which are not available commercially were synthesized as shown in the Scheme 3. 1g was prepared by the treatment of 4-fluoro-3-nitro-aniline (7) with 6% methanolic KOH in absolute methanol at room temperature to afford the required 1g in moderate yields. The dinitro aniline 1j was synthesized starting from 4-fluorobenzoic acid (8) by nitration with fuming nitric acid and 30% sulfuric acid to get 3,5-dinitro-4-fluorobenzoic acid (9) which on treatment with 20% oleum and ethylene dichloride in presence of sodium azide afforded 3,5-dinitro-4-fluoro aniline (10). Treatment of 10 with 6% methanolic KOH in absolute methanol gave the required 3,5-dinitro-4-methoxy aniline (1j) in moderate yields (Scheme 3).24 Commercially not available chlorosulfonylacetic acid ethyl ester (2) was in turn made as shown in Scheme 4. Treatment of ethyl bromoacetate (11) with sodium sulfite gave sodium ethoxycarbonylmethanesulfonate (12) which on reaction with PCl5 at 100 °C resulted in 2 in moderate yields.25 The 2 was also prepared directly by the esterification of chlorosulfonyl acetyl chloride (13) with absolute ethanol in diethyl ether in 55% yield.26
Scheme 1.

Synthesis of (E)-N-Aryl-2-arylethenesulfonamides (6) from N-Arylsulfonylacetic Acidsa
a Reagents and conditions: (a) Et3N, DCM, room temp, 3 h, 80%; (b) 10% NaOH, H2O, room temp, 3 h, 84%; (c) C6H5CH2NH2, AcOH, reflux, 8 h, 50%; (d) piperidine, benzoic acid, toluene, reflux, 4 h, 60%.
Scheme 5.

Synthesis of (E)-N-Aryl-2-arylethenesulfonamides (6) from Phenacyl N-Arylsulfonesa
a Reagents and conditions: (a) SOCl2, DCM, room temp, 6 h, 81% or (COCl)2, DMF, anhydrous CH2Cl2, room temp, 12 h, 67%; (b) AlCl3, DCM, room temp, 4 h, 80%; (c) NaBH4, THF, room temp, 3 h, 88%; (d) p-TSA, toluene, reflux, 3 h, 78%.
Scheme 6.

Synthesis of (E)-N-Aryl-2-arylethenesulfonamides (6) from (E)-Styryl sulfonyl chloridea
a Reagents and conditions: (a) SO2Cl2, DMF, 0 °C to room temp, 3 h, 90%; (b) Et3N, DCM, room temp, 5 h, 85%; (c) pyridine, room temp, 6 h, 80%.
Scheme 2.
Conversion of 3-Nitro and 3,5-Dinitro N-Aryl-2-arylethenesulfonamides to corresponding Amines (6) a
aReagents and conditions: (a) Sodium hydrosulfite, acetone / water (2:1), 50 °C, 30 min, 40%; (b) Iron powder, MeOH / AcOH (2:1), 80 °C, 2 h, 55%.
Scheme 3.
Synthesis of 4-Methoxy-3-nitro Aniline (1g) and 4-Methoxy-3,5-dinitrophenyl Amine (1j) a
aReagents and conditions: (a) absolute methanol, 6% methanolic KOH, room temp, 30 min, 50%; (b) fuming H2SO4 (30%), HNO3 (90%), 95 °C, 3 h, 70%; (c) fuming H2SO4 (20%), ClCH2CH2Cl, NaN3, reflux, 1 h, 70%.
Scheme 4.
Synthesis of Chlorosulfonylacetic acid ethyl ester (2)a
a Reagents and conditions: (a) Na2SO3, H2O, EtOH, 50 °C, 30 min, 70 %; (b) PCl5, 100 °C, 45 min, 85%; (c) EtOH, diethyl ether, 0 °C, 3 h, 55%.
Alternatively, the sulfonamides 6 were also prepared by acylation of the acids 4d, 4g or 4k by thionyl chloride in DCM to get the corresponding acid chlorides 14a-c which on Friedel-Crafts acylation with 1,3,5-trimethoxy benzene (15) afforded 2-aryl-2-oxoethanesulfonic acid aryl amides (16a-c). Sequential reduction of 16 with sodium borohydride and subsequent dehydration with p-toluenesulfonic acid (p-TSA) afforded the desired product 6 in moderate yields (Scheme 5).27 To explore for the simple reaction conditions, less number of steps to obtain the targeted compounds and better overall yields, sulfonamides 6 were also prepared by the condensation of anilines (1a-c) with 2-arylethenesulfonyl chlorides (19a-c) in the presence of triethylamine in DCM in good yields. The sulfonyl chlorides 19 were in turn made by the addition of sulfuryl chloride to styrenes (18a-c)27c in DMF at 0 °C to room temperature to get the desired product in quantitative yields (Scheme 6). The method described in Scheme 6 is superior to the other two methods as it involves less steps, cheaper chemicals and relatively higher yields.
To enhance the bioavailability of the active sulfonamide (E)-2-(2,4,6-trimethoxyphenyl)-2-(3-hydroxy-4-methoxyphenyl)ethanesulfonamide (6p), we explored to make its ester analog 6q. Condensation of 3-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-3-methylbutanoic acid (20) with 6p in the presence of EDAC and DMAP in dichloromethane resulted in ester (E)-2-methoxy-5-(2-(2,4,6-trimethoxyphenyl)vinylsulfonamido)phenyl 2-(2,5-dimethyl-3,6-dioxo-cyclohexa-1,4-dienyl)-2-methylpropanoate (6q) in moderate yields (Scheme 7). The benzoquinone ester 20 in turn was synthesized from 2,5-Dimethyl[1,4]benzoquinone (21) in presence of 3,3-dimethylacrylic acid, aqueous sodiumhydrosulfite and methane sulfonic acid in diethyl ether at 85 °C to yield 6-Hydroxy-4,4,5,8-tetramethylchroman-2-one (22) in 59% yield. N-Bromosuccinimide ring opening reaction of 22 afforded the target compound 20 in moderate yields (Scheme 8).28
Scheme 7.

Synthesis of (E)-2-Methoxy-5-(2-(2,4,6-trimethoxyphenyl)vinylsulfonamido)phenyl 2-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-2-methylpropanoate (6q)
Scheme 8.
Synthesis of 3-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-3-methylbutanoic acid (20)a
a Reagents and conditions: (a) diethyl ether, aq. Na2S2O4, (CH3)2C=CHCOOH, CH3SO3H, 85 °C, 3 h, 81%; (b) CH3CN, acetone, water, NBS, room temp, 30 min, 60%.
To further enhance the bioavailability and water solubility of the active sulfonamide (E)-2-(2,4,6-trimethoxyphenyl)-2-(3-hydroxy-4-methoxyphenyl)ethenesulfonamide 6p, its disodium phosphate prodrug was synthesized in two steps as shown in the Scheme 9. Phosphorylation of the phenolic group in 6p employing phosphorous oxychloride under basic conditions gave 3-O-phosphate 23. Treatment of the phosphate 23 with 25% aq. sodium hydroxide in ethylene glycol dimethyl ether yielded disodium O-phosphate 6r (Scheme 9).29
Scheme 9.

Synthesis of Sodium (E)-2-Methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfon-amido) phenyl phosphate (6r)a
aReagents and conditions: (a) phosphorus oxychloride, THF, triethylamine, 0 °C-room temp, 3 h; (b) water, room temperature,18 h, 69%; (c) 25% aq.NaOH, CH3OCH2CH2OCH3, 0 °C-room temp, 3 h, 85%.
To further enhance the bioavailability and water solubility of the active sulfonamide 6t, several 3- amino substituted esters and acids were made by the reaction of α-bromo esters in the presence of mild base sodium acetate in ethanol to give amine esters (24) which on subsequent hydrolysis with sodium hydroxide in ethanol afforded the corresponding acids 25 (Scheme 10).30
Scheme 10.
Synthesis of amine esters and acids of (E)-N-(3-Amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6t)
Structure-Activity Relationships (SAR)
The newly synthesized compounds were tested for their in vitro cytotoxicity activity against two different human cell lines derived from human prostate (DU145) and leukemic (K562) cancers. The study results are presented in Table 1. These studies reveal that the cytotoxicity of the N-aryl-2-arylethenesulfonamide (6) is totally dependent on the nature and position of the substituents present on the two aromatic rings. For the purpose of structure-activity relationship, we have selected few compounds from a library of more than 500 arylethenesulfonamides synthesized in our laboratory. The cytotoxicity data (Table 1) clearly shows that the molecules are less active when the aromatic rings are without any substitution or mono substituted with halogens or with a combination of a methoxy and a halogen atom (6a-6d). The improvement in the cytotoxicity profile was observed when each aromatic ring has a methoxy group on it (6e, 6f). Based on this observation, additional methoxy groups were added on the styryl aromatic ring keeping a methoxy group constantly at the 4th position of the aryl sulfamyl ring to evaluate the influence of these groups on the cytotoxicity of the molecules (6g-6i). From the cytotoxicity data (Table 1) it is clear that 2,4 and 2,6-dimethoxy substitutions (6g, 6h) moderately enhanced the potency of the molecules while 2,4,6-trimethoxy substitution (6i) enhanced the cytotoxicity potency to several folds higher in the molecule. Altering the positions of the methoxy groups from 2,4,6 to 3,4,5 (6j) resulted in the loss of the cytotoxicity indicating that the optimum activity could be achieved only when the substitution pattern is 2,4,6 on the styryl aromatic ring. Also to evaluate the importance of the location of 2,4,6-trimethoxy groups on styryl aromatic ring, we switched these methoxy groups on to sulfamyl aromatic ring and tested the resulting molecule (6k) for the cytotoxicity. The loss of cytotoxicity activity in 6k clearly shows that the trimethoxy groups not only should be on the styryl aromatic ring but also at 2,4,6- positions. Further to make 6i more water soluble we replaced the methoxy group with a hydroxyl group at the 4th position of the 2,4,6-trimethoxy styryl ring and the resulted molecule (6l) lost the activity indicating that hydrophobic group at that position is required for the activity of the molecule. When fluorine atoms are introduced in place of 2,4,6-trimethoxy groups on 6i, the resulting molecules (6m, 6n) lost their cytotoxic potential on cancer cells showing that the methoxy groups at those positions are critical for the activity of the molecule. After fixing the styryl aromatic ring with 2,4,6-trimethoxy groups to attain the best activity, we then focused our attention on to aryl sulfamyl ring of the molecule. Addition of a fluorine atom at the third position of the sulfamyl ring in 6i retained the activity of the molecule (6o) indicating that the meta position in the ring is vulnerable and can be used for introducing groups that enhance water solubility and bioavailability without affecting the potency of the molecule. Replacing the fluorine in 6o with a hydroxyl group resulted in a potent molecule (6p) that has 5–7 folds higher cytotoxicity than 6o. Conversion of 6p into dimethyl quinone prodrug (6q) enhanced the cytotoxicity of the molecule compared to 6p probably due to better cell permeability compared to 6p. But the solubility of 6q in aqueous buffers has not much improved compared to 6p. Also introduction of the hydroxyl group on the sulfamyl ring created the possibility of producing a highly water-soluble disodium phosphate salt (6r) having more or less same potency as that of 6p. Further attempts to enhance the cytotoxicity with an electron withdrawing nitro group at 3rd position of the sulfamyl ring resulted in a molecule (6s) that is several folds less active than 6p, 6q and 6r. Surprisingly reduction of the nitro group (6s) to an electron releasing amino group resulted in the formation of the most active molecule (6t) of this series. Any other alterations and modifications on 6t resulted in molecules (6u-6z and 6aa-6aj) with reduced or total loss of cytotoxicity indicating that the best activity in N-aryl-2-arylethenesulfonamide series could be obtained only when a molecule bears 3-amino, 4-methoxy on the arylsulfamyl ring and 2,4,6-trimethoxy groups on the styryl ring.
Table 1.
In vitro Cytotoxicity of (E)-N-Aryl-2-arylethenesulfonamides (6)
| ||||
|---|---|---|---|---|
| Compd | R | R1 | IC50(μM)a | |
| DU145 | K562 | |||
| 6a | H | H | 10 | 10 |
| 6b | 4-Cl | H | 20 | 20 |
| 6c | 4-F | 4-Br | 10 | 10 |
| 6d | 4-F | 4-OCH3 | 10 | 10 |
| 6e | 4-OCH3 | 2-OCH3 | 5 | 5 |
| 6f | 4-OCH3 | 4-OCH3 | 5 | 5 |
| 6g | 4-OCH3 | 2,4-(OCH3)2 | 15 | 15 |
| 6h | 4-OCH3 | 2,6-(OCH3)2 | 0.375 | 5 |
| 6i | 4-OCH3 | 2,4,6-(OCH3)3 | 0.2 | 0.075 |
| 6j | 4-OCH3 | 3,4,5-(OCH3)3 | 35 | 35 |
| 6k | 2,4,6-(OCH3)3 | 4-OCH3 | 7.5 | 2.5 |
| 6l | 4-OCH3 | 2,6-(OCH3)2,4-OH | 10 | 10 |
| 6m | 4-OCH3 | 2,4,6-F3 | 75 | 15 |
| 6n | 4-OCH3 | 2,3,4,5,6-F5 | 10 | 5.0 |
| 6o | 3-F,4-OCH3 | 2,4,6-(OCH3)3 | 0.2 | 0.075 |
| 6p | 3-OH,4-OCH3 | 2,4,6-(OCH3)3 | 0.03 | 0.015 |
| 6q | 3-OCOCH2C(CH3)2-C6H(CH3)2O2, 4-OCH3 | 2,4,6-(OCH3)3 | 0.009 | 0.008 |
| 6r | 3-OPO(ONa)2,4-OCH3 | 2,4,6-(OCH3)3 | 0.04 | 0.0075 |
| 6s | 3-NO2,4-OCH3 | 2,4,6-(OCH3)3 | 2.5 | 1.0 |
| 6t | 3-NH2,4-OCH3 | 2,4,6-(OCH3)3 | 0.005 | 0.003 |
| 6u | 3-NO2,4-OCH3 | 3,4,5-(OCH3)3 | 75 | 75 |
| 6v | 3-NH2,4-OCH3 | 3,4,5-(OCH3)3 | 35 | 15 |
| 6w | 3-NO2,4-OCH3 | 2,6-(OCH3)2,4-O(CH2)3COOH | 100 | 100 |
| 6x | 3-NH2,4-OCH3 | 2,6-(OCH3)2,4-O(CH2)3COOH | 10 | 10 |
| 6y | 3-OH,4-OCH3 | 2,6-(OCH3)2,4-O(CH2)3COOH | 10 | 10 |
| 6z | 3-NO2,4-F | 2,4,6-(OCH3)3 | 100 | 75 |
| 6aa | 3-NH2,4-F | 2,4,6-(OCH3)3 | 10 | 5 |
| 6ab | 3,5-(NO2)2,4-OCH3 | 2,4,6-(OCH3)3 | 10 | 10 |
| 6ac | 3,5-(NH2)2,4-OCH3 | 2,4,6-(OCH3)3 | 2.5 | 7.5 |
| 6ad | 3-F,4-OCH3 | 4-OCH3 | 5 | 1 |
| 6ae | 3-F,4-OCH3 | 2,3,4,5,6-F5 | 10 | 10 |
| 6af | 3-NO2,4-OCH3 | 2,3,4,5,6-F5 | 100 | 100 |
| 6ag | 3-NH2,4-OCH3 | 2,3,4,5,6-F5 | 75 | 75 |
| 6ah | 2,3,4,5,6-F5 | 3-NO2,4-OCH3 | 100 | 75 |
| 6ai | 2,3,4,5,6-F5 | 3-NH2,4-OCH3 | 35 | 35 |
| 6aj | 2,3,4,5,6-F5 | 2,3,4,5,6-F5 | 35 | 35 |
IC50 values are the compound concentrations (μM) required to inhibit cell proliferation by 50% of tumor cells following 96 h treatment with the tested compound; values represent the mean SD from the dose response curves of two independent experiments and are within 5–10%.
After identifying the most potent molecule (6t) in the series, we made several modifications on the amino group of 6t to make it more water-soluble and bioavailable. Alkylation of the meta-amino group in 6t with methyl bromoacetate and 2-alkyl, aryl substituted bromoacetates produced 3-glycine esters (24a-g) which on hydrolysis resulted in acids (25a-g). All the glycine analogs (25a-g) showed enhanced water solubility (10–20 mg/mL) and excellent cytotoxicity (Table 2) compared to the glycine esters (24a-g).
Table 2.
In vitro cytotoxicity of Amine esters (24) and Acids (25)
| |||
|---|---|---|---|
| Compd | R2 | IC50 (μM)a | |
| DU145 | K562 | ||
| 24a | CH2 | 0.35 | 0.35 |
| 24b | CH(CH3) | 0.1 | 0.30 |
| 24c | C(CH3)2 | 0.2 | 0.30 |
| 24d | CH(C6H5) | 2.5 | 2.5 |
| 24e | CH(C6H44-F) | 2.5 | 2.5 |
| 24f | CH(C6H44-Cl) | 5.0 | 2.5 |
| 24g | CH(C6H44-Br) | 7.5 | 1.0 |
| 25a | CH2 | 0.4 | 0.02 |
| 25b | CH(CH3) | 0.04 | 0.008 |
| 25c | C(CH3)2 | 0.07 | 0.005 |
| 25d | CH(C6H5) | 0.075 | 0.30 |
| 25e | CH(C6H44-F) | 0.075 | 0.02 |
| 25f | CH(C6H44-Cl) | 0.075 | 0.015 |
| 25g | CH(C6H44-Br) | 0.25 | 0.075 |
IC50 values are the compound concentrations (μM) required to inhibit cell proliferation by 50% of tumor cells following 96 h treatment with the tested compound; values represent the mean SD from the dose response curves of two independent experiments and are within 5–10%.
BIOLOGICAL RESULTS AND DISCUSSION
In vitro Anti-tumor Effects of 6i, 6p, 6t, and 25c Compounds
Screening of the novel arylethenesulfonamide compound library of molecules for anti-tumor activity using cell biological assays yielded several candidate molecules that induced dose-dependent growth inhibition and death of tumor cells (Tables 1 & 2). To identify the arylethenesulfonamides with the broadest range of activity in a wider selection of cancer cell types we chose four of the most potent compounds from the series shown in Chart 2 and found high potency with similar GI50 values (selected data shown in Table 3). The appreciable cell killing across multiple tumor types suggests that these compounds are inhibiting an intrinsically important process of tumor cell division. 6t was found to be the most active of the four compounds exhibiting IC50 values of between 0.003 – 0.01 μM (Table 3), and on that basis, we further investigated the mechanism of action of this compound.
Chart 2.

Chemical structures of lead compounds
Table 3.
In vitro cytotoxicity profiles of most active molecules 6i, 6p, 6t and 25c in various tumor cell lines.
| Cell Line | Tumor Type | IC50 (>M)a
|
|||
|---|---|---|---|---|---|
| 6i | 6p | 6t | 25c | ||
| DU145 | Prostate | 0.075 | 0.050 | 0.008 | 0.15 |
| K562 | Leukemia | 0.075 | 0.015 | 0.003 | 0.01 |
| LNCAP | Prostate | 0.100 | 0.050 | 0.008 | 0.10 |
| PC3 | Prostate | 0.100 | 0.025 | 0.008 | N/D |
| SK-MEL-28 | Melanoma | 0.050 | 0.025 | 0.008 | N/D |
| MCF-7 | Breast | 0.050 | 0.025 | 0.004 | 0.08 |
| SK BR-3 | Breast | 0.050 | 0.025 | 0.004 | N/D |
| BT474 | Breast | 0.250 | 0.025 | 0.010 | 0.15 |
| BT20 | Breast | 0.250 | 0.250 | 0.008 | 0.10 |
| T47D | Breast | 0.100 | 0.025 | 0.008 | N/D |
| A431 | Epidermoid | 0.030 | 0.600 | 0.005 | 0.15 |
| HCT116 | Colo-Rectal | 0.100 | 0.010 | 0.004 | N/D |
| HCT15 | Colo-Rectal | 0.100 | 0.025 | 0.008 | 0.20 |
| SKOV3 | Ovarian | 0.100 | 0.025 | 0.008 | N/D |
| OVCAR3 | Ovarian | 0.100 | 0.100 | 0.004 | N/D |
| H187 | NSCLC | 0.100 | 0.010 | 0.004 | N/D |
| N417 | SCLC | 0.050 | 0.010 | 0.004 | 0.03 |
| RF-48 | Gastric | 0.025 | 0.010 | 0.004 | N/D |
| RF-1 | Gastric | 0.050 | 0.010 | 0.004 | N/D |
| MIAPaCa2 | Pancreatic | 0.100 | 0.010 | 0.004 | 0.08 |
| H80 | Glioma | 0.100 | 0.025 | 0.004 | N/D |
| MES-SA | Uterine | 0.050 | 0.025 | 0.004 | N/D |
| NAMALWA | Lymphocytic | 0.100 | 0.025 | 0.008 | N/D |
| DAUDI | Lymphocytic | 0.100 | 0.025 | 0.008 | 0.03 |
| MOLT-4 | Lymphocytic | 0.100 | 0.025 | 0.008 | 0.03 |
| U87 | Gliomab | 0.229 | 0.026 | 0.009 | 0.09 |
GI50 (growth inhibitory concentration-50%). Cells were treated with 6i, 6p, 6t and 25c in a 96 h dose response assay. The GI50 concentration (μM) was determined from duplicate cell counts using a Hemocytometer following trypsinization and trypan blue staining.
Values are IC50 as measured by SRB assay. ND = not determined
Effects of 6t on Cell Cycle Progression of Tumor Cells
The effect of 6t on cell cycle progression in DU145 human prostate cancer cells using fluorescence-activated cell sorting (FACS) analysis indicated that the addition of 6t to tumor cells resulted in gradual accumulation of cells in the G2/M phase of the cell cycle in a concentration-dependent manner. Figure 1 shows that tumor cells accumulated in the G2/M phase of the cell cycle at 0.005 μM concentration of 6t, and a majority of cells showed G2/M arrest at a 0.02 μM concentration, whereas a normal cell cycle distribution was seen in the vehicle treated cells. In addition, treatment of the tumor cells with 6t resulted in an accumulation of cells containing subG1 content of DNA which is an indication of mitotic arrest.31
Figure 1.
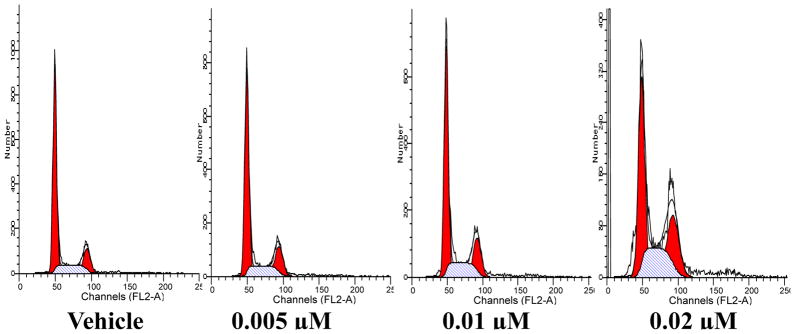
Cell cycle arrest and apoptosis of DU145 cells treated with 6t. DU145 cells were treated with various concentrations of 6t for 24 h and subjected to flow cytometry following propidium iodide staining. DU145 cells were arrested in the G2/M phase of the cell cycle.
Compound 6t Treatment Induces Abnormal Mitotic Spindle Development
We tested if 6t was able to depolymerize cellular microtubules in the same way as other depolymerizing agents. Exposure of DU145 cells to different concentrations of 6t for 24 h led to a complete depolymerization of the microtubule cytoskeleton. Confocal laser microscopy showed that while DMSO-treated cells (vehicle) went through various phases of mitosis without any abnormality, 6t treated cells exhibited profound abnormalities in spindle-formation, resulting in the appearance of abnormal spindles, misalignment of chromosomes and complete loss of co-ordination in mitotic spindle assembly (Figure 2). Some cells were micro-nucleated, and others were arrested in pro-metaphase with a ball or rosette of condensed DNA without a mitotic spindle (type IV spindle).32 The mitotic arrest caused by 6t was accompanied by net microtubule depolymerization. These data suggested that 6t acts as a mitotic inhibitor by blocking cell cycle progression at a time between pro-metaphase and metaphase by its ability to disrupt spindle assembly that led to the caspase activation and apoptosis as evidenced by PARP [Poly(ADP-ribose) polymerase-1] cleavage 33 (Figure 3). Treatment of 6t selectively induced PARP cleavage in the tumor cell line while there was no PARP cleavage in the treated normal cell line (data not shown). These results correlate quite well with those obtained in tubulin assembly experiments.
Figure 2.
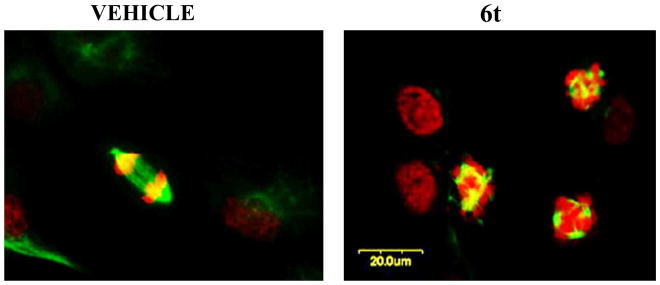
DU145 cells were plated onto glass coverslips and exposed to either 25 nM 6t or DMSO and then fixed, stained with FITC conjugated (green) anti-tubulin antibodies and propidium iodide (red) and analyzed by confocal microscope (Olympus). 6t inhibits the normal formation of mitotic microtubules.
Figure 3.
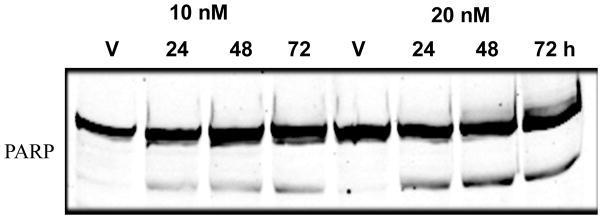
DU145 cells were treated with the indicated concentrations of 6t and harvested at the indicated times. Protein lysates were subjected to western blotting against PARP antibodies. Full length PARP gets cleaved in the 6t treated cells indicating the cells are undergoing apoptosis.
Compound 6t Destabilizes Tubulin Polymerization by Binding to the Colchicine Binding Site on Tubulin
Since chromosomal architecture showed abnormalities in spindle formation, we examined whether 6t effected polymerization of tubulin in vitro. For these assays, the spontaneous polymerization of purified bovine brain tubulin was measured to determine if the antimitotic properties of 6t were due to either stabilization or destabilization of microtubule polymerization. The extent of tubulin polymerization was determined spectrophotometrically by the increasing absorbance at 340 nM. DMSO vehicle is known to slightly stabilize microtubules and produces an intermediate polymerization phenotype.34 As expected, the microtubule stabilizer paclitaxel induced a strong increase in turbidity, whereas nocodazole destabilized growing microtubules such that depolymerization at the (-)-end is faster than polymerization at the (+)-end, giving rise to a net “depolymerization” phenotype and no rise in turbidity (Figure 4A). Attempted polymerization of tubulin in the presence of 6t produced a depolymerization phenotype, suggesting that 6t and related arylethenesulfonamides, 25a and 25c, caused cytotoxicity through destabilization of microtubule polymerization (see Figure 4B). The ability of 6t compounds to compete for known binding sites on tubulin was determined using a mass spectrophotometric (MS) competitive binding assay.35 Three tubulin ligands, corresponding to the 3 binding sites on tubulin, colchicine, vinblastine, and paclitaxel were used for these competitive binding studies. 6t (IC50 = 3.68 μM) specifically competed with colchicine binding to tubulin and with similar avidity as podophylltoxin, (IC50 = 3.71 μM), but it did not compete with either vinblastine or paclitaxel binding to tubulin (Figure 5). Known tubulin binding ligands, podophylltoxin, vincristine and docetaxel effectively competed for the colchicine-, vinblastine- and paclitaxel-tubulin binding sites, respectively, indicating the validity of the MS binding assay.
Figure 4.
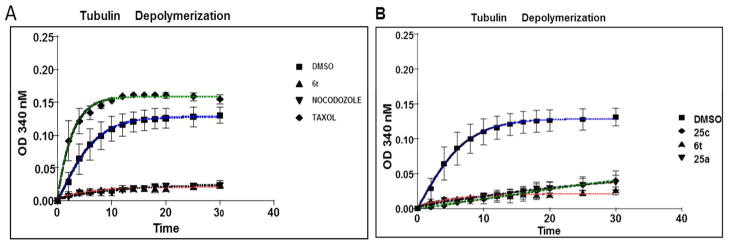
6t inhibit in vitro tubulin polymerization. A. Purified tubulin was incubated in the presence of 10 μM concentration of taxol, nocodazole, 6t and DMSO at 37 °C. The extent of tubulin polymerization was determined by the increasing absorbance at 340 nM over 40 min. 6t and nocodazole (a known tubulin depolymerizer) were able to completely inhibit tubulin polymerization compared to Taxol (tubulin polymerizer) and DMSO suggesting that 6t interfere with normal tubulin polymerization kinetics. Data shown are representative of three independent experiments. B. Purified tubulin was incubated in the presence of 10 μM concentration of 6t, 25a, 25c and DMSO at 37 °C. The extent of tubulin polymerization was determined by the increasing absorbance at 340 nM over 40 min. All three compounds were able to completely inhibit tubulin polymerization suggesting that these compounds interfere with normal tubulin polymerization kinetics. Data shown are representative of three independent experiments.
Figure 5.

Tubulin competitive binding studies. 6t competes with colchicine (A), but not with vinblastine (B) and paclitaxel (C) to bind to tubulin. In panel A and B, tubulin was incubated with colchicine or vinblastine (1.2 μM) in the absence of GTP with increasing concentrations of the test compounds. In panel C, performed microtubules were incubated with paclitaxel (1.2 μM) and 1 mM GTP, and increasing concentration of the test compounds. Podophyllotoxin, vincristine and docetaxel were used as positive controls for competitive binding with colchicine, vinblastine and paclitaxel, respectively.
In Vivo Tubulin Polymerization of 6t
To determine whether this compound also inhibits tubulin polymerization in vivo, we treated human prostate cancer cells (DU145) with increasing concentrations of 6t for 24 h and determined the extent of polymerized tubulin present in cell lysates. This assay, based on differential precipitation of depolymerized (soluble) and polymerized (precipitated) tubulin, detects the abundance of polymerized tubulin (spindle formation) present in normal mitotic cells. Paclitaxel and colchicine were used as controls since paclitaxel promotes tubulin polymerization while colchicine induces de-polymerization of tubulin. Figure 6A shows that 6t caused a concentration-dependent depolymerization of tubulin in treated cells, similar to colchicine. Furthermore, 6t caused tubulin depolymerization even in cells pre-treated with paclitaxel for 24 h that had high levels of polymerized tubulin; colchicine had a similar effect (Figure 6B). These studies clearly indicate that 6t acts as a tubulin-depolymerizing agent and this property explains its ability to induce mitotic arrest of human tumor cells which results in their apoptotic death.
Figure 6.
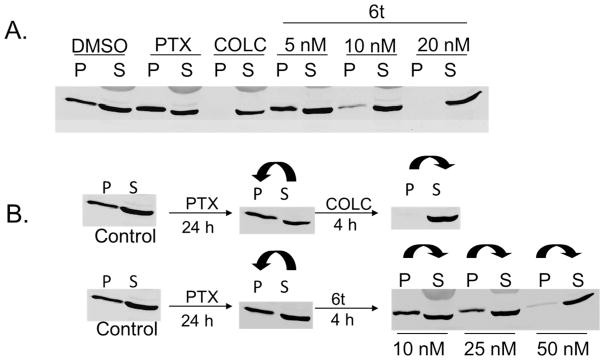
6t inhibits in vivo tubulin polymerization. A. DU145 cells were treated with vehicle (DMSO), paclitaxel (PTX), colchicine (COLC) or the indicated concentrations of 6t for 24 h and lysed in a hypotonic buffer. The pellet and supernatants were resolved by SDS-PAGE. The polymerized tubulin in the pellet (P) and the soluble tubulin in supernatant (S) were analyzed for presence of tubulin by western blotting. Like colchicine, a known tubulin depolymerizer, 6t treatment completely inhibits the polymerization of tubulin. B. DU145 cells were first treated with paclitaxel for 24 h and subsequently treated for 4 h with colchicine or 6t. As expected, paclitaxel treatment induced a shift from soluble to polymerized tubulin (see arrow) while both colchicine and 6t were both effective at depolymerizing paclitaxel induced polymerized tubulin (see arrows).
In vivo Anti-tumor Effects of 6t
6t was next tested for its anti-tumor activity in soft agar and nude mouse assays. For soft agar assays, we used a well-known pancreatic cancer cell line, MIA-Pa-Ca2. Treatment of these cells line with 6t resulted in inhibition of anchorage-independent growth of tumor cells in a dose dependent manner (Figure 7A). In these assays, paclitaxel was used as a positive control, which showed similar potency (data not shown). To examine the anticancer efficacy, we used ER-negative human breast tumor cells (BT-20) grown as xenografts in nude mice. Female athymic (NCr-nu/nu) mice were injected subcutaneously with 0.5–1×107 tumor cells in 0.2 mL of PBS and the tumors allowed to grow for 7–10 days to a size of 100–150 mm prior to treatment. The mice were then paired such that the pairs harbored equal sized tumors which were used to test the therapeutic effects of 6t. Of the pairs, one mouse received the vehicle alone while the second mouse received the compound by intraperitoneal (IP) injection. The tumor size was measured on alternate days for a total of 14 days. Figure 7B shows that 6t readily inhibited tumor growth in this xenograft model system. Of the 8 mice included in each group, 100% of the control mice (placebo administered) showed a doubling of the total tumor volume. On the other hand, most of the mice administered with 6t showed growth arrest or a gradual reduction in their tumor volume, suggesting that this compound could be valuable anti-cancer therapeutic. Measurement of the body weights during the experimental period showed no reduction in body weight indicated the safety profile of the drug.
Figure 7.
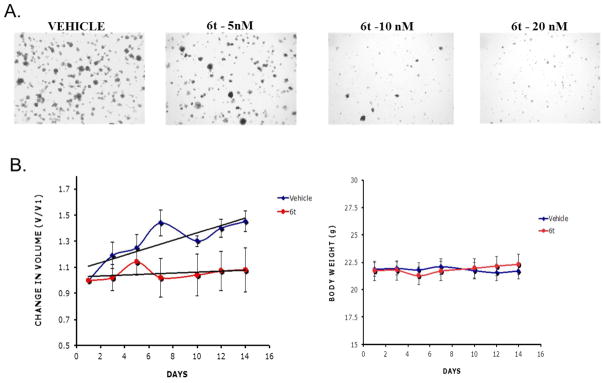
6t Inhibition of tumor growth. A. MIA-PaCa-2 cells were plated in soft agar containing increasing concentrations of 6t in triplicates. After three weeks of growth, the plates were stained for 48-h using 0.05% nitroblue tetrazolium solution. Representative plates were photographed using an Olympus stereoscope mounted with a Sony digital camera system (DKC 5000, Sony Inc). 6t inhibits the semi-solid growth of pancreatic cells indicating activity. B. Human estrogen receptor negative breast cancer cells (BT20) were implanted into the hind quarter of female nude mice. When the tumors became palpable, they were treated with either 20 mg/kg 6t or an equal volume of vehicle every other day for a total of 6 injections. The tumor volumes and body weights were determined and plotted (Blue diamonds: Vehicle; Red circles: 6t) as the average per group along with SEM values (Vehicle: N=7, 6t: N=8). Trend lines were added using linear regression model (Microsoft Excel).
Compounds 6t and 25c are Highly Active Against Drug Resistant Tumor Cell Lines
One major limitation of anticancer drug therapy is intrinsic and acquired multidrug resistance. Tumor expression of the ATP-dependent drug efflux pump, P-glycoprotein (Pgp), is associated with anticancer treatment failure.36 Many microtubulin poisons such as paclitaxel, vincristine, and vinblastine are substrates for the multidrug resistant family members. Drugs that are not substrates for the drug efflux pumps can overcome these mechanisms of resistance and be more efficacious. The ability of the arylethenesulfonamides, specifically 6t and 25c, to overcome drug-induced mechanisms of resistance was tested in pairs of isogenic cell line pairs (see Table 4). In all three pairs of sensitive and drug-resistant cancer cells, 6t and 25c exhibit greatly improved activity with resistance factors < 2. The uterine sarcoma cell line MES-SA and its multidrug resistant subline MES-SA/DX537 has been shown to express high levels of P-glycoprotein and is resistant to a number of drugs including doxorubicin, paclitaxel, vincristine, vinblastine, etoposide, mitoxantrone, dactinomycin, and daunorubucin; in our studies paclitaxel was 190-fold resistant based on GI50 values, whereas both 6t and 25c were nearly equally active. Similar phenomenon was seen in the parental leukemic cell line CEM and its MDR subline CEM/C238 that was selected for resistance to camptothecin and has cross-resistance to etoposide, dactinimycin, bleomycin, mitoxantrone, doxorubicin, and daunorubicin. Finally 6t was equally active in the ovarian carcinoma cell line 2008 and its paclitaxel-resistant clone 2008/17/4, which supports our contention that 6t, based on its activity in multiple drug-resistance cell lines, may be more effective in tumors that exhibit the MDR phenotype.
Table 4.
Evaluation of 6t and 25c against a panel of Multidrug- resistance Human Tumor Cell lines
| compd | cell Line | tumor type | GI50 (nM)a | resistance factor |
|---|---|---|---|---|
| Paclitaxel | MES-SA | sarcoma | 4 | 190 |
| MES-SA / DX5 | resistant sarcoma | 750 | ||
| 6t | MES-SA | sarcoma | 6 | 1.6 |
| MES-SA / DX5 | resistant sarcoma | 10 | ||
|
| ||||
| 25c | MES-SA | sarcoma | 70 | 1.3 |
| MES-SA / DX5 | resistant sarcoma | 90 | ||
|
| ||||
| Camptothecin | CEM | leukemic | 2 | 500 |
| CEM / C2 | resistant leukemic | 1000 | ||
| 6t | CEM | leukemic | 8 | 1 |
| CEM / C2 | resistant leukemic | 8 | ||
|
| ||||
| Paclitaxel | 2008 | ovarian | 3 | 600 |
| 2008 / 17/ 4 | resistant ovarian | 2000 | ||
| 6t | 2008 | ovarian | 6 | 1 |
| 2008 / 17/ 4 | resistant ovarian | 6 | ||
Cytotoxicity results are expressed as GI50 values, the compound concentrations producing 50% cell growth inhibition, and represent the mean ± SD of two independent experiments and are within 5–10%.
Pharmacological and Toxicological data of 6t
A study was conducted to assess the solubility, cytotoxicity, permeability, metabolic stability and protein binding of compound 6t (Table 5). 6t had moderate solubility in aqueous buffer with a limit of complete solubility (as measured by a statistical increase of light scattering over blank) of 62.5 μM measured by nephelometry. 6t was non-toxic to the human adenocarcinoma cell line, Caco-2 with an LD50 of >1000 μM (one hour incubation) in comparison to the negative control compounds, tamoxifen and 2-thiouracil that yielded LD50 values of 343 and >1000 μM, respectively. 6t Was also non-toxic to HepG2 cells, a human hepatocyte cell line, with an LD50 > 1000 μM following a 4 h exposure. Membrane permeability of 6t was determined using MDCK-MDR1 cells, a model for blood-brain barrier (BBB) permeability, as previously reported.39,40 Briefly, confluent MDCK-MDR1 monolayers expressing Pgp were obtained 3–4 days post-seeding and their integrity assessed by measurement of the transepithelial electrical resistance (TEER, Ω · cm2) with a Volt-Ohm Meter (Millicell-ERS, Millipore Corpration, Billerica, MA). After subtraction of the background TEER (i.e. the resistance exhibited by the filter alone) only MDCK-MDR1 cell monolayers that exhibited a TEER > 1000 Ω · cm2 throughout [measured before and after the study] the experiments were used. Drug transport across the cell monolayers was measured in both apical to basolateral (A-B) and basolateral to apical (B-A) directions based on 6t concentrations measured by LC/MS/MS. The starting concentration of 6t was 10 μM and the duration of the studies were 90 min. Apparent permeability values and the efflux ratio (ER) (see Table 5) indicated that 6t underwent passive transcellular diffusion with no appreciable active efflux that is normally indicated by ER values > 30. Metabolic stability of 6t in human, rat and dog S9 liver fractions showed that 6t was most extensively metabolized by human liver S9 followed by rat and dog with 20%, 33% and 57% of parent remaining following 1 h incubations, respectively. Binding of 6t to rat, dog and human plasma was measured by equilibrium dialysis at concentrations of 10 and 50 μM at 37 °C. 6t is extensively bound (>98%) to rat, dog and human plasma proteins.
Table 5.
Pharmacological and Toxicological data of 6t.
| Assay | Parameter | 6t |
|---|---|---|
| Solubility a | Maximum soluble concentration in PBS | 62.5 μM |
| Cytotoxicity b | LD50 to Caco-2 cells | >1000 μM |
| LD50 to HepG2 cells | >1000 μM | |
| Permeability c | A-B Papp x E06 | 9.1 cm/s |
| B-A Papp x E06 | 72 cm/s | |
| ER | 7.9 | |
| S9 Metabolic Stability (1h) c,d | Rat - % remaining | 33% |
| Dog -% remaining | 57% | |
| Human -% remaining | 20% | |
| Plasma Protein Binding (>M) c | Rat -% Bound | 99.73% |
| Dog - % Bound | 98.48% | |
| Human - % bound | 99.67% |
Determined by nephelometry
determined by fluorescence of Alamar Blue dye
quantification of samples was by LC-MS/MS.
S9 liver fractions microsomes (1 mg/mL) incubated for 0, 30 and 60 min with 10 μM concentration of 6t.
In vivo PK profile of 6t
Male ICR mice (N=3 per group) 6–8 weeks of age were used to examine the pharmacokinetics (PK) of 6t. The compound was formulated in NMP:PEG300: water at a volume ratio of 1:4:5 for both administrations at 5 mg/kg by intravenous bolus (IV, 20 μL total volume) and oral gavage (PO, 200 μL total volume). One day prior to drug administrations, a vascular cannula (vinyl tubing of I.D 0.28 mm × O.D 0.61 mm, Scientific Commodities Inc., Lake Havasu City, AZ) was surgically implanted in the right common carotid artery to allow for serial collection of blood (20 μL of blood per time point). For both 6t administrations, blood samples were collected at 2, 5, 15, 30, 60, 120, 240 and 360 min after administration. Plasma sample were prepared by centrifuging the blood samples at 8,000g for 5 min. All plasma samples were stored immediately at −80 °C until analyzed by LC/MS/MS. A protein precipitation method was used for sample preparation. An 80 μL aliquot of acetonitrile containing the internal standard was added to 20 μL of plasma and then thoroughly vortexed for 15 s. After centrifugation, the supernatant was analyzed by LC/MS/MS. The pharmacokinetic parameters were determined using noncompartmental analysis (WinNonlin 6.0, Pharsight Corporation, Mountain View, CA). Evan at this relatively low dose effective concentrations (> than GI50 concentrations) were achieved following single doses.
CONCLUSION
In summary, we have described the synthesis of a new series of tubulin polymerization inhibitors, which induce G2/M-phase cell cycle arrest leading to apoptotic cell death in a wide variety of human tumor cell lines at nanomolar concentrations by disrupting tubulin assembly. Our studies show that the cytotoxic activities of these compounds are completely dependent on the nature and position of the substituents on the two aromatic rings along with a substituted or unsubstituted sulfonamide group. SAR studies revealed that a molecule with an aryl sulfonamide moiety either with 3-hydroxy, 4-methoxy groups or 3-amino, 4-methoxy groups and a styryl ring with methoxy groups at 2, 4 and 6-positions, 6p and 6t, respectively, showed optimum biological activity. The dimethyl quinone ester (6q) and water soluble disodium phosphate (6r) of 6p showed very good activity in both the cell lines tested. The water soluble glycine analog (25c) of 6t showed superior activity among all the 3-amino substituted amino acids tested.
Preliminary mode of action studies demonstrated that the lead compound 6t arrests tumor cell cycle at G2/M-phase and induces apoptotic cell death by microtubule depolymerization and caspase activation. In vivo anti-tumor effect of 6t in soft agar and nude mouse assays showed that this compound demonstrated considerable regression in tumor growth in a xenograft model. The fact that 6t does not appear to be a substrate of P-glycoprotein and other MDR-mediated efflux pumps based on cytotoxicity and cell permeability assays suggests 6t may penetrate the BBB sufficiently to be active in brain tumors, tumors not amenable to therapy by other natural product anti-mitotics. In conclusion, the lead compound 6t possessed oral bioavailability, pharmacological and toxicological characteristics that warrant further development as an exciting new class of anticancer agents.
EXPERIMENTAL SECTION
Chemistry
General Experimental Procedures
Melting points were determined on Electrothermal MEL-Temp 3.0 apparatus and were uncorrected. The proton nuclear resonance (1H NMR) spectra were performed on a Bruker AVANCE 300, 600 (1H, 300, 600 MHz; 13C, 75 MHz), Varian INOVA (400 MHz), and GE (500 MHz). Chemical shifts δ are given in ppm, and the following abbreviations are used: singlet (s), doublet (d), triplet (t), multiplet (m) and broad singlet (br s). Coupling constants (J) were measured in hertz (Hz). All LC/MS data were gathered on an Agilent 1200 LC with Agilent 6410 triple quadrupole mass spectrometer detectors. The compound solution was infused into the electrospray ionization source operating positive and negative modes in methanol/water/trifluoroacetic acid (50:50:0.1% v/v) at 0.4 mL/min. The sample cone (declustering) voltage was set at 100 V. The instrument was externally calibrated for the mass range m/z 100 to m/z 1000. The reactions were followed by TLC (Silica gel,) using chloroform: methanol (9.5:0.5 v/v). The purity of the newly synthesized compounds was determined by LC/MS analysis and was confirmed to be higher than 95% for all compounds. All reagents and solvents were purchased from commercial suppliers and used without further purification unless otherwise stated. Solvents were dried using standard procedures and reactions requiring anhydrous conditions were performed under N2 atmosphere. Reactions were monitored by Thin Layer Chromatography (TLC) on pre-coated silica gel F254 plates (Sigma-Aldrich) with a UV indicator. Yields were of purified product and were not optimized.
General Procedure for the Preparation of 3-Nitro-4-methoxyaniline (1g) (Scheme 3)
To a mixture of 4-fluoro-3-nitroaniline 7 (15.61 g, 100 mmol) in absolute methanol (364 mL) was added 6% methanolic potassium hydroxide (182 mL) at room temperature and stirred for 30 min. After completion of the reaction, the reaction mixture was acidified with concentrated hydrochloric acid and evaporated the methanol under vacuum to dryness. The crude residue on crystallization with aqueous methanol resulted pure 1g. Yield: 50%; pale yellow solid, mp 51–53 °C. 1H NMR (CDCl3, 300 MHz): δ 3.83 (s, 3H, OCH3), 6.28 (br s, 1H, NH2), 7.24 (d, J = 9.0 Hz, 1H, Ar-H), 7.32 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.55 (d, J = 2.7 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 169.0568. Calcd for C7H8N2O3 m/z: 168.0535.
General Procedure for the Preparation of 4-Methoxy-3,5-dinitroaniline (1j) (Scheme 3)
Step 1: Synthesis of 4-Fluoro-3,5-dinitrobenzoic acid (9).24
4-Fluorobenzoic acid 8 (14.0 g, 10 mmol) was added in small portions to a mixture of 30 % oleum (121 g, 121 mmol) and 90% nitric acid (83.4 g, 132 mmol), under stirring at below 25 °C. The resulting clear yellow solution was heated initially to 85 °C and once the initial exothermic subsided, the reaction mixture was heated to 95 °C and maintained at same temperature for 3 h. After completion of the reaction, the mixture was cooled to room temperature and poured onto ice. The solid formed filtered, and washed with cold water, gave pure 9. Yield: 70%; mp 238–240 °C. 1H NMR (DMSO-d6, 300 MHz): δ 8.06 (s, 2H, Ar-H). HRMS found [M-H]− (m/z): 228.9968. Calcd for C7H3FN2O6 m/z: 229.9975.
Step 2: Synthesis of 4-Fluoro-3,5-Dinitroaniline (10)
To a solution of 4-fluoro-3,5-dinitrobenzoic acid, 9 (11.4 g, 49.5 mmol) in 20 % oleum (30 mL) was added ethylene dichloride (40 mL) and continued stirring at below 25 °C. To this sodium azide (3.7 g, 56.9 mmol) was added in small portions and the reaction mixture then heated to reflux and refluxed for 1 h. Once the reaction was completed, cool the reaction mixture to room temperature and separate the ethylene dichloride layer. The acidic solution was poured over ice, the solid formed filtered and washed with water, gave crude 10. Recrystallization from ethylene dichloride gave pure 10. Yield: 70.3%; yellow-orange crystals, mp 149–150 °C. 1H NMR (CDCl3, 500 MHz): δ 6.17 (s, 2H, NH2), 7.52 (d, J = 5.5 Hz, 2H, Ar-H). HRMS found [M+H]+ (m/z): 202.0156. Calcd for C6H4FN3O4 m/z: 201.0186.
Step 3: Synthesis of 4-Methoxy-3,5-Dinitroaniline (1j)
The title compound was obtained from 4-fluoro-3,5-dinitroaniline 10 following the procedure as described in compound 1g. Yield: 50.3%; mp 200–205 °C. 1H NMR (DMSO-d6, 500 MHz): δ 3.77 (s, 3H, OCH3), 6.09 (s, 2H, NH2), 7.34 (s, 2H, Ar-H). HRMS found [M+H]+ (m/z): 214.0409. Calcd for C7H7N3O5 m/z: 213.0386.
General Procedure for the Preparation of Ethyl 2-(Chlorosulfonyl)acetate (2) (Scheme 4)
Method A
Step 1: Synthesis of Sodium 2-Ethoxy-2-oxoethanesulfonate(12).25
To a solution of sodium sulfite (12.6 g, 100 mmol) in water (400 mL) at 18–20 °C was added a mixture of ethyl bromoacetate 11 (16.7 g, 100 mmol) in ethanol (200 mL) drop wise under stirring. After the addition was complete, the reaction mixture was heated to 50 °C and maintained for 30 min. Once the reaction completed, concentrated the reaction mass to dryness with the help of ethanol/benzene mixture (2 X 200 mL, 1:1 v/v) and make sure the water was removed completely. The resulted crude solid was extracted with boiling 2:1 acetic acid/ ethyl acetate (900 mL) and the hot solution was filtered through celite. The filtrate left overnight at room temperature, the solid separated was filtered, washed with cold ethyl acetate, dried under vacuum resulted pure 12 was used without characterization. Yield: 70% as white solid.
Step 2: Synthesis of Ethyl 2-(Chlorosulfonyl)acetate (2)
The mixture of sodium 2-ethoxy-2-oxoethanesulfonate, 12 (17.6 g, 100 mmol) and phosphorus (V) chloride (23.0 g, 110 mmol) was stirred until the reaction mass no longer exothermic. Then the reaction mixture was warmed on a steam bath for 45 min and distills off excess phosphorus (V) chloride under vacuum. To the residue, benzene was added (100 mL), stirred for 10 min and filtered through celite, washed with benzene and removal of solvent under vacuum gave pure 2, as clear oil. Yield: 85%. 1H NMR (CDCl3, 300 MHz): δ 1.29 (t, J = 6.9 Hz, 3H, CH3), 3.94 (s, 2H, CH2), 4.12 (q, J = 7.2 Hz, 2H, OCH2). HRMS found [M+H]+ (m/z): 186.9734. Calcd for C4H7ClO4S m/z: 185.9754.
Method B: Preparation of Ethyl 2-(Chlorosulfonyl)acetate (2).26
To a cooled solution of chlorosulfonylacetyl chloride, 13 (22.2 g, 125 mmol) in anhydrous diethyl ether (115 mL) was added absolute ethanol (5.78 g, 125 mmol) and maintained for 3 h at 0 °C. Removal of diethyl ether under vacuum resulted crude 2. Distillation of the crude resulted pure 2 as an oil. Yield: 55%; bp 123–126 °C (15 mm Hg). The analytical data are in accord with above method A product.
General Procedure for the Preparation of Ethyl Phenylsulfamoyl acetate (3). (Scheme 1)
To a solution of aniline, 1 (10.0 g, 107 mmol) in dichloromethane (150 mL) at 10 °C, was added triethylamine (16.3 g, 161 mmol) dropwise and stirred for 15 min at same temperature. To this ethyl 2-(chlorosulfonyl)acetate, 2 (22.0 g, 118 mmol) dissolved in dichloromethane (25 mL) was added slowly at same temperature. Once the addition is over, the reaction mixture was allowed to warm to room temperature and stirred for 3 h. After completion of reaction, water was added, stirred for 15 min and separated the organic layer, dried over anhydrous sodium sulfate and evaporated under reduced pressure resulted crude 3, as an oil. The crude on silica gel column purification (1:1, ethyl acetate: hexane) resulted pure 3. The following ethyl phenylsulfamoyl acetates 3 were prepared using the above procedure.
Ethyl 2-(N-Phenylsulfamoyl)acetate (3a)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to aniline, 1a yielded the corresponding ethyl 2-(N-phenylsulfamoyl)acetate. Yield: 80%, as oil. 1H NMR (CDCl3, 300 MHz): δ 1.31 (t, J = 6.9 Hz, 3H, CH3), 3.96 (s, 2H, CH2), 4.31 (q, J = 7.2 Hz, 2H, OCH2), 6.85 (br s, 1H, NH), 7.38–7.46 (m, 5H, Ar-H). HRMS found [M+H]+ (m/z): 244.0589. Calcd for C10H13NO4S m/z: 243.0565.
Ethyl 2-(N-(4-Chlorophenyl)sulfamoyl)acetate (3b)
Addition of ethyl 2-(chlorosulfonyl)-acetate, 2 to 4-chloroaniline, 1b yielded the corresponding ethyl 2-(N-(4-chlorophenyl)sulfamoyl)acetate. Yield: 81%; light yellow solid, mp 78–79 °C. 1H NMR (CDCl3, 300 MHz): δ 1.34 (t, J = 6.9 Hz, 3H, CH3), 3.94 (s, 2H, CH2), 4.29 (q, J = 7.2 Hz, 2H, OCH2), 7.05 (br s, 1H, NH), 7.27–7.38 (m, 4H, Ar-H). HRMS found [M+H]+ (m/z): 278.0201. Calcd for C10H12ClNO4S m/z: 277.0176.
Ethyl 2-(N-(4-Fluorophenyl)sulfamoyl)acetate (3c)
Addition of ethyl 2-(chlorosulfonyl)-acetate, 2 to 4-fluoroaniline, 1c yielded the corresponding ethyl 2-(N-(4-fluorophenyl)-sulfamoyl)acetate. Yield: 79%; light brown solid, mp 65–66 °C. 1H NMR (CDCl3, 300 MHz): δ 1.33 (t, J = 7.2 Hz, 3H, CH3), 3.93 (s, 2H, CH2), 4.29 (q, J = 7.2 Hz, 2H, OCH2), 7.03–7.13 (m, 2H, Ar-H), 7.18 (br s, 1H, NH), 7.32–7.40 (m, 2H, Ar-H). HRMS found [M+H]+ (m/z): 262.0500. Calcd for C10H12FNO4S m/z: 261.0471.
Ethyl 2-(N-(4-Methoxyphenyl)sulfamoyl)acetate (3d)
Addition of ethyl 2-(chlorosulfonyl)-acetate, 2 to 4-methoxyaniline, 1d yielded the corresponding ethyl 2-(N-(4-methoxyphenyl)-sulfamoyl)acetate. Yield: 79%; pale yellow sold, mp 74–76 °C. 1H NMR (CDCl3, 300 MHz): δ 1.34 (t, J = 6.9 Hz, 3H, CH3), 3.83 (s, 2H, CH2), 3.86 (s, 3H, OCH3), 4.30 (q, J = 7.2 Hz, 2H, OCH2), 6.82 (br s, 1H, NH), 6.88 (d, J = 9.0 Hz, 2H, Ar-H), 7.14 (d, J = 9.0 Hz, 2H, Ar-H). HRMS found [M+H]+ (m/z): 274.0694. Calcd for C11H15NO5S m/z: 273.0671.
Ethyl 2-(N-(3-Fluoro-4-methoxyphenyl)sulfamoyl)acetate (3e)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to 3-fluoro-4-methoxyaniline, 1e yielded the corresponding ethyl 2-(N-(3-fluoro-4-methoxyphenyl)sulfamoyl)acetate. Yield: 79%; light yellow solid, mp 81–83 °C. 1H NMR (CDCl3, 300 MHz): δ 1.33 (t, J = 7.2 Hz, 3H, CH3), 3.82 (s, 2H, CH2), 3.87 (s, 3H, OCH3), 4.28 (q, J = 7.2 Hz, 2H, OCH2), 6.84 (br s, 1H, NH), 6.92 (d, J = 9.0 Hz, 1H, Ar-H), 7.08 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 7.19 (d, J = 2.4 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 292.0600. Calcd for C11H14FNO5S m/z: 291.0577.
Ethyl 2-(N-(2,4,6-Trimethoxyphenyl)sulfamoyl)acetate (3f)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to 2,4,6-trimethoxyaniline, 1f yielded the corresponding ethyl 2-(N-(2,4,6-trimethoxyphenyl)sulfamoyl)acetate. Yield: 78%; light yellow solid, mp 76–78 °C. 1H NMR (CDCl3, 300 MHz): δ 1.33 (t, J = 7.2 Hz, 3H, CH3), 3.81 (s, 9H, 3 X OCH3), 3.90 (s, 2H, CH2), 4.29 (q, J = 7.2 Hz, 2H, OCH2), 6.04 (s, 2H, Ar-H), 6.79 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 334.0906. Calcd for C13H19NO7S m/z: 333.0882.
Ethyl 2-(N-(4-Methoxy-3-nitrophenyl)sulfamoyl)acetate (3g)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to 3-nitro-4-methoxyaniline, 1g yielded the corresponding ethyl 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetate. Yield: 80%; white crystalline solid, mp 100–102 °C. 1H NMR (CDCl3, 300 MHz): δ 1.36 (t, J = 7.2 Hz, 3H, CH3), 3.94 (s, 2H, CH2), 3.99 (s, 3H, OCH3), 4.32 (q, J = 7.2 Hz, 2H, OCH2), 7.07 (br s, 1H, NH), 7.13 (d, J = 9.0 Hz, 1H, Ar-H), 7.62 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.86 (d, J = 2.7 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 319.0535. Calcd for C11H14N2O7S m/z: 318.0522.
Ethyl 2-(N-(Perfluorophenyl)sulfamoyl)acetate (3h)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to 2,3,4,5,6-pentafluoroaniline, 1h yielded the corresponding ethyl 2-(N-(perfluorophenyl)sulfamoyl)acetate. Yield: 78%; brown liquid. 1H NMR (CDCl3, 300 MHz): δ 1.35 (t, J = 7.2 Hz, 3H, CH3), 3.94 (s, 2H, CH2), 4.32 (q, J = 7.2 Hz, 2H, OCH2), 7.07 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 334.0118. Calcd for C10H8F5NO4S m/z: 333.0094.
Ethyl 2-(N-(4-Fluoro-3-nitrophenyl)sulfamoyl)acetate (3i)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to 4-fluoro-3-nitroaniline, 1i yielded the corresponding ethyl 2-(N-(4-fluoro-3-nitrophenyl)sulfamoyl)acetate. Yield: 78%; light yellow solid, mp 98–100 °C. 1H NMR (CDCl3, 300 MHz): δ 1.35 (t, J = 7.2 Hz, 3H, CH3), 3.92 (s, 2H, CH2), 4.30 (q, J = 7.2 Hz, 2H, OCH2), 7.05 (br s, 1H, NH), 7.32 (d, J = 9.0 Hz, 1H, Ar-H), 7.69 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.80 (d, J = 2.7 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 307.0345. Calcd for C10H11FN2O6S m/z: 306.0322.
Ethyl 2-(N-(4-Methoxy-3,5-dinitrophenyl)sulfamoyl)acetate (3j)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to 4-methoxy-3,5-dinitroaniline, 1j yielded the corresponding ethyl 2-(N-(4-methoxy-3,5-dinitrophenyl)sulfamoyl)acetate. Yield: 79%; light yellow solid, mp 126–128 °C. 1H NMR (DMSO-d6, 300 MHz): δ 1.31 (t, J = 7.2 Hz, 3H, CH3), 3.92 (s, 3H, OCH3), 4.13 (q, J = 7.2 Hz, 2H, OCH2), 4.43 (s, 2H, CH2), 8.04 (s, 2H, Ar-H), 10.90 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 364.0400. Calcd for C11H13N3O9S m/z: 363.0372.
Ethyl 2-(N-(3-Hydroxy-4-methoxyphenyl)sulfamoyl)acetate (3k)
Addition of ethyl 2-(chlorosulfonyl)acetate, 2 to 3-hydroxy-4-methoxyaniline, 1k yielded the corresponding ethyl 2-(N-(3-hydroxy-4-methoxyphenyl)sulfamoyl)acetate. Yield: 79%; light brown solid, mp 116–118 °C. 1H NMR (CDCl3, 300 MHz): δ 1.35 (t, J = 7.2 Hz, 3H, CH3), 3.91 (s, 3H, OCH3), 3.93 (s, 2H, CH2), 4.31 (q, J = 7.2 Hz, 2H, OCH2), 5.63 (br s, 1H, OH), 6.76 (br s, 1H, NH), 6.83 (d, J = 8.4 Hz, 1H, Ar-H), 6.88 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.96 (d, J = 2.4 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 290.0644. Calcd for C11H15NO6S m/z: 289.0620.
General Procedure for the Preparation of Phenylsulfamoyl acetic acid (4). (Scheme 1)
A cooled solution of sodium hydroxide (4.88 g, 122 mmol) in water (122 mL) was added to ethyl 2-(N-phenylsulfamoyl)acetate, 3a (4.15 g, 18.1 mmol) slowly and continued stirring for 3 h at room temperature. After completion of the reaction, the reaction mixture was cooled to 0 °C; concentrated hydrochloric acid was added slowly at 0 °C until the pH of the reaction mixture is in between 3.0–4.0 and stir for 30 min. The solid formed was filtered, washed with cold water and dried under vacuum. The dried product was used without further purification. The following phenylsulfamoyl acetic acids 4 were prepared using the above procedure.
2-(N-Phenylsulfamoyl)acetic acid (4a)
Hydrolysis followed by neutralization of ethyl 2-(N-phenylsulfamoyl) acetate 3a resulted the corresponding 2-(N-phenylsulfamoyl)acetic acid. Yield: 81%: light yellow solid, mp 110–111 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.98 (s, 2H, CH2), 7.38–7.46 (m, 5H, Ar-H), 9.85 (br s, 1H, NH), 12.70 (br s, 1H, COOH). HRMS found [M-H]−(m/z): 214.0276. Calcd for C8H9NO4S m/z: 215.0252.
2-(N-(4-Chlorophenyl)sulfamoyl)acetic acid (4b)
Hydrolysis followed by neutralization of ethyl 2-(N-(4-chlorophenyl)sulfamoyl)acetate, 3b resulted the corresponding 2-(N-(4-chlorophenyl)sulfamoyl)acetic acid. Yield: 84%; white solid, mp 126–128 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.84 (s, 2H, CH2), 7.23 (d, J = 9.0 Hz, 2H, Ar-H), 7.37 (d, J = 9.0 Hz, 2H, Ar-H), 10.32 (br s, 1H, NH). HRMS found [M-H]− (m/z): 247.9833. Calcd for C8H8ClNO4S m/z: 248.9863.
2-(N-(4-Fluorophenyl)sulfamoyl)acetic acid (4c)
Hydrolysis followed by neutralization of ethyl 2-(N-(4-fluorophenyl)sulfamoyl)acetate, 3c resulted the corresponding 2-(N-(4-fluorophenyl)sulfamoyl)acetic acid. Yield; 81%; light brown solid, mp 114–116 °C. 1H NMR (DMSO-d6, 300 MHz): δ 4.06 (s, 2H, CH2), 7.17–7.28 (m, 4H, Ar-H), 10.04 (br s, 1H, NH), 12.70 (br s, 1H, COOH). HRMS found [M-H]− (m/z): 234.0182. Calcd for C8H8FNO4S m/z: 233.0158.
2-(N-(4-Methoxyphenyl)sulfamoyl)acetic acid (4d)
Hydrolysis followed by neutralization of ethyl 2-(N-(4-methoxyphenyl)sulfamoyl)acetate, 3d resulted the corresponding 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid. Yield: 82%; white solid, mp 166–168 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.70 (s, 2H, CH2), 3.73 (s, 3H, OCH3), 6.90 (d, J = 9.0 Hz, 2H, Ar-H), 7.17 (d, J = 9.0 Hz, 2H, Ar-H), 9.76 (br s, 1H, NH). HRMS found [M-H]− (m/z): 246.0381. Calcd for C9H11NO5S m/z: 245.0358.
2-(N-(3-Fluoro-4-methoxyphenyl)sulfamoyl)acetic acid (4e)
Hydrolysis followed by neutralization of ethyl 2-(N-(3-fluoro-4-methoxyphenyl)sulfamoyl)acetate, 3e resulted the corresponding 2-(N-(3-fluoro-4-methoxyphenyl)sulfamoyl)acetic acid. Yield: 82%; light brown sold, mp 176–178 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.77 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 6.98–7.02 (m, 1H, Ar-H), 7.05–7.13 (m, 1H, Ar-H), 7.18 (d, J = 9.0 Hz, 1H, Ar-H), 9.72 (br s, 1H, NH). HRMS found [M-H]− (m/z): 262.0285. Calcd for C9H10FNO5S m/z: 263.0264.
2-(N-(2,4,6-Trimethoxyphenyl)sulfamoyl)acetic acid (4f)
Hydrolysis followed by neutralization of ethyl 2-(N-(2,4,6-trimethoxyphenyl)sulfamoyl)acetate, 3f resulted the corresponding 2-(N-(2,4,6-trimethoxyphenyl)sulfamoyl)acetic acid. Yield: 80%; white solid, mp 150–152 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.81 (s, 9H, 3 X OCH3), 3.90 (s, 2H, CH2), 6.04 (s, 2H, Ar-H), 9.89 (br s, 1H, NH). HRMS found [M-H]− (m/z): 304.0545. Calcd for C11H15NO7S m/z: 305.0569.
2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetic acid (4g)
Hydrolysis followed by neutralization of ethyl 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetate, 3g resulted the corresponding 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetic acid. Yield: 84%; light yellow solid, mp 154–156 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.91 (s, 3H, OCH3), 4.14 (s, 2H, CH2), 7.39 (d, J = 9.0 Hz, 1H, Ar-H), 7.50 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.73 (d, J = 2.7 Hz, 1H, Ar-H), 10.22 (br s, 1H, NH), 12.71 (br s, 1H, COOH). HRMS found [M-H]− (m/z): 289.0232. Calcd for C9H10N2O7S m/z: 290.0209.
2-(N-(Perfluorophenyl)sulfamoyl)acetic acid (4h)
Hydrolysis followed by neutralization of ethyl 2-(N-(perfluorophenyl)sulfamoyl)acetate, 3h resulted the corresponding 2-(N-(perfluorophenyl)sulfamoyl)acetic acid. Yield: 79%; white crystalline solid, mp 142–144 °C. 1H NMR (DMSO-d6, 300 MHz): δ 4.35 (s, 2H, CH2), 10.07 (br s, 1H, NH), 12.71 (br s, 1H, COOH). HRMS found [M-H]− (m/z): 303.9802. Calcd for C8H4F5NO4S m/z: 304.9781.
2-(N-(4-Fluoro-3-nitrophenyl)sulfamoyl)acetic acid (4i)
Hydrolysis followed by neutralization of ethyl 2-(N-(4-fluoro-3-nitrophenyl)sulfamoyl)acetate, 3i resulted the corresponding 2-(N-(4-fluoro-3-nitrophenyl)sulfamoyl)acetic acid. Yield: 78%; light yellow solid, mp 132–134 °C. 1H NMR (DMSO-d6, 500 MHz): δ 4.35 (s, 2H, CH2), 7.47 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.75 (d, J = 9.0 Hz, 1H, Ar-H), 7.85 (d, J = 2.7 Hz, 1H, Ar-H), 10.78 (br s, 1H, NH), 13.26 (br s, 1H, COOH). HRMS found [M-H]− (m/z): 277.0026. Calcd for C8H7FN2O6S m/z: 278.0009.
2-(N-(4-Methoxy-3,5-dinitrophenyl)sulfamoyl)acetic acid (4j)
Hydrolysis followed by neutralization of ethyl 2-(N-(4-methoxy-3,5-dinitrophenyl)sulfamoyl)acetate, 3j resulted the corresponding 2-(N-(4-methoxy-3,5-dinitrophenyl)sulfamoyl)acetic acid. Yield: 79%; brown solid, mp 150–152 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.92 (s, 3H, OCH3), 4.43 (s, 2H, CH2), 8.04 (s, 2H, Ar-H), 10.90 (br s, 1H, NH), 12.98 (br s, 1H, COOH). HRMS found [M-H]− (m/z): 334.0081. Calcd for C9H9N3O9S m/z: 335.0059.
2-(N-(3-Hydroxy-4-methoxyphenyl)sulfamoyl)acetic acid (4k)
Hydrolysis followed by neutralization of ethyl 2-(N-(3-hydroxy-4-methoxyphenyl)sulfamoyl)acetate, 3k resulted the corresponding 2-(N-(3-hydroxy-4-methoxyphenyl)sulfamoyl)acetic acid. Yield: 78%; off-white solid, mp 150–152 °C. 1H NMR (DMSO-d6, 500 MHz): δ 3.78 (s, 2H, CH2), 3.94 (s, 3H, OCH3), 6.61 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.76 (d, J = 2.7 Hz, 1H, Ar-H), 6.82 (d, J = 8.7 Hz, 1H, Ar-H), 9.28 (br s, 1H, NH), 9.52 (br s, 1H, COOH). HRMS found [M-H]− (m/z): 260.0326. Calcd for C9H11NO6S m/z: 261.0307.
General Procedure for the Preparation of (E)-N-Aryl-2-arylethenesulfonamide (6)
Method A (Scheme 1)
A mixture of 2-(N-phenylsulfamoyl)acetic acid 4 (10 mmol), araldehyde 5 (10 mmol), glacial acetic acid (15 mL) was stirred at room temperature for 10 min. A catalytic amount of benzylamine (200 μL) was added and refluxed for about 8 h. After completion of the reaction, the contents were cooled to room temperature and dilute with ethyl acetate. The precipitated solid was filtered and washed with ethyl acetate, the resulted crude 6 on silica gel column purification recrystallized resulted pure 6. If solid was not formed, the diluted reaction mixture with ethyl acetate was washed successively with saturated sodium bicarbonate, dilute hydrochloric acid, and water. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to obtain the desired crude product 6. The crude product on silica gel column purification yielded an analytically pure 6. The following (E)-N-aryl-2-arylethenesulfonamide 6 were prepared using the above procedure.
(E)-N,2-Diphenylethenesulfonamide (6a)
The title compound was obtained from 2-(N-phenylsulfamoyl) acetic acid 4a and benzaldehyde following the procedure as described in method A. Yield, 50%; off white solid, mp 112–114 °C. 1H NMR (CDCl3, 300 MHz): δ 6.58 (br s, 1H, NH), 6.81 (d, J = 15.3 Hz, 1H, =CH), 7.14–7.24 (m, 3H, Ar-H), 7.30–7.37 (m, 2H, Ar-H), 7.38–7.46 (m, 5H, Ar-H), 7.53 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 260.0689. Calcd for C14H13NO2S m/z: 259.0667.
(E)-N-(4-Chlorophenyl)-2-phenylethenesulfonamide (6b)
The title compound was obtained from 2-(N-(4-chlorophenylsulfamoyl)acetic acid 4b and benzaldehyde following the procedure as described in method A. Yield, 49%; white crystalline solid, mp 108–110 °C. 1H NMR (CDCl3, 300 MHz): δ 6.57 (br s, 1H, NH), 6.79 (d, J = 15.3 Hz, 1H, =CH), 7.17 (dd, J = 6.9, 2.4 Hz, 2H, Ar-H), 7.30 (dd, J = 6.6, 2.1 Hz, 2H, Ar-H), 7.37–7.48 (m, 5H, Ar-H), 7.52 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 294.0301. Calcd for C14H12ClNO2S m/z: 293.0277.
(E)-2-(4′-Bromophenyl)-N-(4-fluorophenyl)ethenesulfonamide (6c)
The title compound was obtained from 2-(N-(4-fluorophenylsulfamoyl)acetic acid 4c and 4-bromobenzaldehyde following the procedure as described in method A. Yield, 48%; white crystalline solid, mp 138–140 °C. 1H NMR (CDCl3, 300 MHz): δ 6.46 (br s, 1H, NH), 6.71 (d, J = 15.3 Hz, 1H, =CH), 7.05–7.13 (m, 4H, Ar-H), 7.43–7.46 (m, 4H, Ar-H), 7.49 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 355.9702. Calcd for C14H11BrFNO2S m/z: 354.9678.
(E)-N-(4-Fluorophenyl)-2-(4′-methoxyphenyl)ethenesulfonamide (6d)
The title compound was obtained from 2-(N-(4-fluorophenylsulfamoyl)acetic acid 4c and 4-methoxybenzaldehyde following the procedure as described in method A. Yield, 49%; off white crystalline solid, mp 98–100 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 3H, OCH3), 6.43 (br s, 1H, NH), 6.71 (d, J = 15.3 Hz, 1H, =CH), 6.88–6.92 (m, 2H, Ar-H), 6.98–7.06 (m, 2H, Ar-H), 7.17–7.24 (m, 2H, Ar-H), 7.36–7.40 (m, 2H, Ar-H), 7.41 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 308.0703. Calcd for C15H14FNO3S m/z: 307.0678.
(E)-2-(2′-Methoxyphenyl)-N-(4-methoxyphenyl)ethenesulfonamide (6e)
Method B (Scheme 1)
A mixture of 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid, 4d (5 mmol), 2-methoxy-benzaldehyde (5.5 mmol), benzoic acid (0.30 mmol), and piperidine (0.30 mmol) in toluene (50 mL) was refluxed for 4 h with continuous removal of water using a Dean-Stark water separator. After the reaction completion, the solvent was evaporated. To the residue added methanol and stirred for 10 min. The solid formed was filtered and washed with cold methanol and dried under vacuum yielded product 6e. If no formation of solid observed, concentrated and water was added to the residue and extracted with ethyl acetate. The organic phase was washed with saturated sodium bicarbonate solution, dilute hydrochloric acid, and water and dried over anhydrous sodium sulfate. The organic phase was filtered, and evaporation of the solvent under vacuum yielded a crude product 6. The pure compound 6 was obtained following purification by silica gel flash column chromatography. Yield, 65%; white solid, mp 112–114 °C. 1H NMR (DMSO-d6, 600 MHz): δ 3.61 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 6.79 (d, J = 7.9 Hz, 2H, Ar-H), 6.89 (t, J = 6.8 Hz, 1H, Ar-H), 7.03 (m, 4H, Ar-H), 7.34 (t, J = 6.8 Hz, 1H, Ar-H), 7.41 (d, J = 15.4 Hz, 1H, =CH), 7.55 (d, J = 6.9 Hz, 1H, Ar-H), 9.53 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 320.0849. Calcd for C16H17NO4S m/z: 319.0878.
(E)-N,2-Bis(4-methoxyphenyl)ethenesulfonamide (6f)
The title compound was obtained from 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid (4d) and 4-methoxybenzaldehyde following the procedure as described in 6e method B. Yield, 60%; semi solid. 1H NMR (DMSO-d6, 600 MHz): δ 3.61 (s, 3H, OCH3), 3.70 (s, 3H, OCH3), 6.78 (d, J = 9.0 Hz, 2H, Ar-H), 6.87 (d, J = 8.9 Hz, 2H, Ar-H), 6.92 (d, J = 15.4 Hz, 1H, =CH), 7.03 (d, J = 8.9 Hz, 2H, Ar-H), 7.18 (d, J = 15.4 Hz, 1H, CH=), 7.54 (d, J = 8.7 Hz, 2H, Ar-H), 9.50 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 320.0846. Calcd for C16H17NO4S m/z: 319.0878.
(E)-2-(2′,4′-Dimethoxyphenyl)-N-(4-methoxyphenyl)ethenesulfonamide (6g)
The title compound was obtained from 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid (4d) and 2,4-dimethoxybenzaldehyde following the procedure as described in 6e method B. Yield, 60%; white solid, mp 162–164 °C. 1H NMR (DMSO-d6, 600 MHz): δ 3.61 (s, 3H, OCH3), 3.72 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 6.48 (dd, J = 8.5, 1.5 Hz, 1H, Ar-H), 6.53 (d, J = 1.5 Hz, 1H, Ar-H), 6.78 (d, J = 8.8 Hz, 2H, Ar-H), 6.86 (d, J = 15.5 Hz, 1H, =CH), 7.02 (d, J = 8.8 Hz, 2H, Ar-H), 7.33 (d, J = 15.5 Hz, 1H, CH=), 7.48 (d, J = 8.5 Hz, 1H, Ar-H), 9.42 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 350.1007. Calcd for C17H19NO5S m/z: 349.0984.
(E)-2-(2′,6′-Dimethoxyphenyl)-N-(4-methoxyphenyl)ethenesulfonamide (6h)
The title compound was obtained from 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid (4d) and 2,6-dimethoxybenzaldehyde following the procedure as described in 6e method B. Yield, 65%; white solid, mp 160–162 °C. 1H NMR (DMSO-d6, 600 MHz): δ 3.61 (s, 3H, OCH3), 3.75 (s, 6H, 2 X OCH3), 6.63 (d, J = 8.4 Hz, 2H, Ar-H), 6.79 (d, J = 8.7 Hz, 2H, Ar-H), 7.02 (d, J = 8.8 Hz, 2H, Ar-H), 7.05 (d, J = 15.9 Hz, 1H, =CH), 7.29 (t, J = 8.3 Hz, 1H, Ar-H), 7.51 (d, J = 15.6 Hz, 1H, CH=), 9.41 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 350.0959. Calcd for C17H19NO5S m/z: 349.0984.
(E)-N-(4-Methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6i)
The title compound was obtained from 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid 4d and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 49%; light yellow solid, mp 176–178 °C. 1H NMR (CDCl3, 300 MHz): δ 3.79 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 6.09 (s, 2H, Ar-H), 6.12 (br s, 1H, NH), 6.85 (dd, J = 6.6, 2.1 Hz, 2H, Ar-H), 7.11 (d, J = 15.6 Hz, 1H, =CH), 7.18 (dd, J = 6.6, 2.1 Hz, 2H, Ar-H), 7.81 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 380.1113. Calcd for C18H21NO6S m/z: 379.1090.
(E)-N-(4-Methoxyphenyl)-2-(3′,4′,5′-trimethoxyphenyl)ethenesulfonamide (6j)
The title compound was obtained from 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid 4d and 3,4,5-trimethoxybenzaldehyde following the procedure as described in 6e method B. Yield, 60%; pale yellow solid, mp 66–68 °C. 1H NMR (DMSO-d6, 600 MHz): δ 3.61 (s, 3H, OCH3), 3.65 (s, 3H, OCH3), 3.71 (s, 6H, 2 X OCH3), 6.84 (d, J = 8.9 Hz, 2H, Ar-H), 6.95 (s, 2H, Ar-H), 7.07 (d, J = 8.9 Hz, 2H, Ar-H), 7.15 (d, J = 15.3 Hz, 1H, =CH), 7.19 (d, J = 15.3 Hz, 1H, CH=), 9.58 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 380.1068. Calcd for C18H21NO6S m/z: 379.1090.
(E)-2-(4′-Methoxyphenyl)-N-(2,4,6-trimethoxyphenyl)ethenesulfonamide (6k)
The title compound was obtained from 2-(N-(2,4,6-trimethoxyphenyl)sulfamoyl)acetic acid 4f and 4-methoxybenzaldehyde following the procedure as described in method A. Yield, 49%; off white solid, mp 156–158 °C. 1H NMR (CDCl3, 300 MHz): δ 3.81 (s, 9H, 3 X OCH3), 3.86 (s, 3H, OCH3), 5.84 (br s, 1H, NH), 6.13 (s, 2H, Ar-H), 6.83 (d, J = 15.3 Hz, 1H, =CH), 6.92 (dd, J = 2.1, 6.6 Hz, 2H, Ar-H), 7.32 (d, J = 15.6 Hz, 1H, CH=), 7.42 (dd, J = 2.1, 6.6 Hz, 2H, Ar-H). HRMS found [M+H]+ (m/z): 380.1071. Calcd for C18H21NO6S m/z: 379.1090.
(E)-2-(4′-Hydroxy-2′,6′-dimethoxyphenyl)-N-(4-methoxyphenyl)ethenesulfonamide (6l)
The title compound was obtained from 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid 4d and 2,6-dimethoxy-4-hydroxybenzaldehyde following the procedure as described in 6e method B. Yield, 60%; white solid, mp 146–148 °C. 1H NMR (CDCl3, 300 MHz): δ 3.82 (s, 3H, OCH3), 3.86 (s, 6H, 2 X OCH3), 6.05 (s, 2H, Ar-H), 6.50 (br s, 1H, NH), 6.86 (dd, J = 6.6, 2.1 Hz, 2H, Ar-H), 7.13 (d, J = 15.6 Hz, 1H, =CH), 7.20 (dd, J = 6.6, 2.1 Hz, 2H, Ar-H), 7.82 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M-H]− (m/z): 364.0957. Calcd for C17H19NO6S m/z: 365.0933.
(E)-N-(4-Methoxyphenyl)-2-(2′,4′,6′-trifluorophenyl)ethenesulfonamide (6m)
The title compound was obtained from 2-(N-(4-methoxyphenyl)sulfamoyl)acetic acid 4d and 2,4,6-trifluorobenzaldehyde following the procedure as described in 6e method B. Yield, 60%; pale yellow solid, mp 120–122 °C. 1H NMR (DMSO-d6, 600 MHz): δ 3.62 (s, 3H, OCH3), 6.81 (d, J = 8.9 Hz, 2H, Ar-H), 6.94 (d, J = 15.8 Hz, 1H, =CH), 7.05 (d, J = 8.9 Hz, 2H, Ar-H), 7.06 (d, J = 15.8 Hz, 1H, CH=), 7.28 (t, J = 9.1 Hz, 2H, Ar-H), 9.68 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 344.0511. Calcd for C15H12F3NO3S m/z: 343.0490.
(E)-N-(4-Methoxyphenyl)-2-(perfluorophenyl)ethenesulfonamide (6n)
The title compound was obtained from 2-(N-(4-methoxyphenylsulfamoyl)acetic acid 4d and 2,3,4,5,6-pentafluorobenzaldehyde following the procedure as described in method A. Yield, 48%; light yellow solid, mp 131–133 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 3H, OCH3), 6.43 (b r s, 1H, NH), 6.85 (dd, J = 6.6, 2.1 Hz, 2H, Ar-H), 7.12 (d, J = 15.3 Hz, 1H, =CH), 7.18 (dd, J = 6.6, 2.1 Hz, 2H, Ar-H), 7.81 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 380.0326. Calcd for C15H10F5NO3S m/z: 379.0302.
(E)-N-(3-Fluoro-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6o)
The title compound was obtained from 2-(N-(3-fluoro-4-methoxyphenyl)sulfamoyl)acetic acid 4e and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 49%; light yellow solid, mp 152–154 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 6.10 (s, 2H, Ar-H), 6.24 (br s, 1H, NH), 6.86 – 6.95 (m, 2H, Ar-H), 7.07 (dd, J = 12.0, 2.1 Hz, 1H, Ar-H), 7.09 (d, J = 15.6 Hz, 1H, =CH), 7.85 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 398.1014. Calcd for C18H20FNO6S m/z: 397.0995.
(E)-N-(3-Hydroxy-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6p)
The title compound was obtained from 2-(N-(3-Hydroxy-4-methoxyphenyl)sulfamoyl)acetic acid 4k and 2,4,6-trimethoxybenzaldehyde following the procedure as described in 6e method B. Yield, 64%; light green solid, mp 148–150 °C. 1H NMR (CDCl3, 300 MHz): δ 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 5.63 (br s, 1H, OH), 6.09 (s, 2H, Ar-H), 6.11 (br s, 1H, NH), 6.73 – 6.83 (m, 3H, Ar-H), 7.12 (d, J = 15.6 Hz, 1H, =CH), 7.86 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 396.1062. Calcd for C18H21NO7S m/z: 395.1039.
(E)-2-Methoxy-5-(2-(2,4,6-trimethoxyphenyl)vinylsulfonamido)phenyl 2-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-2-methylpropanoate (6q). (Scheme 7)
Step 1: Preparation of 3-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-3-methylbutanoic acid (20). (Scheme 8).29
Stage 1: Preparation of 6-Hydroxy-4,4,5,8-tetramethylchroman-2-one (22)
To a solution of 2,5-Dimethylbenzoquinone 21 (10 g, 73.5 mmol) in diethyl ether (300 mL) was added an aqueous solution of sodium hydrosulfite (178.0 g, 870 mmol in 150 mL water). The mixture was shaken until the ether layer became colorless. The ether layer was separated and the aqueous layer was extracted with diethyl ether (3 X 200 mL). The combined ether layer were washed with brine (2 X 150 mL) and dried over anhydrous sodium sulfate. Evaporation of solvent under vacuum resulted dihydroquinone as a white solid. The above dihydroquinone (10.03 g, 72.6 mmol) was mixed with 3,3-dimethylacrylic acid (8.0 g, 80 mmol) and methanesulfonic acid (111 mL) and stirred at room temperature for 10 min and heated to 85 °C for 3 h. After completion of reaction, cooled to room temperature and poured onto ice and extracted with ethyl acetate (4 X 100 mL). The combined organic layers were washed with saturated sodium bicarbonate solution (2 X 100 mL), water (3 X 100 mL) and dried over anhydrous sodium sulfate. Evaporation of solvent under vacuum resulted crude 22, which on recrystallization with ethyl acetate and hexane (1:1) resulted pure 22. Yield, 81%; light brown solid, mp 194–196 °C. 1H NMR (CDCl3, 400 MHz): δ 1.64 (s, 6H, 2 X CH3), 2.39 (s, 3H, CH3), 2.51 (s, 3H, CH3), 2.74 (s, 2H, CH2), 5.10 (bs, 1H, OH), 6.75 (s, 1H, Ar-H). HRMS found [M+H]+ (m/z): 221.1112. Calcd for C13H16O3 m/z: 220.1099.
Stage 2: Preparation of 3-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-3-methylbutanoic acid (20)
To a solution of 6-Hydroxy-4,4,5,8-tetramethylchroman-2-one, 22 (3.0 g, 13.6 mmol) in a mixture of acetonitrile (90 mL), acetone (10 mL), and water (90 mL) was added N-bromosuccinimide (2.45 g, 13.6 mmol) in portions at room temperature under stirring and maintained for 30 min. After completion of reaction, evaporated the solvent under reduced pressure and extracted with diethyl ether (300 mL). The ethereal solution was washed with saturated NaHCO3 (3 X 300 mL), and the combined aqueous phase was washed with diethyl ether (3 X 100 mL). After acidification of the aqueous solution with concentrated HCl to pH 2–3, the aqueous solution was extracted with diethyl ether (2 X 200 mL), dried over anhydrous Na2SO4. Evaporation of solvent under vacuum resulted as yellow oily residue, 20. Yield: 63%. 1H NMR (CDCl3, 300 MHz): δ 1.37 (s, 6H, 2 X CH3), 1.89 (s, 3H, CH3), 2.04 (s, 3H, CH3), 2.94 (s, 2H, CH2), 6.38 (s, 1H, Ar-H). HRMS found [M-H]− (m/z): 235.1061. Calcd for C13H16O4 m/z: 236.1049.
Step 2: (E)-2-Methoxy-5-(2-(2,4,6-trimethoxyphenyl)vinylsulfonamido)phenyl 2-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-2-methylpropanoate (6q)
To a solution of (E)-N-(3-hydroxy-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide, 6p (0.3 g, 0.76 mmol) in anhydrous dichloromethane (30 mL) containing 3-(2,5-dimethyl-3,6-dioxocyclohexa-1,4-dienyl)-3-methylbutanoic acid, 20 (0.18 g, 0.76 mmol) were added 1-ethyl-3-(3-dimethylaminopropylcarbodiimide hydrochloride (EDAC, 0.44 g, 2.3 mmol) and 4-dimethylamino pyridine (DMAP, 0.07 g, 0.58 mmol). The mixture was stirred at room temperature for 12 h. After completion of reaction, the reaction mixture was filtered and evaporated the filtrate under vacuum, resulted crude 6q. The crude product was chromatographed over silica gel column and eluted successively with chloroform and chloroform-methanol mixture with increasing polarity. The product containing fractions were combined and concentrated under vacuum, afforded pure 6q. Yield: 69%, white solid, mp 90–92 °C. 1H NMR (CDCl3, 400 MHz): δ 1.45 (s, 6H, 2 X CH3), 1.85 (s, 3H, CH3), 2.07 (s, 3H, CH3), 3.14 (s, 2H, CH2), 3.64 (s, 3H, OCH3), 3.74 (s, 6H, 2 X OCH3), 3.77 (s, 3H, OCH3), 6.00 (s, 2H, Ar-H), 6.14 (br s, 1H, NH), 6.30 (d, J = 1.6 Hz, 1H, Ar-H), 6.75 (d, J = 8.8 Hz, 1H, Ar-H), 6.81 (d, J = 2.7 Hz, 1H, Ar-H), 6.96 (dd, J = 2.7, 8.8 Hz, 1H, Ar-H), 7.00 (d, J = 15.5 Hz, 1H, =CH), 7.75 (d, J = 15.5 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 614.1997. Calcd for C31H35NO10S m/z: 613.1982.
Sodium (E)-2-Methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenyl phosphate (6r) (Scheme 9)
Preparation of (E)-2-methoxy-5-(2-(2′,4′,6′-trimethoxy-phenyl) vinylsulfonamido)phenyl dihydrogen phosphate (23)
To a cooled solution of phosphorus oxychloride (0.5 mL, 5.3 mmol) in tetrahydrofuran (2 mL) was added a filtered solution of (E)-N-(3-hydroxy-4-methoxyphenyl)-2-(2-(2′,4′,6′-trimethoxyphenyl)ethane-sulfonamide 6p (0.5 g, 1.3 mmol) in tetrahydrofuran (5 mL) and triethylamine (1.23 mL, 16.7 mmol) drop wise at 0 °C. After addition completion, the reaction mixture stirred at room temperature for 3 h. After completion of the reaction, monitored by TLC, the reaction mixture poured onto crushed ice (15.0 g) and resulting solution was stirred at room temperature for 18 h. Removal of tetrahydrofuran under vacuum at 40 °C resulted semi solid, which on stirring for 1 h resulted solid. The solid was filtered, and taken in acidic water (15 mL, pH 4–6) and stirred for 1 h at room temperature; the purified solid was filtered and dried under vacuum at room temperature. Yield, 69%; yellow solid, mp 158–160 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.69 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.83 (s, 6H, 2 X OCH3), 6.26 (s, 2H, Ar-H), 6.83–6.85 (dd, J = 2.0, 8.7 Hz, 1H, Ar-H), 6.92 (d, J = 8.8 Hz, 1H, Ar-H), 6.98 (d, J = 15.5 Hz, 1H, =CH), 7.23 (d, J = 2.1 Hz, 1H, Ar-H), 7.58 (d, J = 15.6 Hz, 1H, CH=), 9.48 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 476.0726. Calcd for C18H22NO10PS m/z: 475.0702.
Preparation of Sodium (E)-2-Methoxy-5-(2-(2,4,6-trimethoxyphenyl)vinylsulfonamido)-phenyl phosphate (6r)
To an ice-cold stirred solution of (E)-2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenyl dihydrogen phosphate, 23 (0.2 g, 0.42 mmol) in 1,2-dimethoxyethane (3 mL) was added 25% sodium hydroxide solution (0.17 μL) and the reaction mixture was stirred for 3 h at room temperature. The solvent was decanted and the crude product was dried under vacuum. The crude solid was triturated with dry ethyl acetate (2 mL), filtered and washed with dry ethyl acetate (2 mL) to get pure 6r. Yield, 85%; pale yellow solid, mp 182–184 °C. 1H NMR (D2O, 300 MHz): δ 3.63 (s, 3H, OCH3), 3.68 (s, 9H, 3 × OCH3), 5.98 (s, 2H, Ar-H), 6.66–6.68 (dd, J = 2.1, 8.7 Hz, 1H, Ar-H), 6.78 (d, J = 8.7 Hz, 1H, Ar-H), 6.91 (d, J = 2.1 Hz, 1H, Ar-H), 7.06 (d, J = 15.8 Hz, 1H, =CH), 7.43 (d, J = 15.7 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 476.0501. Calcd for C18H20NNa2O10PS m/z: 519.0341.
(E)-N-(4-methoxy-3-nitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6s)
The title compound was obtained from 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetic acid 4g and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 49%; yellow solid, mp 172–174 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.83 (s, 9H, 3 X OCH3), 3.86 (s, 3H, OCH3), 6.28 (s, 2H, Ar-H), 6.95 (d, J = 15.3 Hz, 1H, =CH), 7.34 (d, J = 9.0 Hz, 1H, Ar-H), 7.42 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.61 (d, J = 15.6 Hz, 1H, CH=), 7.65 (d, J = 2.7 Hz, 1H, Ar-H), 9.94 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 425.0963. Calcd for C18H20N2O8S m/z: 424.0940.
(E)-N-(3-Amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6t) (Scheme 2)
Method A
The (E)-N-(4-Methoxy-3-nitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)-ethanesulfonamide 6s (0.65 mg, 1.5 mmol) was dissolved in acetone/water (40:20 mL) and heated to 50 °C. After 30 min sodium hydrosulfite (5.27 g, 30.0 mmol) was added slowly, and temperature was maintained at 50 °C for 30 min. After completion of reaction, the contents were cooled to room temperature, water was added, and the product was isolated by extraction with ethyl acetate. The organic phase was washed with water (3 X 100 mL), brine (50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to obtain the desired crude product 6t. The pure compound 6t was obtained following purification by silica gel flash column chromatography. Yield, 40%; yellow solid, mp 164–166 °C. 1H NMR (CDCl3, 300 MHz): δ 3.82 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 6.07 (br s, 1H, NH), 6.09 (s, 2H, Ar-H), 6.53 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.67 – 6.69 (m, 2H, Ar-H), 7.12 (d, J = 15.6 Hz, 1H, =CH), 7.85 (d, J = 15.6 Hz, 1H, CH=). 13C NMR (CDCl3, 75 MHz): δ 163.3, 161.2, 145.1, 136.8, 133.1, 130.2, 123.1, 112.3, 110.6, 109.7, 104.0, 90.4, 55.8, 55.7, 55.4. HRMS found [M+H]+ (m/z): 395.1216. Calcd for C18H22N2O6S m/z: 394.1199.
Method B
A solution of (E)-N-(4-Methoxy-3-nitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)-ethanesulfonamide 6s (0.65 mg, 1.5 mmol) in methanol/acetic acid (2:1, 30 mL) was heated to 60 °C and iron powder (0.42 g, 7.5 mmol) was added and continued stirring at 80 °C for 2 h. After completion of the reaction, the contents were cooled to 0 °C, DCM (30 mL) was added and neutralized the acetic acid with 10% sodium hydroxide solution (10 mL). The contents were stirred for 15 min and separated the organic phase, washed with water, brine and dried over anhydrous sodium sulfate. Removal of the solvent under reduced pressure gave crude product which on purification by silica gel flash column chromatography (ethyl acetate/hexane) afforded pure 6t. Analytical data are the same as obtained by method A.
(E)-N-(4-methoxy-3-nitrophenyl)-2-(3′,4′,5′-trimethoxyphenyl)ethenesulfonamide (6u)
The title compound was obtained from 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetic acid 4g and 3,4,5-trimethoxybenzaldehyde following the procedure as described in 6e, method B. Yield, 75%; pale yellow solid, mp 170–172 °C. 1H NMR (DMSO-d6; 300 MHz): 3.67 (s, 3H, OCH3), 3.79 (s, 6H, 2 × OCH3), 3.86 (s, 3H, OCH3), 7.05 (s, 2H, Ar-H), 7.30 (d, J = 15.4 Hz, 1H, =CH), 7.33 (d, J = 9.2 Hz, 1H, Ar-H), 7.37 (d, J = 15.4 Hz, 1H, =CH), 7.44 (dd, J = 9.1, 2.6 Hz, 1H, Ar-H), 7.67 (d, J= 2.6 Hz, 1H, Ar-H), 10.17 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 425.0969. Calcd for C18H20N2O8S m/z: 424.0940.
(E)-N-(3-Amino-4-methoxyphenyl)-2-(3′,4′,5′-trimethoxyphenyl)ethenesulfonamide (6v)
The title compound was obtained by the reduction of above (E)-N-(4-methoxy-3-nitrophenyl)-2-(3′,4′,5′-trimethoxyphenyl)ethanesulfonamide, 6u following the procedure as described for compound 6t, method B. Yield, 55%; white solid, mp 132–134 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.67 (s, 3H, OCH3), 3.68 (s, 3H, OCH3), 3.79 (s, 6H, 2 X OCH3), 4.76 (br s, 2H, NH2), 6.34 (d, J = 6.8 Hz, 1H, Ar-H), 6.52 (s, 1H, Ar-H), 6.65 (d, J = 7.8 Hz, 1H, Ar-H), 7.00 (s, 2H, Ar-H), 7.15 (d, J = 15.2 Hz, 1H, =CH), 7.25 (d, J = 15.2 Hz, 1H, CH=), 9.43 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 395.1221. Calcd for C18H22N2O6S m/z: 394.1199.
(E)-4-(3′,5′-dimethoxy-(4-(2-(N-(3-nitro-4-methoxyphenyl)sulfamoyl)vinyl)phenoxy)-butanoic acid (6w)
The title compound was obtained from 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetic acid 4g and 4-(4-formyl-3,5-dimethoxy)butyric acid following the procedure as described in 6e, method B. Yield, 63%; pale yellow solid, mp 182–184 °C. 1H NMR (CDCl3, 300 MHz): δ 2.11–2.18 (m, 2H, CH2), 2.61 (t, J = 7.1Hz, 2H, CH2), 3.83 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 4.08 (t, J = 6.1Hz, 2H, CH2), 6.09 (s, 2H, Ar-H), 6.24 (br s, 1H, NH), 7.12 (d, J=15.6 Hz, 1H, =CH), 7.15 (d, J = 8.7 Hz, 1H, Ar-H), 7.43 (dd, J = 8.7, 2.3 Hz, 1H, Ar-H), 7.64 (d, J = 2.2 Hz, 1H, Ar-H), 7.78 (d, J=15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 497.1173. Calcd for C21H24N2O10S m/z: 496.1152.
(E)-4-(4-(2-(N-(3-Amino-4-methoxyphenyl)sulfamoyl)vinyl)-3′,5′-dimethoxyphenoxy)-butanoic acid (6×)
The title compound was obtained by the reduction of above (E)-4-(3′,5′-dimethoxy-(4-(2-(N-(3-nitro-4-methoxyphenyl)sulfamoyl)vinyl)phenoxy)butanoic acid, 6w following the procedure as described for compound 6t, method B. Yield, 51%; light yellow solid, mp 164–166 °C. 1H NMR (CDCl3, 300 MHz): δ 2.04–2.10 (m, 2H, CH2), 2.53 (t, J = 7.1 Hz, 2H, CH2), 3.75 (s, 6H, 2 X OCH3), 3.77 (s, 3H, OCH3), 4.00 (t, J = 6.0 Hz, 2H, CH2), 6.02 (s, 2H, Ar-H), 6.20 (br s, 1H, NH), 6.66–6.70 (m, 3H, Ar-H), 7.02 (d, J = 15.6 Hz, 1H, =CH), 7.77 (d, J=15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 467.1432. Calcd for C21H26N2O8S m/z: 466.1410.
(E)-4-(4-(2-(N-(3-Hydroxy-4-methoxyphenyl)sulfamoyl)vinyl)-3′,5′-dimethoxyphenoxy)-butanoic acid (6y)
The title compound was obtained from 2-(N-(3-Hydroxy-4-methoxy-phenyl) sulfamoyl)acetic acid 4k and 4-(4-formyl-3,5-dimethoxy)butyric acid following the procedure as described in 6e, method B. Yield, 60%; orange red solid, mp 116–118 °C. 1H NMR (CDCl3, 300 MHz): δ 2.01–2.08 (m, 2H, CH2), 2.41 (t, J = 7.1 Hz, 2H, CH2), 3.83 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 4.10 (t, J = 6.1 Hz, 2H, CH2), 6.09 (s, 2H, Ar-H), 6.24 (br s, 1H, NH), 7.06 (d, J = 8.7 Hz, 1H, Ar-H), 7.33 (dd, J = 2.4, 8.7 Hz, 1H, Ar-H), 7.42 (d, J = 15.6 Hz, 1H, =CH), 7.78 (d, J = 15.6 Hz, 1H, CH=), 7.84 (d, J = 2.4 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 468.1273. Calcd for C21H25NO9S m/z: 467.1250.
(E)-N-(4-fluoro-3-nitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide (6z)
The title compound was obtained from 2-(N-(4-Fluoro-3-nitrophenyl)sulfamoyl)acetic acid 4i and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 49%; light yellow solid, mp 196–198 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.83 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 6.28 (s, 2H, Ar-H), 6.98 (d, J = 15.3 Hz, 1H, =CH), 7.47 – 7.52 (m, 1H, Ar-H), 7.58 (d, J = 9.0 Hz, 1H, Ar-H), 7.71 (d, J = 15.6 Hz, 1H, CH=), 7.87 (dd, J = 6.6, 2.4 Hz, 1H, Ar-H), 10.35 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 413.0761. Calcd for C17H17FN2O7S m/z: 412.0740.
(E)-N-(3-Amino-4-fluorophenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6aa)
The title compound was obtained by the reduction of above (E)-N-(4-fluoro-3-nitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide, 6z following the procedure as described for compound 6t, method B. Yield, 54%; light brown solid, mp 154–156 °C. 1H NMR (CDCl3, 300 MHz): δ 3.83 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 6.09 (s, 2H, Ar-H), 6.30 (br s, 1H, NH), 6.44 – 6.49 (m, 1H, Ar-H), 6.74 (dd, J = 7.8, 2.4 Hz, 1H, Ar-H), 6.88 (dd, J = 9.0, 1.8 Hz, 1H, Ar-H), 7.11 (d, J = 15.6 Hz, 1H, =CH), 7.87 (d, J = 15.3 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 383.1016. Calcd for C17H19FN2O5S m/z: 382.0999.
(E)-N-(4-Methoxy-3,5-dinitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6ab)
The title compound was obtained from 2-(N-(4-Methoxy-3,5-dinitrophenyl)sulfamoyl)acetic acid 4j and 2,4,6-trimethoxybenzaldehyde following the procedure as described in method A. Yield, 49%; yellow solid, mp 200–202 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.84 (s, 3H, OCH3), 3.86 (s, 6H, 2 X OCH3), 3.88 (s, 3H, OCH3), 6.29 (s, 2H, Ar-H), 7.04 (d, J = 15.6 Hz, 1H, =CH), 7.79 (d, J = 15.6 Hz, 1H, CH=), 7.97 (s, 2H, Ar-H), 10.73 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 470.0811. Calcd for C18H19N3O10S m/z: 469.0791.
(E)-N-(3,5-Diamino-4-methoxyphenyl)-2-(2,4,6-trimethoxyphenyl)ethenesulfonamide (6ac)
The title compound was obtained by the reduction of above (E)-N-(4-methoxy-3,5-dinitrophenyl)-2-(2,4,6-trimethoxyphenyl)ethenesulfonamide, 6ab following the procedure as described for compound 6t, method A. Yield, 40%; brown solid, mp 160–162 °C. 1H NMR (DMSO-d6, 500 MHz): δ 3.47 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.81 (s, 6H, 2 X OCH3), 4.61 (br s, 4H, 2 X NH2), 5.79 (s, 2H, Ar-H), 6.25 (s, 2H, Ar-H), 6.96 (d, J = 15.5 Hz, 1H, =CH), 7.56 (d, J = 15.5 Hz, 1H, CH=), 9.00 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 410.1329. Calcd for C18H23N3O6S m/z: 409.1308.
(E)-N-(3-Fluoro-4-methoxyphenyl)-2-(4′-methoxyphenyl)ethenesulfonamide (6ad)
The title compound was obtained from 2-(N-(3-fluoro-4-methoxyphenyl)sulfamoyl)acetic acid 4e and 4-methoxybenzaldehyde following the procedure as described in method A. Yield, 49%; light brown solid, mp 122–124 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 6.50 (br s, 1H, NH), 6.64 (d, J = 15.3 Hz, 1H, =CH), 6.86 – 6.96 (m, 4H, Ar-H), 7.08 (dd, J = 12.0, 2.1 Hz, 1H, Ar-H), 7.37–7.41 (m, 2H, Ar-H), 7.42 (d, J = 15.3 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 338.0800. Calcd for C16H16FNO4S m/z: 337.0784.
(E)-N-(3-Fluoro-4-methoxyphenyl)-2-(perfluorophenyl)ethenesulfonamide (6ae)
The title compound was obtained from 2-(N-(3-fluoro-4-methoxyphenyl)sulfamoyl)acetic acid 4e and 2,3,4,5,6-pentafluorobenzaldehyde following the procedure as described in method A. Yield, 48%; light yellow solid, mp 113–115 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 3H, OCH3), 6.23 (br s, 1H, NH), 6.73 (d, J = 8.7 Hz, 1H, Ar-H), 6.83 (dd, J = 2.4, 8.7 Hz, 1H, Ar-H), 6.91 (d, J = 15.3 Hz, 1H, =CH), 7.06 (d, J = 8.7 Hz, 1H, Ar-H), 7.82 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 398.0222. Calcd for C15H9F6NO3S m/z: 397.0207.
(E)-N-(4-Methoxy-3-nitrophenyl)-2-(perfluorophenyl)ethenesulfonamide (6af)
The title compound was obtained from 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetic acid 4g and 2,3,4,5,6-pentafluorobenzaldehyde following the procedure as described in method A. Yield, 49%; light yellow solid, mp 102–104 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.83 (s, 3H, OCH3), 6.30 (br s, 1H, NH), 7.32 (d, J = 15.6 Hz, 1H, =CH), 7.41 (d, J = 2.7 Hz, 1H, Ar-H), 7.60 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H). 7.76 (d, J = 15.6 Hz, 1H, CH=), 7.86 (d, J = 2.7 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 425.0172. Calcd for C15H9F5N2O5S m/z: 424.0152.
(E)-N-(3-Amino-4-methoxyphenyl)-2-(perfluorophenyl)ethenesulfonamide (6ag)
The title compound was obtained by the reduction of above (E)-N-(4-methoxy-3-nitrophenyl)-2-(perfluorophenyl)ethenesulfonamide, 6af following the procedure as described for compound 6t, method B. Yield, 54%; yellow solid, mp 86–88 °C. 1H NMR (CDCl3, 300 MHz): δ 3.84 (s, 3H, OCH3), 6.38 (br s, 1H, NH), 6.47 (dd, J = 2.7, 8.7 Hz, 1H, Ar-H), 6.73 (d, J = 2.7 Hz, 1H, Ar-H), 7.03 (d, J = 2.7 Hz, 1H, Ar-H), 7.14 (d, J = 15.6 Hz, 1H, =CH), 7.49 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 395.0424. Calcd for C15H11F5N2O3S m/z: 394.0411.
(E)-2-(4′-Methoxy-3′-nitrophenyl)-N-(perfluorophenyl)ethenesulfonamide (6ah)
The title compound was obtained from 2-(N-(perfluorophenyl)sulfamoyl)acetic acid 4h and 4-methoxy-3-nitrobenzaldehyde following the procedure as described in method A. Yield, 49%; light brown solid, mp 160–162 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 3H, OCH3), 7.07 (d, J = 9.0 Hz, 1H, Ar-H), 7.21 (d, J = 15.3 Hz, 1H, =CH), 7.33 (br s, 1H, NH), 7.48 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.61 (d, J = 15.6 Hz, 1H, CH=), 7.73 (d, J = 2.7 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 425.0168. Calcd for C15H9F5N2O5S m/z: 424.0152.
(E)-2-(3′-Amino-4′-methoxyphenyl)-N-(perfluorophenyl)ethenesulfonamide (6ai)
The title compound was obtained by the reduction of above (E)-2-(4′-methoxy-3′-nitrophenyl)-N-(perfluorophenyl)ethenesulfonamide, 6ah following the procedure as described for compound 6t, method A. Yield, 41%; pale yellow solid, mp 98–100 °C. 1H NMR (CDCl3, 300 MHz): δ 3.83 (s, 3H, OCH3), 7.04 (d, J = 8.7 Hz, 1H, Ar-H), 7.27 (d, J = 15.6 Hz, 1H, =CH), 7.29 (br s, 1H, NH), 7.42 (dd, J = 2.4, 8.7 Hz, 1H, Ar-H), 7.62 (d, J = 15.6 Hz, 1H, CH=), 7.69 (d, J = 2.7 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 395.0426. Calcd for C15H11F5N2O3S m/z: 394.0411.
(E)-N,2-Bis(perfluorophenyl)ethenesulfonamide (6aj)
The title compound was obtained from 2-(N-(perfluorophenyl)sulfamoyl)acetic acid 4h and 2,3,4,5,6-pentafluorobenzaldehyde following the procedure as described in method A. Yield, 49%; off white solid, mp 138–140 °C. 1H NMR (CDCl3, 400 MHz): δ 6.14 (br s, 1H, NH), 7.24 (d, J = 15.8 Hz, 1H, =CH), 7.44 (d, J = 15.8 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 439.9746. Calcd for C14H3F10NO2S m/z: 438.9725.
General Procedure for the preparation of (E)-N-Aryl-2-arylethenesulfonamides (6)
Method C (Scheme 5)
Step 1: General Procedure for the preparation of 2-(N-(phenyl)sulfamoyl)acetyl chloride (14)
Method 1
To a solution of 2-(N-(4-methoxy-phenyl) sulfamoyl)acetic acid, 4d (2.0 g, 8.2 mmol) in anhydrous methylene chloride (100 mL), was added thionyl chloride (4.0 mL) in portions at room temperature. The resulting solution was stirred at room temperature for 6 h. After reaction completion, monitor by TLC, the reaction mixture was concentrated under reduced pressure, and the residue was dissolved in toluene and then reconcentrated to give the crude acid chloride, which was used in next step without further purification.
Method 2
To a solution of 2-(N-(4-methoxy-phenyl)sulfamoyl)acetic acid, 4d (2.0 g, 8.2 mmol) in anhydrous methylene chloride (100 mL), was added oxalyl chloride (1.6 mL, 18.0 mmol) drop wise at room temperature and continue stirring for 20 min. Then added 5 drops of anhydrous N,N-dimethylformamide to the reaction mixture. The mixture was stirred at room temperature for 12 h. After completion, the reaction mixture concentrated under reduced pressure and the residue triturated with hexane: diethyl ether, 1:1 to give solid which was isolated by filtration. The crude phenylsulfamoyl acetyl chloride was used in next step without purification. The following phenylsulfamoyl acetyl chlorides 14 were prepared using one of the methods.
2-(N-(4-Methoxyphenyl)sulfamoyl)acetyl chloride (14a)
Chlorination of 2-(N-(4-methoxyphenyl) sulfamoyl)acetic acid, 4d with thionyl chloride as described in method A yielded the corresponding 2-(N-(4-methoxyphenyl)sulfamoyl)acetyl chloride. Yield: 80%; white solid, mp 147–149 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.73 (s, 3H, OCH3), 4.29 (s, 2H, CH2), 6.90 (d, J = 9.0 Hz, 2H, Ar-H), 7.17 (d, J = 9.0 Hz, 2H, Ar-H), 9.76 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 264.0037. Calcd for C9H10ClNO4S m/z: 263.0019.
2-(N-(4-Methoxy-3-nitrophenyl)sulfamoyl)acetyl chloride (14b)
Chlorination of 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetic acid, 4g with thionyl chloride as described in method A yielded the corresponding 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetyl chloride. Yield: 78%; pale yellow solid, mp 136–138 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.91 (s, 3H, OCH3), 4.44 (s, 2H, CH2), 7.39 (d, J = 9.0 Hz, 1H, Ar-H), 7.50 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.73 (d, J = 2.7 Hz, 1H, Ar-H), 10.22 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 308.9896. Calcd for C9H9ClN2O6S m/z: 307.9870.
2-(N-(3-Hydroxy-4-methoxyphenyl)sulfamoyl)acetyl chloride (14c)
Chlorination of 2-(N-(3-hydroxy-4-methoxyphenyl)sulfamoyl)acetic acid, 4k with oxalyl chloride as described in method B yielded the corresponding 2-(N-(3-hydroxy-4-methoxyphenyl)sulfamoyl)acetyl chloride. Yield: 67%; light yellow solid, mp 129–131 °C. 1H NMR (DMSO-d6, 500 MHz): δ 3.94 (s, 3H, OCH3), 4.38 (s, 2H, CH2), 6.61 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.76 (d, J = 2.7 Hz, 1H, Ar-H), 6.82 (d, J = 8.7 Hz, 1H, Ar-H), 9.28 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 280.0000. Calcd for C9H10ClNO5S m/z: 278.9968.
Step 2: General Procedure for the preparation of Phenacyl N-Arylsulfone (16)
To a cooled solution of anhydrous aluminium chloride (1.85 g, 13.91 mmol) and 1,3,5-trimethoxy benzene, 15 (15.91 g, 94.60 mmol) in anhydrous dichloromethane (100 mL) was added 2-(N-(4-methoxyphenyl)sulfamoyl)acetyl chloride, 14a (5.98 g, 22.68 mmol) in portions at 0 °C over a period of 30 min and continued stirring at room temperature for 4 h. After completion of reaction, the reaction mixture quenched by addition of water, extracted with methylene chloride and washed with water. The organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum to obtain the desired product, which is used in next step without further purification.
N-(4-Methoxyphenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide (16a)
Friedel-Crafts alkylation of 2-(N-(4-methoxyphenyl)sulfamoyl)acetyl chloride, 14a with excess 1,3,5-trimethoxybenzene (15) yielded the corresponding N-(4-methoxyphenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide. Yield: 80%; off white solid, mp 148–150 °C. 1H NMR (CDCl3, 300 MHz): δ 3.73 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.97 (s, 2H, CH2), 6.09 (s, 2H, Ar-H), 6.22 (br s, 1H, NH), 6.90 (d, J = 8.6 Hz, 2H, Ar-H), 7.32 (d, J = 8.6 Hz, 2H, Ar-H). HRMS found [M+H]+ (m/z): 396.1060. Calcd for C18H21NO7S m/z: 395.1039.
N-(4-Methoxy-3-nitrophenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide (16b)
Friedel-Crafts alkylation of 2-(N-(4-methoxy-3-nitrophenyl)sulfamoyl)acetyl chloride, 14b with excess 1,3,5-trimethoxybenzene (15) yielded the corresponding N-(4-methoxy-3-nitrophenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide. Yield: 78%; light yellow solid, mp 160–162 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.76 (s, 3H, OCH3), 3.87 (s, 6H, 2 X OCH3), 3.89 (s, 3H, OCH3), 4.86 (s, 2H, CH2), 6.09 (s, 2H, Ar-H), 7.41 (d, J = 9.0 Hz, 1H, Ar-H), 7.56 (dd, J = 2.7, 9.0 Hz, 1H, Ar-H), 7.78 (d, J = 2.7 Hz, 1H, Ar-H), 9.62 (br s, 1H, NH). HRMS found [M+H]+ (m/z): 441.0916. Calcd for C18H20N2O9S m/z: 440.0890.
N-(3-Hydroxy-4-methoxyphenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide (16c)
Friedel-Crafts alkylation of 2-(N-(3-hydroxy-4-methoxyphenyl)sulfamoyl)acetyl chloride, 14c with excess 1,3,5-trimethoxybenzene (15) yielded the corresponding N-(3-hydroxy-4-methoxyphenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide. Yield: 77%; white solid, mp 158–160 °C. 1H NMR (CDCl3, 300 MHz): δ 3.73 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.61 (s, 2H, CH2), 6.16 (s, 2H, Ar-H), 6.27 (br s, 1H, NH), 6.61 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.76 (d, J = 2.7 Hz, 1H, Ar-H), 6.82 (d, J = 8.7 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 412.1015. Calcd for C18H21NO8S m/z: 411.0988.
Step 3: General Procedure for the preparation of 2-Hydroxy-N-Aryl-2-arylethane-sulfonamide (17)
To a cooled solution of N-aryl-2-oxo-2-arylethanesulfonamide, 16 (10 mmol) in anhydrous tetrahydrofuran (40 mL) under N2 atmosphere, was added NaBH4 (10 mmol) slowly at 0 °C. The reaction mixture was maintained at room temperature for 3 h. After completion of the reaction, the contents were poured onto crushed ice. The solid that separated out was filtered, washed with cold water, and dried under vacuum resulted 17. The following - hydroxy-N-Aryl-2-arylethane-sulfonamide 17 were prepared using above the method.
2-Hydroxy-N-(4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide (17a)
The title compound was obtained by the reduction of N-(4-methoxyphenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide, 16a following the procedure as described for compound 17. Yield, 88%; white solid, mp 162–164 °C. 1H NMR (CDCl3, 300 MHz): δ 3.49 (dd, J = 9.9, 4.6 Hz, 2H, CH2-CH), 3.73 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.97 (m, 1H, CH-OH), 5.34 (d, J = 4.2 Hz, 1H, CH2-CH), 6.11 (s, 2H, Ar-H), 6.20 (br s, 1H, NH), 6.92 (d, J = 8.6 Hz, 2H, Ar-H), 7.36 (d, J = 8.6 Hz, 2H, Ar-H). HRMS found [M+H]+ (m/z): 398.1219. Calcd for C18H23NO7S m/z: 397.1195.
2-Hydroxy-N-(4-methoxy-3-nitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide (17b)
The title compound was obtained by the reduction of N-(4-methoxy-3-nitrophenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide, 16b following the procedure as described for compound 17. Yield, 85%; white solid, mp 168–170 °C. 1H NMR (CDCl3, 300 MHz): δ 3.52 (dd, J = 9.9, 4.6 Hz, 2H, CH2-CH), 3.76 (s, 3H, OCH3), 3.86 (s, 6H, 2 X OCH3), 3.88 (s, 3H, OCH3), 4.99 (m, 1H, CH-OH), 5.39 (d, J = 4.2 Hz, 1H, CH2-CH), 6.14 (s, 2H, Ar-H), 6.19 (br s, 1H, NH), 7.42 (d, J = 9.0 Hz, 1H, Ar-H), 7.69 (dd, J = 8,7, 1.8 Hz, 1H, Ar-H), 7.93 (d, J = 1.8 Hz, 1H, Ar-H). HRMS found [M+H]+ (m/z): 443.1057. Calcd for C18H22N2O9S m/z: 442.1046.
2-Hydroxy-N-(3-hydroxy-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethane-sulfonamide (17c)
The title compound was obtained by the reduction of N-(3-hydroxy-4-methoxyphenyl)-2-oxo-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide, 16c following the procedure as described for compound 17. Yield, 86%; white solid, mp 152–154 °C. 1H NMR (CDCl3, 300 MHz): δ 3.43 (dd, J = 9.9, 4.6 Hz, 2H, CH2-CH), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 5.05 (m, 1H, CH-OH), 5.63 (br s, 1H, OH), 6.02 (d, J = 4.2 Hz, 1H, CH2-CH), 6.09 (s, 2H, Ar-H), 6.19 (br s, 1H, NH), 6.73–6.83 (m, 3H, Ar-H). HRMS found [M+H]+ (m/z): 414.1163. Calcd for C18H23NO8S m/z: 413.1144.
Step 4: General Procedure for the preparation of (E)-N-Aryl-2-arylethenesulfonamide (6). (Scheme 5)
p-Toluenesulfonic acid (1 mmol) was added in one portion to a mixture of 2-hydroxy-N-Aryl-2-arylethanesulfonamide, 17 (5 mmol) in anhydrous toluene (25 mL) at room temperature and under N2 atmosphere. The temperature was raised to 120 °C, and the mixture was refluxed for 3 h using a Dean-Stark water separator. After completion of the reaction, the reaction mixture was concentrated under reduced pressure and then quenched by the addition of water (25 mL). The aqueous layer was neutralized with a saturated aqueous solution of sodium hydrogen carbonate and extracted with dichloromethane (3 X 25 mL). The combined organic extracts were washed with brine (2 X 25 mL), dried over Na2SO4, filtered and the solvent was evaporated under reduced pressure to afford crude product, which on recrystallization in 2-propanol afforded the desired product 6. The following (E)-N-aryl-2-arylethenesulfonamides were prepared using the above procedure.
(E)-N-(4-Methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide (6i)
The title compound was obtained by the dehydration of 2-hydroxy-N-(4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethanesulfonamide, 17a following the procedure as described for compound 6 in scheme 4. Yield, 56%; light yellow solid, mp 176–178 °C. Analytical data are the same as 6i obtained by method A in scheme 1.
(E)-N-(4-methoxy-3-nitrophenyl)-2-(2,4,6-trimethoxyphenyl)ethenesulfonamide (6s)
The title compound was obtained by the dehydration of 2-hydroxy-N-(4-methoxy-3-nitrophenyl)-2-(2′,4′,6′-trimethoxyphenyl)-ethanesulfonamide, 17b following the procedure as described for compound 6 in scheme 4. Yield, 58%; yellow solid, mp 172–174 °C. Analytical data are the same as 6s obtained by method A in scheme 1.
(E)-N-(3-Hydroxy-4-methoxyphenyl)-2-(2,4,6-trimethoxyphenyl)ethenesulfonamide (6p)
The title compound was obtained by the dehydration of 2-hydroxy-N-(3-hydroxy-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)-ethanesulfonamide, 17c following the procedure as described for compound 6 in scheme 4. Yield, 55%; light green solid, mp 148–150 °C. Analytical data are the same as 6p obtained by method B in scheme 1.
General Procedure for the preparation of (E)-N-Aryl-2-arylethenesulfonamides (6)
Method D (Scheme 6)
Step 1: General Procedure for the synthesis of (E)-Styryl sulfonyl chloride 19. (Scheme 6)
The following (E)-Styryl sulfonyl chloride, 19 were prepared according to the procedure reported in the literature.27c
(E)-2-Phenylethenesulfonyl chloride (19a)
Chlorosulfonylation of styrene 18a with sulfuryl chloride resulted the corresponding (E)-2-phenylethenesulfonyl chloride. The yield of this reaction was 85% giving a white solid with a melting point of 86–88 °C. 1H NMR (CDCl3, 300 MHz): δ 7.26 (d, J = 15.6 Hz, 1H, = CH), 7.34 – 7.63 (m, 5H, Ar-H), 7.70 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 202.9878. Calcd for C8H7ClO2S m/z: 201.9855.
(E)-2-(4-Bromophenyl)ethenesulfonyl chloride (19b)
Chlorosulfonylation of 4-bromostyrene 18b with sulfuryl chloride resulted the corresponding (E)-2-(4-bromophenyl)ethenesulfonyl chloride. The yield of this reaction was 89% giving a white solid with a melting point of 98–100 °C. 1H NMR (CDCl3, 300 MHz): δ 7.11 (d, J = 15.6 Hz, 1H, = CH), 7.52 (d, J = 8.4 Hz, 2H, Ar-H), 7.59 (d, J = 8.4 Hz, 2H, Ar-H), 7.78 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 280.8982. Calcd for C8H6BrClO2S m/z: 279.8960.
(E)-2-(4-Methoxyphenyl)ethenesulfonyl chloride (19c)
Chlorosulfonylation of 4-methoxystyrene 18c with sulfuryl chloride resulted the corresponding (E)-2-(4-methoxyphenyl)ethenesulfonyl chloride. The yield of this reaction was 79% giving a white solid with a melting point of 82–84 °C. 1H NMR (CDCl3, 300 MHz): δ 3.85 (s, 3H, OCH3), 6.94 (d, J = 8.7 Hz, 2H, Ar-H), 7.16 (d, J = 15.3 Hz, 1H, = CH), 7.62 (d, J = 8.7 Hz, 2H, Ar-H),7.41 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 232.9986. Calcd for C9H9ClO3S m/z: 231.9961.
Step 2: General Procedure for the Preparation of (E)-N-aryl-2-arylethenesulfonamide (6). (Scheme 6)
Method 1
To a solution of phenylethenesulfonyl chloride, 19 (0.5 g, 2.5 mmol) and triethylamine (2.6 g, 25 mmol) in dichloromethane (15 mL) at room temperature, was added a solution of aniline, 1a (0.235 g, 2.5 mmol) in dichloromethane (10 mL) drop wise over a period of 15 min and continued stirring for 5 h. After completion of reaction, water was added, stirred for 15 min and separated the organic layer, dried over anhydrous sodium sulfate and evaporated under reduced pressure resulted crude 6. The crude on silica gel column purification resulted pure 6.
Method 2
To a solution of phenylethenesulfonyl chloride, 19 (0.5 g, 2.5 mmol) in pyridine (5 mL) at room temperature, was added aniline, 1a (0.235 g, 2.5 mmol) over a period of 5 min and continued stirring for 6 h. After completion of reaction, water was added, stirred for 30 min and the solid separated was filtered, washed with cold water and dried under vacuum resulted crude 6. The crude on silica gel column purification resulted pure 6. The following (E)-N-Aryl-2-arylethenesulfonamide, 6 were prepared using one of the above methods.
(E)-N,2-Diphenylethenesulfonamide (6a)
Condensation of (E)-2-phenylethenesulfonyl chloride, 19a with aniline, 1a resulted the corresponding (E)-N,2-diphenylethenesulfonamide. The yield of this reaction was 83%, giving a white solid with a melting point 112–114 °C. The analytical data are the same as 6a obtained by method A in scheme 1.
(E)-N-(4-Chlorophenyl)-2-phenylethenesulfonamide (6b)
Condensation of (E)-2-phenylethenesulfonyl chloride, 19a with 4-chloroaniline, 1b resulted the corresponding (E)-N-(4-chlorophenyl)-2-phenylethenesulfonamide. The yield of this reaction was 80%, giving a white crystalline solid with a melting point 108–110 °C. The analytical data are the same as 6b obtained by method A in scheme 1.
(E)-2-(4′-Bromophenyl)-N-(4-fluorophenyl)ethenesulfonamide (6c)
Condensation of (E)-2-(4-bromophenyl)ethenesulfonyl chloride, 19b with 4-fluoroaniline, 1c resulted the corresponding (E)-2-(4′-bromophenyl)-N-(4-fluorophenyl)ethenesulfonamide. The yield of this reaction was 79%, giving a white solid with a melting point 138–140 °C. The analytical data are the same as 6c obtained by method A in scheme 1.
(E)-N-(4-Fluorophenyl)-2-(4′-methoxyphenyl)ethenesulfonamide (6d)
Condensation of (E)-2-(4-methoxyphenyl)ethenesulfonyl chloride, 19c with 4-fluoroaniline, 1c resulted the corresponding (E)-N-(4-fluorophenyl)-2-(4′-methoxyphenyl)ethenesulfonamide. The yield of this reaction was 78%, giving a white solid with a melting point 98–100 °C. The analytical data are the same as 6d obtained by method A in scheme 1.
General Procedure for the Preparation of Sulfonamido Amino Esters (24). (Scheme 10)
To a solution of sodium acetate (41.0 g, 500 mmol) dissolved in ethanol (250 mL) was added methyl 2-bromoacetate (76.4 g, 500 mmol) and refluxed for 10 min. To the cooled reaction mixture compound (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxy-phenyl)ethenesulfonamide, 6t (39.44 g, 100 mmol) was added and then refluxed for 48 h. After completion of the reaction, the reaction mixture was concentrated under vacuum and poured into ice-water. The solid formed was filtered, washed with water, finally with cold 2-propanol and dried under vacuum, (24) which was used in next step without further purification. The following amino esters were prepared using the above procedure.
(E)-Methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-acetate (24a)
The title compound was obtained by the alkylation of (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide 6t with methyl 2-bromoacetate following the procedure as described for compound 24. Yield, 70%; light brown solid, mp 142–144 °C. 1H NMR (CDCl3, 300 MHz): δ 3.78 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.84 (s, 6H, 2 × OCH3), 3.85 (s, 3H, OCH3), 3.91 (s, 2H, CH2), 4.74 (t, J = 5.4 Hz, 1H, NH), 6.09 (s, 2H, Ar-H), 6.44 (d, J = 2.1 Hz, 1H, Ar-H), 6.49 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.67 (d, J = 8.4 Hz, 1H, Ar-H), 7.12 (d, J = 15.6 Hz, 1H, =CH), 7.86 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 467.1429. Calcd for C21H26N2O8S m/z: 466.1410.
(E)-Methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-propanoate (24b)
The title compound was obtained by the alkylation of (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide 6t with methyl 2-bromo-propionate following the procedure as described for compound 24. Yield, 66%; off white solid, mp 146–148 °C. 1H NMR (CDCl3, 300 MHz): δ 1.47 (d, J = 6.9 Hz, 3H, CH3), 3.73 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.12 (br s, 1H, CH), 4.75 (br s, 1H, NH), 6.09 (s, 2H, Ar-H), 6.10 (s, 1H, NH), 6.46 (d, J = 2.4 Hz, 1H, Ar-H), 6.49 (dd, J = 2.4, 8.1 Hz, 1H, Ar-H), 6.66 (d, J = 8.1 Hz, 1H, Ar-H), 7.12 (d, J = 15.6 Hz, 1H, =CH), 7.84 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 481.1591. Calcd for C22H28N2O8S m/z: 480.1566.
(E)-Methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-2-methylpropanoate (24c)
The title compound was obtained by the alkylation of (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide 6t with methyl 2-bromo2-methylpropionate following the procedure as described for compound 24. Yield, 65%; brown solid, mp 140–142 °C. 1H NMR (CDCl3, 300 MHz): δ 1.55 (s, 6H, 2 X CH3), 3.71 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.69 (br s, 1H, NH), 6.02 (br s, 1H, NH), 6.08 (s, 2H, Ar-H), 6.33 (d, J = 2.4 Hz, 1H, Ar-H), 6.51 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.66 (d, J = 8.7 Hz, 1H, Ar-H), 7.12 (d, J = 15.6 Hz, 1H, =CH), 7.82 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 495.1719. Calcd for C23H30N2O8S m/z: 494.1723.
(E)-Methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-2-phenylacetate (24d)
The title compound was obtained by the alkylation of (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide 6t with methyl α-bromo-phenylacetate following the procedure as described for compound 24. Yield, 63%; light yellow solid, mp 138–140 °C. 1H NMR (CDCl3, 300 MHz): δ 3.71 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.85 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 4.69 (d, J = 6.0 Hz, 1H, CH), 5.83 (d, J = 6.3 Hz, 1H, NH), 6.09 (s, 2H, Ar-H), 6.34 (d, J = 2.4 Hz, 1H, Ar-H), 6.41 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.57 (d, J = 8.7 Hz, 1H, Ar-H), 7.09 (d, J = 15.6 Hz, 1H, =CH), 7.25–7.33 (m, 3H, Ar-H), 7.46 (d, J = 8.7 Hz, 2H, Ar-H), 7.83 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 543.1747. Calcd for C27H30N2O8S m/z: 542.1723.
(E)-Methyl 2-(4″-Fluorophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfon-amido) phenylamino)acetate (24e)
The title compound was obtained by the alkylation of (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide 6t with methyl 2-bromo-2- (4-fluorophenyl)acetate following the procedure as described for compound 24. Yield, 64%; off white solid, mp 124–126 °C. 1H NMR (CDCl3, 300 MHz): δ 3.69 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.83 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.78 (d, J = 5.5 Hz, 1H, CH), 5.98 (br s, 1H, NH), 6.09 (s, 2H, Ar-H), 6.23 (d, J = 2.1 Hz, 1H, Ar-H), 6.39 (dd, J = 2.1, 8.1 Hz, 1H, Ar-H), 6.63 (d, J = 8.1 Hz, 1H, Ar-H), 7.12 (d, J = 8.4 Hz, 2H, Ar-H), 7.17 (d, J = 15.6 Hz, 1H, =CH), 7.37 (d, J = 8.4 Hz, 2H, Ar-H), 7.86 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 561.1653. Calcd for C27H29FN2O8S m/z: 560.1629.
(E)-Methyl 2-(4″-Chlorophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfon-amido) phenylamino)acetate (24f)
The title compound was obtained by the alkylation of (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide 6t with methyl 2-bromo-2- (4-chlorophenyl)acetate following the procedure as described for compound 24. Yield, 64%; light yellow solid, mp 144–146 °C. 1H NMR (CDCl3, 300 MHz): δ 3.71 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.85 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 4.86 (s, 1H, CH), 6.04 (br s, 1H, NH), 6.11 (s, 2H, Ar-H), 6.27 (d, J = 2.4 Hz, 1H, Ar-H), 6.39 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.63 (d, J = 8.4 Hz, 1H, Ar-H), 7.01 (d, J = 15.6 Hz, 1H, =CH), 7.11 (d, J = 8.4 Hz, 2H, Ar-H), 7.24 (d, J = 8.4 Hz, 2H, Ar-H), 7.94 (d, J = 15.6 Hz, 1H, CH=). HRMS found [M+H]+ (m/z): 577.1361. Calcd for C27H29ClN2O8S m/z: 576.1333.
(E)-Methyl 2-(4″-Bromophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfon-amido) phenylamino)acetate (24g)
The title compound was obtained by the alkylation of (E)-N-(3-amino-4-methoxyphenyl)-2-(2′,4′,6′-trimethoxyphenyl)ethenesulfonamide 6t with methyl 2-bromo-2- (4-bromophenyl)acetate following the procedure as described for compound 24. Yield, 62%; light brown solid, mp 150–152 °C. 1H NMR (CDCl3, 300 MHz): δ 3.68 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.82 (s, 6H, 2 X OCH3), 3.85 (s, 3H, OCH3), 4.89 (s, 1H, CH), 6.07 (br s, 1H, NH), 6.09 (s, 2H, Ar-H), 6.27 (d, J = 1.8 Hz, 1H, Ar-H), 6.44 (dd, J = 1.8, 8.1 Hz, 1H, Ar-H), 6.50 (d, J = 8.1 Hz, 1H, Ar-H), 6.95 (d, J = 15.6 Hz, 1H, =CH), 7.18 (d, J = 8.4 Hz, 2H, Ar-H), 7.86 (d, J = 15.6 Hz, 1H, CH=), 7.91 (d, J = 8.4 Hz, 2H, Ar-H). HRMS found [M+H]+ (m/z): 621.0851. Calcd for C27H29BrN2O8S m/z: 620.0828.
General Procedure for the Preparation of Sulfonamido Amino Acids (25). (Scheme 10)
To a solution of sulfonamido amine ester 24 (46.6 g, 100 mmol) in ethanol (200 mL), 20% aqueous sodium hydroxide solution (100 mL) was added slowly with vigorous stirring. The reaction mixture was stirred at 60 °C for 1 h. After completion of the reaction, the solvent was removed under vacuum and the remainder was acidified with dilute hydrochloric acid to pH 4. The solid that formed was filtered, washed with water and dried to get the sulfonamido amino acid 25.
(E)-2-(2-Methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)acetic acid (25a)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)acetate, 24a following the procedure as described for compound 25. Yield, 50%; pale yellow solid, mp 110–112 °C. 1H NMR(DMSO-d6, 300 MHz): δ 3.71 (s, 2H, CH2), 3.82 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.87 (s, 6H, 2 X OCH3), 6.26 (s, 2H, Ar-H), 6.48 (d, J = 1.8 Hz, 1H, Ar-H), 6.65 (dd, J = 1.8, 8.1 Hz, 1H, Ar-H), 6.86 (d, J = 8.1 Hz, 1H, Ar-H), 6.94 (d, J = 15.6 Hz, 1H, =CH), 7.60 (d, J = 15.6 Hz, 1H, CH=), 8.99 (br s, 1H, NH). HRMS found [M-H]− (m/z): 451.1209. Calcd for C20H24N2O8S m/z: 452.1253.
(E)-2-(2-Methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-propanoic acid (25b)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)propanoate, 24b following the procedure as described for compound 25. Yield, 57%; light brown solid, mp 116–118 °C. 1H NMR (DMSO-d6, 300 MHz): δ 1.28 (d, J = 6.9 Hz, 3H, CH3), 3.69 (s, 3H, OCH3), 3.71 (d, J = 6.9 Hz, 1H, CH), 3.83 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 4.77 (br s, 1H, NH), 6.28 (s, 2H, Ar-H), 6.34 (d, J = 2.4 Hz, 1H, Ar-H), 6.51 (d, J = 2.7 Hz, 1H, Ar-H), 6.66 (dd, J = 2.1, 8.1 Hz, 1H, Ar-H), 6.95 (d, J = 15.6 Hz, 1H, =CH), 7.59 (d, J = 15.6 Hz, 1H, CH=), 9.15 (br s, 1H, NH). HRMS found [M-H]− (m/z): 465.1386. Calcd for C21H26N2O8S m/z: 466.1410.
(E)-2-(2-Methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-2-methylpropanoic acid (25c)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-2-methylpropanoate, 24c following the procedure as described for compound 25. Yield, 61%; light yellow solid, mp 122–124 °C. 1H NMR (DMSO-d6, 300 MHz): δ 1.44 (s, 6H, 2 X CH3), 3.72 (s, 3H, OCH3), 3.82 (s, 9H, 3 X OCH3), 6.27 (s, 2H, Ar-H), 6.34 (dd, J = 2.4, 8.4 Hz 1H, Ar-H), 6.45 (d, J = 2.4 Hz, 1H, Ar-H), 6.70 (d, J = 8.7 Hz, 1H, Ar-H), 6.95 (d, J = 15.6 Hz, 1H, =CH), 7.57 (d, J = 15.6 Hz, 1H, CH=), 9.24 (br s, 1H, NH). 13C NMR (DMSO-d6, 75 MHz): δ 176.6, 163.1, 160.6, 143.7, 135.3, 131.1, 130.6, 123.9, 109.8, 108.6, 105.6, 102.5, 90.9, 56.0, 55.9, 55.5, 55.4, 25.2. HRMS found [M-H]− (m/z): 479.1539. Calcd for C22H28N2O8S m/z: 480.1566.
(E)-2-(2-Methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-2-phenylacetic acid (25d)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)phenylamino)-2-phenylacetate, 24d following the procedure as described for compound 25. Yield, 60%; white solid, mp 124–126 °C. 1H NMR (CDCl3, 300 MHz): δ 3.79 (s, 6H, 2 X OCH3), 3.81 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 4.86 (d, J = 6.0 Hz, 1H, CH), 6.28 (s, 2H, Ar-H), 6.32 (d, J = 2.4 Hz, 1H, Ar-H), 6.42 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 6.59 (d, J = 8.7 Hz, 1H, Ar-H), 7.14 (d, J = 15.6 Hz, 1H, =CH), 7.22–7.26 (m, 3H, Ar-H), 7.38 (d, J = 8.4 Hz, 2H, Ar-H), 7.69 (d, J = 15.6 Hz, 1H, CH=), 9.20 (br s, 1H, NH). HRMS found [M-H]− (m/z): 527.1541. Calcd for C26H28N2O8S m/z: 528.1566.
(E)-2-(4″-Fluorophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)-phenylamino)acetic acid (25e)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(4″-fluorophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)-phenylamino)acetate, 24e following the procedure as described for compound 25. Yield, 48%; light yellow solid, mp 102–104 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.79 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 4.72 (d, J = 5.5 Hz, 1H, CH), 6.29 (s, 2H, Ar-H), 6.33 (d, J = 2.1 Hz, 1H, Ar-H), 6.69 (dd, J = 2.1, 8.1 Hz, 1H, Ar-H), 6.73 (d, J = 8.1 Hz, 1H, Ar-H), 7.09 (d, J = 8.4 Hz, 2H, Ar-H), 7.17 (d, J = 15.6 Hz, 1H, =CH), 7.32 (d, J = 8.4 Hz, 2H, Ar-H), 7.66 (d, J = 15.6 Hz, 1H, CH=), 9.04 (br s, 1H, NH). HRMS found [M-H]− (m/z): 545.1427. Calcd for C26H27FN2O8S m/z: 546.1472.
(E)-2-(4″-Chlorophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)-phenylamino)acetic acid (25f)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(4″-chlorophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)-phenylamino)acetate, 24f following the procedure as described for compound 25. Yield, 60%; brown solid, mp 108–110 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.78 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.87 (s, 3H, OCH3), 4.89 (s, 1H, CH), 6.18 (d, J =2.1 Hz, 1H, Ar-H), 6.30 (s, 2H, Ar-H), 6.36 (dd, J = 2.4, 8.7 Hz, 1H, Ar-H), 6.73 (d, J=8.7 Hz, 1H, Ar-H), 6.87 (d, J = 15.6 Hz, 1H, =CH), 7.20 (d, J = 8.4 Hz, 2H, Ar-H), 7.58 (d, J = 15.6 Hz, 1H, CH=), 7.94 (d, J = 8.4 Hz, 2H, Ar-H), 9.14 (br s, 1H, NH). HRMS found [M-H]− (m/z): 561.1156. Calcd for C26H27ClN2O8S m/z: 562.1177.
(E)-2-(4″-Bromophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)-phenylamino)acetic acid (25g)
The title compound was obtained by the hydrolysis of (E)-methyl 2-(4″-bromophenyl)-2-(2-methoxy-5-(2-(2′,4′,6′-trimethoxyphenyl)vinylsulfonamido)-phenylamino)acetate, 24g following the procedure as described for compound 25. Yield, 70%; light brown solid, mp 138–140 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.83 (s, 3H, OCH3), 3.84 (s, 6H, 2 X OCH3), 3.86 (s, 3H, OCH3), 4.96 (s, 1H, CH), 6.31 (s, 2H, Ar-H), 6.41 (d, J = 2.1 Hz, 1H, Ar-H), 6.69 (dd, J = 2.1, 8.1 Hz, 1H, Ar-H), 6.91 (d, J = 8.1 Hz, 1H, Ar-H), 7.14 (d, J = 15.6 Hz, 1H, =CH), 7.39 (d, J = 8.4 Hz, 2H, Ar-H), 7.57 (d, J = 8.4 Hz, 2H, Ar-H),7.61 (d, J = 15.6 Hz, 1H, CH=), 9.12 (br s, 1H, NH). HRMS found [M-H]− (m/z): 605.0647. Calcd for C26H27BrN2O8S m/z: 606.0671.
Biology
Tissue Culture and Reagents
Paclitaxel (Taxol), Nocodazole, Colchicine, Tamoxifen and 2-Thiouracil was purchased from Sigma-Aldrich (St. Louis, MO). Cell lines were purchased from ATCC (Manassas, VA). Cell lines were routinely grown in DMEM or RPM1 (CellGro, Manassas, VA) supplemented with 10% fetal bovine serum (Cell Generation, CO) and 1 unit/mL Penicillin-Streptomycin (GIBCO-Life Technologies, Inc., Gaithersburg, MD).
Cytotoxicity Assay
We have tested a number of tumor cell lines using a dose response end point assay system. The cells were grown in either DMEM or RPMI supplemented with 10% fetal bovine serum and 1 unit/mL penicillin-streptomycin solution. The tumor cells were plated into twelve-well dishes at a cell density of 2.5 ×104 cells/mL/well, and compounds were added 24 h later at various concentrations. Cell counts were determined from duplicate wells after 96 h of treatment. The total number of viable cells was determined by trypan blue exclusion.
Flow Cytometry
Cancer cell line DU145 (human prostate tumor) cells, were grown in DMEM (Cellgro) supplemented with 10% fetal bovine serum and 1 unit/mL penicillin-streptomycin. The cells were plated onto 100 mm2 dishes at a cell density of 1.0 × 106 cells/dish, and 24 h later, they were treated with increasing concentration of 6t. The cells were harvested 24 h after treatment. The cells were removed from the plate by trypsin digestion and combined with the non-attached cells found in the medium. The cell pellets were washed in phosphate buffered saline (PBS), and fixed in ice cold 70% ethanol for at least 24 h. The fixed cells were then washed with room temperature PBS and stained with propidium iodide (50 μg/mL) and RNase A (0.5 mg) for 30 min at 37 °C. The stained cells were then analyzed on a Becton-Dickinson (BD) (FACScan) flow cytometer and the data analyzed by cell cycle analysis software (Modfit, BD).
PARP Western
DU145 cells were plated at a density of 1.0 × 106 cells per 100 mm2 plate and treated 24 h later with either DMSO or increasing concentrations of 6t. The cells were collected the indicated time points and cell pellets were frozen. The frozen cell pellets were lysed in 0.1 % Triton X-100 based lysis buffer containing protease inhibitors. Equal amounts of total cellular protein was then resolved on a 10%-SDS-polyacrylamide gel. The gels were transferred onto nitrocellulose paper (S/S). Licor’s (Nebraska) Odyssey western detection procedure was used to visualize PARP cleavage. Following hybridization to anti- PARP antibodies (BD), the blot was treated with secondary antibodies labeled with IRDye 800 (Licor) and developed using the Odyssey scanner.
Fluorescent Tubulin Staining
DU145 cells were grown on glass coverslips and then treated with 0.025 μM 6t. The treated coverslips were harvested after 24 h and washed with room temperature PBS and fixed in freshly prepared 4% paraformaldehyde. The fixed cells were then treated with PBS containing 10 % FBS/0.1%Triton X-100 for 45 min at room temperature. The coverslips were washed and then treated with monoclonal anti alpha-tubulin antibody conjugated with FITC (DM1A, Sigma) for 1 h at 37 °C. The coverslips were washed and then treated with PBS containing 1 μg/mL Propidium iodide for 5 min at room temperature. The coverslips were washed and mounted onto slides using Prolong Gold Antifade solution (Molecular Probes, Invitrogen, CA). The stained cells were analyzed by confocal microscopy using the inverted Olympus microscope FluoView system with a 60× objective. The image was generated by XYZ-sectioning (35-0.2 μM sections) and compiling the scans using the software provided.
In vivo Tubulin Polymerization
DU145 cells were plated in 24 well dish at a cell number of 1.0 × 104 cells per well in 1 mL completed DMEM. Compounds were added the following day at the desired concentration. Following the 24 h paclitaxel treatment, compounds were added to the cells for 4 h. The cells were lysed in 100 μL hypotonic lysis buffer:1 mM MgCl2, 2 mM EGTA, Tris-HCL (pH 6.6), 0.5% NP40, 1–2 mM PMSF, 2 μg/mL Aprotinin, 2 μg/mL Leupeptin and the lysates were spun for 10 min at 14,000 rpm at room temperature. To the supernatant, 25 μL of 4× sample buffer was added and the pellet was resuspended pellet in 100 μL hypotonic buffer and add 25 μL sample buffer. The samples were resolved by 10% SDS-PAGE. The blot was hybridized using a monoclonal antibody to alpha tubulin (Sigma clone DM1A).
Soft Agar Assay
The soft agar plates were prepared as described in Cosenza, et al.41 Briefly, Noble bottom agar (0.8%) was plated onto 60 mm tissue culture plates. Exponentially growing MIA-PaCa-2 cells (1.0 × 105) were mixed with growth medium with various concentrations of each compound and mixed with Noble agar to a final concentration of 0.4%. Each concentration was plated in triplicates. The top agar was allowed to harden and the plates were then incubated at 5% CO2 at 37 °C for 3 weeks. The plates were then stained with 0.05% nitroblue tetrazolium (NBT) solution and representative plates were photographed using an Olympus stereoscope mounted with a Sony digital camera system (DKC5000, Sony Inc).
Nude Mouse Assay
Female athymic (NCr-nu/nu, Taconic) nude mice were injected with 0.5–1.0 ×107 BT20 cells subcutaneously in the hind leg using a 1 mL tuberculin syringe equipped with a 27–1/2 gauge needle. Approximately 14 days later, mice were paired (N=7:Vehicle and N=8:6t) such that each group harbored tumors with an average volume of approximately 100–150 mm3. The intraperitoneal injections were performed using a 1 mL tuberculin syringe equipped with a 27–1/2 gauge needle. The animals were treated 3 times a week for 2 weeks with 20 mg/kg of 6t dissolved in DMSO and 0.1 mL of 6t solution in DMSO was injected in mice. Tumor measurements (two dimensions) were done 3× a week using traceable digital vernier calipers (Fisher). Tumor volume was calculated using the following equation: V= (L x (S2)π/6, where L is the longer and S is the shorter of the two dimensions. Change in tumor volume was determined by dividing the starting volume (V) by the determined volume (V1) following treatment. Body weight was determined during each measurement. The animals were observed for signs of toxicity. There was no body weight loss of more than 10% in any group nor were there any animal deaths. All studies were performed under the guidelines of Temple University IACUC.
Competitive Mass Spectrometry Binding Assay
Competitive mass spectrometry binding studies were conducted as previously described.34 Colchicine, vinblastine and paclitaxel (1.2 μM for each) were incubated with porcine brain tubulin (1.0 mg/mL) in the incubation buffer [80 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 2.0 mM magnesium chloride (MgCl2), 0.5 mM ethylene glycol tetraacetic acid (EGTA), pH 6.9] at 37°C for 1h. To study the colchicine and vinblastine binding sites, the incubations were performed under conditions which did not contain GTP because these ligands prefer to bind to dimeric tubulin. For paclitaxel binding studies, preformed microtubules in 100 μL incubation buffer were prepared by pre-incubating tubulin in the presence of GTP (1 mM) for 1h. Varying concentrations (0.1–100 μM) of podophyllotoxin, vincristine, and docetaxel (positive controls for each binding sites) and 6t (0.1–100 μM) were used to compete with the binding of colchicine- vinblastine- and paclitaxel-tubulin binding, and after a 1h incubation, the filtrate was obtained by ultrafiltration (molecular size cutoff of 30kDa, Microcon, Bedford, MA) and analyzed for either colchicine, vinblastine or paclitaxel concentrations by mass spectrometry. The ability of 6t to inhibit the binding of each ligand was expressed as a percentage of control binding in the absence of any competitor. Each experiment was performed in triplicate. The IC50 values of 6t and podophylltoxin were calculated by fitting the Hill equation to the measured percentage binding as a function of the log of concentration using nonlinear regression (GraphPad Prism Software, San Diego, CA).
Figure 8.

Plasma 6t concentrations (mean ± SD) following either administration of 5 mg/kg as an IV bolus or oral gavage to normal mice. The resultant IV PK parameters were; total clearance = 2.48 ± 0.19 L/h/kg, volume of distribution at steady-state = 2.13 ± 0.46 L/kg and an elimination half-life of 1.7 h. Oral bioavailability equaled 34% based on ratio of the AUCpo = 0.70 ± 0.42 μg-h/mL to the AUCiv = 2.04 ± 0.17 μg-h/mL.
Acknowledgments
Authors are thankful to Onconova Therapeutics Inc., Newtown, PA and Mount Sinai School of Medicine, New York for the financial assistance and interest in this project. Partial support from NIH grant CA127963.
ABBREVATIONS USED
- ADME-Tox
Adsorption, distribution, metabolism, and excretion - Toxicology
- MDR
Multidrug resistance
- P-gp
P-glycoprotein
- BBB
Blood-Brain Barrier
- FACS
Fluorescence activated cell sorting
- PARP
poly(ADP-ribose)polymerase
- FITC
Fluorescein Isothiocyanate
Footnotes
The authors declare the following competing financial interest(s): Dr. E. P. Reddy is a stockholder, Board member, grant recipient and paid consultant of Onconova Therapeutics Inc. Dr. M. V. R. Reddy is a stock holder and paid consultant of Onconova Inc. Dr. S. Cosenza is a paid consultant of Onconova Therapeutics Inc.
References
- 1.(a) McIntosh JR, Grishchuk E, West RR. Chromosome-Microtubule Interactions During Mitosis. Annu Rev Cell Dev Bi. 2002;18:193–219. doi: 10.1146/annurev.cellbio.18.032002.132412. [DOI] [PubMed] [Google Scholar]; (b) Gundersen GG, Cook TA. Microtubules and signal transduction. Curr Opin Cell Biol. 1999;11:81–94. doi: 10.1016/s0955-0674(99)80010-6. [DOI] [PubMed] [Google Scholar]; (c) Nogales E. Structural Insights Into Microtubule Function. Annu Rev Biochem. 2000;69:277–302. doi: 10.1146/annurev.biochem.69.1.277. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shi Q, Chen K, Morris-Natschke SL, Lee KH. Recent progress in the development of tubulin inhibitors as antimitotic antitumor agents. Curr Pharm Design. 1998;4:219–248. [PubMed] [Google Scholar]
- 3.(a) Leggans EK, Duncan KK, Barker TJ, Schleicher KD, Boger DL. A Remarkable Series of Vinblastine Analogues Displaying Enhanced Activity and an Unprecedented Tubulin Binding Steric Tolerance: C20′ Urea Derivatives. J Med Chem. 2013;56:628–639. doi: 10.1021/jm3015684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schleicher KD, Sasaki Y, Tam A, Kato D, Duncan KK, Boger DL. Total Synthesis and Evaluation of Vinblastine Analogues Containing Systematic Deep-Seated Modifications in the Vindoline Subunit Ring System: Core Redesign. J Med Chem. 2013;56:483–495. doi: 10.1021/jm3014376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neuss N, Neuss MN. The Therapeutic Use of Bisindole Alkaloids from Catharanthus. In: Brossi A, editor. The Alkaloids. Vol. 37. Academic Press; San Diego: 1990. pp. 229–240. [Google Scholar]; (d) Pearce HL. Medicinal Chemistry of Bisindole Alkaloids from Catharanthus. In: Brossi A, editor. The Alkaloids. Vol. 37. Academic Press; San Diego: 1990. pp. 145–204. [Google Scholar]; (e) Kuehne ME, Marko I. Syntheses of Vinblastine-Type Alkaloids. In: Brossi A, editor. The Alkaloids. Vol. 37. Academic Press; San Diego: 1990. pp. 77–132. [Google Scholar]
- 4.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 5.Kuppens IELM. Current State of the Art of New Tubulin Inhibitors in the Clinic. Curr Clin Pharmacol. 2006;1:57–70. doi: 10.2174/157488406775268200. [DOI] [PubMed] [Google Scholar]
- 6.(a) Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]; (b) Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol. 1998;10:123–130. doi: 10.1016/s0955-0674(98)80095-1. [DOI] [PubMed] [Google Scholar]; (c) Wilson L, Panda D, Jordan MA. Modulation of microtubule dynamics by drugs: a paradigm for the actions of cellular regulators. Cell Struct Funct. 1999;24:329–335. doi: 10.1247/csf.24.329. [DOI] [PubMed] [Google Scholar]; (d) Panda D, Jordan MA, Chu KC, Wilson L. Differential effects of vinblastine on polymerization and dynamics at opposite microtubule ends. J Biol Chem. 1996;271:29807–29812. doi: 10.1074/jbc.271.47.29807. [DOI] [PubMed] [Google Scholar]; (e) Amos LA. What tubulin drugs tell us about microtubule structure and dynamics. Semin Cell Dev Biol. 2011;22:916–926. doi: 10.1016/j.semcdb.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 7.(a) Jordan MA. Mechanism of action of antitumor drugs that interacts with microtubules and tubulin. Curr Med Chem: Anti-Cancer Agents. 2002;2:1–17. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]; (b) Jordan MA, Thrower D, Wilson L. Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 1991;51:2212–2222. [PubMed] [Google Scholar]; (c) Acharya BR, Choudhury D, Das A, Chakrabarti G. Vitamin K3 disrupts the microtubule networks by tubulin binding: A novel mechanism of its anti-proliferative activity. Biochemistry. 2009;48:6963–6974. doi: 10.1021/bi900152k. [DOI] [PubMed] [Google Scholar]
- 8.(a) Cragg GM, Grothaus PG, Newman DJ. Impact of Natural Products on Developing New Anti-Cancer Agents. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]; (b) Laura Cosentino L, Redondo-Horcajo M, Zhao Y, Santos AR, Chowdury KF, Vinader V, Abdallah QMA, Abdel-Rahman H, Fournier-Dit-Chabert J, Shnyder SD, Loadman PM, Fang W, Diaz JF, Barasoain I, Burns PA, Pors K. Synthesis and Biological Evaluation of Colchicine B-Ring Analogues Tethered with Halogenated Benzyl Moieties. J Med Chem. 2012;55:11062–11066. doi: 10.1021/jm301151t. [DOI] [PubMed] [Google Scholar]; (c) Ballatore C, Brunden KR, Huryn DM, Trojanowski JQ, Lee VM, Smith AB., III Microtubule Stabilizing Agents as a Potential Treatment for Alzheimer’s Disease and Related Neurodegenerative Tauopathies. J Med Chem. 2012;55:8979–8996. doi: 10.1021/jm301079z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu J, Zheng C, Ren X, Zhou F, Li W, Zhu J, Lv J, Zhou Y. Synthesis and Biological Evaluation of 1-Benzylidene-3,4-dihydronaphthalen-2-one as a New Class of Microtubule-Targeting Agents. J Med Chem. 2012;55:5720–5733. doi: 10.1021/jm300596s. [DOI] [PubMed] [Google Scholar]; (e) Abad A, Lopez-Perez JL, Olmo ED, Garcia-Fernandez LF, Francesch A, Trigili C, Barasoain I, Andreu JM, Diaz JF, Feliciano AS. Synthesis and Antimitotic and Tubulin Interaction Profiles of Novel Pinacol Derivatives of Podophyllotoxins. J Med Chem. 2012;55:6724–6737. doi: 10.1021/jm2017573. [DOI] [PubMed] [Google Scholar]; (f) Regina GL, Bai R, Rensen WM, Cesare ED, Coluccia A, Piscitelli F, Famiglini V, Reggio A, Nalli M, Pelliccia S, Pozzo ED, Costa B, Granata I, Porta A, Maresca B, Soriani A, Iannitto ML, Santoni A, Li J, Cona MM, Chen F, Ni Y, Brancale A, Dondio G, Vultaggio S, Varasi M, Mercurio C, Martini C, Hamel E, Lavia P, Novellino E, Silvestri R. Toward Highly Potent Cancer Agents by Modulating the C-2 Group of the Arylthioindole Class of Tubulin Polymerization Inhibitors. J Med Chem. 2013;56:123–149. doi: 10.1021/jm3013097. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yi X, Zhong B, Smith KM, Geldenhuys WJ, Feng Y, Pink JJ, Dowlati A, Xu Y, Zhou A, Su B. Identification of a Class of Novel Tubulin Inhibitors. J Med Chem. 2012;55:3425–3435. doi: 10.1021/jm300100d. [DOI] [PubMed] [Google Scholar]; (h) Lee H, Chang J, Nien C, Kuo C, Shih K, Wu C, Chang C, Lai W, Liou J. 5-Amino-2-aroylquinolines as Highly Potent Tubulin Polymerization Inhibitors. Part 2. The Impact of Bridging Groups at Position C-2. J Med Chem. 2011;54:8517–8525. doi: 10.1021/jm201031f. [DOI] [PubMed] [Google Scholar]; (i) Regina GL, Bai R, Rensen W, Coluccia A, Piscitelli F, Gatti V, Bolonesi A, Lavecchia A, Granata I, Porta A, Bruno M, Soriani A, Iannitto ML, Mariani M, Santoni A, Brancale A, Ferlini C, Dondio G, Varasi M, Mercurio C, Hamel E, Lavia P, Novellino E, Silvestri R. Design and Synthesis of 2-Heterocyclyl-3-arylthio-1H-indoles as Potent Tubulin Polymerization and Cell Growth Inhibitors with Improved Metabolic Stability. J Med Chem. 2011;54:8394–8406. doi: 10.1021/jm2012886. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Fortin F, Wei L, Moreau E, Lacroix J, Cokte M, Petitclerc E, Kotra LP, C-Gaudreault R. Design, Synthesis, Biological Evaluation, and Structure–Activity Relationships of Substituted Phenyl 4-(2-Oxoimidazolidin-1-yl)benzenesulfonates as New Tubulin Inhibitors Mimicking Combretastatin A-4. J Med Chem. 2011;54:4559–4580. doi: 10.1021/jm200488a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Chekler ELP, Kiselyov AS, Ouyang X, Chen X, Pattaropong V, Wang Y, Tuma MC, Doody JF. Discovery of Dual VEGFR-2 and Tubulin Inhibitors with in Vivo Efficacy. ACS Med Chem Lett. 2010;1:488–492. doi: 10.1021/ml1001568. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Giannakakou P, Sackett D, Fojo T. Tubulin/microtubules: still a promising target for new chemotherapeutic agents. J Natl Cancer Inst. 2000;92:182–183. doi: 10.1093/jnci/92.3.182. [DOI] [PubMed] [Google Scholar]; (m) Mooberry SL, Weiderhold KN, Dakshanamurthy S, Hamel E, Banner EJ, Kharlamova A, Hempel J, Gupton JT, Brown ML. Identification and characterization of a new tubulin binding tetrasubstituted brominated pyrrole. Mol Pharmacol. 2007;72:132–140. doi: 10.1124/mol.107.034876. [DOI] [PubMed] [Google Scholar]
- 9.(a) Deeken JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]; (b) Dumontet C, Duran GE, Steger KA, Beketic-Oreskovic L, Sikic BI. Resistance mechanisms in human sarcoma mutants derived by single-step exposure to paclitaxel (Taxol) Cancer Res. 1996;56:1091–1097. [PubMed] [Google Scholar]; (c) Matesanz R, Barasoain I, Yang CG, Wang L, Li X, de Ines C, Coderch C, Gago F, Barbero JJ, Andreu JM, Fang WS, Diaz JF. Optimization of taxane binding to microtubules: binding affinity dissection and incremental construction of a high affinity analog of paclitaxel. Chem Biol. 2008;15:573–585. doi: 10.1016/j.chembiol.2008.05.008. [DOI] [PubMed] [Google Scholar]; (d) Miller DS, Bauer B, Hartz AMS. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. 2008;60:196–209. doi: 10.1124/pr.107.07109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Pellegrini F, Budman DR. Tubulin function, action of antitubulin drugs, and new drug development. Cancer Invest. 2005;23:264–273. doi: 10.1081/cnv-200055970. [DOI] [PubMed] [Google Scholar]; (b) Rice A, Liu Y, Michaelis ML, Himes RH, Georg GI, Audus KL. Chemical modification of paclitaxel (Taxol) reduces P-glycoprotein interactions and increases permeation across the blood-brain barrier in vitro and in situ. J Med Chem. 2005;48:832–838. doi: 10.1021/jm040114b. [DOI] [PubMed] [Google Scholar]; (c) Tong YG, Zhang XW, Geng MY, Yue JM, Xin XL, Tian F, Shen X, Tong LJ, Li MH, Zhang C, Li WH, Lin LP, Ding J. Pseudolarix acid B, a new tubulin-binding agent, inhibits angiogenesis by interacting with a novel binding site on tubulin. Mol Pharmacol. 2006;69:1226–1233. doi: 10.1124/mol.105.020537. [DOI] [PubMed] [Google Scholar]; (d) Mulligan JM, Greene LM, Cloonan S, Mc Gee MM, Onnis V, Campiani G, Fattorusso C, Lawler M, Williams DC, Zisterer DM. Identification of tubulin as the molecular target of proapoptotic pyrrolo-1,5-benzoxazepines. Mol Pharmacol. 2006;70:60–70. doi: 10.1124/mol.105.021204. [DOI] [PubMed] [Google Scholar]; (e) Beyer CF, Zhang N, Hernandez R, Vitale D, Lucas J, Nguyen T, Discafani C, Ayral-Kaloustian S, Gibbons JJ. TTI-237, a novel microtubule-active compound with in vivo antitumor activity. Cancer Res. 2008;68:2292–2300. doi: 10.1158/0008-5472.CAN-07-1420. [DOI] [PubMed] [Google Scholar]; (f) Liberatore AM, Coulomb H, Pons D, Dutruel O, Kasprzyk PG, Carlson M, Nelson AS, Newman SP, Stengel C, Auvray P, Hesry V, Foll B, Narboux N, Morlais D, Le Moing M, Bernetiere S, Dellile R, Camara J, Ferrandis E, Bigg DC, Prevost GP. IRC-083927 is a new tubulin binder that inhibits growth of human tumor cells resistant to standard tubulin binding agents. Mol Cancer Ther. 2008;7:2426–2434. doi: 10.1158/1535-7163.MCT-08-0208. [DOI] [PubMed] [Google Scholar]
- 11.(a) Xie F, Zhao H, Li D, Chen H, Quan H, Shi X, Lou L, Hu Y. Synthesis and Biological Evaluation of 2,4,5-Substituted Pyrimidines as a New Class of Tubulin Polymerization Inhibitors. J Med Chem. 2011;54:3200–3205. doi: 10.1021/jm101388d. [DOI] [PubMed] [Google Scholar]; (b) Kemnitzer W, Sirisoma N, May C, Tseng B, Drewe J, Cai SX. Discovery of 4-anilino-N-methylthieno[ 3,2-d]pyrimidines and 4-anilino-N-methylthieno[2,3-d]pyrimidines as potent apoptosis inducers. Bioorg Med Chem Lett. 2009;19:3536–3540. doi: 10.1016/j.bmcl.2009.04.145. [DOI] [PubMed] [Google Scholar]; (c) Sirisoma N, Pervin A, Nguyen B, Crogan-Grundy C, Kasibhatla S, Tseng B, Drewe J, Cai SX. Discovery of substituted 4-anilino-2-arylpyrimidines as a new series of apoptosis inducers using a cell- and caspase-based high throughput screening assay. 2. Structure–activity relationships of the 2-aryl group. Bioorg Med Chem Lett. 2009;19:2305–2309. doi: 10.1016/j.bmcl.2009.02.074. [DOI] [PubMed] [Google Scholar]; (d) Lee J, Kim SJ, Choi H, Kim YH, Lim IT, Yang HM, Lee CS, Kang HR, Ahn SK, Moon SK, Kim DH, Lee S, Choi NS, Lee KJ. Identification of CKD-516: A Potent Tubulin Polymerization Inhibitor with Marked Antitumor Activity against Murine and Human Solid Tumors. J Med Chem. 2010;53:6337–6354. doi: 10.1021/jm1002414. [DOI] [PubMed] [Google Scholar]
- 12.(a) Chaplin DJ, Horsman MR, Siemann DW. Current developments status of small-molecule vascular disrupting agents. Curr Opin Invest Dr. 2006;7:522–528. [PubMed] [Google Scholar]; (b) Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J Med Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]; (c) Li Q, Sham HL. Discovery and development of mitotic agents that inhibit polymerization for the treatment of cancers. Expert Opin Ther Pat. 2002;12:1663–1702. [Google Scholar]; (d) Nam NH. Combretastatin A4 analogues as antimitotic antitumor agents. Curr Med Chem. 2003;10:1697–1722. doi: 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]; (e) Hsieh HP, Liou JP, Mahindroo N. Pharmaceutical design of antimitotic agents based on combretastatin. Curr Pharm Design. 2005;11:1655–1677. doi: 10.2174/1381612053764751. [DOI] [PubMed] [Google Scholar]; (f) Mahindroo N, Liou JP, Chang JY, Hsieh HP. Antitubulin agents for the treatment of cancer- a medicinal chemistry updates. Expert Opin Ther Pat. 2006;16:647–691. [Google Scholar]; (g) Tron GC, Pagliai F, Del Grosso E, Genazzani AA, Sorba G. Synthesis and cytotoxic evaluation of combretafurazans. J Med Chem. 2005;48:3260–3268. doi: 10.1021/jm049096o. [DOI] [PubMed] [Google Scholar]; (h) Pirali T, Busacca S, Beltrami L, Imovilli D, Pagliai F, Miglio G, Massarotti A, Verotta L, Tron GC, Sorba G, Genazzani AA. Synthesis and cytotoxicity evaluation of combretafuran, potential scaffolds for dual-action antitumor agents. J Med Chem. 2006;49:5372–5376. doi: 10.1021/jm060621o. [DOI] [PubMed] [Google Scholar]; (i) Zhang Q, Peng Y, Wang XI, Keenan SM, Arora S, Welsh WJ. Highly potent triazole-based tubulin polymerization inhibitors. J Med Chem. 2007;50:749–754. doi: 10.1021/jm061142s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West CML, Price P. Combretastatin A4 phosphate. Anti-Cancer Drug. 2004;15:179–87. doi: 10.1097/00001813-200403000-00001.(b) www.clinicaltrials.gov
- 14.(a) Ohsumi K, Hatanaka T, Nakagawa R, Fukuda Y, Morinaga Y, Suga Y, Nihei Y, Ohishi K, Akiyama Y, Tsuji T. Synthesis and antitumor activities of amino acid prodrugs of aminocombretastatins. Anti-Cancer Drug Des. 1999;14:539–548. [PubMed] [Google Scholar]; (b) Hori K, Saito S. Microvascular mechanisms by which the combretastatin A-4 derivative AC7700 (AVE8062) induces tumour blood flow stasis. Brit J Cancer. 2003;89:1334–1344. doi: 10.1038/sj.bjc.6601261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Davis PD, Dougherty GJ, Blakey DC, Galbraith SM, Tozer GM, Holder AL, Naylor MA, Nolan J, Stratford MRL, Chaplin DJ, Hill SA. ZD6126: A novel vascular-targeting agent that causes selective destruction of tumor vasculature. Cancer Res. 2002;62:7247–7253. [PubMed] [Google Scholar]; (b) Micheletti G, Poli M, Borsotti P, Martinelli M, Imberti B, Taraboletti G, Giavazzi R. Vasculartargeting activity of ZD6126, a novel tubulin-binding agent. Cancer Res. 2003;63:1534–1537. [PubMed] [Google Scholar]
- 16.(a) Koyanagi N, Nagasu T, Fujita F, Watanabe T, Tsukahara K, Funahashi Y, Fujita M, Taguchi T, Yoshino H, Kitoh K. In vivo tumor growth inhibition produced by a novel sulfonamide, E7010, against rodent and human tumors. Cancer Res. 1994;54:1702–1706. [PubMed] [Google Scholar]; (b) Segreti JA, Polakowski JS, Koch KA, Marsh KC, Bauch JL, Rosenberg SH, Sham HL, Cox BF, Reinhart GA. Tumor selective antivascular effects of the novel antimitotic compound ABT-751: An in vivo rat regional hemodynamic study. Cancer Chemoth Pharma. 2004;54:273–281. doi: 10.1007/s00280-004-0807-0. [DOI] [PubMed] [Google Scholar]
- 17.Shan B, Medina JC, Santha E, Frankmoelle WP, Chou T-C, Learned RM, Narbut MR, Stott D, Wu P, Jaen JC, Rosen T, Timmermans PBMWM, Beckmann H. Selective, covalent modification of a-tubulin residue Cys-239 by T138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proc Natl Acad Sci USA. 1999;96:5686–5691. doi: 10.1073/pnas.96.10.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medina JC. Preparation of benzenesulfonamides and benzamides regulating low-density lipoprotein (LDL) receptor expression and of inhibiting abnormal cell proliferation. 9936391. WO. 1999
- 19.(a) Li Q, Sham H, Woods KW, Steiner BA, Gwaltney SL, II, Barr KJ, Imade HM, Rosenberg S. Preparation of sulfonamides as cell proliferation inhibitors. 6521658. US. 2003; (b) Prinz H. Recent advances in the field of tubulin polymerization inhibitors. Expert Rev Anticanc. 2002;2:695–708. doi: 10.1586/14737140.2.6.695. [DOI] [PubMed] [Google Scholar]
- 20.(a) Luo Y, Hradil VP, Frost DJ, Rosenberg SH, Gordon GB, Morgan SJ, Gagne GD, Cox BF, Tahir SK, Fox GB. ABT-751, a novel tubulin-binding agent, decreases tumor perfusion and disrupts tumor vasculature. Anti-cancer Drug. 2009;20:483–492. doi: 10.1097/CAD.0b013e32832c0acf. [DOI] [PubMed] [Google Scholar]; (b) Mauer AM, Cohen EE, Ma PC, Kozloff MF, Schwartzberg L, Coates AI, Qian J, Hagey AE, Gordon GB. A phase II study of ABT-751 in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:631–636. doi: 10.1097/JTO.0b013e318174e01f. [DOI] [PubMed] [Google Scholar]; (c) Rustin GJ, Shreeves G, Nathan PD, Gaya A, Ganesan TS, Wang D, Boxall J, Poupard L, Chaplin DJ, Stratford MR, Balkissoon J, Zweifel M. A Phase Ib trial of CA4P (combretastatin A-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer. Brit J Cancer. 2010;102:1355–1360. doi: 10.1038/sj.bjc.6605650. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kuppens IELM. Current State of the Art of New Tubulin Inhibitors in the Clinic. Current Clinical Pharmacology. 2006;1:57–70. doi: 10.2174/157488406775268200. [DOI] [PubMed] [Google Scholar]
- 21.(a) Reddy MVR, Reddy S. Synthesis of α, β-Unsaturated Suflones. Acta Chim Hung. 1984;115:269–271. [Google Scholar]; (b) Reddy DB, Reddy NS, Reddy MVR, Balasubramanyam S. Preparation of styryl benzyl sulfones and 1, 2-bis- (styrylsulfonylmethyl)-4,5-dimethylbenzenes. Org Prep Proc Int. 1988;20:205–212. [Google Scholar]
- 22.Reddy AK, Lohray BB, Bhushan V, Reddy AS, Mamidi NVSR, Reddy PP, Saibaba V, Reddy NJ, Suryaprakash A, Misra P, Vikramadithan RK, Rajagopalan R. Novel Antidiabetic and Hypolipidemic Agents.5. Hydroxyl versus Benzyloxy Containing Chroman Derivatives. J Med Chem. 1999;42:3265–3278. doi: 10.1021/jm9805541. [DOI] [PubMed] [Google Scholar]
- 23.Pallela VR, Mallireddigari MR, Cosenza SC, Akula B, Subbaiah DRCV, Reddy EP, Reddy MVR. Hydrothiolation of benzyl mercaptan to arylacetylene: application to the synthesis of (E) and (Z)-isomers of ON 01910·Na (Rigosertib®), a phase III clinical stage anti-cancer agent. Org Biomol Chem. 2013;11:1964–1977. doi: 10.1039/c3ob27220f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen AT, Chafin AT, Christian SL. Nitrocarbons. 4. Reaction of Polynitrobenzenes with Hydrogen Halides. Formation of Polynitrohalobenzenes. J Org Chem. 1984;49:4575–4580. [Google Scholar]
- 25.Oliver JE, DeMilo AB. A Knoevenagel-Type Synthesis of Styrene-ω-sulfonanilides. Synthesis. 1975:321–322. [Google Scholar]
- 26.(a) Long DD, Termin AP. Ring-closing metathesis to a divergent endocyclic sulfonamide template. Tetrahedron Lett. 2000;41:6743–6747. [Google Scholar]; (b) Berre pALe, Etienne A, Desmazieres B. Acides α-sulfocarboxyliques et derives. V.-Sulfamoylcarboxyesters et carboxamides acycliques. Thiazetidine-1,2 one-3 dioxydes-1,1. Bull Soc Chim Fr. 1975:807–811. [Google Scholar]
- 27.(a) Reddy MVR, Reddy S, Reddy DB. Facile method for the synthesis of 2-(arylsulfonyl)-1-phenyl-3-aryl-2-propen-1-ones. Sulfur Lett. 1987;7:43–48. [Google Scholar]; (b) Russell Llyod B, Anthony D, Chantal Renee F, Richard Francis L. Novel Knoevenagel condensation of a β-keto sulfone and a β-carboalkoxy sulfone. Sulfur Lett. 1999;23:11–31. [Google Scholar]; (c) Reddy MM, Venkat RP, Reddy EP, Reddy MVR. Sequential Reduction and Dehydration of Phenacyl- (E)-Styryl Sulfones to Unsymmetrical (E, E)- Bis(styryl) Sulfones. Synthesis. 2005:3639–3643. [Google Scholar]; (d) Touati A, Cazaux L. Synthesis of sulfonamides, sulfonates and thiosulfonates which are inhibitors of coniferyl alcohol dehydrogenase. J Soc Alger Chim. 1996;6:39–52. [Google Scholar]
- 28.Zheng Ailian, Shan Daxian, Binghe Wang A. Redox-Sensitive Resin Linker for the Solid Phase Synthesis of C-Terminal Modified Peptides. J Org Chem. 1999;64:156–161. doi: 10.1021/jo981528d. [DOI] [PubMed] [Google Scholar]
- 29.Reddy S. Process for preparing (E)-Styryl benzylsulfone compounds and uses thereof for treating proliferative disorders. 2010/0152491. US. 2010:A1.
- 30.Reddy MVR, Venkatapuram P, Mallireddigari MR, Pallela VR, Cosenza SC, Robell KA, Akula B, Hoffman BS, Reddy EP. Discovery of a Clinical Stage Multi Kinase Inhibitor Sodium (E)-2-{2-Methoxy-5-[(2′, 4′, 6′-trimethoxystyrylsulfonyl)-methyl]phenylamino}acetate (ON 01910.Na): Synthesis, Structure Activity Relationship and Biological Activity. J Med Chem. 2011;54:6254–6276. doi: 10.1021/jm200570p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;10:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 32.Jordan MA, Thrower D, Wilson L. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J Cell Sci. 1992;102:401–416. doi: 10.1242/jcs.102.3.401. [DOI] [PubMed] [Google Scholar]
- 33.Soldani C, Scovassi AI. Poly (ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321–328. doi: 10.1023/a:1016119328968. [DOI] [PubMed] [Google Scholar]
- 34.Robinson J, Engelborghs Y. Tubulin polymerization in dimethyl sulfoxide. J Biol Chem. 1982;257:5367–5371. [PubMed] [Google Scholar]
- 35.Li CM, Lu Y, Ahn S, Narayanan R, Miller DD, Dalton JT. Competitive mass spectrometry binding assay for characterization of three binding sites of tubulin. J Mass Spectrom. 2010;45:1160–1166. doi: 10.1002/jms.1804. [DOI] [PubMed] [Google Scholar]
- 36.Lee Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 37.Harker WG, Sikic BI. Multidrug (pleiotropic) resistance in doxorubicin selected variants of the human sarcoma cell lines MES-SA. Cancer Res. 1985;45:4091–4096. [PubMed] [Google Scholar]
- 38.Fujimori A, Arker WG, Kohlhagen G, Hoki Y, Pommier Y. Mutation at the catalytic site of topoisomerase 1 in CEM/C2, a human leukemia cell line resistant to camptothecin. Cancer Res. 1995;55:1339–1346. [PubMed] [Google Scholar]
- 39.Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood-brain barrier. Int J Pharm. 2005;288:349–359. doi: 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Lv H, Zhang X, Sharma J, Reddy MVR, Reddy EP, Gallo JM. Integrated pharmacokinetic-driven approach to screen candidate anticancer drugs for brain tumor chemotherapy. AAPS J. 2013;15:250–257. doi: 10.1208/s12248-012-9428-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cosenza SC, Baker SJ, Reddy EP. In: Methods for oncogenic detection. In Cell Growth, Differentiation and Senescence. Studzinski GP, editor. Oxford University Press; 1999. pp. 161–176. [Google Scholar]