

Background: Transport of localized mRNAs encoding yeast membrane proteins to the bud is coordinated with segregation of the endoplasmic reticulum.
Results: The localized RNA-binding protein She2p associates with membranes of the endoplasmic reticulum in a curvature-dependent manner.
Conclusion: She2p is an RNA- and lipid-binding protein.
Significance: Direct targeting of mRNAs via membrane-associated RNA-binding proteins might facilitate their local translation.
Keywords: Endoplasmic Reticulum (ER), Membrane, RNA Binding Protein, RNA Transport, Yeast
Abstract
Localization of mRNAs contributes to the generation and maintenance of cellular asymmetry in a wide range of organisms. In Saccharomyces cerevisiae, the so-called locasome complex with its core components Myo4p, She2p, and She3p localizes more than 30 mRNAs to the yeast bud tip. A significant fraction of these mRNAs encodes membrane or secreted proteins. Their localization requires, besides the locasome, a functional segregation apparatus of the cortical endoplasmic reticulum (ER), including the machinery that is involved in the movement of ER tubules into the bud. Colocalization of RNA-containing particles with these tubules suggests a coordinated transport of localized mRNAs and the cortical ER to the bud. Association of localized mRNAs to the ER requires the presence of the locasome component She2p. Here we report that She2p is not only an RNA-binding protein but can specifically bind to ER-derived membranes in a membrane curvature-dependent manner in vitro. Although it does not contain any known curvature recognizing motifs, the protein shows a binding preference for liposomes with a diameter resembling that of yeast ER tubules. In addition, membrane binding depends on tetramerization of She2p. In an in vivo membrane-tethering assay, She2p can target a viral peptide GFP fusion protein to the cortical ER, indicating that a fraction of She2p associates with the ER in vivo. Combining RNA- and membrane-binding features makes She2p an ideal coordinator of ER tubule and mRNA cotransport.
Introduction
Localization of mRNA is widely used to spatially control gene expression and to generate cellular asymmetry (1, 2). Generally, messenger ribonucleoprotein (mRNP)3 particles containing translationally silent transcripts are transported from perinuclear areas to their peripheral destination, where their translation is activated (2, 3).
In budding yeast, mRNA localization has been studied mainly using ASH1 mRNA as a model RNA. ASH1 encodes a transcriptional repressor and cell fate determinant and is localized to the tip of the mature bud (the daughter cell). Its local translation at the tip of the daughter cell ensures the repression of the HO endonuclease after cell division only in the daughter cell (4).
The RNA-binding protein She2p is a core component of mRNPs containing ASH1 (5, 6). She2p binds to ASH1 cotranscriptionally (7) and escorts it to the cytoplasm (8, 9). After nuclear export, this primary mRNP complex binds to the myosin motor protein Myo4p via the associated She3p protein (10, 11). Myo4p is required for the active translocation of localized mRNPs to the bud tip (4). In addition to the Myo4p-She3p-She2p core complex, three additional RNA-binding proteins (Loc1p, Puf6p, and Khd1p) are needed for efficient ASH1 mRNA localization and translational silencing of the mRNA (12–14).
Besides ASH1, more than 30 additional mRNAs are localized in a Myo4p- and She2p-dependent way to the bud tip (15–17). Many of these mRNAs encode membrane or secreted proteins that are translocated into the ER. In contrast to ASH1, the localization of these mRNAs depends on additional proteins, like reticulons, Aux1p, Srp101, or Sec3p, that are also required for proper segregation of the cortical endoplasmic reticulum (cER, also called plasma membrane-attached ER) to the bud (15, 18, 19).
Cortical ER segregation begins during S phase when tubular structures emanate from the perinuclear ER and move in a Myo4p/She3p-dependent manner into the growing bud (20, 21). Shaping of the tubular ER involves two protein families, the reticulons (Rtns) and DP1/Yop1p (22), that help to generate the high curvature required to form tubular membrane structures of 30- to 40-nm diameter in yeast or 50-nm diameter in mammals (21, 23). These tubular structures move in an actin-dependent manner into the bud, followed by spreading of the cER along the bud cortex (20). Movement, but not spreading, of the cER along the plasma membrane is required for the localization of a subset of mRNAs to the bud tip (18). These mRNAs encode membrane proteins and are expressed at the time when ER tubules move into the bud. Cotransport of mRNAs with the ER might, therefore, support an efficient synthesis at the cortical ER.
Although the RNA-binding protein She2p is not essential for cER segregation (24), it is necessary for association of some of its target mRNAs with ER tubules (19). She2p has been shown to cofractionate with the ER, but the nature of the association remained obscure. Here we demonstrate that She2p can specifically bind to ER-derived microsomes in vitro even in the absence of other components of the locasome or associated mRNAs. Furthermore, it can bind to protein-free liposomes with a preference for highly curved membranes. In vivo, it can direct a viral peptide tag to the cortical ER, consistent with the idea that She2p is not only an RNA- but also a membrane-binding protein that could directly link its target mRNAs to ER membranes with high curvature, as present in the cER.
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
Plasmids for expression of GST fusions to wild-type She2p and She2p Asn-36 → Ser/Arg-63 → Lys have been described (8, 25). To generate expression vectors for She2p mutants Ser-120 → Tyr and Lys-130 → Tyr, site-directed mutagenesis of pGEX-TEV-SHE2 was performed using the oligonucleotide pair RJO2003 and RJO2004 (Ser-120 → Tyr) or RJO2035 and RJO2036 (Lys-130 → Tyr), respectively. Generation of GST-She2pΔhelixE has been described elsewhere (26). The GFP-TGB3(25–52) plasmid allowing the expression of GFP fused to a peptide from the plant potexvirus TGB3 protein (27) was a gift from Dr. Chao-Wen Wang. To introduce the SHE2 or MS2 coat protein (MS2-CP) coding regions, they were amplified with primers RJO4320, RJO4321, RJO4405, and RJO4406 and cloned between GFP and the signal sequence of TGB3(25–52) to create the corresponding fusion proteins. The resulting plasmids RJP1848 and RJP1850 were transformed into BY4741 WT yeast cells.
Purification of Recombinant She2p
GST was expressed from plasmid pET-23a and purified according to the instructions of the manufacturer (GE Healthcare). Wild-type She2p and all She2p mutants were expressed as GST fusion proteins in Escherichia coli BL21(DE3)/pRIL (Invitrogen). Purification to > 95% homogeneity was reached using standard chromatography techniques (25, 28). The GST tag was removed by cleavage with tobacco etch virus protease (Invitrogen) or, in the case of She2pΔhelixE, by cleavage using PreScission protease (GE Healthcare). After addition of glycerol to 20%, She2p was quickly cooled in liquid N2 and stored at −80 °C.
Yeast Subcellular Fractionation and Sucrose Gradient Centrifugation
Cell lysis was performed from yeast spheroplasts generated from strains BY4742 (SHE2) or RJY2053 (she2Δ::kanMX4) to maximally preserve subcellular integrity. Velocity gradient centrifugation of cell lysates on 18–60% sucrose gradients was performed as described (19, 24, 29). In brief, cells corresponding to 400 A600 units were harvested, spheroplasted, lysed with a needle, and cleared from cell debris as described above. 1 ml of the homogenate (corresponding to 66 A600 units) was then loaded onto a linear 18–60% gradient of sucrose in 20 mm Hepes-KOH (pH 6.8), 140 mm potassium acetate, and 1 mm magnesium acetate. Gradients were spun in a SW40 rotor for 2.3 h at 38,000 × g. 12 × 1-ml fractions were collected starting close to the bottom of the gradient. The remaining pellet was resuspended in 1 ml of lysis buffer. Fractions were precipitated with TCA and resuspended in 100 μl of SDS sample buffer. 20 μl of these were used for Western blot analysis, except for the top three fractions, where only 7 μl were used to avoid overloading of the gel. Western blots were performed using ECL, scanned, and quantified by using ImageJ. For in vitro binding experiments, yeast lysate was prepared from the she2Δ strain RJY2053. 1 ml of lysate (corresponding to 66 A600 units) was preincubated with recombinant She2p or She2p mutant protein (50 pmol protein/binding reaction) for 30 min on ice. The suspension was loaded on an 18–60% gradient and treated as described above.
Quick subcellular fraction of yeast cells to separate heavy membranes (nuclei and ER) from free polysomes and cytosolic proteins was performed according to Frey et al. (30). In short, yeast cells were lysed with glass beads, and three centrifugation steps at 4 °C were performed (20 min at 6000 × g to pellet heavy membranes, 20 min at 18, 000 × g to pellet light membranes and heavy polysomes, and 20 min at 20,0000 × g to pellet ribosomes).
To follow the distribution of She2p or marker proteins, commercial antibodies against GFP (Covance, Princeton); Dpm1 (Molecular Probes); Pgk1p (Invitrogen); GST (Novagen); maltose binding protein (Novagen); or custom antibodies against Sec61p, She2p, OM45p, or Mcr1p were used in Western blot analyses.
In Vitro Binding Assay with Flotation-purified ER Vesicles or Mitochondria
Mitochondria of yeast cells were isolated by differential centrifugation as described previously (31). Further purification of mitochondria was achieved via a self-generated Percoll or sucrose step gradients (32). Preparation of yeast microsomes from the she2Δ strains RJY2053 (she2Δ) or RJY2370 (she2Δ HMG1-GFP::URA3) was essentially performed as described previously (33, 34). In brief, yeast spheroplasts were lysed by Dounce homogenization with a tight-fitting pestle. To collect a membrane fraction enriched in microsomes (yeast rough membranes), 15 ml of lysate was loaded onto a 15-ml 1 m sucrose cushion, followed by 10-min centrifugation at 3000 × g and 4 °C. The upper layer was recovered avoiding the interface, and membranes were pelleted (21, 000 × g, 10 min, 4 °C). The membrane pellet was resuspended in buffer 88 (20 mm Hepes-KOH (pH 6.8), 150 mm potassium acetate (pH 7.4), 250 mm sorbitol, and 5 mm magnesium acetate), pelleted again, and resuspended in 2 ml of buffer 88 to a final concentration of about A260 = 40/ml (corresponding to a protein concentration of 10–12 mg/ml). For flotation purification, membranes were mixed with 2.5 m sucrose in flotation buffer (50 mm Hepes-KOH (pH 7.5), 5 mm magnesium acetate, 150 mm potassium acetate, and 1.5 mm DTT). This mixture was layered at the bottom of an ultraclear SW55 tube and carefully covered by a cushion of 1.9 m sucrose in flotation buffer (cushion II), followed by a second cushion of flotation buffer only (cushion I). Centrifugation was carried out at 4 °C for 90 min at 240,000 × g, and the membrane fraction was collected at the interface between cushions I and II. If binding assays were performed with protease-treated membranes, yeast rough membranes were adjusted to 10 mm CaCl2 and treated for 30 min at 37 °C with a combination of 2 mg/ml pronase E and 2 mg/ml proteinase K. Subsequently, proteases were inhibited with an excess of EGTA, EDTA, and 1 mm PMSF. Membrane preparations were washed two times with buffer 88, and the same buffer was used to resuspend membranes to a final concentration of about A260 = 40/ml. The size distribution of prepared membrane vesicles was verified by dynamic light scattering in a Zetasizer Nano ZS (Malvern Instruments, Herrenberg, Germany). For the binding assay, flotation-purified ER membranes and pure mitochondria corresponding to 100 μg of protein were diluted with 2 volumes of flotation buffer and mixed with recombinant protein (10 pmol She2p). After 15 min of incubation on ice, the binding mixture was then layered over a 400-μl sucrose cushion (1.2 m sucrose in flotation buffer). Subsequent to centrifugation (60 min at 530,000 × g and 4 °C), 200 μl of the supernatant and of the bottom fraction were collected. The pellet was resuspended in 200 μl of binding assay buffer, and all samples were TCA-precipitated, analyzed by SDS-PAGE, and immunoblotted using antibodies against GST, maltose binding protein, She2p, Sec61p, OM45p, and Mcr1p.
Flotation Assay with Protein-free Liposomes
Liposomes were prepared from lipid mixtures as described (35). For most binding studies, including analysis of liposome size dependence, a phospholipid extract from soybeans was used (Sigma) that contains as main phospholipids 55% phosphatidylcholine (PC) and 25% phosphatidylethanolamine (PE). To investigate the influence of lipid headgroup packing, increasing amounts of DOPC (1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine) were added to yield final concentrations of 20% or 40% DOPC in the lipid mixture. To compare protein binding to liposomes with or without phosphatidylserine (PS) and phosphatidylinositol (PI), a lipid mixture resembling lipid composition of the yeast ER (36, 37) (40% PC, 24% PE, 10% PS, 10% PI, and 16% ergosterol) or a mixture containing 55% PC, 36% PE, and 20% ergosterol was used. Lipids were dissolved in chloroform, and organic solvent was removed by evaporation under an N2 atmosphere. Lipids were completely dissolved to a final total lipid concentration of 10 mg/ml in degassed liposome buffer (20 mm Hepes (pH 7.4) and 100 mm NaCl), and the emulsion was passed 21 times through a polycarbonate filter membrane mounted in a mini extruder (Avanti Polar Lipids) to create unilamellar liposomes. Liposomes with different diameters were generated using filter membranes with 30-nm, 80-nm, 200-nm, or 400-nm pore size. Liposome size distribution was verified by dynamic light scattering in a Zetasizer Nano ZS (Malvern Instruments). Liposomes were used immediately for flotation assays or stored at 4 °C for a maximum of 1 week. 50 μl of liposomes were mixed with 50 pmol She2p, GST, MBP-Mim1p, or OM45p in 190 μl of binding buffer (50 mm Hepes/KOH, 150 mm potassium acetate, 1 mm magnesium acetate, 1 mm EDTA, and 1 mm DTT) and incubated for 15 min on ice. 40 μl of the sample were kept as input control. 200 μl were mixed with 3 ml of binding buffer containing 70% sucrose and added to the bottom of a SW40 polycarbonate tube. The sample was then covered with three cushions of 3 ml of binding buffer containing 50, 40, and 0% sucrose. After centrifugation to equilibrium (70,000 × g for 4 h at 4 °C), the liposome-containing interface between the 40 and 0% sucrose cushions was harvested, precipitated by TCA, and dissolved in 45 μl of SDS sample buffer. Flotation samples and input controls were analyzed as described above. She2p signals were analyzed densitometrically using ImageJ.
For the RNA competition assay, She2p was preincubated with 500 pmol (10× excess) or 1 nmol (20× excess) of in vitro-transcribed ASH1 E3 localization element (28) for 15 min at room temperature and 10 min on ice in binding buffer containing RNasin. Subsequently, liposomes were added, and the mixture was treated as described above. Input control and flotation samples were either processed for Western blot analysis or phenol/chloroform-extracted for RNA recovery. After ethanol precipitation, RNA was resuspended in 25 μl of Tris/EDTA (pH 7.5) and applied to 2% agarose gel electrophoresis and ethidium bromide staining.
Microscopy
Yeast cells were scraped from a fresh selective plate, resuspended in 2 ml of synthetic complete medium, grown for 3–4 h at 30 °C, dropped onto a thin agarose layer of synthetic complete medium, and covered with a coverslip. Images were captured as image stacks on a Zeiss AxioObserver.Z1 fluorescence microscope operated by AxioVision software (Zeiss) and deconvolved using the same software package. Imaging data were analyzed using AxioVision Rel 4.8.
RESULTS
Oligomerization, but Not RNA-binding of She2p, Is Required for ER Association
Because localization of mRNAs such as WSC2 depends on functional cER segregation but is independent of cotranslational targeting (18), localization might require mRNA attachment to the ER via RNA-binding proteins like its cognate partner She2p. We have demonstrated previously that a fraction of She2p comigrates with ER-derived membranes (microsomes) in sucrose gradient centrifugation (19). However, a further analysis of, e.g., the amino acids of She2p that mediate this association has been hampered by the distinctive behavior of several She2p mutants because their expression in yeast often results in retention in the nucleus or nucleolus (8, 9, 19). To identify potential binding sites in She2p, we therefore devised an in vitro assay to study wild-type and mutant She2p proteins for their association with the ER.
When added to a lysate from a she2Δ yeast strain, bacterially expressed She2p cofractionates with the ER marker protein Sec61p to dense fractions of a sucrose gradient (Fig. 1A), which is similar to the pattern seen with endogenous She2p (19). In contrast to Sec61p, She2p can also be detected in the top fraction (16 ± 1.5% of total She2p present in all fractions), implying that not all She2p added to the lysate is associated with the ER. This distribution can also be detected with endogenous She2p (19), suggesting that recombinant She2p retains the ability to associate with microsomes. We next tested for cofractionation of a mutant protein, She2p Lys-130 → Tyr, that is impaired in tetramerization of She2p. Tetramer formation is crucial for RNA target selection but does not diminish general RNA binding (28). Compared with wild-type She2p, where 63% of total She2p cofractionates with Sec61p, the Lys-130 → Tyr mutant shows a reduced cofractionation because only 20% of She2p Lys-130 → Tyr can be found in gradient fractions containing the Sec61p ER marker (fractions 1–3). In contrast, about 30% of She2p Lys-130 → Tyr accumulates as free protein in the top gradient (compared with 14% in case of wild-type She2p), and a large part of it is found in the pellet of the gradient, presumably because the protein is partially aggregating (28). A reduction of She2p cofractionation with the ER was also observed in the case of the mutant protein She2p Ser-120 → Tyr (Fig. 2), which carries a mutation in the She2p homodimerization interface (25). However, this reduction was not as strong as in the Lys-130 → Tyr mutant. In contrast to these mutants, deletion of helix E, which is involved in target mRNA selection (26), does not significantly affect She2p Sec61p comigration (Fig. 2). Similar observations have been made for the She2p Asn-36 → Ser/Arg-63 → Lys mutant that is impaired in RNA binding (19). We conclude from these studies that the correct tertiary structure of She2p, but not its RNA binding ability, is required for proper association with the ER.
FIGURE 1.

Recombinant She2p but not She2p(L130Y) cofractionates with yeast microsomes in vitro. A, recombinant She2p comigrates with the ER in sucrose velocity gradient centrifugation. Top panel, bacterially expressed She2p was incubated with a cell extract from strain RJY2053 (she2Δ) before separation on a linear 18–60% sucrose gradient. Aliquots of 12 fractions (with fraction 1 as the bottom and 12 as the top fraction) and the pellet (P) were analyzed by Western blotting against an ER integral membrane protein (Sec61p) and She2p. Bottom panel, relative amounts of She2p and Sec61p in the corresponding fractions (100% = total amount of She2p or Sec61p detected in all fractions). Mean ± S.D. from three experiments are displayed. B, reduced comigration with the ER of the tetramerization mutant She2p-L130Y. Top panel, Western blot analyses against recombinant She2p-L130Y and Sec61p after gradient centrifugation as described above. Bottom panel, quantification as described in A.
FIGURE 2.

She2p-S120Y and She2p(ΔhelixE) mutant proteins that affect RNA-binding comigrate with the ER in sucrose velocity gradients. A, recombinant She2p comigrates with the ER in sucrose velocity gradient centrifugation. Bacterially expressed She2p was incubated with a cell extract from strain RJY2053 (she2Δ) before separation on a linear 18–60% sucrose gradient. Relative amounts of She2p and Sec61p in the corresponding fractions (with fraction 1 as the top fraction) of a sucrose density gradient are displayed. Numbers are given in percent of total She2p or Sec61p detected in all fractions. Mean ± S.D. from three experiments are shown. B, comigration with the ER of the mutant She2p-S120Y. Quantitative results are shown as described in A. C, comigration with the ER of the mutant She2p-S120Y. Quantitative results are shown as described in A.
Recombinant She2p Binds to ER Membranes but not to Mitochondria
Next, we wanted to address whether the comigration of She2p and ER marker proteins reflect an association of She2p with the ER and whether She2p could also bind to other organelles. To test this, we implemented a second in vitro binding assay. Membrane vesicles were purified from yeast lysates via a two-step procedure, yielding a membrane fraction enriched in ER microsomes (see “Experimental Procedures” and Ref. 33). Purified She2p or GST as a control was added to these microsomes, and the mixture was subjected to centrifugation through a 1.2 m sucrose cushion, thus separating unbound She2p in the supernatant from membrane-bound She2p in the pellet (Fig. 3A). In the presence of microsomal membranes, She2p but not GST penetrated the cushion. When adding increasing amounts of She2p to purified membranes, we observed that binding was saturable and, thus, might involve a binding partner on the membrane (Fig. 3B).
FIGURE 3.

Recombinant She2p binds to ER microsomes but not to mitochondria. A, pelleting of purified microsomes with recombinant She2p and GST. She2p or GST were mixed with ER membranes or buffer before centrifugation through a 1.2 m sucrose cushion. Equivalent amounts of the pellet (Pel), supernatant (Sup), or input material were analyzed by Western blotting against She2p, GST, or Sec61p. She2p is detectable in the pellet only in the presence of membranes. Spurious amounts of GST can be seen in the pellet fraction. B, the indicated amounts of She2p were incubated with 40 μl of microsomes (corresponding to 40 μg of membrane-associated protein). Input and bound She2p were analyzed by quantitative Western blotting, and the ratio of bound She2p signal versus input was quantified. The mean ratio (in percent) and S.D. of three experiments is displayed. C, purified microsomes or yeast mitochondria were mixed with She2p and pelleted through a sucrose cushion. She2p copellets only with microsomes. Sec61p served as an ER marker, whereas Mcr1p indicated mitochondria. In the absence of membranes, She2p can only be detected in the supernatant. Inp, input.
To test whether this binding is specific for the ER, we isolated microsomes or mitochondria from yeast (see “Experimental Procedures”) and added She2p to each before subjecting them to membrane pelleting as described above. As described above, addition of ER-derived membranes leads to pelleting of She2p, as indicated by the presence of both She2p and the ER marker Sec61p in the pellet (Fig. 3C, center rows). In contrast, when microsomes are replaced by purified mitochondria, only little pelleting of She2p is detected, although the mitochondrial marker protein Mcr1p is efficiently pelleted (Fig. 3C, right column). This indicates that ER-derived membranes carry a component that allows a specific association of She2p.
Protease-treated Microsomes Retain She2p-binding Properties
To obtain additional evidence for a specific binding partner on the ER membrane, we treated the membrane fraction with protease before addition of She2p. Protease treatment results in removal of the cytoplasmic epitope recognized by the anti-Sec61p antibody, revealing that proteolysis was successful (Fig. 4A). Increasing amounts of protease for membrane treatment reduces but does not abrogate copelleting of She2p with membranes (Fig. 4B). Even after maximal protease treatment, more than 40% of added She2p can be detected in the membrane pellet fraction. Because She2p can potentially form large multimers in vitro (28), this pellet fraction could correspond to pelleted multimers. To rule out this possibility, She2p was mixed with a membrane-deprived 100,000 × g supernatant of a cell lysate (S100). In this case, only very little She2p could be detected in the pellet (Fig. 4A), suggesting that its presence in the pellet fraction is not due to formation of large multimers.
FIGURE 4.

Protease treatment of membranes reduces, but does not inhibit, binding of She2p. A, pelleting assay of recombinant She2p and protease-treated microsomes. She2p was either incubated with membranes (membr.), membranes treated with a combination of pronase E and proteinase K (treat. membr.), or a supernatant depleted of membranes (S100). After the binding reaction, all three samples were analyzed as described above. P, pellet; S, supernatant. B, protease treatment of microsomes results in only partial loss of She2p binding capacity. Identical quantities of microsomes were treated with increasing protease amounts before addition of She2p and membrane pelleting. Even at high protease concentration, > 40% of She2p can be pelleted. Error bars indicate mean values from four experiments.
She2p Binds to Protein-free Liposomes
Our previous results suggest that binding of She2p to membranes requires proteins for optimal binding but that protease-free membranes could also serve as a binding interface for She2p. We therefore prepared artificial liposomes from soybean lipids containing as major lipids 55% PC and 25% PE. These artificial liposomes were equilibrated with 70% sucrose, covered with cushions of 40% sucrose and buffer lacking sucrose, and used in a flotation experiment adapted from Kanai et al. (38) (see “Experimental Procedures”). The rationale of this experiment is that because of their density liposomes will float to the interphase between the 0 and 40% sucrose cushion together with proteins binding to their lipids. Fig. 5 shows that She2p, when mixed with liposomes before flotation, cofloats to sucrose cushion interphase (Fig. 5A, top row). In contrast, GST does not float in the presence or absence of liposomes (Fig. 5A, bottom panel). Similar results were obtained when purified glycerophospholipids reflecting ER-like lipid composition (36, 37) (40% PC, 24% PE, 10% PS, 10% PI, and 16% ergosterol) were used (data not shown). To show that the flotation assay results reflect membrane association, we used the mitochondrial outer membrane protein OM45p (encoded by the ORF YIL136w), which can bind to mitochondrial membranes and liposomes in vitro (39). The detection of OM45p in the floated fraction indicates that the assay reflects binding of the added protein to liposomes. In summary, She2p can directly bind to liposomes and, therefore, shares features with peripheral membrane proteins.
FIGURE 5.

She2p binds to protein-free liposomes. A, flotation assay of recombinant proteins added to artificial liposomes (200-nm average diameter). Like the outer mitochondrial membrane protein OM45p, She2p, but not GST, floats to the top of a sucrose gradient. Flotation of OM45p and She2p depends on the presence of liposomes. B, She2p interacts with liposomes deprived of phosphatidylserine and phosphatidylinositol. She2p was floated (Flot.) with either ER-like liposomes as in A or with vesicles lacking the phospholipids PS and PI. No difference in coflotation is detectable. C, She2p binding to liposomes inversely correlates to liposome size. Flotation was performed as described under “Experimental Procedures” with liposomes of the indicated diameter. Error bars display the average (av.) ratio of floated protein versus input in artificial units (a.u.). Binding to liposomes with a 30-nm diameter is set to 100 a.u. for She2p, OM45p, or Mim1p. Although She2p flotation decreases with increasing liposome size, relative binding of OM45p or Mim1p to liposomes is unchanged over a large liposome size range. Error bars indicate S.D. in quantitation of three independent flotation experiments. D, lipid headgroup packing does not influence She2p binding to liposomes. A flotation assay with liposomes of 30-nm and 80-nm diameter and a varying percentage of headgroup packing increasing phospholipid DOPC was performed as described. Error bars display the average ratio of floated protein versus input as described in C. Binding to 30-nm liposomes is set to 100%. Relative to 80-nm liposomes without DOPC, liposomes with 20% DOPC or 40% DOPC liposomes do not significantly bind more She2p. Error bars indicate S.D. in the quantitation of three independent flotation experiments.
To address the question whether the lipid content of liposomes influences She2p binding, we prepared liposomes with varying lipid content. Because She2p carries a large number of positively charged amino acids at the surface (25), we were especially interested to see whether phospholipids with a negative net charge, like PS and PI, increase She2p binding. Compared with liposomes with an ER-like lipid content of PI and PS (see above), no difference in She2p binding was detectable (Fig. 5B). We conclude that the negatively charged phospholipids PS and PI are dispensable for She2p binding to liposomes.
The specific interaction of peripheral proteins with membranes can also be mediated by recognition of the membrane curvature (50). To test this idea, we generated protein-free liposomes of a different diameter, ranging from 30 nm to 400 nm (see “Experimental Procedures”). The rationale was that smaller liposomes generated by this method have a higher membrane curvature than larger ones. Equal amounts of phospholipids were used for the preparation of differently sized liposomes, which were then mixed with She2p and used in parallel flotation experiments. As control proteins, we used two mitochondrial membrane proteins, OM45p and Mim1p (40). Western blot analysis and quantification of the She2p signals in the floated fraction and the input revealed an increase of bound She2p with decreasing liposome size. When binding of She2p to liposomes with a 30-nm diameter was arbitrarily set to 100% (Fig. 5C), 80-nm liposomes bound 73% (± 9%, n = 3) of She2p, 200-nm liposomes only 50% (± 13%), and liposomes with an average diameter of 400 nm only 30% (± 14.5%). No such preference could be detected for OM45p that showed similar binding to liposomes of various size, whereas Mim1p binding was strongest to larger liposomes but did not significantly vary between 30-nm and 200-nm liposomes. These data suggest that binding of She2p to membranes, in contrast to OM45p or Mim1p, depends on liposome curvature or phospholipid headgroup packing, which is also altered by liposome size. To distinguish between these possibilities, we prepared liposomes of 80-nm diameter containing 20 and 40% DOPC, an inverted cone-shaped phospholipid that allows higher packing in curved membranes (41). If lipid packing were important, one would expect an increase in membrane interaction of She2p because of incorporation of DOPC in liposomes. However, although quantification of the She2p Western blot signals in the floated fraction and the input revealed a modest increase in average binding (9%) compared with 80-nm liposomes without DOPC, this indicates that the increased interaction of She2p with smaller liposomes might indeed mainly be due to curvature increase (Fig. 5D).
Liposome Association of an RNA Localization Element Occurs via She2p
A previous analysis demonstrated that copelleting of localized mRNAs with the ER is altered in cells lacking She2p, indicating that She2p is a crucial factor for their association with membranes (15). Our flotation assay allowed us to address whether She2p is not only required but also sufficient for this association. If binding of She2p to RNA targets and lipids could occur simultaneously, they should show no competition. We therefore tested whether a cognate RNA, the ASH1 E3 localization element, competes with liposomes for She2p binding. In two independent experiments, a 10-fold or 20-fold molar excess of an in vitro-transcribed E3 localization element was added to a mixture of liposomes and She2p before flotation. In parallel controls, the E3 RNA was omitted (“mock”). Flotation of She2p remained unperturbed, even in the presence of a 20-fold excess of E3 (Fig. 6), indicating that an RNA localization element does not compete with liposomes for She2p binding. Analysis of RNA extracted from floated fractions by agarose gel electrophoresis and ethidium bromide staining revealed the presence of E3 RNA in the floated fraction. These results demonstrate that She2p can simultaneously bind to liposomes and a localization element and suggest that She2p mediates the observed association of localized mRNAs with the ER (15, 19).
FIGURE 6.

Simultaneous binding of She2p to liposomes and an RNA localization element. Recombinant She2p was incubated with only binding buffer (mock) and a 10-fold and 20-fold molar excess of in vitro-transcribed ASH1 E3 element, followed by incubation with liposomes and flotation as described in Fig. 3. Top panel, Western blot analysis against She2p. Bottom panel, ethidium bromide-stained gel of RNA extracted from floated fractions (Flot.) and input.
She2p Targets a Viral Peptide to the ER
Our in vitro analysis indicates that She2p can bind to ER-derived membranes. These results are supported by previous observations that mRNPs of mRNAs associated with She2p colocalize with ER tubules during budding (19). To address whether She2p can bind the ER in vivo, we implemented a new assay on the basis of the observation that expression of an N-terminal peptide from bamboo mosaic potexvirus protein Tgb3 results in its targeting to the ER when fused to a membrane protein (27). This assay would allow us to detect membrane binding even when this is transient or when only a subfraction of She2p is membrane-associated. If expressed in yeast, the potexvirus Tgb3 protein is targeted to the cortical ER. The targeting is due to a signal at its N terminus (comprising amino acids 25–52). The signal peptide, when expressed alone, results in cytoplasmic localization, but a fusion of the signal peptide with a membrane protein results in targeting to the cortical ER (27). We therefore wondered whether She2p can adopt the role of the membrane protein and target a Tgbp3(aa25–52)-GFP fusion to the cell cortex by providing the missing membrane targeting information. As a control to test whether this is a general feature of other RNA-binding proteins, we used the coat protein from bacteriophage MS2 (42). This fusion protein showed the same diffuse cytoplasmic distribution as Tgbp3(aa25–52)-GFP (Fig. 7A). In contrast, fusion of She2p results in redistribution of Tgbp3(aa25–52)-GFP to foci in the bud and mother cell (Fig. 7A, left panel). A majority of these foci (73%, n = 55) colocalize or overlap with the endoplasmic reticulum in the mother cell and at the bud tip (Fig. 7B), suggesting that She2p might target the viral peptide to ER membranes. Colocalization of similar foci with ER structures has been reported from other fusion proteins with Tgb3p (27). To independently test for ER targeting, we performed subcellular fractionation by differential centrifugation of cell lysates (30) containing either Tgbp3(aa25–52)-GFP or She2p-Tgbp3(aa25–52)-GFP (Fig. 7B). The majority of Tgbp3(aa25–52)-GFP is detected in fraction S200, containing cytosolic proteins like Pgk1p (30). In contrast, She2p-Tgbp3(aa25–52)-GFP is enriched in fraction P6, which contains heavy membranes, including the ER, as seen by the ER marker Dpm1p (30). These results indicate that She2p has the potential to target a reporter peptide to ER membranes in vivo and, therefore, strongly supports our in vitro analysis.
FIGURE 7.
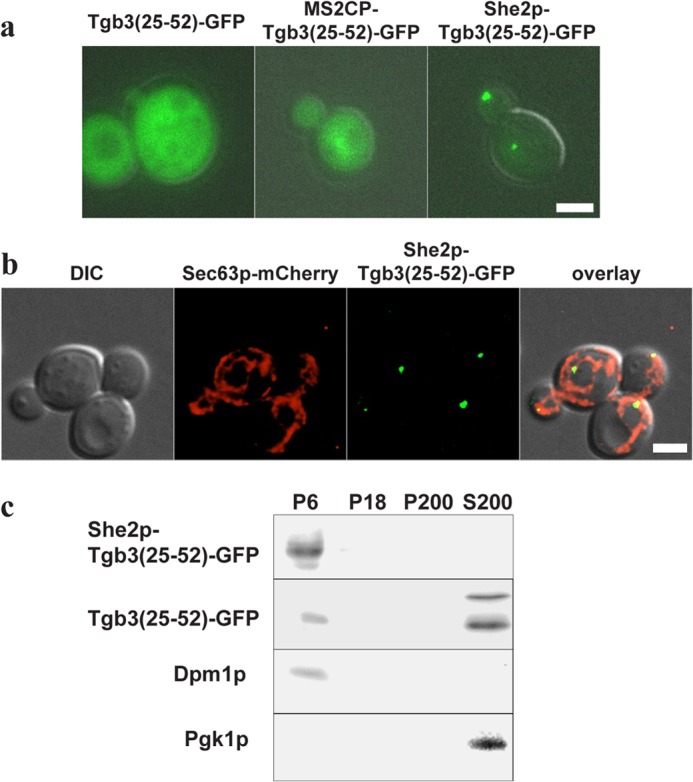
She2p mediates binding of a viral peptide to yeast cortical ER. A, GFP fused to an amino-terminal peptide of bamboo potexvirus protein TGB3 (Tgb3(aa25–52)) accumulates in the cytoplasm of yeast cells. Fusion of She2p, but not of MS2 coat protein, allows formation of distinct particles (left panel). Scale bar = 2 μm. B, She2p-Tgb3(aa25–52) particles colocalize with the ER. first panel, differential interference contrast image (DIC). Second panel, Sec63-mCherry. Third panel, She2p-Tgb3(aa25–52)-GFP. Fourth panel, overlay of differential interference contrast image, mCherry, and GFP. Images represent single frames from a deconvolved z stack to ensure better resolution in the z axis. Scale bar = to 2 μm. C, in subcellular fractionation, She2p-Tgb3(25–52)-GFP accumulates in fraction P6 that contains the ER, as shown by the presence of the ER marker Dpm1p. In contrast, Tgb3(25–52)-GFP is mainly detected in a cytosolic fraction (S200), as indicated by the presence of phosphoglycerokinase (Pgk1p).
DISCUSSION
In budding yeast, more than 30 mRNAs localized to the growing bud encode membrane or secreted proteins. Localization of these mRNAs does not only require the locasome components Myo4p, She3p, and She2p (17) but also proteins that are crucial for early steps during the segregation of the cortical ER (15, 18, 19). Cortical ER segregation is initiated by the movement of tubular ER structures into the bud, which, among other factors, requires Myo4p and She3p but not She2p (24). However, localizing mRNPs comigrate with ER tubules during movement (19), and because mRNAs in these mRNPs are bound by She2p, it might nevertheless be present on these tubules. Consistently, localized mRNAs only cofractionate with membranes in subcellular fractionation studies when She2p is present in these cells (15), suggesting that She2p might mediate the binding of these mRNAs with ER membranes.
Although the association of mRNAs to the ER independent of targeting via the signal recognition particle is a widespread phenomenon (43–45), the role of RNA-binding proteins in targeting mRNA to the ER is not well understood. Targeting could involve RNA-binding membrane proteins like the ER protein p180 (46) or cytoplasmic RNA-binding proteins that direct mRNAs to the ER (47). In budding yeast, the RNA-binding proteins Scp160p (30), Whi3p (48), Bfr1p (49), and She2p at least partially colocalize or cofractionate with the ER. Although the exact mechanism of ER association of Whi3p is unknown, colocalization of Scp160p and Bfr1p with the ER depends on translation and might, therefore, be indirectly mediated via nascent peptide chains (30, 49). In contrast, She2p cofractionates with the ER even after the disruption of polysomes or degradation of mRNA (15, 19) and, therefore, represents a likely candidate for a protein mediating the association of specific mRNAs to the ER in vivo. The observations that She2p can simultaneously bind to an RNA localization element and to membranes (Fig. 6) and that it is required for mRNA-ER cofractionation (15) are consistent with this idea. So far, She2p has been visualized in cells only in a crescent at the bud tip (5), as a diffuse signal in the cytoplasm (8), or in the nucleus (8, 9), but colocalization with the ER has not been reported. However, it can target a viral peptide from the bamboo potexvirus Tgb3 protein to the ER, a feature that has so far been demonstrated for membrane or membrane-associated proteins (27). This suggests that at least a fraction of She2p is able to temporarily associate with membranes in vivo, which allows the peptide to be transferred to the ER.
How does She2p bind to ER membranes? Two lines of evidence suggest that binding occurs both via lipids and a protein component. Because She2p can bind to protein-free liposomes, it is able to directly interact with lipids. However, She2p binding to protease-treated ER membranes is significantly reduced, and binding occurs to ER membranes but not to mitochondrial membranes with a different protein composition. ER-specific versus mitochondrial binding cannot be explained by the smaller size of ER-derived vesicles as compared with vesicles from mitochondrial preparations because dynamic light scattering indicated that mitochondrial vesicles are, on average, half the size of microsomal vesicles (data not shown). Binding also does not require membranes with a specific lipid composition because omission of phospholipids with a negative net charge, like phosphatidylserine or phosphatidylinositol, does not change the binding behavior. Instead, we observed a preferential binding of She2p for liposomes of a small diameter as compared with larger ones. This increase could result from a change in curvature, which suggests that She2p recognizes membrane shape. Alternatively, it might result from an overall larger surface area of the sum of all small liposomes or, as the density of lipid headgroup changes with liposome size, She2p binding might also depend on lipid headgroup density. Because She2p binding to 80-nm liposomes drops by about 30% as compared with 30 nm, but the surface area of the outer leaflet decreases by only 10%, a simple difference in total binding area cannot account for the observed difference. Our results that incorporation into liposomes of DOPC, a cone-shaped lipid with a small headgroup, does not significantly alter binding efficiency argue against lipid density recognition. Consistent with curvature recognition, mRNPs containing She2p colocalize with tubular ER structures (19) that, in yeast, have an average diameter of 30–40 nm (21), which reflects the observed binding preference in our flotation assays. Furthermore, Δyop1Δrtn1 mutant yeast cells that contain bloated ER tubules with an increased diameter (21) are defective in localization of mRNAs whose localization depends on ER segregation (18). Interestingly, these bloated tubules are still segregated into the bud (21) but might no more associate with RNPs containing She2p.
According to both sequence and protein structure analysis, She2p does not contain known motifs present in membrane-shape recognizing peripheral membrane proteins (50, 51) or other membrane-binding motifs. Because we used a bacterially expressed She2p in our binding assays, it is also less likely that other posttranslational modifications, like phosphorylation, are required for association of She2p with the ER. It thus remains open how the protein associates with the ER or liposomes. Tetramerization of She2p seems to be involved in membrane attachment because the fraction of She2p that comigrates with the ER in sucrose gradients is reduced in a She2p mutant carrying the Leu-130 → Tyr mutation that disrupts tetramer formation (28). Although this mutation also diminishes RNA binding, other mutations that disturb RNA binding, like the deletion of helix E (26), do not alter She2p-ER comigration. Tetramerization, therefore, seems to serve RNA and membrane binding, in the latter case possibly by providing a larger interaction interface of She2p with components of the cytoplasmic face of the ER. In addition, not only ER tubules but other ER structures, like the central cisternal ER and plasma membrane-attached ER (21, 52) as well as small vesicles, exhibit high membrane curvature, suggesting an additional mechanism to specifically direct She2p and bound mRNAs to ER tubules. This might involve yet-unknown protein adapters on the ER and is corroborated by our finding that protease treatment of microsomes reduces the fraction of She2p interacting with microsomal membranes.
Our study strongly suggests that, despite the absence of recognizable lipid interaction domains, the RNA-binding protein She2p has an unexpected membrane binding property, which is consistent with its requirement for the association of localized mRNAs with ER. It not only raises the question about the molecular basis of this association but also whether the proposed direct linking of specific mRNAs to membranes via She2p is a mechanism used by other RNA-binding proteins involved in targeting of specific mRNAs to the ER, like rice OsTudorSN (53) or Xenopus laevis Vg1RBP/Vera (54).
Acknowledgments
We thank T.-G. Du in generating the She2p Ser-120 → Tyr mutant; D. Niessing and M. Müller for providing plasmid pGEX-SHE2-ΔhE; C.-W. Wang for the GFP-TGB3(25–52) expression plasmid; E. Merklinger, R. Singhal, M. Sinzel, and D. Rapaport for providing purified OM45p, MBP-Mim1p, and purified mitochondria; and J. Bauer for help during dynamic light scattering.
This work was supported by Deutsche Forschungsgesellschaft Grant DFG JA696/7-1 (to R. P. J.).
- mRNP
- messenger ribonucleoprotein
- ER
- endoplasmic reticulum
- cER
- cortical endoplasmic reticulum
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- DOPC
- 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine
- PS
- phosphatidylserine
- PI
- phosphatidylinositol
- aa
- amino acids.
REFERENCES
- 1. Du T.-G., Schmid M., Jansen R.-P. (2007) Why cells move messages. The biological functions of mRNA localization. Semin. Cell Dev. Biol. 18, 171–177 [DOI] [PubMed] [Google Scholar]
- 2. Martin K. C., Ephrussi A. (2009) mRNA localization. Gene expression in the spatial dimension. Cell 136, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Besse F., Ephrussi A. (2008) Translational control of localized mRNAs. Restricting protein synthesis in space and time. Nat. Rev. Mol. Cell Biol. 9, 971–980 [DOI] [PubMed] [Google Scholar]
- 4. Long R. M., Singer R. H., Meng X., Gonzalez I., Nasmyth K., Jansen R. P. (1997) Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science 277, 383–387 [DOI] [PubMed] [Google Scholar]
- 5. Böhl F., Kruse C., Frank A., Ferring D., Jansen R. P. (2000) She2p, a novel RNA-binding protein tethers ASH1 mRNA to the Myo4p myosin motor via She3p. EMBO J. 19, 5514–5524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Long R. M., Gu W., Lorimer E., Singer R. H., Chartrand P. (2000) She2p is a novel RNA-binding protein that recruits the Myo4p-She3p complex to ASH1 mRNA. EMBO J. 19, 6592–6601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen Z., St-Denis A., Chartrand P. (2010) Cotranscriptional recruitment of She2p by RNA pol II elongation factor Spt4-Spt5/DSIF promotes mRNA localization to the yeast bud. Genes Dev. 24, 1914–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du T.-G., Jellbauer S., Müller M., Schmid M., Niessing D., Jansen R.-P. (2008) Nuclear transit of the RNA-binding protein She2 is required for translational control of localized ASH1 mRNA. EMBO Rep. 9, 781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen Z., Paquin N., Forget A., Chartrand P. (2009) Nuclear shuttling of She2p couples ASH1 mRNA localization to its translational repression by recruiting Loc1p and Puf6p. Mol. Biol. Cell 20, 2265–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heuck A., Fetka I., Brewer D. N., Hüls D., Munson M., Jansen R.-P., Niessing D. (2010) The structure of the Myo4p globular tail and its function in ASH1 mRNA localization. J. Cell Biol. 189, 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hodges A. R., Krementsova E. B., Trybus K. M. (2008) She3p binds to the rod of yeast myosin V and prevents it from dimerizing, forming a single-headed motor complex. J. Biol. Chem. 283, 6906–6914 [DOI] [PubMed] [Google Scholar]
- 12. Gu W., Deng Y., Zenklusen D., Singer R. H. (2004) A new yeast PUF family protein, Puf6p, represses ASH1 mRNA translation and is required for its localization. Genes Dev. 18, 1452–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Irie K., Tadauchi T., Takizawa P. A., Vale R. D., Matsumoto K., Herskowitz I. (2002) The Khd1 protein, which has three KH RNA-binding motifs, is required for proper localization of ASH1 mRNA in yeast. EMBO J. 21, 1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Long R. M., Gu W., Meng X., Gonsalvez G., Singer R. H., Chartrand P. (2001) An exclusively nuclear RNA-binding protein affects asymmetric localization of ASH1 mRNA and Ash1p in yeast. J. Cell Biol. 153, 307–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aronov S., Gelin-Licht R., Zipor G., Haim L., Safran E., Gerst J. E. (2007) mRNAs encoding polarity and exocytosis factors are cotransported with the cortical endoplasmic reticulum to the incipient bud in Saccharomyces cerevisiae. Mol. Cell Biol. 27, 3441–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oeffinger M., Wei K. E., Rogers R., DeGrasse J. A., Chait B. T., Aitchison J. D., Rout M. P. (2007) Comprehensive analysis of diverse ribonucleoprotein complexes. Nat. Meth. 4, 951–956 [DOI] [PubMed] [Google Scholar]
- 17. Shepard K. A., Gerber A. P., Jambhekar A., Takizawa P. A., Brown P. O., Herschlag D., DeRisi J. L., Vale R. D. (2003) Widespread cytoplasmic mRNA transport in yeast. Identification of 22 bud-localized transcripts using DNA microarray analysis. Proc. Natl. Acad. Sci. U.S.A. 100, 11429–11434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fundakowski J., Hermesh O., Jansen R.-P. (2012) Localization of a subset of yeast mRNAs depends on inheritance of endoplasmic reticulum. Traffic 13, 1642–1652 [DOI] [PubMed] [Google Scholar]
- 19. Schmid M., Jaedicke A., Du T.-G., Jansen R.-P. (2006) Coordination of endoplasmic reticulum and mRNA localization to the yeast bud. Curr. Biol. 16, 1538–1543 [DOI] [PubMed] [Google Scholar]
- 20. Du Y., Ferro-Novick S., Novick P. (2004) Dynamics and inheritance of the endoplasmic reticulum. J. Cell Sci. 117, 2871–2878 [DOI] [PubMed] [Google Scholar]
- 21. West M., Zurek N., Hoenger A., Voeltz G. K. (2011) A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 193, 333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 [DOI] [PubMed] [Google Scholar]
- 23. Hu J., Shibata Y., Voss C., Shemesh T., Li Z., Coughlin M., Kozlov M. M., Rapoport T. A., Prinz W. A. (2008) Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 319, 1247–1250 [DOI] [PubMed] [Google Scholar]
- 24. Estrada P., Kim J., Coleman J., Walker L., Dunn B., Takizawa P., Novick P., Ferro-Novick S. (2003) Myo4p and She3p are required for cortical ER inheritance in Saccharomyces cerevisiae. J. Cell Biol. 163, 1255–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Niessing D., Hüttelmaier S., Zenklusen D., Singer R. H., Burley S. K. (2004) She2p is a novel RNA binding protein with a basic helical hairpin motif. Cell 119, 491–502 [DOI] [PubMed] [Google Scholar]
- 26. Müller M., Heym R. G., Mayer A., Kramer K., Schmid M., Cramer P., Urlaub H., Jansen R.-P., Niessing D. (2011) A cytoplasmic complex mediates specific mRNA recognition and localization in yeast. PLoS Biol. 9, e1000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu C.-H., Lee S.-C., Wang C.-W. (2011) Viral protein targeting to the cortical endoplasmic reticulum is required for cell-cell spreading in plants. J. Cell Biol. 193, 521–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Müller M., Richter K., Heuck A., Kremmer E., Buchner J., Jansen R.-P., Niessing D. (2009) Formation of She2p tetramers is required for mRNA binding, mRNP assembly, and localization. RNA 15, 2002–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barrowman J., Sacher M., Ferro-Novick S. (2000) TRAPP stably associates with the Golgi and is required for vesicle docking. EMBO J. 19, 862–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frey S., Pool M., Seedorf M. (2001) Scp160p, an RNA-binding, polysome-associated protein, localizes to the endoplasmic reticulum of Saccharomyces cerevisiae in a microtubule-dependent manner. J. Biol. Chem. 276, 15905–15912 [DOI] [PubMed] [Google Scholar]
- 31. Daum G., Gasser S. M., Schatz G. (1982) Import of proteins into mitochondria. Energy-dependent, two-step processing of the intermembrane space enzyme cytochrome b2 by isolated yeast mitochondria. J. Biol. Chem. 257, 13075–13080 [PubMed] [Google Scholar]
- 32. Graham J. M. (2001) Purification of a Crude Mitochondrial Fraction by Density-Gradient Centrifugation, John Wiley & Sons, Inc., Hoboken, NJ: [DOI] [PubMed] [Google Scholar]
- 33. Brodsky J. L., Schekman R. (1993) A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 123, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rothblatt J. A., Meyer D. I. (1986) Secretion in yeast. Reconstitution of the translocation and glycosylation of α-factor and invertase in a homologous cell-free system. Cell 44, 619–628 [DOI] [PubMed] [Google Scholar]
- 35. Qbadou S., Tien R., Soll J., Schleiff E. (2003) Membrane insertion of the chloroplast outer envelope protein, Toc34. Constrains for insertion and topology. J. Cell Sci. 116, 837–846 [DOI] [PubMed] [Google Scholar]
- 36. Tuller G., Nemec T., Hrastnik C., Daum G. (1999) Lipid composition of subcellular membranes of an FY1679-derived haploid yeast wild-type strain grown on different carbon sources. Yeast 15, 1555–1564 [DOI] [PubMed] [Google Scholar]
- 37. Schneiter R., Brügger B., Sandhoff R., Zellnig G., Leber A., Lampl M., Athenstaedt K., Hrastnik C., Eder S., Daum G., Paltauf F., Wieland F. T., Kohlwein S. D. (1999) Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J. Cell Biol. 146, 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanai Y., Dohmae N., Hirokawa N. (2004) Kinesin transports RNA. Isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525 [DOI] [PubMed] [Google Scholar]
- 39. Merklinger E., Gofman Y., Kedrov A., Driessen A. J., Ben-Tal N., Shai Y., Rapaport D. (2012) Membrane integration of a mitochondrial signal-anchored protein does not require additional proteinaceous factors. Biochem. J. 442, 381–389 [DOI] [PubMed] [Google Scholar]
- 40. Waizenegger T., Schmitt S., Zivkovic J., Neupert W., Rapaport D. (2004) Mim1, a protein required for the assembly of the TOM complex of mitochondria. EMBO Rep. 6, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brouillette C. G., Segrest J. P., Ng T. C., Jones J. L. (1982) Minimal size phosphatidylcholine vesicles. Effects of radius of curvature on head group packing and conformation. Biochemistry 21, 4569–4575 [DOI] [PubMed] [Google Scholar]
- 42. Peabody D. S., Lim F. (1996) Complementation of RNA binding site mutations in MS2 coat protein heterodimers. Nucleic Acids Res. 24, 2352–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pyhtila B., Zheng T., Lager P. J., Keene J. D., Reedy M. C., Nicchitta C. V. (2008) Signal sequence- and translation-independent mRNA localization to the endoplasmic reticulum. RNA 14, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reid D. W., Nicchitta C. V. (2012) Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J. Biol. Chem. 287, 5518–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hermesh O., Jansen R.-P. (2013) Take the (RN)A-train. Localization of mRNA to the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2519–2525 [DOI] [PubMed] [Google Scholar]
- 46. Cui X. A., Zhang H., Palazzo A. F. (2012) p180 promotes the ribosome-independent localization of a subset of mRNA to the endoplasmic reticulum. PLoS Biol. 10, e1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kraut-Cohen J., Gerst J. E. (2010) Addressing mRNAs to the ER. Cis sequences act up! Trends Biochem. Sci. 35, 459–469 [DOI] [PubMed] [Google Scholar]
- 48. Colomina N., Ferrezuelo F., Wang H., Aldea M., Garí E. (2008) Whi3, a developmental regulator of budding yeast, binds a large set of mRNAs functionally related to the endoplasmic reticulum. J. Biol. Chem. 283, 28670–28679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lang B. D., Li Am, Black-Brewster H. D., Fridovich-Keil J. L. (2001) The brefeldin A resistance protein Bfr1p is a component of polyribosome-associated mRNP complexes in yeast. Nucleic Acids Res. 29, 2567–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 51. Madsen K. L., Bhatia V. K., Gether U., Stamou D. (2010) BAR domains, amphipathic helices and membrane-anchored proteins use the same mechanism to sense membrane curvature. FEBS Lett. 584, 1848–1855 [DOI] [PubMed] [Google Scholar]
- 52. Shibata Y., Shemesh T., Prinz W. A., Palazzo A. F., Kozlov M. M., Rapoport T. A. (2010) Mechanisms determining the morphology of the peripheral ER. Cell 143, 774–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang C., Washida H., Crofts A. J., Hamada S., Katsube-Tanaka T., Kim D., Choi S.-B., Modi M., Singh S., Okita T. W. (2008) The cytoplasmic-localized, cytoskeletal-associated RNA binding protein OsTudor-SN. Evidence for an essential role in storage protein RNA transport and localization. Plant J. 55, 443–454 [DOI] [PubMed] [Google Scholar]
- 54. Deshler J. O., Highett M. I., Abramson T., Schnapp B. J. (1998) A highly conserved RNA-binding protein for cytoplasmic mRNA localization in vertebrates. Curr. Biol. 8, 489–496 [DOI] [PubMed] [Google Scholar]