

Abstract
The Proteomics Standards Initiative has recently released the mzIdentML data standard for representing peptide and protein identification results, for example, created by a search engine. When a new standard format is produced, it is important that software tools are available that make it straightforward for laboratory scientists to use it routinely and for bioinformaticians to embed support in their own tools. Here we report the release of several open-source Java-based software packages based on mzIdentML: ProteoIDViewer, mzidLibrary, and mzidValidator. The ProteoIDViewer is a desktop application allowing users to visualize mzIdentML-formatted results originating from any appropriate identification software; it supports visualization of all the features of the mzIdentML format. The mzidLibrary is a software library containing routines for importing data from external search engines, post-processing identification data (such as false discovery rate calculations), combining results from multiple search engines, performing protein inference, setting identification thresholds, and exporting results from mzIdentML to plain text files. The mzidValidator is able to process files and report warnings or errors if files are not correctly formatted or contain some semantic error. We anticipate that these developments will simplify adoption of the new standard in proteomics laboratories and the integration of mzIdentML into other software tools. All three tools are freely available in the public domain.
The Proteomics Standards Initiative (PSI)1 recently released the mzIdentML standard data format (stable version 1.1) for reporting peptide and protein identifications in order to improve capabilities for data sharing and make it simpler for bioinformatics groups to focus development on a single, comprehensive file format (1). The format is represented in XML and is formally defined by the combination of the XML Schema Definition and a separate mapping file describing where controlled vocabulary (CV) terms must be used within the format. A core part of the standard captures lists of peptide-spectrum matches (PSMs) with associated scores or measures, described by CV terms. Each PSM should be linked to the spectrum that was searched in a separate file, such as represented in the PSI's mzML standard (2). The PSM captures the modifications identified, again using CV terms sourced from Unimod (3) or the PSI-MOD ontology (4). In an mzIdentML file, each PSM is linked to all protein sequences from which the peptide could have been derived (using the enzyme specified in the search). The mzIdentML standard also has a separate section capturing protein identification results in a two-level hierarchy. The top level comprises groups of proteins representing a putatively detected “isoform”, with each group containing a list of individual proteins (entries in the database searched to be specific) for which there is ambiguity regarding which of those entities was actually identified, typically because of the existence of shared peptides (i.e. the well-known protein inference problem (5)). Note that in this context an “isoform” is simply a group of accessions from the source database and could be the result of database errors (duplications, sequencing errors) as well as related biological entities. The format also contains structures for describing the search parameters in a standard way, sourcing CV terms from the PSI-MS CV for enzyme descriptors, score thresholds, and so on (6).
The format thus contains some complexity (accurately reflecting the nature of the source data), and tools are needed focused on both end users and informaticians. At present, there is support for mzIdentML export from Mascot version 2.4 (7), Phenyx (via a patch from the GeneBio developers) (8), Scaffold (9), PEAKS (10), MSGF+, MyriMatch (11), and the Trans-Proteomic Pipeline (12) via ProteoWizard (13). In addition, there is the capability for the input and export of mzIdentML in OpenMS (14) and file format converters developed for Sequest within Proteome Discoverer (Thermo Scientific) in the ProCon software. To support other developers, our groups have developed a Java application programming interface (API) for reading and writing mzIdentML files, called jmzIdentML (15). The current state of mzIdentML implementations is summarized on the PSI website.
We are introducing here a suite of three open-source tools for mzIdentML, developed in Java. Firstly, we have built a graphical user interface for the standard, called the ProteoIDViewer, focused on both end users and developers. Secondly, we have created the mzidLibrary, a set of routines that can be built into a pipeline for manipulating or post-processing data, including tools for converting the output formats of the popular free search engines OMSSA (16) (OMX or comma-separated value (CSV) files) and X!Tandem (17) (tandem XML files) into mzIdentML. In addition to a command line mode, the mzidLibrary has a simple graphical interface allowing the routines to be called by end users in a straightforward manner. Selected tools, such as the format converters, are also embedded directly in the ProteoIDViewer. Thirdly, we have developed the mzidValidator, a graphical interface for checking that files are correctly formatted and use appropriate CV terms correctly. It is primarily intended as a developer tool, but it also is aimed at end users who encounter problems with files from a particular source. An online validator has also been developed by the OpenMS team, but this is restricted to small files because of the requirements for uploading to the Web interface. All the tools we have developed are open source, released with a permissive license, and the code is maintained in subversion repositories at Google Code, allowing issues, bugs, and feature requests to be logged and documented. We demonstrate the utility of the toolset by using publicly available data created by the 2008 Proteome Informatics Research Group (iPRG) study of the Association of Biomolecular Resource Facilities.
EXPERIMENTAL PROCEDURES
All three tools (ProteoIDViewer, mzidLibrary, and mzidValidator) are built on top of jmzIdentML (15), which provides file reading and writing capabilities, converting XML-formatted files into Java objects and vice versa. The API uses an indexing strategy called the xxIndex, in which the byte positions of objects in the file are stored, allowing fast, random access to the objects file without the need to load large files into memory.
ProteoIDViewer
The ProteoIDViewer was constructed using Java Swing components designed in NetBeans. It was designed to process files using jmzidentML and load protein, peptide-level, or spectrum-level data into tabular structures, with appropriate links created between data represented on different panels. Graphical displays were developed on top of external libraries incorporating graphs for decoy database results using JFreeChart and an interactive spectrum viewer based on an open-source library developed by Lennart Martens's group (18).
mzidLibrary
The mzidLibrary contains a set of routines for file manipulation based on previously published algorithms or software, such as FDRScore and the associated method for combining multiple search engine results to increase sensitivity (19), the exponentially modified Protein Abundance Index (emPAI) protocol for quantitation based on spectral counting (20), and file format converters developed on top of two open-source APIs produced by Martens's group, namely, the OMSSA Parser (21) and the XTandem Parser (22), which can read the OMSSA XML format (OMX files) and X!Tandem XML files, respectively.
Code was created to connect the APIs over the OMSSA and X!Tandem formats to objects within jmzIdentML. In addition, a new module was created in the mzidLibrary for processing OMSSA CSV-formatted files, because there is a large memory overhead associated with processing large search results in the OMSSA OMX format, and the OMSSA CSV results files are considerably smaller and easier to handle. The OMSSA CSV parser created here requires an additional CSV file containing the search metadata, required for a valid mzIdentML file (the format of the input parameters file is described in the mzidLibrary user guide, available from the project website), as these details are not provided in the results output. The mzidLibrary also contains routines for setting identification thresholds in the file and retrieving additional data about protein sequences from the FASTA file (database) that was searched. Lastly, the mzidLibrary contains new software for performing protein inference, called ProteoGrouper, for which the algorithm is described here for the first time (see below).
ProteoGrouper Algorithm
The ProteoGrouper algorithm performs the following steps:
- ProteoGrouper first requires that all the following parameters be set:
- The PSM score (accession of the corresponding CV term) to be used for producing a protein score. If multiple search engine results have been combined, the results must be present in a single list in mzIdentML (<SpectrumIdentificationList>) with a comparable score, identified by one CV term.
- Whether a log transformation should be performed on the PSM score, which will be summed to create a protein-level score (i.e. to convert an e-value, p value, or false discovery rate (FDR)-based score into a positive number (true | false)).
- Whether only PSMs with ‘passThreshold’ = “true” or all PSMs in the file should be included (true | false).
<ProteinDetectionHypothesis> (PDH) elements in mzIdentML (representing single protein database entries) are constructed by referencing all PSMs (that pass the threshold, if specified) that have a mapping to the current protein accession.
A PDHScore is created for each PDH by summing the PSM scores according to the user parameters.
- An implicit ordering is determined for all PDH elements and is used for assigning “razor peptides” to proteins within a group. A razor peptide is one that can be assigned to more than one protein and thus is assigned to the protein with the most independent evidence, following the terminology in Ref. 23. The order of PDHs is determined by the following:
- Highest number of distinct peptides.
- Highest protein score.
- Alphabetical order.
Any protein that (i) has any “unique peptides” or (ii) has been assigned razor peptides is scored as a putatively detected isoform (a “representative protein”) and forms a new protein group (<ProteinAmbiguityGroup> element in mzIdentML). Unique peptides are defined as those that can be mapped to only a single protein database accession, allowing for I/L ambiguity that cannot typically be differentiated by MS/MS. Additional ambiguous mappings will be introduced in later releases.
All subset or same-set proteins with regard to the representative protein are placed in the same group.
Any protein that is multiply subsumed by one or more representative proteins is placed in the group with the representative protein with which it shares the most peptides. In the case of a tie, the multiply subsumed protein is placed into the group that has the strongest evidence, according to the criteria given above.
Data Set Used for Benchmarking
Here we demonstrate the use of the tools within the mzidLibrary, in particular the ProteoGrouper, using the iPRG 2008 study materials. These data were created by the iPRG and distributed to study participants to see how well different groups could perform protein identification on a standard input data set. Briefly, peptides derived from the tryptic digestion of proteins from mouse liver samples were labeled with iTRAQ® reagents (although quantification was not the focus of the analysis). Methyl methanethiosulfonate alkylation was performed and peptides were separated via cation exchange chromatography into 13 fractions. The fractions were analyzed on a QTRAP® instrument to produce 29 mass spectra files, available in various formats (Mascot generic format, dta, etc.). The files were then analyzed by different iPRG members and study participants using a variety of search engines and software for performing protein inference. There was a three-level hierarchy to the results reported: “cluster”, “isoform” and “accession”. “Cluster” indicates a group of putatively detected isoforms sharing at least some peptides in common but with independent evidence for individual isoforms being identifiable. An isoform is the core unit of biological identification, potentially containing a group of database accessions, for example, as a result of same set or subset relationships among group members. An accession is one entry in the source database that was searched. Manual analysis was performed to classify results into the following categories:
Class 1: Research Group (RG) consensus multi-detection clusters (numbered 101–116). For these, iPRG determined the exact number of identifiable isoforms within each cluster, and all clusters contained more than one isoform.
Class 2: Debatable multi-detection clusters (numbered 201–211); depending upon the approach taken, each cluster may contain one or more isoforms.
Class 3: RG consensus single-detection clusters (301–482); each cluster contains exactly one isoform.
Classes 4 and 5: Non-consensus detections by RG and non-RG, respectively; isoforms in these classes were identified by research group members or study participants without general consensus on their identification. For more details, consult the resources available from the Association of Biomolecular Resource Facilities.
In the answer key provided by the iPRG after the study, the total number of identifications (isoforms) expected (true positives) from classes 1, 2, and 3 is 254 (the number of database accessions reported within these isoforms is not analyzed). If too many isoforms are reported for any of the clusters, they are counted as false positives.
We searched the iPRG data set (Mascot generic format files) using Mascot version 2.3 (Matrix Science, UK), OMSSA version 2.1.9 (16), and X!Tandem (version tandem-win-11–12-01–1) (24) with the following parameters (following the iPRG guidelines): precursor tolerance, 0.9 Da; fragment tolerance, 0.6 Da; fixed modifications, methyl methanethiosulfonate on cysteine and iTRAQ4plex labeling on lysine and N termini; variable modifications, acetylation of protein N termini, Gln-> pyro-Glu on N-terminal asparagine, and oxidation of methionine; one missed cleavage allowed. Searches were performed against the database constructed specifically by iPRG 2008 (iPRG2008_FASTA_fixed_w_forw+rev_Decoys, downloaded from the Association of Biomolecular Resource Facilities website). Results were post-processed in the mzidLibrary, which includes the ProteoGrouper algorithm, using the pipeline described below.
mzidLibrary Pipeline Constructed for Processing the iPRG Data Set
To demonstrate the use of the mzidLibrary and benchmark the ProteoGrouper algorithm, we constructed a pipeline of routines using a batch file to access the command-line mode (Fig. 1). As described above, spectra files were searched in Mascot, X!Tandem, and OMSSA. Results were exported from Mascot in mzIdentML format (using an in-house adaptation of the mascot_dat2.pl script, as full mzIdentML version 1.1 export is provided only for Mascot 2.4). Results from OMSSA (OMX format) and X!Tandem (XML format) were obtained and converted to mzIdentML using the Omssa2mzid and Tandem2mzid converters. Next, the three results mzIdentML files were combined into a single mzIdentML file and re-scored using the CombineSearchEngines routine.
Fig. 1.
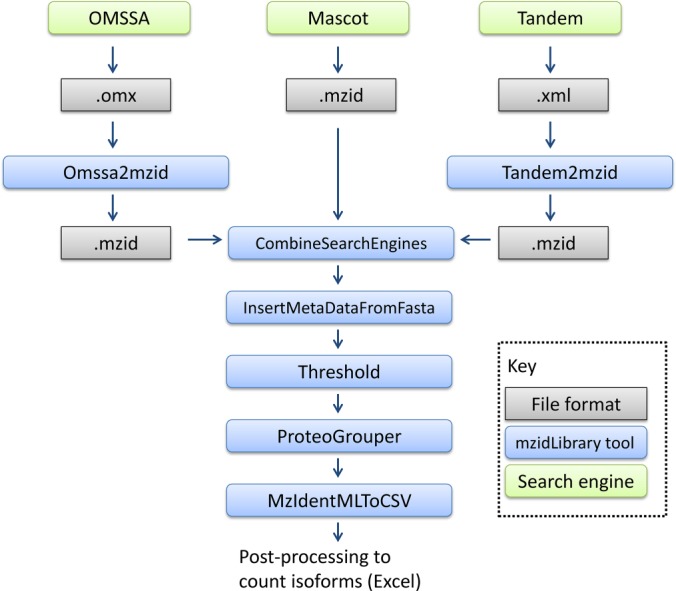
The pipeline of mzidLibrary tools constructed for testing different routines and performing benchmarking using the iPRG 2008 data set.
The mzidLibrary contains a routine called InsertMetaDataFromFasta for retrieving protein sequences and protein description lines from a FASTA file and inserting these details into <DBSequence> elements (modeling protein database entries in mzIdentML), in case search engines do not natively export this level of detail. We created an artificial FASTA file containing the assigned iPRG cluster identifiers for database entries and called this routine for inserting these additional identifiers into the file the “description line” for each classified protein to simplify downstream post-processing. Next, we used the Threshold routine to set the “passThreshold” attribute in the mzIdentML file to “true” for any PSMs (modeled in a structure called <SpectrumIdentificationItem> in mzIdentML) with a combined FDRScore ≤ 0.01. For details of this score, see Ref. 19, which reports identifications produced from different search engines with FDR ≅ 0.01 across the final set.
The ProteoGrouper was subsequently run with the following parameters: “requireSIIsToPassThreshold:true” (only PSMs passing the threshold were used for inference), cvAccForSIIScore: “MS:1002125” (this is the accession of the “combined FDRScore” term, which is used for protein-level scoring), and “logTransScore:true”. The mzIdentML output file from the ProteoGrouper was converted to various CSV formats (PSMs only, representative proteins only, protein groups) using Mzid2Csv. These outputs were manually analyzed and compared with the reported iPRG 2008 results in Microsoft Excel. Manual analysis consisted of ordering results by “PAG:score” and selecting proteins with protein FDR = 0%, followed by the use of a PivotTable to count the number of accessions mapped to each cluster identifier.
We also performed the same analysis on the Mascot results only, using the following workflow: FalseDiscoveryRate to calculate “FDRScore” (instead of “combined FDRScore” as used for multiple search engines above; PSI-MS CV accession “MS:1001874”); Threshold to set “passThreshold” = true for FDRScores ≤ 0.01; ProteoGrouper (“requireSIIsToPassThreshold:true”, cvAccForSIIScore: “MS:1001874”, “logTransScore:true”); and Mzid2Csv and post-processing as described above.
The mzIdentML outputs were also visualized in the ProteoIDViewer and checked with the mzidValidator to ensure that they were structurally and semantically valid.
mzidValidator
The mzidValidator was developed as a basic Java graphical user interface, building an extension of the PSI validator framework (25). For the Minimum Information about a Proteomics Experiment (MIAPE) Mass Spectrometry Informatics validation, a new mapping rule file was created containing additional and more restrictive rules, requiring CV terms to be presented in different elements of the file than in the standard mzIdentML mapping file. Both semantic and MIAPE mapping rules files are available in the final downloadable package for advanced users who might want to test some extra validations in their mzIdentML files by modifying them.
RESULTS
The ProteoIDViewer
The ProteoIDViewer was developed for use by laboratory scientists, and thus bioinformatics support is not required in order for a user to get started. The viewer can be simply downloaded and installed with no complex setup procedures. The ProteoIDViewer aims at providing intuitive and useful views of peptides/proteins and the search metadata and supports various approaches, including the use of multiple search engines. The viewer provides several different methods for visualizing data (Fig. 2).
Fig. 2.

Screenshots of ProteoIDViewer. A, the “Protein View” panel with protein groups, individual accessions, and further details about each protein identified. B, the “Peptide View” panel, showing a listing of all PSMs, including spectrum visualization and annotation of fragment products identified (if present in the file. C, the “Global Statistics” panel, showing graphs plotting statistics estimated from decoy database searching.
Protein View: all protein groups identified, as well as individual proteins within those groups and links to the supporting peptide evidence for each identification.
Spectrum View: ranked identifications for each input spectrum, including peptide fragmentation data.
Peptide View: all peptides identified, as well as the possible protein mappings for each peptide.
Global View: global statistics include the total count of PSMs and proteins identified, statistics such as FDR if a decoy database search has been performed, and graphs showing various statistics.
Protein Database View: all protein sequence records contained within the file.
Protocols View: the search parameters for the peptide and protein identification stages.
The ProteoIDViewer uses a mandatory mzIdentML file containing the identification results and an optional file containing the spectra that were searched (mzML and Mascot generic format are currently supported). The viewer is also able to load other identification file formats (using mzidLibrary's parsers) currently supporting OMSSA OMX and X!Tandem XML files. These files are converted automatically to mzIdentML format and are loaded in the viewer. The first tab (Fig. 2A) is the “Protein View” tab that contains all protein groups and the individual proteins identified within those groups. In addition, it displays any additional information about the protein sequences from the source database, such as the protein description and protein sequence, assuming these are present in mzIdentML (apart from the source database accession, these attributes are all optional). The Protein View provides a panel containing all the PSMs that were used to infer the presence of a given protein.
The second tab is the Spectrum View, which contains the list of all spectra that were searched and all ranked PSMs. When the user clicks on a spectrum, a separate panel displays the spectrum (Fig. 2B). If a user clicks one of the PSMs made by searching the spectrum, the fragmentation products identified by the search engine will be annotated on top of the spectrum (assuming these optional details are present in the mzIdentML file). We imagine that this feature will be particularly useful for researchers aiming to fulfill journal reporting guidelines, such as those of Molecular and Cellular Proteomics, which require that protein identifications based on a single PSM be supported by a graphical display of the fragmentation products. The third tab is “Peptide Summary View”, which is similar to the “Spectrum View” tab but provides entry into the data via a list of all peptides identified, rather than by spectra. The “Protein Database View” tab contains all database protein sequences that are stored in the mzIdentML file. This tab contains the sequence identifier, the accession, the sequence, and the protein description (assuming these details are present in the file). The “Global Statistics” tab provides summaries of the data contained within the file, such as the total number of spectra for which a peptide identification is provided, the total number of PSMs, the total PSMs above and below the threshold defined in the file, the total number of protein groups, total proteins, and so on. The view also provides access to routines in the mzidLibrary for estimating the count of false positive and true positive PSMs and the FDR, if a decoy database search has been performed. Decoy identifications are identified using the isDecoy attribute in the file or using a regular expression set in the viewer, in case the mzIdentML export software did not correctly identify decoys. The tab produces three graphs: FDR versus native score (e.g. −log(e-value)), true positives versus false positives, and true positives versus q-value (Fig. 2C). The last tab is “Protocols View,” which displays the parameters used within the peptide identification and protein identification stages.
The ProteoIDViewer has been tested with file sizes of up to 200 MB, and it can accept both native mzIdentML and gzip compressed files (which is the PSI-recommended method for storing and transferring mzIdentML files).
The mzidLibrary
The mzidLibrary has a set of functions that can be used to manipulate mzIdentML files so that proteome informatics developers can easily build mzIdentML support into their own software. We also have created a simple graphical user interface, so the software can be used by lab scientists directly (supplementary File S4, supplementary Fig. S1). At present the library contains a number of routines that may be useful in different contexts, as summarized in Table I. The library also contains a number of smaller or developing projects that do not yet form part of the public release. We also expect that other developers who use the software might wish to contribute new routines to the library. The functionality in the current release is described below.
Table I. The routines contained within the mzidLibrary release reported here. All routines must specify the input and output files and have an additional option for compressing the output using the gzip protocol. Most routines have an additional “verbose” mode for debugging output or providing more detail about the run.
| Tool | Description | Parameters |
|---|---|---|
| InsertMetaData-FromFasta | Extracts protein sequences and description lines from a FASTA file and inserts them into an mzIdentML file | Location of FASTA file |
| Regular expression (regex) to split the accession from the description line | ||
| FalseDiscovery-Rate | Calculates FDR, q-value, and FDRScore (19) when a decoy database search has been done | Optional regex for decoy hits (if the isDecoy attribute is not set in the file) |
| Ratio of targets to decoys | ||
| Accession of CV term for score in file to use for ordering | ||
| Scores are ordered low to high | ||
| CombineSearch-Engines | Re-scores and combines PSMs from two or three search engines to produce a single output (19) | Regex for decoy hits (as above) |
| Ratio of targets to decoys | ||
| - Locations of input files and identifiers for the type of search engine | ||
| Threshold | Sets the passThreshold attribute on PSMs or proteins, according to the entered value; optionally, it can remove PSMs that fall below the threshold (see main text) | Threshold is for PSMs or proteins |
| Score type to be used for setting the threshold | ||
| Accession of CV term for score in the file to use for thresholding | ||
| Scores are ordered low to high | ||
| Omssa2mzid | Converts OMSSA OMX format to mzIdentML | Include fragmentation products |
| Regex for decoy hits | ||
| Csv2mzid | Converts results in OMSSA CSV format to mzIdentML | Regex for decoy hits |
| Accession of CV term for score in file to use for ordering (in case the CSV file is not from OMSSA) | ||
| Location of the file containing search metadata | ||
| Tandem2mzid | Converts Tandem XML format to mzIdentML | Include fragmentation products |
| Regex for decoy hits | ||
| Whether identifiers start from 0 (mzML file searched) or from 1 (other peak lists were searched) | ||
| Options for capturing additional metadata difficult to parse from file (database file format, peak list file format) | ||
| Mzid2Csv | Exports from mzIdentML format to various CSV formats | Type of export to perform: one row per PSM (no protein information), proteins with details of all PSMs, one row per protein or one row per protein group |
| AddEmpaiToMzid | Calculates pseudo-quantitative abundance values, based on spectral counting (see main text) | Location of FASTA file |
| Regular expression to split the accession from the description line | ||
| ProteoGrouper | Performs protein inference from the PSMs (see main text) | Use only PSMs with ‘passThreshold’ = true |
| Accession of CV term for score in file to use for ordering | ||
| Scores are ordered low to high | ||
| If the score should be log transformed |
Threshold
Both the list of PSMs and the protein list within mzIdentML have the capability to include identifications deemed to have passed a given threshold and those that have not (via the attribute “passThreshold” = “true | false”). The inclusion of PSMs or proteins below the threshold allows the calculation of global statistics and provides the option for consumers of the files to produce different sets of different results using alternative tools. Additionally, many protein inference algorithms may include weak PSMs in protein results, even though the PSM would not typically pass a PSM quality threshold. The code traverses the appropriate PSM or protein list, setting the “passThreshold” attribute based on the parameters entered. Optionally, the routine can also remove PSMs (and associated data) that fall below the threshold in order to reduce the file size. The setting of a threshold on the PSM list should be used with care, however, because if a protein list is also already present in the file, changes to the PSM list will not be reflected in the proteins contained in the protein list (i.e. Threshold does not perform a new protein inference process).
Omssa2mzid, Csv2mzid, and Tandem2mzid
One of the anticipated benefits of mzIdentML is that bioinformatics developers should be able to focus on new algorithm development rather than file format conversions. Both OMSSA and X!Tandem are popular free search engines that do not, at this stage, support native export of mzIdentML. With this in mind, we have developed software for converting the native output of these search engines into mzIdentML. Both packages create lists of PSMs but do not perform protein inference, so the converters also create only PSM lists and not protein lists in mzIdentML. However, as described below, ProteoGrouper can be applied to create protein lists. Omssa2mzid accepts the OMSSA OMX format, and Csv2mzid accepts the OMSSA CSV format (requiring an additional, manually created file containing search metadata). In order for the conversion from the OMSSA OMX format to be successful, the search must have been performed in OMSSA with the following option “-w” (include spectra and search parameters in search results). This option inserts the search parameters into the OMX file that are required for conversion to valid mzIdentML. The Tandem2mzid module converts the native XML output of X!Tandem to mzIdentML. In order for this conversion to be successful, the user also must have set the X!Tandem option “output, parameters = yes” when performing the search, again so that the metadata can be correctly inserted in the resulting mzIdentML file.
Mzid2Csv
In order to support users wishing to import their results easily into spreadsheet or statistical software, we developed a routine for flattening the XML structure into a CSV file. Additionally, some bioinformatics developers may want to post-process results from mzIdentML without having to read the XML structure, particularly programmers not proficient in Java (jmzIdentML) or C++ (ProteoWizard). The software can export four views on the same file: a list of PSMs only (one row per PSM), a list of protein groups plus all supporting PSMs (one row per PSM), a list of all identified proteins (one row per protein), and a list of identified protein groups showing the group representative only (one row per protein group).
AddEmpaiToMzid
The emPAI protocol generates pseudo-quantitative abundance values for proteins, based on a scheme to normalize for the number of peptides in a given protein (16). An emPAI value is calculated as 10observed/observable − 1, where “observed” is the number of unique parent ions identified for a given protein (i.e. the count of distinct peptides identified, allowing for different charge states and modifications) and “observable” is the number of peptide sequences in a theoretical digest that could be analyzed by the instrument. Implementations differ in terms of how the observed and observable counts are generated. Our implementation uses the following rules, following the source protocol as closely as possible: (i) the observed count is generated for a given protein from all unique calculated (theoretical) m/z values of PSMs that have ‘passThreshold’ = “true” (thus a modified and unmodified peptide is counted twice), and any peptide that can be matched to multiple proteins is counted for every protein; (ii) the observable count is generated by first calculating the lowest and highest Mr (relative mass) values from PSMs in the entire file; and (iii) for each protein in the FASTA database, an in silico digest is performed assuming 100% tryptic digestion, and the number of peptides is counted with Mr between the lowest and highest Mr values.
To demonstrate this functionality in the mzidLibrary, the results for the iPRG 2008 data set analyzed by Mascot are presented in supplementary File S1, exported directly from Mascot as a CSV (using an edited version of the mascot_dat2.pl script to export observed and observable values as calculated by Mascot) and analyzed by AddEmpaiToMzid. The emPAI values calculated differ between the two implementations; for example, the emPAI value for the top hit Cntm.P00760 TRY1_BOVIN is 2.1 in the native Mascot output and 4.62 from AddEmpaiToMzid. We believe that our implementation produces results closer to the intended original protocol. For example, both Mascot and AddEmpaiToMzid calculate “observed” for Cntm.P00760 TRY1_BOVIN as 9, but the calculations differ greatly for “observable” (12 (AddEmpaiToMzid) versus 18.32 (Mascot native)). The Mascot implementation uses a rule based on protein mass and amino acid composition to estimate observable values. As shown in supplementary File S1 (sheet “TRY1 observable analysis”), for Cntm.P00760 TRY1_BOVIN, Mascot produced an inaccurate estimate for observable peptides (18.32), as there are only 17 tryptic peptides in total in this protein, of which 12 have the realistic potential to be analyzed via MS. For this protein, this means that Mascot is underestimating the emPAI value by more than 2-fold. In some proteins, the native Mascot implementation and our implementation also differed in the “observed” count, because we take only those PSMs with ‘passThreshold’ = “true”, whereas Mascot uses PSMs above the “Mascot homology threshold” for inclusion. In this implementation, ‘passThreshold’ = “true” is based on the more stringent “Mascot identity threshold” although, as noted above, the ‘passThreshold’ attribute can be altered using the Threshold routine.
ProteoGrouper
The so-called protein inference problem in proteomics arises because the link from a peptide identification to the source protein is lost in most workflows. Many peptides could be assigned to more than one protein, and thus software is required to determine the optimal mapping and assign a set of proteins to groups where ambiguity remains or where a protein contains the same peptide identifications as another protein (or a subset thereof). A variety of software is available for performing protein inference, either embedded in commercial software or open source (e.g. 26–30). However, there is currently limited or no support in these tools for mzIdentML. As a result, we have encoded a protein inference algorithm within ProteoGrouper allowing lists of PSMs in mzIdentML to be converted into protein groups/lists, for example, following a search with OMSSA, X!Tandem, or MSGF+, which do not natively support protein inference.
In order to benchmark the ProteoGrouper, we post-processed the combined results from Mascot, OMSSA, and X!Tandem and followed the iPRG scoring scheme for true and false positives. Using the parameters stated in “Experimental Procedures”, the ProteoGrouper made 237 true positive protein isoform identifications and only two false positive identifications (supplementary File S2). We have plotted these results alongside those obtained by the research group members and study participants in the original study (Fig. 3). The results in Fig. 3 demonstrate that the ProteoGrouper is performing high-quality protein inference (almost all detectable isoforms are observed) without incurring substantial false positives. It should also be noted that this is an imperfect benchmark, as it is not possible to differentiate between the quality of the PSMs and the protein inference algorithm. In the original study, the best results were obtained with Paragon and ProGroup (AB Sciex), which may be optimized to work with data that were generated on an AB Sciex instrument. To test the quality of the inference alone, we re-ran the ProteoGrouper over the PSMs exported from Mascot only, resulting in 218 true positives and 1 false positive (supplementary File S3). The ProteoGrouper results compare favorably with the results from a recently published protein grouping algorithm from Matrix Science that used the same data set and search parameters (30), for which the authors reported 219 true positives and 4 false positives (annotated for comparison in Fig. 3). The complete set of results, including all input, intermediate, and output files, is available online in the mzIdentML Library.
Fig. 3.
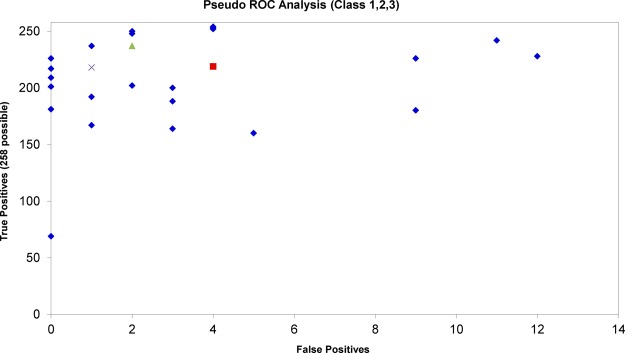
A pseudo–receiver operating characteristic (ROC) plot showing the results from ProteoGrouper for post-processing the iPRG 2008 data sets searched in Mascot, OMSSA, and X!Tandem combined (triangles) and in Mascot alone (X) compared with results from the other research group members and study participants. The results produced by Mascot with a new “protein family” algorithm (30) are represented by the square for comparison.
The ProteoGrouper has the advantage of being able to accept PSMs from any search engine result that can be converted to mzIdentML, allowing it to be used with well-established software or search engines under development to produce results suitable for publication. Our benchmarking demonstrates that it is able to perform high-quality protein inference without incurring substantial false positives or reporting protein isoforms with no independent evidence.
The mzidValidator
In order to ensure the consistency of the information contained in mzIdentML files, a new tool has been developed as an extension of the original PSI semantic validator framework (25). The original framework was designed as a generic mechanism not only for checking the XML syntax, but also to enforce rules regarding the use of valid CV terms—for example, it could be used to verify that the terms exist in the resource and that they are used in the correct location of a document. In addition, the framework allows the definition of “object rules” consisting of Java classes that check certain elements in the file in a more versatile way.
Here we report the development of the implementation of the validation framework for mzIdentML, called the mzidValidator (supplementary File S4, supplementary Fig. S2). The mzidValidator allows the user to select an mzIdentML file and run validation—either standard “semantic” validation or more stringent MIAPE validation. The basic semantic validation checks whether files are valid against the schema and contain the correct CV terms only. After validation has been performed, a set of messages are reported to the user about any errors detected in the file. The MIAPE validation option is used to check the compliance of the files with the MIAPE guidelines (31) and, specifically for mzIdentML, the MIAPE Mass Spectrometry Informatics module (32).
To tackle mzIdentML MIAPE Mass Spectrometry Informatics validation, new CV terms were added to the PSI-MS CV, new rules were defined in a new mapping file, and some additional object rules were required. We have thus produced a stand-alone tool mainly aimed at developers of mzIdentML exporters that enables semantic or MIAPE validation, depending on the user selection. As all code is open source, mzIdentML validation can be easily included in third-party tools, including commercial packages. The current version of the mzidValidator can be downloaded from the PSI group repository.
DISCUSSION
Data standards produced by PSI are mostly represented in XML format. XML is generally preferred by PSI because it is an industry standard specification, there are many high-quality tools available for manipulating XML files, and the structure of the format can be formally defined with an XML Schema Definition file. However, XML formats are not straightforward for developers to work with immediately and cannot be easily visualized by end users without bespoke tools. We have developed the ProteoIDViewer to help end users and developers start working with files in mzIdentML format. The viewer has the advantage of being able to work with data from any search engine capable of exporting data into the mzIdentML format. As there is now a route for direct export or format conversion from almost any search engine, the ProteoIDViewer has the potential to function as a universal viewer for peptide and protein identification data.
The data analysis in a typical proteomics workflow involves many steps after a search engine has performed the core function of making PSMs. We have added routines covering several common post-processing steps to the mzidLibrary, so that search engines that do not offer these capabilities can be used to produce “final” lists of proteins suitable for publication. We will continue to add new routines to the library, and we encourage other developers to contribute code as well. Lastly, in order to cope with an evolving vocabulary of terms, the PSI standards use an additional mapping framework to ensure that valid terms are provided within files. As a result, checking that files are correct is not simply a matter of running a standard XML Schema validator available in a variety of tools. We have developed the mzidValidator to assist developers as they are building mzIdentML export capability to ensure that their files are valid, and to help end users check that files are correct if they encounter files that do not function in certain tools.
We anticipate that our tools will make it simpler for bioinformatics groups to build mzIdentML support into their tools and improve overall adoption of the new standard. The tools should also assist proteomics researchers in comparing outputs from a variety of software producing peptide and protein identifications, and thus enable benchmarking of different approaches, which at present is difficult to achieve.
Supplementary Material
Acknowledgments
We thank the iPRG for the provision of high-quality data sets for benchmarking purposes; these resources are vital for supporting tool development. We also thank Lennart Martens and his group for providing various tools in the open source domain, which we have re-used in our tools.
Footnotes
* This work was funded by BBSRC grants to A.R.J. (BB/G010781/1, BB/H024654/1, BB/K004123/1). J.A.V. and F.R. are funded by the Wellcome Trust (Grant No. WT085949MA). J.A.V. is also funded by the EU FP7 project “ProteomeXchange” (Grant No. 260558). P.L. was funded by grants from the Netherlands Proteomics Centre (NPC) and the Netherlands BioInformatics Centre (NBIC), both part of the Netherlands Genomics Initiative (NGI).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- API
- application programming interface
- CSV
- comma-separated value
- CV
- controlled vocabulary
- emPAI
- exponentially modified Protein Abundance Index
- FDR
- false discovery rate
- iPRG
- Proteome Informatics Research Group
- MIAPE
- Minimum Information about a Proteomics Experiment
- MS
- mass spectrometry
- PDH
- protein detection hypothesis
- PSI
- Proteomics Standards Initiative
- PSM
- peptide spectrum match
- RG
- Research Group
- XML
- Extensible Markup Language.
REFERENCES
- 1. Jones A. R., Eisenacher M., Mayer G., Kohlbacher O., Siepen J., Hubbard S., Selley J., Searle B., Shofstahl J., Seymour S., Julian R., Binz P.-A., Deutsch E. W., Hermjakob H., Reisinger F., Griss J., Vizcaino J. A., Chambers M., Pizarro A., Creasy D. (2012) The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics 11, M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martens L., Chambers M., Sturm M., Kessner D., Levander F., Shofstahl J., Tang W. H., Römpp A., Neumann S., Pizarro A. D., Montecchi-Palazzi L., Tasman N., Coleman M., Reisinger F., Souda P., Hermjakob H., Binz P.-A., Deutsch E. W. (2011) mzML—a community standard for mass spectrometry data. Mol. Cell. Proteomics 10, R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Creasy D. M., Cottrell J. S. (2004) Unimod: protein modifications for mass spectrometry. Proteomics 4, 1534–1536 [DOI] [PubMed] [Google Scholar]
- 4. Montecchi-Palazzi L., Beavis R., Binz P.-A., Chalkley R. J., Cottrell J., Creasy D., Shofstahl J., Seymour S. L., Garavelli J. S. (2008) The PSI-MOD community standard for representation of protein modification data. Nat. Biotechnol. 26, 864–866 [DOI] [PubMed] [Google Scholar]
- 5. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 6. Mayer G., Montecchi-Palazzi L., Ovelleiro D., Jones A. R., Binz P.-A., Deutsch E. W., Chambers M., Kallhardt M., Levander F., Shofstahl J., Orchard S., Antonio Vizcaíno J., Hermjakob H., Stephan C., Meyer H. E., Eisenacher M. (2013) The HUPO proteomics standards initiative—mass spectrometry controlled vocabulary. Database (Oxford) 2013, 10.1093/database/bat009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 8. Colinge J., Masselot A., Giron M., Dessingy T., Magnin J. (2003) OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 3, 1454–1463 [DOI] [PubMed] [Google Scholar]
- 9. Searle B. C. (2010) Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 10, 1265–1269 [DOI] [PubMed] [Google Scholar]
- 10. Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 11. Tabb D. L., Fernando C. G., Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sturm M., Bertsch A., Gropl C., Hildebrandt A., Hussong R., Lange E., Pfeifer N., Schulz-Trieglaff O., Zerck A., Reinert K., Kohlbacher O. (2008) OpenMS—an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reisinger F., Krishna R., Ghali F., Ríos D., Hermjakob H., Antonio Vizcaíno J., Jones A. R. (2012) jmzIdentML API: a Java interface to the mzIdentML standard for peptide and protein identification data. Proteomics 12, 790–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 17. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 18. Barsnes H., Vaudel M., Colaert N., Helsens K., Sickmann A., Berven F. S., Martens L. (2011) compomics-utilities: an open-source Java library for computational proteomics. BMC Bioinformatics 12, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jones A. R., Siepen J. A., Hubbard S. J., Paton N. W. (2009) Improving sensitivity in proteome studies by analysis of false discovery rates for multiple search engines. Proteomics 9, 1220–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 21. Barsnes H., Huber S., Sickmann A., Eidhammer I., Martens L. (2009) OMSSA parser: an open-source library to parse and extract data from OMSSA MS/MS search results. Proteomics 9, 3772–3774 [DOI] [PubMed] [Google Scholar]
- 22. Muth T., Vaudel M., Barsnes H., Martens L., Sickmann A. (2010) XTandem parser: an open-source library to parse and analyse X!Tandem MS/MS search results. Proteomics 10, 1522–1524 [DOI] [PubMed] [Google Scholar]
- 23. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 24. Fenyo D., Beavis R. C. (2003) A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 75, 768–774 [DOI] [PubMed] [Google Scholar]
- 25. Montecchi-Palazzi L., Kerrien S., Reisinger F., Aranda B., Jones A. R., Martens L., Hermjakob H. (2009) The PSI semantic validator: a framework to check MIAPE compliance of proteomics data. Proteomics 9, 5112–5119 [DOI] [PubMed] [Google Scholar]
- 26. Tabb D. L., McDonald W. H., Yates J. R. (2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 28. Zhang B., Chambers M. C., Tabb D. L. (2007) Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Slotta D. J., McFarland M. A., Markey S. P. (2010) MassSieve: panning MS/MS peptide data for proteins. Proteomics 10, 3035–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koskinen V. R., Emery P. A., Creasy D. M., Cottrell J. S. (2011) Hierarchical clustering of shotgun proteomics data. Mol. Cell. Proteomics 10, M110.003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taylor C. F., Paton N. W., Lilley K. S., Binz P. A., Julian R. K., Jr., Jones A. R., Zhu W., Apweiler R., Aebersold R., Deutsch E. W., Dunn M. J., Heck A. J., Leitner A., Macht M., Mann M., Martens L., Neubert T. A., Patterson S. D., Ping P., Seymour S. L., Souda P., Tsugita A., Vandekerckhove J., Vondriska T. M., Whitelegge J. P., Wilkins M. R., Xenarios I., Yates J. R., 3rd, Hermjakob H. (2007) The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 25, 887–893 [DOI] [PubMed] [Google Scholar]
- 32. Binz P. A., Barkovich R., Beavis R. C., Creasy D., Horn D. M., Julian R. K., Jr., Seymour S. L., Taylor C. F., Vandenbrouck Y. (2008) Guidelines for reporting the use of mass spectrometry informatics in proteomics. Nat. Biotechnol. 26, 862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.