

Abstract
The molecular responses of macrophages to copper-based nanoparticles have been investigated via a combination of proteomic and biochemical approaches, using the RAW264.7 cell line as a model. Both metallic copper and copper oxide nanoparticles have been tested, with copper ion and zirconium oxide nanoparticles used as controls. Proteomic analysis highlighted changes in proteins implicated in oxidative stress responses (superoxide dismutases and peroxiredoxins), glutathione biosynthesis, the actomyosin cytoskeleton, and mitochondrial proteins (especially oxidative phosphorylation complex subunits). Validation studies employing functional analyses showed that the increases in glutathione biosynthesis and in mitochondrial complexes observed in the proteomic screen were critical to cell survival upon stress with copper-based nanoparticles; pharmacological inhibition of these two pathways enhanced cell vulnerability to copper-based nanoparticles, but not to copper ions. Furthermore, functional analyses using primary macrophages derived from bone marrow showed a decrease in reduced glutathione levels, a decrease in the mitochondrial transmembrane potential, and inhibition of phagocytosis and of lipopolysaccharide-induced nitric oxide production. However, only a fraction of these effects could be obtained with copper ions. In conclusion, this study showed that macrophage functions are significantly altered by copper-based nanoparticles. Also highlighted are the cellular pathways modulated by cells for survival and the exemplified cross-toxicities that can occur between copper-based nanoparticles and pharmacological agents.
Manufactured nanoparticles are more and more widely used in more and more consumer products, ranging from personal care products to tires and concrete. Among the nanoparticles, metals and metal oxides represent an important part of the total production and are used in water treatment, as antibacterials, in antifouling paints, and in microelectronics. These varied uses in turn pose the problem of the toxicological evaluation of these nanoparticles (1, 2), and especially of the long-term effects that often come not from simple cell mortality but from altered cellular functions.
Macrophages are one of the cell types that deserve special attention in toxicology, because of the variety of their functions. Altered cytokine production can lead to adverse long-term effects, as documented, for example, in the case of asbestos (3). Other dysfunctions of the innate immune system can lead to deregulation of the immune responses and to severe adverse effects, such as a higher incidence of tumors (4).
It is therefore not surprising that the immunotoxicology of nanoparticles is a developing field (5–7), and several studies have been devoted to macrophages' response to nanoparticles. However, most of these studies have been limited to the effect of nanoparticles on cell viability and on cytokine production (e.g. 8–11), although some also studied oxidative stress (12–14) and sometimes other functional parameters (15–17). Very few studies have used the analytical power of proteomics to go deeper into the mechanisms of the response to nanoparticles or metals (reviewed in Ref. 18). A few exceptions are studies on, for example, carbon-based nanoparticles (19) and titanium dioxide (20, 21).
Most of the toxicological studies in this field have been focused on a few nanoparticles used either as health products, such as iron oxide (15, 17, 22), or in a variety of consumer products, such as silver (13, 14), silica (9, 12), and titanium dioxide (11, 16, 20, 21).
However, many other nanoparticles are being used more and more in industrial applications without extensive toxicological testing. Good examples are indium-tin oxide, used in electronic screens, which appears to be toxic (23), and the copper-based nanoparticles used in high-performance batteries (24), in water depollution (25), and as bactericides as a replacement for nano-silver. Copper and copper oxide induce a strong toxicity (26, 27), coupled with inflammation (28), oxidative stress (29), and genotoxicity (30), at least in epithelial cells.
In light of these various effects, we decided to use a combination of a proteomics approach and targeted approaches to address in more molecular detail the responses of macrophages to copper-based nanoparticles (i.e. both metallic copper and copper II oxide).
MATERIALS AND METHODS
Nanoparticles
Metallic copper and copper oxide nanoparticles (<50 nm) were purchased from Sigma-Aldrich (catalog numbers 684007 and 544868, respectively). They were dispersed in water as a 5.5% w/v suspension via sonication for 60 min in a cup-horn instrument (BioBlock Scientific, Strasbourg, France) under 5 °C thermostated water circulation. A one-tenth volume of 10% w/v PVP40 solution was added under sterile conditions, and the particles were coated for 1 h under constant agitation. The actual size of the particles was determined after dilution in water or in complete culture medium by means of dynamic light scattering using a Wyatt Dynapro Nanostar instrument or a Malvern HS 3000 instrument; the latter instrument was also used to determine the zeta potential.
The morphology of the samples was observed via SEM. A 200 mesh carbon grid was dipped in the nanoparticle suspensions and dried under air before imaging.
The amount of coating attached to the inorganic nanoparticles was evaluated based on weight loss (from about 10 mg of sample) after annealing under air using a thermogravimetric analysis device (Setaram, Caluire, France). The temperature cycle consisted of heating at a rate of 10 °C/min up to 600 °C followed by a dwelling time of 30 min and natural cooling.
Zirconium oxide nanoparticles (<100 nm) were purchased from Sigma-Aldrich as a 10% (w/v) dispersion in water (catalog number 643025). Prior to use, they were diluted by mixing one volume of dispersion with one volume of 2% w/v PVP40 for 1 h under constant agitation. The actual size of the final dispersion in complete culture medium was determined as for the copper nanoparticles.
Nanoparticle Dissolution in Culture Medium
Nanoparticles were added at 5, 10, or 20 μg/ml to complete culture medium (RPMI 1640 + 10% fetal bovine serum) in cell culture six-well plates containing 2 ml culture medium per well. In some experiments, conditioned medium (i.e. complete cell culture medium that had been in contact with the cells for at least 24 h) was used in place of fresh complete culture medium. Known concentrations of copper chloride were also added to complete culture medium in control wells and incubated under the same conditions. The plates were incubated for 24 h in a cell culture incubator at 37 °C and 5% CO2 atmosphere. The culture medium was recovered and centrifuged at 270,000 × g for 45 min to sediment the nanoparticles (31). The concentration of copper ions was then determined using the Zincon method (32). Briefly, 1 ml of supernatant was acidified with trichloroacetic acid (7.5% w/v final concentration) to precipitate proteins and release bound copper ions. This precipitation step was carried out for 30 min on ice. The precipitated proteins were eliminated via centrifugation (15,000 × g for 15 min), and the supernatants were collected. After neutralization by the addition of 0.3 volumes of 2 m Tris, Zincon was added (50 μm final concentration) and the copper concentration was determined via absorbance at 600 nm, using the copper chloride in medium points to build the standard curve.
Cell Culture
Cell Lines
The mouse macrophage cell line RAW264.7 was obtained from the European Cell Culture Collection (Salisbury, UK). The cells were cultured in RPMI 1640 medium + 10% fetal bovine serum. Cells were seeded every 2 days at 200,000 cells/ml and harvested at 1,000,000 cells/ml.
Primary Macrophages
Primary mouse macrophages were obtained from bone marrow essentially as described by Schleicher and Bogdan (33) and Tarakanova et al. (34). Briefly, bone marrow was aseptically collected from femurs and tibias of 4-week-old C57Bl6 mice that had been sacrificed by means of cervical dislocation. The marrow plugs were dissociated by repeated flushing in the culture medium (DMEM 65% v/v, L929 cell culture supernatant 20% v/v, fetal bovine serum 10% v/v, and horse serum 5% v/v) and plated at 4 million cells/ml on bacteriology culture dishes. An equal volume of medium was added after 4 days in culture. After 7 days of culture, the cells were scraped and replated at 1 million cells/ml in the same medium, except that the L929 supernatant was lowered to 10% v/v and DMEM was increased to 75% v/v. Mature macrophages were obtained after an additional 3 days and were viable and stable for up to 2 weeks, provided that the culture medium was renewed every 3 days.
Cell Treatment and Viability Assessment
Cells were treated with variable concentrations of nanoparticles added directly into the culture medium. After the cells had been in the presence of the nanoparticles or ions for 24 h, their viability was measured via trypan blue exclusion. For treatment with inhibitors, cells were pretreated for 6 h with 100 μm buthionine sulfoximine, 200 nm rotenone, or 200 nm antimycin A. Copper oxide nanoparticles (or copper chloride) were then added for a further 18 h before collection of the cells and viability measurements.
Neutral Red Uptake Assay
This assay was performed essentially as described in Ref. 35. Briefly, cells grown in 12-well plates and treated or not with nanoparticles were incubated for 1 h with 40 μg/ml neutral red (final concentration, added from a 100× stock solution in 50% ethanol water). At the end of the incubation period, the medium was discarded and the cell layer was rinsed twice with PBS1 (5 min per rinse). The PBS was removed, and 1 ml of elution solution (50% ethanol and 1% acetic acid in water) was added per well. The dye was eluted for 15 min under constant agitation, and the dye concentration was read spectrophotometrically at 540 nm. The results were expressed as a percentage of the control cells (untreated with nanoparticles).
Phagocytosis Activity Measurement
The phagocytic activity was measured using fluorescent latex beads (1-μm diameter, green-labeled, catalog number L4655 from Sigma), essentially as described in Ref. 36. Briefly, the beads were pre-incubated at a final concentration of 55 mg/ml for 30 min at 37 °C in a 1:1 (v/v) PBS/horse serum solution. They were then incubated with the cells (5.5 mg/l final concentration) for 2 h at 37 °C. The cells were then harvested, washed twice with cold PBS, and incubated for 5 min on ice with a 0.25 m d-sorbitol solution in order to remove beads adhering to the cell surface. After another wash with cold PBS, cells were analyzed via flow cytometry on a FACScalibur instrument (BD Biosciences) using fluorescein excitation/emission filters and the live cell selection as described above.
Transmission Electron Microscopy
For transmission electron microscopy, fixation was initiated by adding an equal volume of fixative solution, previously warmed to 37 °C, to the cells after treatment with copper oxide nanoparticles for 24 h. The fixative solution contained 5% glutaraldehyde (Electron Microscopy Sciences, Euromedex, Strasbourg, France) in a 0.1 m sodium cacodylate buffer (Merck, Darmstadt, Germany) (305 mOsm, pH 7.3). After 10 min, the mixture was centrifuged, the supernatant was discarded, and the pellet was resuspended in the fixative solution containing 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer for 45 min at room temperature. The cells were then washed in 0.1 m sodium cacodylate buffer, postfixed for 1 h at 4 °C with 1% osmium tetroxide (Merck) in the same buffer, and stained for 1 h at 4 °C in 4% uranyl acetate. After further washing in distilled water, the cells were dehydrated in graded (50%, 70%, 80%, 95%, and 100%) ethanol solutions, incubated for 1 h in Epon (Electron Microscopy Sciences):absolute alcohol (1:1, v/v) and then overnight in Epon, and embedded in Epon. Ultrathin sections stained with lead citrate (Leica, Bron, France) and uranyl acetate (Merck) were examined under a Philips CM 120 BioTwin electron microscope (120 kV).
Intracellular Glutathione Measurements
Intracellular glutathione levels were analyzed via the monochlorobimane technique (37), with some modifications (38). Briefly, the cells were harvested, centrifuged for 5 min, and then labeled for 5 min with a 50 μm monochlorobimane solution diluted in PBS at 37 °C. The reaction was stopped for 5 min on ice in the dark, and the cells were then washed twice with cold PBS. Finally, the cells were analyzed via flow cytometry on a MoFlo instrument (Beckman Coulter) using a UV laser for excitation and reading the emission at 450 nm. The analysis was restricted to live cells only.
Mitochondrial Transmembrane Potential Assessment
The mitochondrial transmembrane potential was assessed based on Rhodamine 123 uptake (39). Briefly, cells were treated with 10 μg/ml Rhodamine 123 in complete culture medium for 30 min at 37 °C. Cells were then harvested, rinsed twice in cold PBS, and analyzed via flow cytometry on a FACScalibur instrument (BD Biosciences), using fluorescein excitation/emission filters. First the live cells were selected on the basis of size and granularity, and then the fluorescence of these live cells was measured using the fluorescein channel.
NO Production
The cells were grown to confluence in a six-well plate and then treated or not with copper nanoparticles, copper oxide nanoparticles, or copper chloride. After 24 h of treatment, half of the wells were treated with 1 μg/ml LPS (from salmonella), and arginine monohydrochloride was added to all the wells (5 mm final concentration) to give a high concentration of substrate for the nitric oxide synthases. After 24 h of incubation, the cell culture medium was recovered and centrifuged at 10,000 × g for 10 min to remove cells and debris, and the nitrite concentration was read at 540 nm after the addition of an equal volume of Griess reagent and incubation at room temperature for 30 min.
Proteomics
Two-dimensional Gel Electrophoresis
Sample Preparation
The cells were collected by scraping and then washed three times in PBS. The cells were then washed once in TSE buffer (Tris-HCl 10 mm pH 7.5, sucrose 0.25 m, EDTA 1 mm), and the volume of the cell pellet was estimated. The pellet was resuspended in its own volume of TSE buffer. Then 4 volumes (respective to the cell suspension just prepared) of concentrated lysis buffer (urea 8.75 m, thiourea 2.5 m, CHAPS 5% w/v, tris(carboxyethyl)phospine-HCl 6.25 mm, spermine base 12.5 mm) were added and the solution was left to extract at room temperature for 1 h. The nucleic acids were then pelleted via ultracentrifugation (270,000 × g at room temperature for 1 h), and the protein concentration in the supernatant was determined via a dye-binding assay (40). Carrier ampholytes (Pharmalytes pH 3–10) were added to a final concentration of 0.4% (w/v), and the samples were kept frozen at −20 °C until use.
Isoelectric Focusing
Homemade 160-mm-long 4–8 linear pH gradient gels (41) were cast according to published procedures (42). Strips were cut to 4-mm widths and rehydrated overnight with the sample, diluted in a final volume of 0.6 ml of rehydration solution (7 m urea, 2 m thiourea, 4% CHAPS, 0.4% carrier ampholytes (Pharmalytes 3–10)) and 100 mm dithiodiethanol (43, 44).
The strips were then placed in a Multiphor plate (GE Healthcare), and isoelectric focusing was carried out with the following electrical parameters: 100 V for 1 h, then 300 V for 3 h, then 1000 V for 1 h, then 3400 V up to 60 to 70 kVh. After isoelectric focusing, the gels were equilibrated for 20 min in Tris 125 mm, HCl 100 mm, SDS 2.5%, glycerol 30%, and urea 6 m (45). They were then transferred on top of the SDS gels and sealed in place with 1% agarose dissolved in Tris 125 mm, HCl 100 mm, SDS 0.4%, and 0.005% (w/v) bromphenol blue.
SDS Electrophoresis and Protein Detection
Ten-percent gels (160 × 200 × 1.5 mm) were used for protein separation. The Tris taurine buffer system (46) was used and operated at an ionic strength of 0.1 and a pH of 7.9. The final gel composition was thus Tris 180 mm, HCl 100 mm, acrylamide 10% (w/v), and bisacrylamide 0.27%. The upper electrode buffer was Tris 50 mm, taurine 200 mm, SDS 0.1%. The lower electrode buffer was Tris 50 mm, glycine 200 mm, SDS 0.1%. The gels were run at 25 V for 1 h and then 12.5 W per gel until the dye front reached the bottom of the gel. Detection was carried out with fast silver staining (47).
Image Analysis
The gels were scanned after silver staining on a flatbed scanner (Epson Perfection V750), using a 16-bit grayscale image acquisition. The gel images were then analyzed using the Delta 2D software (v 3.6). Four gels coming from four independent cultures were used for each experimental group. Spots that were never expressed above 100 ppm of the total spots were first filtered out. Then, significantly varying spots were selected on the basis of their Student t test p value between the treated and the control groups. Spots showing a p value lower than 0.05 were selected. However, two additional steps of statistical validation were used. On a global point perspective, the Storey and Tibshirani approach (48) was used to evaluate the false discovery rate, as described previously (49). Then each of the selected spots was tested for significance by the Mann–Whitney ranking test to rule out any bias that might exist in the t test because of its underlying hypotheses.
Mass Spectrometry
The spots selected for identification were excised from silver-stained gels and destained with ferricyanide/thiosulfate on the same day as silver staining in order to improve the efficiency of the identification process (50, 51). In-gel digestion was performed with an automated protein digestion system, the MassPrep Station (Waters, Milford, MA). The gel plugs were washed twice with 50 μl of 25 mm ammonium hydrogen carbonate (NH4HCO3) and 50 μl of acetonitrile. The cysteine residues were reduced by 50 μl of 10 mm dithiothreitol at 57 °C and alkylated by 50 μl of 55 mm iodoacetamide. After dehydration with acetonitrile, the proteins were cleaved in gel with 10 μl of 12.5 ng/μl of modified porcine trypsin (Promega, Madison, WI) in 25 mm NH4HCO3. The digestion was performed overnight at room temperature. The generated peptides were extracted with 30 μl of 60% acetonitrile in 0.1% formic acid. Acetonitrile was evaporated under vacuum before nano-LC-MS/MS analysis.
Nano-LC-MS/MS analysis was performed using a nano-LC-QTOF-MS system and a nano-LC-IT-MS system. The nano-LC-QTOF-MS system was composed of a nanoACQUITY Ultra-Performance-LC (Waters Corporation, Milford, MA) coupled to a SynaptTM High Definition Mass SpectrometerTM (Waters Corporation, Milford, MA). The system was fully controlled by MassLynx 4.1 SCN639 (Waters Corporation, Milford, MA).
The nano-LC system was composed of an ACQUITY UPLC® BEH130 C18 column (250 mm × 75 μm with a 1.7-μm particle size; Waters Corporation, Milford, MA) and a Symmetry C18 precolumn (20 mm × 180 μm with a 5-μm particle size; Waters Corporation, Milford, MA). The solvent system consisted of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). 4 μl of sample were loaded into the enrichment column over 3 min at 5 μl/min with 99% of solvent A and 1% of solvent B. Elution of the peptides was performed at a flow rate of 300 nl/min with an 8%–35% linear gradient of solvent B over 9 min.
The tandem mass spectrometer was equipped with a Z-spray ion source and a lock mass system. The capillary voltage was set at 2.8 kV, and the cone voltage at 35 V. Mass calibration of the TOF was achieved using fragment ions from Glu-fibrino-peptide B in the 50–2000 m/z range. Online correction of this calibration was performed with Glu-fibrino-peptide B as the lock-mass. During the analysis, the ion (M+2H)2+ at m/z 785.8426 was used to calibrate MS data, and the fragment ion (M+H)+ at m/z 684.3469 was used to calibrate MS/MS data. The system was operated in data-dependent acquisition mode with automatic switching between MS (0.5 s/scan in the m/z range of 150–1700) and MS/MS modes (0.5 s/scan in the m/z range of 50–2000). The two most abundant peptides (intensity threshold: 20 counts/s), preferably doubly and triply charged ions, were selected on each MS spectrum for further isolation and collision-induced dissociation fragmentation using the collision energy profile. Fragmentation was performed using argon as the collision gas.
Mass data collected during analysis were processed and converted into .pkl files using ProteinLynx Global Server 2.3 (Waters Corporation, Milford, MA). A normal background subtraction type was used for both MS and MS/MS with a 5% threshold and polynomial correction of order 5. Smoothing was performed on MS/MS spectra (Savitsky-Golay, two iterations, window of three channels). De-isotoping was applied for both MS (medium de-isotoping) and MS/MS (fast de-isotoping).
The nano-LC-IT-MS system was composed of an Agilent 1200 series nanoLC-Chip system (Agilent Technologies, Palo Alto, CA) coupled to an amaZon ion trap (Bruker Daltonics GmbH, Bremen, Germany). The system was fully controlled by HyStar 3.2 and trapControl 7.0 (Bruker Daltonics, Bremen, Germany).
The chip was composed of a Zorbax 300SB-C18 analytical column (43 mm × 75 μm with a 5-μm particle size) and a Zorbax 300SB-C18 enrichment column (40 nl, 5 μm). The solvent system consisted of 2% acetonitrile, 0.1% formic acid in water (solvent A) and 2% water, 0.1% formic acid in acetonitrile (solvent B). 4 μl of sample were loaded into the enrichment column at a flow rate of 3.75 μl/min with solvent A. Elution of the peptides was performed at a flow rate of 300 nl/min with an 8%–40% linear gradient of solvent B over 7 min.
For tandem MS experiments, the system was operated in data-dependent acquisition mode with automatic switching between MS and MS/MS. The voltage applied to the capillary cap was optimized to −1850 V. The MS scanning was performed in the standard enhanced resolution mode at a scan rate of 8100 m/z per second. The mass range was 250–1500 m/z. The ion charge control was 200,000, and the maximum accumulation time was 200 ms. A total of two scans were averaged to obtain an MS spectrum, and the rolling average was 1. The six most abundant precursor ions with an isolation width of 4 m/z were selected on each MS spectrum for further isolation and fragmentation. The MS/MS scanning was performed in the ultrascan mode at a scan rate of 32,500 m/z per second. The mass range was 100–2000 m/z. The ion charge control was 300,000. A total of two scans were averaged to obtain an MS/MS spectrum.
Mass data collected during analysis were processed and converted into .mgf files using DataAnalysis 4.0 (Bruker Daltonics GmbH, Bremen, Germany). A maximum of 1700 compounds were detected with an intensity threshold of 150,000. A charge deconvolution was applied on the MS full scan and the MS/MS spectra with abundance cutoffs of 5 and 2%, respectively, and with maximum charge states of 3 and 2, respectively.
For protein identification, the MS/MS data were interpreted using a local Mascot server with the MASCOT 2.4.0 algorithm (Matrix Science, London, UK) against UniProtKB/SwissProt (version 2012 08, 537,505 sequences). The research was carried out in all species. A maximum of one trypsin missed cleavage was allowed. Spectra from QTOF were searched with a mass tolerance of 15 ppm for MS and 0.05 Da for MS/MS data, and spectra from the ion trap were searched with a mass tolerance of 0.3 Da in MS and MS/MS modes. Carbamidomethylation of cysteine residues and oxidation of methionine residues were specified as variable modifications. Protein identifications were validated with at least two peptides with Mascot ion scores above 20 or with one peptide with a Mascot ion score above 30 and five consecutive fragment ions in the MS/MS spectrum.
RESULTS AND DISCUSSION
Nanoparticle Characterization in Culture Medium
Upon sonication, copper and copper oxide nanoparticles could be dispersed to a microaggregate size of approximately 250 nm. These microaggregates were stable upon dilution in pure water, but we observed an immediate aggregation over the micron size when the nanoparticles were diluted in culture medium, even when the medium contained fetal bovine serum. Unfortunately, this aggregation resulted in immediate sedimentation, leading to a very uneven exposure of the cell layer to the particles in the cell culture dish.
All of the undesirable phenomena were prevented when the copper and copper oxide nanoparticles were coated with polyvinylpyrrolidone (PVP) prior to dilution in the cell culture medium. PVP has been used in the preparation of stable copper nanoparticles via aqueous routes (52–54), such as for conductive inks (55). PVP is also used for the stabilization of silver nanoparticles (56, 57) and in commercial nanoparticle dispersions (e.g. silver nanoparticles from Buhler). It is also classified as a biocompatible polymer (58, 59) and does not activate the macrophages by itself (60). Indeed, it induced no biological response in either our proteomic experiments or the targeted ones (data not shown).
Light scattering and scanning electron microscopy experiments showed that coating with PVP did not significantly change the microaggregation in water. However, the macroaggregation observed in culture media for noncoated particles was prevented, and the average diameter was kept close to 250 nm (Fig. 1). The process of coating with PVP significantly changed the zeta potential, which was measured at 0 mV instead of the classical 30 mV of a water suspension of CuO (61).
Fig. 1.

Characterization of the nanoparticles after dispersion by sonication and coating with PVP40. A, size distribution of copper nanoparticles after sonication, coating with PVP40, and dilution in RPMI 1640 medium containing 10% fetal calf serum. B, SEM image of copper nanoparticles. C, size distribution of copper oxide nanoparticles after sonication, coating with PVP40, and dilution in RPMI 1640 medium containing 10% fetal calf serum. D, SEM image of copper oxide nanoparticles.
Zirconium oxide nanoparticles had the same aggregation property in culture media, which was prevented as well by PVP coating. The resulting average size was 180 nm as measured by light scattering.
Dissolution studies, carried out in complete cell culture medium and in conditioned cell culture medium (both without cells), revealed that approximately 60% of the copper present initially in the nanoparticles was dissolved in the culture medium, and that dissolution was linear in the 5–20 μg/ml range. The dissolution data were similar whether fresh or conditioned medium was used and whether uncoated or PVP-coated nanoparticles were used. These dissolution figures are quite close to others measured recently under the same conditions (i.e. dispersion in water and then dilution in complete cell culture medium) (31).
Determination of Effective Copper Doses
In order to carry out a proteomics study, it was necessary to determine the dose that would allow us to obtain the best compromise between viability and biological effect. We therefore decided to use an LD20, which is a dose leading to a cell mortality of 20%, knowing that the mortality of the control culture was ∼5%. As nanoparticles are known to interfere with many viability estimation methods (62), we chose a very conservative and robust dye exclusion method for this critical step (63).
The results show that the LD20 was obtained at 10 μg/ml for the Cu and CuO nanoparticles for the RAW264 cell line, whereas it was obtained at 5 μg/ml for primary macrophages (Fig. 2). A copper ion concentration of 0.2 mm corresponded to the LD20 for primary macrophages, whereas it induced no more than 10% mortality for the cell lines.
Fig. 2.
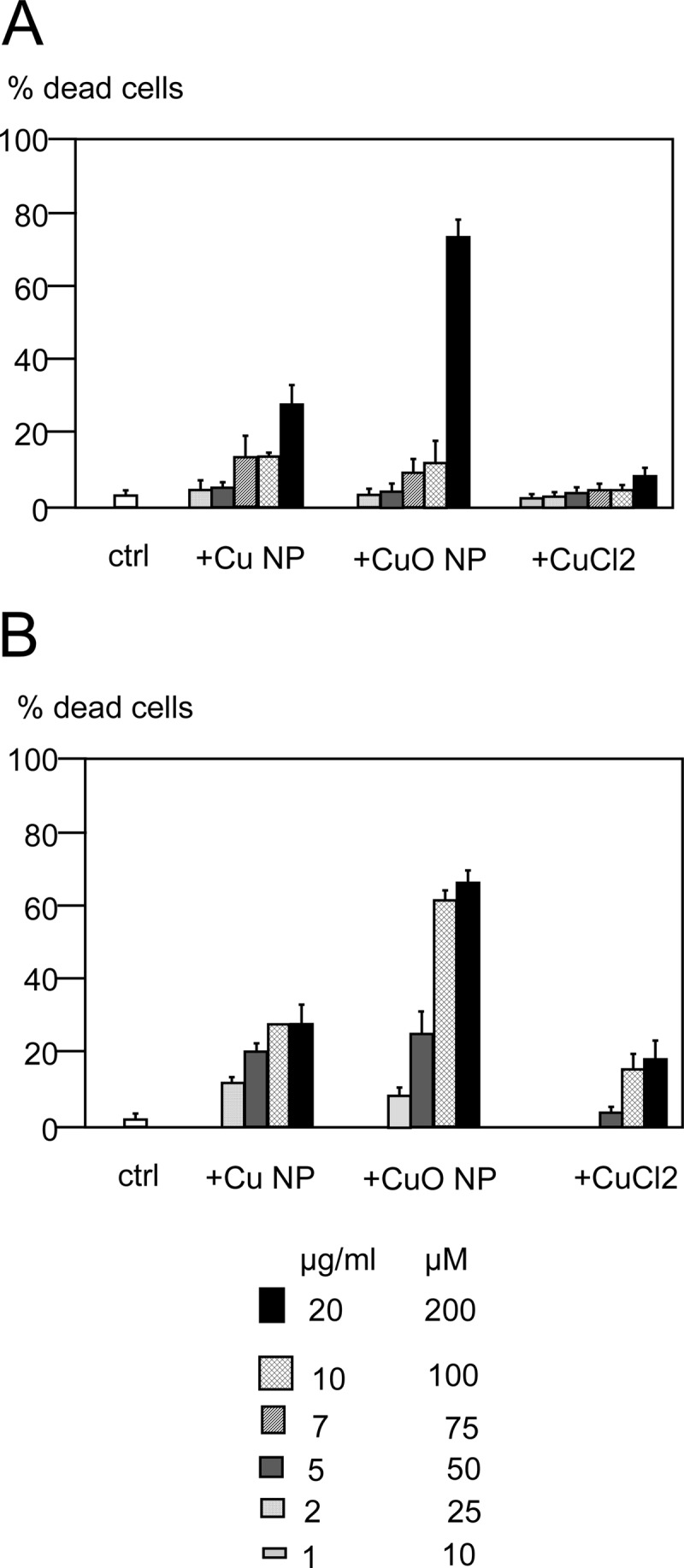
Determination of the viability of the cells after treatment with copper nanoparticles, copper oxide nanoparticles, and copper(II) chloride. The viability was measured via trypan blue exclusion. A, RAW264 cells. B, primary macrophages derived from bone marrow.
We therefore fixed our working concentrations at LD20 for the nanoparticles and at 0.2 mm for the copper ions.
Determination of Nanoparticle Fate in Cells
In order to determine the fate of the nanoparticles in the presence of macrophages, transmission electron microscopy was performed on ultrathin sections of cells. These experiments showed that nanoparticles were present in vacuolar structures that were identified as multivesicular bodies and lysosomes (Fig. 3). These results were completely in line with what can be expected from professional phagocytic cells, as macrophages are. Moreover, these results demonstrate that macrophages seem to ingest nanoparticle aggregates present in the culture medium and break them down within the endosomes/lysosomes. The first step was obviously the disaggregation of the aggregates into the nominal nanoparticles. However, the disaggregation did not seem to stop at this stage, as much smaller copper oxide particles were present (Fig. 3). This observation is consistent with an intracellular dissolution of the copper oxide nanoparticles, which could be expected in an acidic organelle such as the lysosome. Macrophages are known to be able to uncoat nanoparticles (17), and this is further confirmed in our case by the existence of eroded nanoparticles within the cells. This size distribution was observed at both 4 h and 24 h of treatment with the nanoparticles. Thus, it can be concluded that the intracellular solubilization of nanoparticles, which seems important for their biological effects (64), is kept even with coated nanoparticles. Overall, the coating process does not seem to interfere with either extracellular or intracellular processes.
Fig. 3.

Transmission electron microscopy analysis of nanoparticle-treated cells. RAW264 cells were imaged via transmission electron microscopy after treatment with copper oxide nanoparticles for 4 h (panels A, C, and D) and 24 h (panel B). The copper nanoparticles are concentrated in multivesicular bodies and in lysosomes, as expected for professional phagocytic cells. Cells treated for 24 h show much larger, fusioned lysosomes than cells treated for only 4 h. Various sizes of copper oxide can be seen, from the 200- to 300-nm aggregates present in the medium (thick arrows) to the 30- to 50-nm sizes of the initial particles (thin arrows) and even smaller sizes such as 10 nm (arrowheads), as can be expected from the intracellular dissolution of the nanoparticles.
Proteomic Analyses
The proteomic analyses were carried out on the RAW264 cell line, and typical gels are shown in Fig. 4. More than 1400 reliable spots were detected by the image analysis software, and they were further analyzed for quantitative variations. As with all omics strategies, the issue of multiple testing must be taken into account. In other terms, positive hits will always be obtained when many molecules are screened simultaneously, as in many comparative omic screens, simply because of the statistical noise. Most classical statistical corrections for multiple testing, such as the Bonferroni correction, are not suited to omics data, in which relatively low replicate numbers are available, and are in fact much too conservative (65). We therefore used the t test distribution approach, pioneered by Storey and Tibshirani (48) for transcriptomics and first applied to proteomics by Karp and coworkers (49). The distribution of the p values (t test) for the three tested conditions (nanoparticular Cu, nanoparticular CuO, and Cu ion) is shown in supplemental Fig. S1. This approach showed us that we could not expect any really significant results with a 25-μm dose of copper ion, which has no effect on the cells. However, significant results could be expected for a higher dose of copper ion, corresponding to the LD20, as well as for copper and, even more so, for copper oxide nanoparticles at the LD20.
Fig. 4.

Proteomic analysis via two-dimensional electrophoresis. Top: A, total cell extracts of RAW264 cells were separated via two-dimensional gel electrophoresis. The first dimension covered a pH range of 4–8, and the second dimension a range of 15–200 kDa. Total cellular proteins (150 μg) were loaded on the first-dimension gel, and the spots were detected via silver staining. A, gel obtained from control cells. B, gel obtained from copper-nanoparticle-treated cells. C, gel obtained from cells treated with copper oxide nanoparticles. The arrows point to spots that show reproducible and statistically significant changes between the control condition and the treatments with Cu or CuO nanoparticles. The numbering of the spots is explained in Table I. Rectangles A through D point to areas with a high density of selected spots, shown in a different figure. Gels coming from cells treated with copper chloride have been omitted for clarity reasons. Bottom: Insets from gels displayed in the upper portion, shown at a higher magnification for more clarity in spot pointing.
We therefore focused our analysis on spots that showed consistent variation between the Cu and CuO treatments, and these spots are indicated in Fig. 4. These spots were then identified via mass spectrometry, and the results are shown in Table I, with more details in supplemental Tables S1 and S2. Some neighboring spots or spots corresponding to other proteins of interest were also identified. On a global level, in addition to the classical metabolic modulation (66, 67), some cellular compartments/functions were highlighted by this proteomic analysis, such as cytoskeleton, cell signaling, mitochondrion, and oxidative stress response, especially in the glutathione system.
Table I. Differentially expressed proteins identified in the proteomic screen.
| Spot number | Protein name | Protein accession numbers | Protein molecular weight (Da) | Cu/control (ratio/p value) | CuO/control (ratio/p value) | Cu2+/control (ratio/p value) | ZrO2/control (ratio/p value) | Number of unique peptides |
|---|---|---|---|---|---|---|---|---|
| Oxidative stress | ||||||||
| 1 | Superoxide dismutase [Cu-Zn] | P08228 | 15,924 | 0.40/0.08 | 0.33/0.05 | 1.09/0.72 | 0.824/0.06 | 7 |
| 2 | Peroxiredoxin-6 | O08709 | 24,854 | 0.91/0.42 | 0.75/0.04 | 0.91/0.55 | 1.016/0.64 | 5 |
| 3 | Peroxiredoxin-1 | P35700 | 22,160 | 2.8/0.04 | 3.3/0.03 | 1.0/1.0 | 1.17/0.21 | 6 |
| 4 | Peroxiredoxin-1 | P35700 | 22,160 | 1.28/0.21 | 2.1/0.002 | 1.56/0.01 | 0.921/0.25 | 7 |
| 5 | Heme oxygenase 1 | P14901 | 32,911 | 3.8/0.05 | 3.8/0.02 | 0.81/0.52 | 0.805/0.82 | 1 |
| 6 | Heme oxygenase 1 | P14901 | 32,911 | 1.47/0.12 | 1.61/0.02 | 1.00/0.97 | 0.732/0.25 | 2 |
| 7 | Heme oxygenase 1 | P14901 | 32,911 | 3.2/0.05 | 1.49/0.23 | 5.35/0.02 | 2 | |
| 8 | Heme oxygenase 1 | P14901 | 32,911 | 1.96/0.001 | 1.93/0.02 | 2.25/0.04 | 1.265/0.05 | 4 |
| 9 | Heme oxygenase 1 | P14901 | 32,911 | 1.7/0.22 | 2.1/0.11 | 1.16/0.75 | 1.049/0.70 | 1 |
| 10 | S-formylglutathione hydrolase | Q9R0P3 | 31,302 | 1.47/0.03 | 1.84/0.03 | 1.23/0.41 | 0.958/0.70 | 2 |
| 11 | Glutamate-cysteine ligase regulatory subunit | O09172 | 30,517 | 1.3/0.04 | 1.35/0.01 | 2.38/0.002 | 0.929/0.15 | 3 |
| Mitochondrion | ||||||||
| 12 | NADH dehydrogenase iron-sulfur protein 8, mitochondrial | Q8K3J1 | 24,039 | 0.97/0.6 | 0.6/0.002 | 0.98/0.96 | 0.89/0.14 | 8 |
| 13 | NADH dehydrogenase iron-sulfur protein 3, mitochondrial | Q9DCT2 | 30,131 | 1.55/0.03 | 1.1/0.5 | 0.8/0.15 | 0.86/0.06 | 4 |
| 14 | Cytochrome b-c1 complex subunit 1, mitochondrial | Q9CZ13 | 52,834 | 1.4/0.005 | 1.4/0.007 | 0.72/0.11 | 1.142/0.21 | 6 |
| 15 | Elongation factor Tu, mitochondrial | Q8BFR5 | 49,491 | 1.3/0.27 | 1.6/0.05 | 1.16/0.31 | 0.797/0.06 | 3 |
| 16 | Glutamate dehydrogenase 1, mitochondrial | P26443 | 61,320 | 1.016/0.92 | 1.345/0.1 | 1.1/0.56 | 0.99/0.88 | 2 |
| 17 | Glutamate dehydrogenase 1, mitochondrial | P26443 | 61,320 | 1.32/0.01 | 1.44/0.04 | 0.77/0.19 | 1.095/0.82 | 2 |
| Cell signaling | ||||||||
| 18 | Rho GDP-dissociation inhibitor 2 | Q61599 | 22,833 | 0.73/0.02 | 0.70/0.01 | 1.048/0.74 | 0.973/0.48 | 3 |
| 19 | Rho GDP-dissociation inhibitor 2 | Q61599 | 22,833 | 0.77/0.25 | 0.606/0.07 | 0.94/0.8 | 0.902/0.02 | 3 |
| 20 | Rho GDP-dissociation inhibitor 1 | Q99PT1 | 23,390 | 0.67/0.004 | 0.73/0.04 | 0.70/0.09 | 0.947/0.50 | 4 |
| 21 | Rho GDP-dissociation inhibitor 1 | Q99PT1 | 23,390 | 081/0.07 | 0.78/0.02 | 0.95/0.54 | 0.977/0.40 | 7 |
| 22 | Inositol monophosphatase 1 | O55023 | 30,437 | 0.66/0.003 | 0.67/0.01 | 0.97/0.74 | 1.065/0.46 | 8 |
| 23 | 14–3-3 protein zeta/delta | P63101 | 27,772 | 0.67/0.06 | 0.4/0.05 | 0.80/0.25 | 0.933/0.16 | 10 |
| 24 | 14–3-3 protein beta/alpha | Q9CQV8 | 28,069 | 0.77/0.11 | 0.66/0.04 | 0.84/0.55 | 0.971/0.80 | 2 |
| 25 | 14–3-3 protein gamma | P61982 | 28,285 | 0.805/0.004 | 0.77/0.06 | 0.74/0.60 | 1.088/0.42 | 5 |
| 26 | 14–3-3 protein gamma | P61982 | 28,285 | 0.77/0.2 | 0.49/0.015 | 0.88/0.66 | 1.09/0.32 | 7 |
| 27 | 14–3-3 protein beta/alpha | Q9CQV8 | 28,069 | 0.89/0.45 | 0.91/0.67 | 0.91/0.68 | 0.889/0.25 | 4 |
| 28 | Guanine nucleotide-binding protein G(i) subunit alpha-2 | P08752 | 40,472 | 0.6/0.01 | 0.55/0.005 | 0.89/0.23 | 0.882/0.50 | 4 |
| 29 | Guanine nucleotide-binding protein G(i) subunit alpha-2 | P08752 | 40,472 | 0.8/0.06 | 0.78/0.11 | 0.88/0.29 | 0.924/0.32 | 5 |
| 30 | Serine-threonine kinase receptor-associated protein | Q9Z1Z2 | 38,425 | 0.75/0.02 | 0.65/0.01 | 0.98/0.94 | 0.946/0.52 | 4 |
| Central metabolism | ||||||||
| 31 | Malate dehydrogenase, cytoplasmic | P14152 | 36,512 | 0.61/0.045 | 0.62/0.01 | 0.9/0.53 | 0.906/0.19 | 5 |
| 32 | Malate dehydrogenase, cytoplasmic | P14152 | 36,512 | 0.79/0.2 | 0.63/0.03 | 1.077/0.61 | 0.88/0.11 | 9 |
| 33 | Alpha-enolase | P17182 | 47,124 | 0.96/0.65 | 0.82/0.05 | 0.87/0.54 | 0.76/0.08 | 3 |
| 34 | Alpha-enolase | P17182 | 47,124 | 0.9/0.3 | 0.8/0.05 | 0.85/0.36 | 0.942/0.64 | 3 |
| 35 | Galactokinase | Q9R0N0 | 42,158 | 0.77/0.015 | 0.5/0.001 | 0.91/0.33 | 1.011/0.85 | 5 |
| 36 | l-lactate dehydrogenase A chain | P06151 | 36,481 | 0.8/0.03 | 0.85/0.11 | 0.8/0.01 | 1.066/0.23 | 7 |
| Protein production and folding | ||||||||
| 37 | Eukaryotic translation initiation factor 5A-1 | P63242 | 16,815 | 0.84/0.2 | 0.43/0.01 | 1.07/0.50 | 0.874/0.14 | 2 |
| 38 | Elongation factor 1-beta | O70251 | 24,676 | 0.7/0.06 | 0.5/0.015 | 0.7/0.45 | 1.023/0.87 | 5 |
| 39 | Elongation factor 1-beta | O70251 | 24,676 | 1.1/0.57 | 0.8/0.5 | 0.66/0.45 | 1.14/0.23 | 2 |
| 40 | Elongation factor 1-delta | P57776 | 31,275 | 0.70/0.07 | 0.48/0.01 | 1.033/0.93 | 1.167/0.21 | 4 |
| 41 | Elongation factor 1-delta | P57776 | 31,275 | 0.79/0.16 | 0.7/0.1 | 0.97/0.94 | 1.059/0.27 | 4 |
| 42 | 60S acidic ribosomal protein P0 | P14869 | 34,256 | 1.98/0.05 | 2.3/0.001 | 2.6/0.007 | 0.814/0.27 | 2 |
| 43 | Endoplasmic reticulum resident protein 29 | P57759 | 28,807 | 1.33/0.08 | 1.78/0.01 | 0.88/0.46 | 0.875/0.80 | 5 |
| 44 | Peptidyl-prolyl cis-trans isomerase A | P17742 | 17,954 | 0.66/0.23 | 0.5/0.02 | 1.23/0.57 | 0.828/0.20 | 2 |
| 45 | Peptidyl-prolyl cis-trans isomerase A | P17742 | 17,954 | 0.5/0.03 | 0.65/0.15 | 0.88/0.34 | 0.993/0.92 | 1 |
| Cytoskeleton and intracellular traffic | ||||||||
| 46 | Tropomyosin alpha-3 chain | Q63610 | 28,989 | 0.5/0.07 | 0.45/0.05 | 0.87/0.30 | 1.15/0.24 | 6 |
| 47 | Tropomyosin alpha-3 chain | Q63610 | 28,989 | 0.84/0.49 | 0.825/0.45 | 1.03/0.67 | 0.845/0.03 | 12 |
| 48 | Vimentin | P20152 | 53,671 | 0.9/0.22 | 0.77/0.05 | 0643/0.13 | 0.944/0.40 | 8 |
| 49 | V-type proton ATPase subunit H | Q8BVE3 | 55,838 | 0.77/0.09 | 0.69/0.01 | 1.054/0.80 | 0.835/0.12 | 2 |
| 50 | Transmembrane emp24 domain-containing protein 2 | Q9R0Q3 | 22,687 | 1.55/0.04 | 1.6/0.01 | 1.13/0.77 | 1.135/0.26 | 3 |
| Miscellaneous | ||||||||
| 51 | Chloride intracellular channel protein 1 | Q9Z1Q5 | 26,996 | 0.79/0.09 | 0.71/0.03 | 1.09/0.58 | 0.85/0.14 | 4 |
| 52 | Chloride intracellular channel protein 1 | Q9Z1Q5 | 26,996 | 0.86/0.23 | 0.7/0.03 | 0.98/0.57 | 0.982/0.61 | 5 |
| 53 | EF-hand domain-containing protein D2 | Q4FZY0 | 26,742 | 0.87/0.21 | 0.69/0.008 | 1.025/0.84 | 1.038/0.62 | 2 |
| 54 | Proliferating cell nuclear antigen | P17918 | 28,768 | 0.75/0.06 | 0.66/0.02 | 0.99/0.95 | 0.984/0.85 | 2 |
| 55 | Farnesyl pyrophosphate synthase | Q920E5 | 40,565 | 0.8/0.001 | 0.8/0.04 | 0.8/0.14 | 0.984/0.87 | 2 |
| 56 | Poly(rC)-binding protein 1 | O19048 | 37,480 | 0.76/0.02 | 0.79/0.003 | 1.2/0.24 | 1.084/0.46 | 1 |
| 58 | Ubiquitin thioesterase OTUB1 | Q7TQI3 | 31,253 | 0.77/0.04 | 0.66/0.04 | 1.02/0.83 | 1.015/0.80 | 2 |
| 59 | Adenosylhomocysteinase | P50247 | 47,671 | 0.47/0.004 | 0.65/0.02 | 0.9/0.51 | 0.958/0.47 | 11 |
| 60 | Adenosylhomocysteinase | P50247 | 47,671 | 0.77/0.01 | 0.78/0.008 | 1.14/0.03 | 1.056/0.65 | 21 |
| Other proteins | ||||||||
| 61 | Peptidyl-prolyl cis-trans isomerase A | P17742 | 17,954 | 0.75/0.15 | 0.78/0.18 | 1.012/0.95 | 0.981/0.74 | 13 |
| 62 | Superoxide dismutase [Mn], mitochondrial | P09671 | 24,585 | 1.17/0.22 | 1.08/0.75 | 0.38/0.11 | 1.347/0.53 | 4 |
| 63 | Peroxiredoxin-2 | Q61171 | 21,761 | 0.92/0.62 | 0.70/0.23 | 1.03/0.72 | 1.02/0.73 | 7 |
| 64 | Peroxiredoxin-3 | P20108 | 28,109 | 1.41/0.10 | 1.30/0.22 | 1.13/0.12 | 0.976/0.73 | 6 |
| 65 | Ferritin light chain 1 | P29391 | 20,785 | 1.29/0.09 | 1.31/0.25 | 1.62/0.025 | 1.18/0.05 | 9 |
| 66 | Eukaryotic initiation factor 4A-III | P38919 | 46,824 | 1.046/0.75 | 1.13/0.38 | 0.97/0.88 | 0.933/0.55 | 20 |
| 67 | Elongation factor 1-gamma | Q9D8N0 | 50,043 | 1.004/0.98 | 0.952/0.73 | 1.0/0.99 | 0.964/0.65 | 16 |
Note: The accession numbers are those of the SwissProt Database.
In order to check the specificity of the proteomic variations observed, we used zirconium oxide nanoparticles. These particles show very weak cytotoxicity (68) and were indeed nontoxic to RAW264 cells up to 100 μg/ml. As the density of zirconium oxide (5.68) is close to that of copper oxide (6.31), treatment of the cells with an equal concentration in weight corresponded to equal concentrations in particle numbers, which is an important parameter for phagocytic cells. Furthermore, as zirconium oxide nanoparticles are also dispersed with PVP, this control integrated any possible effect of absorbed PVP.
Treatment with 10 μg/ml zirconium oxide for 24 h induced very few detectable changes in our proteomic screen (supplemental Table S3 and supplemental Fig. S2), and none of these changes corresponded to the ones induced by copper and copper oxide nanoparticles (Table I). This showed the specificity of our proteomic results. We therefore decided to test specifically these functions in targeted analyses on both the RAW264 cell line and primary macrophages derived from bone marrow. As the proteomic effects were more pronounced with CuO nanoparticles, we primarily used these nanoparticles for the targeted experiments, although some experiments were also carried out with copper nanoparticles.
Characterization of the General Stress Response
Quite frequently, proteomic studies identify a general cellular stress response module that encompasses proteins involved in central metabolism and protein production and folding (67). However, in the case of the response of macrophages to copper-based nanoparticles, we did not observe the classical induction in central metabolism proteins. In contrast, we observed a small decrease in malate dehydrogenase, galactokinase, and lactate dehydrogenase. We also did not observe changes in the heat shock proteins, in contrast to what has been described at the mRNA level in lung cells exposed to copper oxide nanoparticles (31) or at the protein level for cells exposed to gold nanoparticles (69). However, and consistent with the latter publication, we also observed a strong induction of the ribosomal protein P0.
Characterization of the Oxidative Stress Response
Oxidative stress has been observed in many cell types and with many nanoparticles (reviewed, for example, in Ref. 70). However, cellular responses to oxidative stress are not well characterized. The cellular oxidative stress response system is quite complex and involves several classes of enzymes, including catalases, peroxidases (including peroxiredoxins), superoxide dismutases, and enzymes producing and using glutathione.
Our proteomic screen revealed that the heme oxygenase was very strongly induced, with a complex pattern of modifications. For the more classically observed peroxiredoxins (66, 67), we could determine by using the t-butyl hydroperoxide oxidation test (71) that the induced form of Prx1 was indeed the oxidized form, whereas the native form of Prx6 was induced in our experiments. Other peroxiredoxins, such as Prx2 and the mitochondrial Prx3, remained unchanged, as shown in Table I. The induction of Prx1 in response to gold nanoparticles has been described recently (69), but the status of the other peroxiredoxins had not been investigated previously. In addition, Prx1 is known to be inducible by heavy metals, as is heme oxygenase (72). Furthermore, our results are consistent with those obtained for Prx6 with oxidative stress in lung cells (73) and in myoblasts (74).
As for the superoxide dismutases, we observed a decrease in copper-zinc superoxide dismutase. The response of copper-zinc superoxide dismutase seems to be variable, as various studies have described an increase (75), a decrease (76), or no change (29) in response to metallic nanoparticles. We also observed no change for the mitochondrial manganese superoxide dismutase, although an increase in manganese superoxide dismutase has been described at the mRNA level (not at the protein level) in lung cells exposed to copper oxide nanoparticles (31).
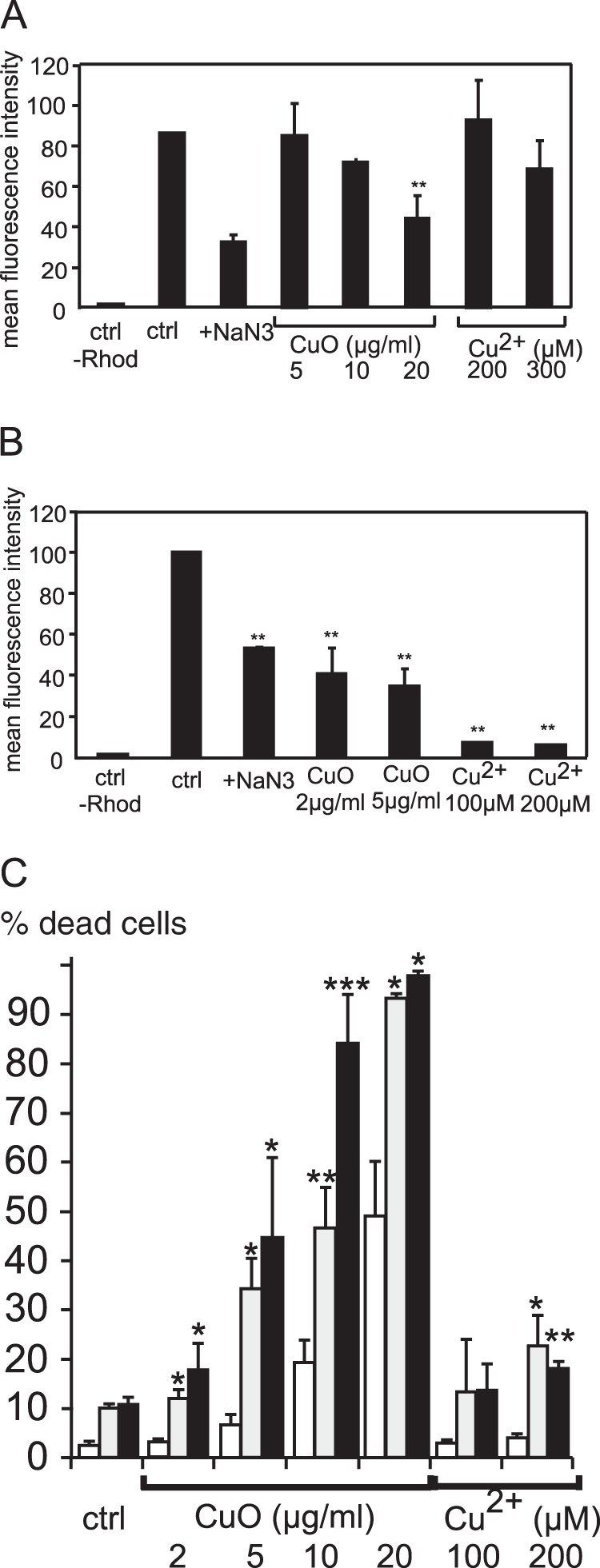
Finally, regarding the glutathione-dependent oxidative stress response system, we observed an increase in formyl glutathione hydrolase and a strong induction of the regulatory subunit of the glutamate-cysteine ligase, the activating subunit of the limiting enzyme in glutathione biosynthesis (77). This suggested that the cells responding to copper-based nanoparticles made a strong effort to increase their glutathione level, and we tested the intracellular levels of reduced glutathione with a chlorobimane conjugation test (37). Relative to the positive control represented with diethyl maleate, which destroys reduced glutathione through alkylation, copper ion and copper-based nanoparticles induced a more subtle effect (Fig. 5). A subpopulation of cells with low levels of reduced glutathione was induced, especially by copper nanoparticles. However, most of the cells had normal reduced glutathione levels, so that the average for the whole cell population was not significantly changed in most cases. In addition, the glutathione reductase was not induced by copper treatment (Fig. 5).
Fig. 5.

Analysis of the glutathione-based antioxidant system. This figure shows the variation of the intracellular levels of reduced glutathione (GSH), estimated through conjugation with monochlorobimane (MCB) to form a fluorescent adduct and analysis by a MoFlo cell sorter. Top: distribution of the MCB-GSH adduct fluorescence in control cells (A) and in cells treated with diethyl maleate (B), which acts as a negative control by reacting with reduced glutathione before the cells are treated with monochlorobimane. This experiment allowed us to define the GSH-high and GSH-low cells based on their fluorescence. Middle: variation in intracellular glutathione levels upon treatment with copper nanoparticles, copper oxide nanoparticles, or copper ion. Three independent experiments were conducted per condition. C, variation in the percentage of GSH-high cells in RAW264 cells, showing the appearance of a population of GSH-low cells, especially in cells treated with copper nanoparticles. D, variation in the percentage of GSH-high cells in primary macrophages, showing the appearance of a population of GSH-low cells, especially in cells treated with copper nanoparticles. E, variation in the glutathione reductase activity in RAW264 cells treated with copper oxide nanoparticles or with copper ion. *p < 0.05; **p < 0.01; ***p < 0.001 (Mann–Whitney test). F, cell viability upon the inhibition of glutathione biosynthesis. Control cells (white bars) or cells pretreated with buthionine sulfoximine to inhibit glutathione biosynthesis (gray bars) were treated with various doses of copper oxide nanoparticles. Viability was measured via trypan blue exclusion. *p < 0.05; **p < 0.01; ***p < 0.001 (Mann–Whitney test).
Primary macrophages, however, were more sensitive and showed a decrease in their free glutathione levels of copper ions and of the two-copper-based nanoparticles. These results were consistent with those observed previously with copper oxide nanoparticles in lung cells (29, 30) and with silver in liver cells (75, 78). In the case of copper-based nanoparticles, this is also consistent with the well-known role of glutathione as a copper buffer in cells (79). Consistent with this role, a decrease in free glutathione levels was also observed when the cells were treated with copper ions.
However, the RAW264.7 cells appeared less sensitive to copper oxide and copper ions, as if the strong induction of glutamate-cysteine ligase were sufficient in this case to restore close-to-normal reduced glutathione levels. To verify this hypothesis, we treated RAW264.7 cells with nontoxic doses of buthionine sulfoximine, an inhibitor of glutamate-cysteine ligase. This induced a marked hypersensitivity of the cells to copper-based nanoparticles, as shown in Fig. 5F.
Overall, these proteomics-based results show the fine tuning of the cellular response to oxidative stress, as exemplified by the differential response of the three cytosolic peroxidoxins (Prx1, Prx2, and Prx6) and the response of the glutathione-dependent system.
Characterization of the Mitochondrial Response
Mitochondrial function is sometimes tested in response to nanoparticle exposure by testing the main parameter of mitochondrial transmembrane potential. A decrease in mitochondrial transmembrane potential has been observed in response to copper-based nanoparticles or to copper ions in liver (78), in lung cells (80), and in macrophages (81). We also observed such a decrease in our study using a Rhodamine 123 uptake test (39), as shown in Fig. 6. Both nanoparticles and copper ions induced a dose-dependent decrease in the mitochondrial transmembrane potential, and this effect was more pronounced in the primary macrophages than in the cell line.
Fig. 6.
Analysis of mitochondrial function. The top two panels of this figure show the amount of rhodamine internalized in the cells, expressed as a percentage of the fluorescence in control cells (three independent experiments). Fluorescence measurements were carried out on a FACScalibur cell sorter. A, results obtained with RAW264 cells. B, results obtained with primary macrophages derived from bone marrow. *p < 0.05; **p < 0.01 (Mann–Whitney test). C, cell survival after treatment with inhibitors of the mitochondrial respiratory chain and subsequent treatment with copper oxide nanoparticles. White bars: control cells; gray bars: cells treated with 200 nm rotenone; black bars: cells treated with 200 nm antimycin A. *p < 0.05; **p < 0.01; ***p < 0.001 (Student's t test).
Little is known, however, about the molecular events that could be related to the observed decrease in the mitochondrial transmembrane potential, which could even be due to a change in the plasma membrane potential. However, our proteomics screen suggested some possible mechanisms. First of all, we did not observe any change in the two major mitochondrial antioxidant proteins, superoxide dismutase 2 and peroxiredoxin 3. This suggests that there is no massive mitochondrial oxidative stress. However, we observed an increase in the levels of mit-EF-Tu. As this protein is a translation factor for the intramitochondrial synthesis of the few respiratory chain proteins encoded by the mitochondrial genome, this result suggested an increase in the synthesis of the proteins of the oxidative phosphorylation chain, which was corroborated by an increase in one nuclear-coded subunit of the NADH dehydrogenase and one of the ubiquinol-cytochrome c oxidoreductase complex. However, the synthesis of another subunit of NADH dehydrogenase containing an iron-sulfur cluster (Ndufs8) was decreased. This suggests a toxic mechanism in which copper ions pumped into the mitochondria may alter the function of redox-sensitive proteins containing iron-sulfur clusters and, thus, alter both electron transfer and proton pumping through the inner mitochondrial membrane, leading to the observed decrease in the transmembrane potential. In this context, the observed increases in mit-EF-Tu, Uqcrc1, and Ndufs3 would represent an attempt of the cells to restore a normal mitochondrial transmembrane potential, which is observed at nontoxic doses of nanoparticles in RAW264 cells. In order to test this hypothesis, we analyzed cell survival after treatment with subtoxic doses of inhibitors targeting the two respiratory complexes highlighted by proteomics, namely, Complex I and Complex III, inhibited, respectively, by rotenone and antimycin A. The results show the synergistic toxicity of mitochondrial inhibitors and copper oxide nanoparticles (Fig. 6).
Changes in the Cytoskeleton, Intracellular Trafficking, and Phagocytosis
Our proteomic screen pointed to changes in the cytoskeleton induced by copper-based nanoparticles. However, not all types of cytoskeletal proteins were altered by the nanoparticles. The tubulin cytoskeleton did not show any change in our screen, and besides vimentin, mostly the actin-myosin cytoskeleton was altered, either directly (decrease in tropomyosins) or indirectly (decrease in Rho GDI inhibitors, which is functionally equivalent to an increase in the activity of the Rho proteins). Rho proteins control specific modulations of the actin cytoskeleton (e.g. the formation of stress fibers) that have been associated with decreased phagocytic capacity in macrophages stressed by hyperoxia (82).
A decrease in a subunit of the proton pump also pointed to a possible phagocytic deficiency. This prompted us to assay phagocytosis, which is a key macrophage function. CuO nanoparticles had no effect at moderately toxic doses in cell lines, and an inhibition of phagocytosis was apparent only at a toxic dose (20 μg/ml) (Fig. 7). Copper nanoparticles were more efficient at inhibiting phagocytosis, whereas copper ions had no effect. In primary macrophages, which were more sensitive to nanoparticles in all our tests, both Cu and CuO nanoparticles were efficient in inhibiting phagocytosis, whereas Cu ions had no effect. Interestingly, tropomyosin, Rho GDI inhibitors, and proton pump subunits were not modulated by copper ions in our proteomic screen.
Fig. 7.
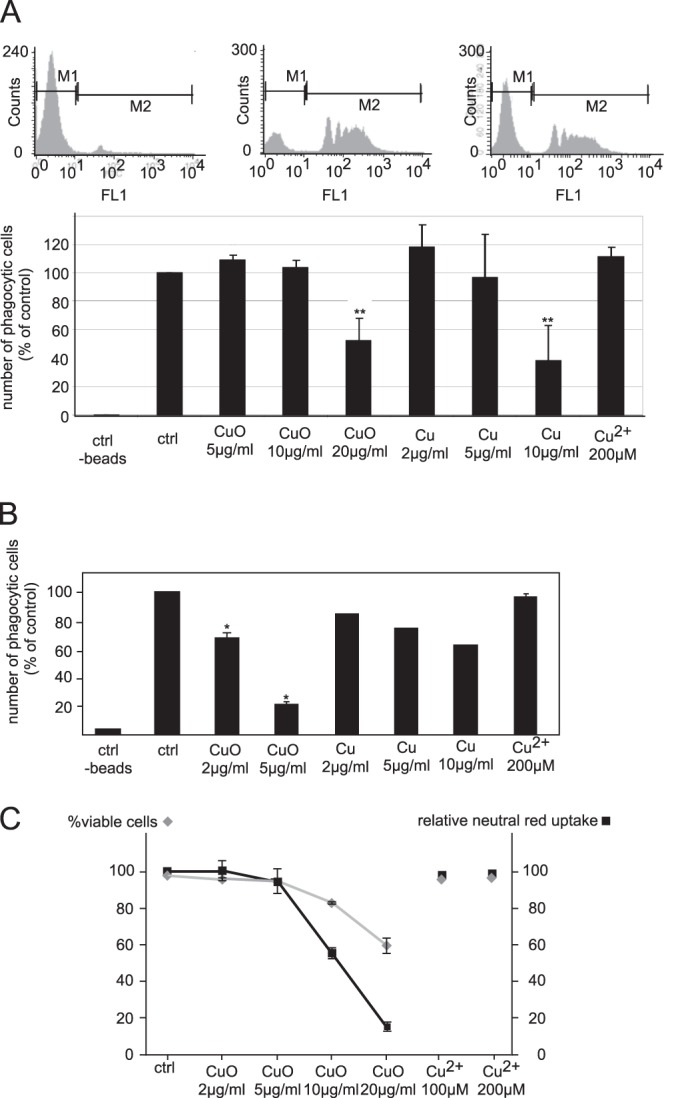
Analysis of the phagocytic capacity. This figure shows the determination of the phagocytic properties of the cells through the ingestion of fluorescent beads and analysis with a FACScalibur cell sorter. Top panel: distribution of the fluorescence of the cells, showing the negative cells (control at 4 °C) and the positive cells (control at 37 °C), as well as the raw data obtained for cells treated with copper oxide nanoparticles for 24 h prior to treatment with the fluorescent beads. Middle panels: variation of the phagocytic capacity of the cells under different conditions, expressed as a percentage of the control cells. Three independent experiments were conducted. A, results obtained with RAW264 cells. B, results obtained with primary macrophages derived from bone marrow (one experiment only for copper nanoparticles). The results show the strong effect of copper and copper oxide nanoparticles on the phagocytic capacity of the cells, and the absence of effect of the copper ion. *p < 0.05; **p < 0.01 (Mann–Whitney test). Bottom panel: C, comparative variation of cell viability and neutral red uptake capacity. This figure shows that the lysosomal functionality of the cells was altered by the copper oxide nanoparticles (but not the ion), as the neutral red uptake decreased more steeply than the overall cell viability.
To further study the functional effects of the observed decrease in the proton pump, we performed a classical neutral red uptake assay. Although classically used as a general viability assay (35), neutral red uptake is indeed an assay of lysosomal function, because the acidic conditions cause neutral red to be retained inside the lysosomes.
Fig. 7C shows that neutral red uptake decreased more severely upon treatment with copper oxide nanoparticles than should be expected from a simple decrease in cell viability, showing a specific effect on lysosomes and validating the proteomically observed decrease in the proton pump.
NO Production
In addition to phagocytosis, the production of cytokines and NO is a key function of macrophages. We therefore tested the production of NO in two different setups. In the first setup, we tested whether nanoparticles could directly induce the production of NO, as has been shown to occur with silver nanoparticles (13). In the second setup, we tested the modulation of the LPS-induced production of NO by a previous treatment with nanoparticles (Fig. 8). Although Cu nanoparticles, CuO nanoparticles, and copper ions did not induce the production of NO by themselves, all three moderately inhibited LPS-induced NO production in primary macrophages. However, only copper ions were able to reduce the LPS-induced NO production in the case of the RAW264 cell line. These results on NO production could be linked, at least indirectly, with the decrease in S-adenosylhomocysteine hydrolase observed in our proteomic screen. Inhibition of S-adenosylhomocysteine hydrolase has been shown to reduce proinflammatory properties (83), and this can be linked, at least in part to a decrease in adenosine concentrations, as adenosine is a potent activator of NO production in macrophages (84). In this pathway, a reduced level of S-adenosylhomocysteine hydrolase would suggest a reduced ability to produce NO, at least in response to LPS, and this is exactly what was observed when NO production was specifically tested.
Fig. 8.
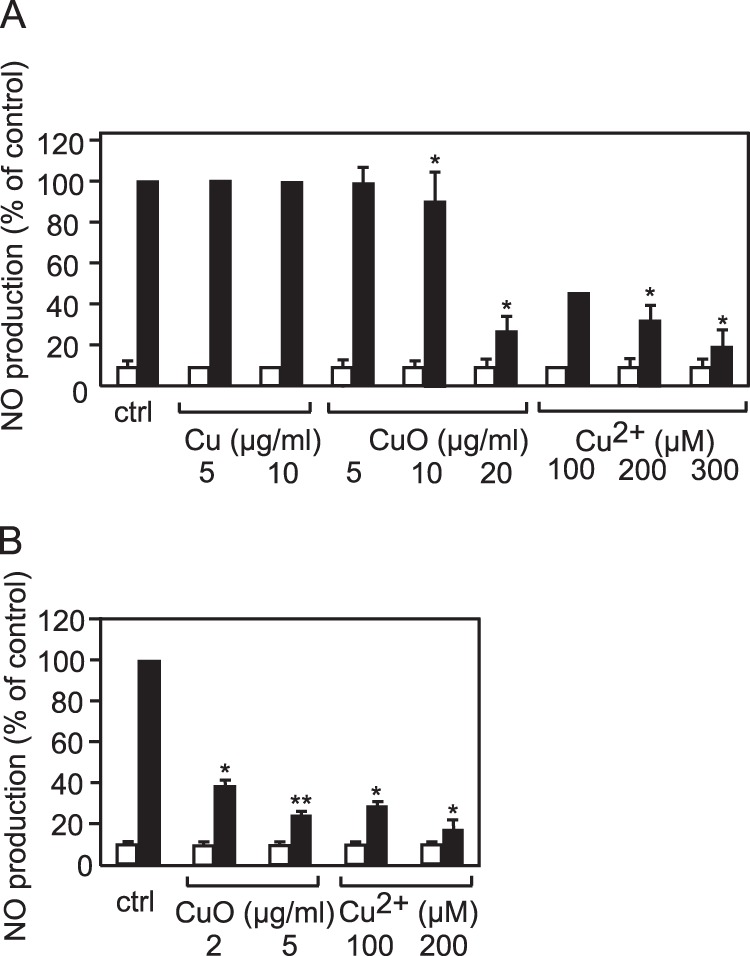
Analysis of nitrous oxide production. This figures shows the amount of NO produced by the cells. White bars: without secondary treatment with LPS. Black bars: with secondary treatment with LPS. A, RAW264 cells. B, primary macrophages derived from bone marrow. *p < 0.05; **p < 0.01 (Mann–Whitney test).
Overall, what can be explained regarding the role of copper ions in the toxicity of nanoparticles? Since the demonstration of an important copper ion release from copper-based nanoparticles in biological media (85), it generally has been thought that most of the nanoparticle toxicity arises from the copper ion itself, and not directly from the nanoparticles. However, transcriptomic studies (31), as well as our work presented here, show that extracellular copper ions can account for only a part of the cellular responses to copper-based nanoparticles. In our case, extracellular copper ions induced general cellular stress responses (mitochondrial or glutathione dependent) and altered some specific cellular responses (e.g. nitric oxide production), but not all. For example, phagocytosis, a key function of macrophages, was altered by the nanoparticles but not by the free ions. We could, however, rule out a mechanical explanation of this inhibition, as titanium dioxide nanoparticles did not inhibit phagocytosis (data not shown).
These differences between the responses to the ion itself and to the nanoparticles may be the result of the existence of two different copper channels at the plasma membrane (Ctr1) and at the lysosomal membrane (Ctr2) (86). Thus, the real intracellular concentration and, even more subtly, the local copper concentrations within the cell can differ depending on the mode of entry. This might be the case for highly phagocytic cells such as macrophages, for which the lysosome-dependent entry can be much more important than the plasma-membrane-dependent entry.
CONCLUDING REMARKS
Despite its limited depth, estimated at about 500 proteins (1500 spots, for an average of 3 spots per protein (87)), the proteomics screen used here allowed us to point out several cellular functions that were modified upon the treatment of macrophages with copper-based nanoparticles and/or copper ions. Interestingly, copper treatment was associated with a reduction in protein levels, suggesting either a decrease in their synthesis or an increase in their degradation. The fact that levels of some copper-binding proteins (e.g. copper-zinc superoxide dismutase and S-adenosylhomocysteine hydrolase) diminished suggests that an excess of intracellular copper could lead to protein destructuration and then degradation.
Moreover, the increase in the glutamate cysteine ligase subunit and of some mitochondrial proteins shows how proteomics can point to cellular resistance mechanisms even when they are successful, so that no apparent change can be detected in the downstream parameter (e.g. glutathione levels).
In the frame of macrophage physiology, the uptake of copper does not bring about activation of the macrophages, with the associated increase in the production of inflammatory mediators, as described for SiO2 an TiO2 nanoparticles (4, 88). It rather brings a depression of core macrophage activities such as NO production and phagocytosis, which in turn can have consequences for the overall efficiency of the immune system.
Finally, our results also show that even at nonlethal doses, nanoparticles can act synergistically with other toxicants to induce cell mortality and dysfunction.
Supplementary Material
Acknowledgments
S.T. thanks the Université Joseph Fourier for a Ph.D. fellowship.
Footnotes
* Financial support from the CEA toxicology program (Nanobiomet and Nanostress grants) and the support of the Labex SERENADE (11-LABX-0064) are gratefully acknowledged.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- LPS
- lipopolysaccharide
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- PBS
- phosphate buffered saline
- PVP
- polyvinyl pyrrolidone.
REFERENCES
- 1. Seaton A., Tran L., Aitken R., Donaldson K. (2010) Nanoparticles, human health hazard and regulation. J. R. Soc. Interface 7 Suppl 1, S119-S129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Teow Y., Asharani P. V., Hande M. P., Valiyaveettil S. (2011) Health impact and safety of engineered nanomaterials. Chem. Commun. (Camb.) 47, 7025–7038 [DOI] [PubMed] [Google Scholar]
- 3. Hamilton J. A. (1980) Macrophage stimulation and the inflammatory response to asbestos. Environ. Health Perspect. 34(FEB), 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moon E. Y., Yi G. H., Kang J. S., Lim J. S., Kim H. M., Pyo S. (2011) An increase in mouse tumor growth by an in vivo immunomodulating effect of titanium dioxide nanoparticles. J. Immunotoxicol. 8, 56–67 [DOI] [PubMed] [Google Scholar]
- 5. Dobrovolskaia M. A., Germolec D. R., Weaver J. L. (2009) Evaluation of nanoparticle immunotoxicity. Nat. Nanotechnol. 4, 411–414 [DOI] [PubMed] [Google Scholar]
- 6. Zolnik B. S., Gonzalez-Fernandez A., Sadrieh N., Dobrovolskaia M. A. (2010) Nanoparticles and the immune system. Endocrinology 151, 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Di Gioacchino M., Petrarca C., Lazzarin F., Di Giampaolo L., Sabbioni E., Boscolo P., Mariani-Costantini R., Bernardini G. (2011) Immunotoxicity of nanoparticles. Int. J. Immunopathol. Pharmacol. 24(1 Suppl), 65S–71S [PubMed] [Google Scholar]
- 8. Vamanu C. I., Cimpan M. R., Hol P. J., Sornes S., Lie S. A., Gjerdet N. R. (2008) Induction of cell death by TiO2 nanoparticles: studies on a human monoblastoid cell line. Toxicol. In Vitro 22, 1689–1696 [DOI] [PubMed] [Google Scholar]
- 9. Choi J., Zhang Q., Reipa V., Wang N. S., Stratmeyer M. E., Hitchins V. M., Goering P. L. (2009) Comparison of cytotoxic and inflammatory responses of photoluminescent silicon nanoparticles with silicon micron-sized particles in RAW 264.7 macrophages. J. Appl. Toxicol. 29, 52–60 [DOI] [PubMed] [Google Scholar]
- 10. Scheel J., Weimans S., Thiemann A., Heisler E., Hermann M. (2009) Exposure of the murine RAW 264.7 macrophage cell line to hydroxyapatite dispersions of various composition and morphology: Assessment of activation and stress response. Toxicol. In Vitro 23, 531–538 [DOI] [PubMed] [Google Scholar]
- 11. Morishige T., Yoshioka Y., Tanabe A., Yao X. L., Tsunoda S., Tsutsumi Y., Mukai Y., Okada N., Nakagawa S. (2010) Titanium dioxide induces different levels of IL-1 beta production dependent on its particle characteristics through caspase-1 activation mediated by reactive oxygen species and cathepsin B. Biochem. Biophys. Res. Commun. 392, 160–165 [DOI] [PubMed] [Google Scholar]
- 12. Park E.-J., Park K. (2009) Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol. Lett. 184, 18–25 [DOI] [PubMed] [Google Scholar]
- 13. Park E.-J., Yi J., Kim Y., Choi K., Park K. (2010) Silver nanoparticles induce cytotoxicity by a Trojan-horse type mechanism. Toxicol. In Vitro 24, 872–878 [DOI] [PubMed] [Google Scholar]
- 14. Park J., Lim D. H., Lim H. J., Kwon T., Choi J. S., Jeong S., Choi I. H., Cheon J. (2011) Size dependent macrophage responses and toxicological effects of Ag nanoparticles. Chem. Commun. 47, 4382–4384 [DOI] [PubMed] [Google Scholar]
- 15. Hsiao J. K., Chu H. H., Wang Y. H., Lai C. W., Chou P. T., Hsieh S. T., Wang J. L., Liu H. M. (2008) Macrophage physiological function after superparamagnetic iron oxide labeling. NMR Biomed. 21, 820–829 [DOI] [PubMed] [Google Scholar]
- 16. Palomaki J., Karisola P., Pylkkanen L., Savolainen K., Alenius H. (2010) Engineered nanomaterials cause cytotoxicity and activation on mouse antigen presenting cells. Toxicology 267, 125–131 [DOI] [PubMed] [Google Scholar]
- 17. Yang C. Y., Tai M. F., Lin C. P., Lu C. W., Wang J. L., Hsiao J. K., Liu H. M. (2011) Mechanism of cellular uptake and impact of ferucarbotran on macrophage physiology. PLoS One 6, e25524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luque-Garcia J. L., Cabezas-Sanchez P., Camara C. (2011) Proteomics as a tool for examining the toxicity of heavy metals. Trends Analyt. Chem. 30, 703–716 [Google Scholar]
- 19. Haniu H., Matsuda Y., Usui Y., Aoki K., Shimizu M., Ogihara N., Hara K., Okamoto M., Takanashi S., Ishigaki N., Nakamura K., Kato H., Saito N. (2011) Toxicoproteomic evaluation of carbon nanomaterials in vitro. J. Proteomics 74, 2703–2712 [DOI] [PubMed] [Google Scholar]
- 20. Cha M.-H., Rhim T., Kim K. H., Jang A.-S., Paik Y.-K., Park C.-S. (2007) Proteomic identification of macrophage migration-inhibitory factor upon exposure to TiO2 particles. Mol. Cell. Proteomics 6, 56–63 [DOI] [PubMed] [Google Scholar]
- 21. Jeon Y. M., Park S. K., Kim W. J., Ham J. H., Lee M. Y. (2011) The effects of TiO(2) nanoparticles on the protein expression in mouse lung. Mol. Cell. Toxicol. 7, 283–289 [Google Scholar]
- 22. Singh N., Jenkins G. J., Asadi R., Doak S. H. (2010) Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano. Rev. 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lison D., Laloy J., Corazzari I., Muller J., Rabolli V., Panin N., Huaux F., Fenoglio I., Fubini B. (2009) Sintered indium-tin-oxide (ITO) particles: a new pneumotoxic entity. Toxicol. Sci. 108, 472–481 [DOI] [PubMed] [Google Scholar]
- 24. Guo K., Pan Q., Wang L., Fang S. (2002) Nano-scale copper-coated graphite as anode material for lithium-ion batteries. J. Appl. Electrochem. 32, 679–685 [Google Scholar]
- 25. Ben-Moshe T., Dror I., Berkowitz B. (2009) Oxidation of organic pollutants in aqueous solutions by nanosized copper oxide catalysts. Appl. Catal. B 85, 207–211 [Google Scholar]
- 26. Karlsson H. L., Cronholm P., Gustafsson J., Moller L. (2008) Copper oxide nanoparticles are highly toxic: a comparison between metal oxide nanoparticles and carbon nanotubes. Chem. Res. Toxicol. 21, 1726–1732 [DOI] [PubMed] [Google Scholar]
- 27. Prabhu B. M., Ali S. F., Murdock R. C., Hussain S. M., Srivatsan M. (2010) Copper nanoparticles exert size and concentration dependent toxicity on somatosensory neurons of rat. Nanotoxicology 4, 150–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cho W. S., Duffin R., Poland C. A., Howie S. E., MacNee W., Bradley M., Megson I. L., Donaldson K. (2010) Metal oxide nanoparticles induce unique inflammatory footprints in the lung: important implications for nanoparticle testing. Environ. Health Perspect. 118, 1699–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fahmy B., Cormier S. A. (2009) Copper oxide nanoparticles induce oxidative stress and cytotoxicity in airway epithelial cells. Toxicol. In Vitro 23, 1365–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahamed M., Siddiqui M. A., Akhtar M. J., Ahmad I., Pant A. B., Alhadlaq H. A. (2010) Genotoxic potential of copper oxide nanoparticles in human lung epithelial cells. Biochem. Biophys. Res. Commun. 396, 578–583 [DOI] [PubMed] [Google Scholar]
- 31. Hanagata N., Zhuang F., Connolly S., Li J., Ogawa N., Xu M. (2011) Molecular responses of human lung epithelial cells to the toxicity of copper oxide nanoparticles inferred from whole genome expression analysis. ACS Nano 5, 9326–9338 [DOI] [PubMed] [Google Scholar]
- 32. Sabel C. E., Neureuther J. M., Siemann S. (2010) A spectrophotometric method for the determination of zinc, copper, and cobalt ions in metalloproteins using Zincon. Anal. Biochem. 397, 218–226 [DOI] [PubMed] [Google Scholar]
- 33. Schleicher U., Bogdan C. (2009) Generation, culture and flow-cytometric characterization of primary mouse macrophages. Methods Mol. Biol. 531, 203–224 [DOI] [PubMed] [Google Scholar]
- 34. Tarakanova V. L., Leung-Pineda V., Hwang S., Yang C. W., Matatall K., Basson M., Sun R., Piwnica-Worms H., Sleckman B. P., Virgin H. W. T. (2007) Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Repetto G., del Peso A., Zurita J. L. (2008) Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 3, 1125–1131 [DOI] [PubMed] [Google Scholar]
- 36. Abel G., Szollosi J., Fachet J. (1991) Phagocytosis of fluorescent latex microbeads by peritoneal macrophages in different strains of mice: a flow cytometric study. Eur. J. Immunogenet. 18, 239–245 [DOI] [PubMed] [Google Scholar]
- 37. Rice G. C., Bump E. A., Shrieve D. C., Lee W., Kovacs M. (1986) Quantitative analysis of cellular glutathione by flow cytometry utilizing monochlorobimane: some applications to radiation and drug resistance in vitro and in vivo. Cancer Res. 46(12 Pt 1), 6105–6110 [PubMed] [Google Scholar]
- 38. Aude-Garcia C., Villiers C., Candeias S. M., Garrel C., Bertrand C., Collin V., Marche P. N., Jouvin-Marche E. (2011) Enhanced susceptibility of T lymphocytes to oxidative stress in the absence of the cellular prion protein. Cell. Mol. Life Sci. 68, 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson L. V., Walsh M. L., Chen L. B. (1980) Localization of mitochondria in living cells with rhodamine 123. Proc. Natl. Acad. Sci. U.S.A. 77, 990–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 41. Gianazza E., Celentano F., Magenes S., Ettori C., Righetti P. G. (1989) Formulations for immobilized pH gradients including pH extremes. Electrophoresis 10, 806–808 [DOI] [PubMed] [Google Scholar]
- 42. Rabilloud T., Valette C., Lawrence J. J. (1994) Sample application by in-gel rehydration improves the resolution of two-dimensional electrophoresis with immobilized pH gradients in the first dimension. Electrophoresis 15, 1552–1558 [DOI] [PubMed] [Google Scholar]
- 43. Rabilloud T., Adessi C., Giraudel A., Lunardi J. (1997) Improvement of the solubilization of proteins in two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 18, 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luche S., Diemer H., Tastet C., Chevallet M., Van Dorsselaer A., Leize-Wagner E., Rabilloud T. (2004) About thiol derivatization and resolution of basic proteins in two-dimensional electrophoresis. Proteomics 4, 551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gorg A., Postel W., Weser J., Gunther S., Strahler J. R., Hanash S. M., Somerlot L. (1987) Elimination of point streaking on silver stained two-dimensional gels by addition of iodoacetamide to the equilibration buffer. Electrophoresis 8, 122–124 [Google Scholar]
- 46. Tastet C., Lescuyer P., Diemer H., Luche S., van Dorsselaer A., Rabilloud T. (2003) A versatile electrophoresis system for the analysis of high- and low-molecular-weight proteins. Electrophoresis 24, 1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chevallet M., Luche S., Rabilloud T. (2006) Silver staining of proteins in polyacrylamide gels. Nat. Protoc. 1, 1852–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Storey J. D., Tibshirani R. (2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karp N. A., McCormick P. S., Russell M. R., Lilley K. S. (2007) Experimental and statistical considerations to avoid false conclusions in proteomics using differential in-gel electrophoresis. Mol. Cell. Proteomics 6, 1354–1364 [DOI] [PubMed] [Google Scholar]
- 50. Gharahdaghi F., Weinberg C. R., Meagher D. A., Imai B. S., Mische S. M. (1999) Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: A method for the removal of silver ions to enhance sensitivity. Electrophoresis 20, 601–605 [DOI] [PubMed] [Google Scholar]
- 51. Richert S., Luche S., Chevallet M., Van Dorsselaer A., Leize-Wagner E., Rabilloud T. (2004) About the mechanism of interference of silver staining with peptide mass spectrometry. Proteomics 4, 909–916 [DOI] [PubMed] [Google Scholar]
- 52. Haas I., Shanmugam S., Gedanken A. (2006) Pulsed sonoelectrochemical synthesis of size-controlled copper nanoparticles stabilized by poly(N-vinylpyrrolidone). J. Phys. Chem. B 110, 16947–16952 [DOI] [PubMed] [Google Scholar]
- 53. Yu W., Xie H., Chen L., Li Y., Zhang C. (2009) Synthesis and characterization of monodispersed copper colloids in polar solvents. Nanoscale Res. Lett. 4, 465–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Engels V., Benaskar F., Jefferson D. A., Johnson B. F., Wheatley A. E. (2010) Nanoparticulate copper–routes towards oxidative stability. Dalton Trans. 39, 6496–6502 [DOI] [PubMed] [Google Scholar]
- 55. Lee Y., Choi J. R., Lee K. J., Stott N. E., Kim D. (2008) Large-scale synthesis of copper nanoparticles by chemically controlled reduction for applications of inkjet-printed electronics. Nanotechnology 19(41), 415604. [DOI] [PubMed] [Google Scholar]
- 56. El Badawy A. M., Scheckel K. G., Suidan M., Tolaymat T. (2012) The impact of stabilization mechanism on the aggregation kinetics of silver nanoparticles. Sci. Total Environ. 429, 325–331 [DOI] [PubMed] [Google Scholar]
- 57. Jovanovic Z., Radosavljevic A., Kacarevic-Popovic Z., Stojkovska J., Peric-Grujic A., Ristic M., Matic I. Z., Juranic Z. D., Obradovic B., Miskovic-Stankovic V. (2013) Bioreactor validation and biocompatibility of Ag/poly(N-vinyl-2-pyrrolidone) hydrogel nanocomposites. Colloids Surf. B Biointerfaces 105, 230–235 [DOI] [PubMed] [Google Scholar]
- 58. Rogero S. O., Malmonge S. M., Lugao A. B., Ikeda T. I., Miyamaru L., Cruz A. S. (2003) Biocompatibility study of polymeric biomaterials. Artif. Organs 27, 424–427 [DOI] [PubMed] [Google Scholar]
- 59. Liu X., Xu Y., Wu Z., Chen H. (2013) Poly(N-vinylpyrrolidone)-modified surfaces for biomedical applications. Macromol. Biosci. 13, 147–154 [DOI] [PubMed] [Google Scholar]
- 60. Risbud M., Bhonde M., Bhonde R. (2001) Chitosan-polyvinyl pyrrolidone hydrogel does not activate macrophages: potentials for transplantation applications. Cell Transplant 10, 195–202 [PubMed] [Google Scholar]
- 61. Guedes M., Ferreira J. M., Ferro A. C. (2009) A study on the aqueous dispersion mechanism of CuO powders using Tiron. J. Colloid Interface Sci. 330, 119–124 [DOI] [PubMed] [Google Scholar]
- 62. Kroll A., Pillukat M. H., Hahn D., Schnekenburger J. (2009) Current in vitro methods in nanoparticle risk assessment: limitations and challenges. Eur. J. Pharm. Biopharm. 72, 370–377 [DOI] [PubMed] [Google Scholar]
- 63. Diaz B., Sanchez-Espinel C., Arruebo M., Faro J., de Miguel E., Magadan S., Yague C., Fernandez-Pacheco R., Ibarra M. R., Santamaria J., Gonzalez-Fernandez A. (2008) Assessing methods for blood cell cytotoxic responses to inorganic nanoparticles and nanoparticle aggregates. Small 4, 2025–2034 [DOI] [PubMed] [Google Scholar]
- 64. Studer A. M., Limbach L. K., Van Duc L., Krumeich F., Athanassiou E. K., Gerber L. C., Moch H., Stark W. J. (2010) Nanoparticle cytotoxicity depends on intracellular solubility: comparison of stabilized copper metal and degradable copper oxide nanoparticles. Toxicol. Lett. 197, 169–174 [DOI] [PubMed] [Google Scholar]
- 65. Diz A. P., Carvajal-Rodriguez A., Skibinski D. O. (2011) Multiple hypothesis testing in proteomics: a strategy for experimental work. Mol. Cell. Proteomics 10, M110.004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Petrak J., Ivanek R., Toman O., Cmejla R., Cmejlova J., Vyoral D., Zivny J., Vulpe C. D. (2008) Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics 8, 1744–1749 [DOI] [PubMed] [Google Scholar]
- 67. Wang P., Bouwman F. G., Mariman E. C. (2009) Generally detected proteins in comparative proteomics–a matter of cellular stress response? Proteomics 9, 2955–2966 [DOI] [PubMed] [Google Scholar]
- 68. Brunner T. J., Wick P., Manser P., Spohn P., Grass R. N., Limbach L. K., Bruinink A., Stark W. J. (2006) In vitro cytotoxicity of oxide nanoparticles: comparison to asbestos, silica, and the effect of particle solubility. Environ. Sci. Technol. 40, 4374–4381 [DOI] [PubMed] [Google Scholar]
- 69. Tsai Y. Y., Huang Y. H., Chao Y. L., Hu K. Y., Chin L. T., Chou S. H., Hour A. L., Yao Y. D., Tu C. S., Liang Y. J., Tsai C. Y., Wu H. Y., Tan S. W., Chen H. M. (2011) Identification of the nanogold particle-induced endoplasmic reticulum stress by omic techniques and systems biology analysis. ACS Nano 5, 9354–9369 [DOI] [PubMed] [Google Scholar]
- 70. Horie M., Kato H., Fujita K., Endoh S., Iwahashi H. (2012) In vitro evaluation of cellular response induced by manufactured nanoparticles. Chem. Res. Toxicol. 25, 605–619 [DOI] [PubMed] [Google Scholar]
- 71. Chevallet M., Wagner E., Luche S., van Dorsselaer A., Leize-Wagner E., Rabilloud T. (2003) Regeneration of peroxiredoxins during recovery after oxidative stress: only some overoxidized peroxiredoxins can be reduced during recovery after oxidative stress. J. Biol. Chem. 278, 37146–37153 [DOI] [PubMed] [Google Scholar]
- 72. Sato H., Ishii T., Sugita Y., Tateishi N., Bannai S. (1993) Induction of a 23 kDa stress protein by oxidative and sulfhydryl-reactive agents in mouse peritoneal macrophages. Biochim. Biophys. Acta 1148, 127–132 [DOI] [PubMed] [Google Scholar]
- 73. Kim H. S., Manevich Y., Feinstein S. I., Pak J. H., Ho Y. S., Fisher A. B. (2003) Induction of 1-cys peroxiredoxin expression by oxidative stress in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 285, L363–L369 [DOI] [PubMed] [Google Scholar]
- 74. Baraibar M. A., Hyzewicz J., Rogowska-Wrzesinska A., Ladouce R., Roepstorff P., Mouly V., Friguet B. (2011) Oxidative stress-induced proteome alterations target different cellular pathways in human myoblasts. Free Radic. Biol. Med. 51, 1522–1532 [DOI] [PubMed] [Google Scholar]
- 75. Kim S., Choi J. E., Choi J., Chung K. H., Park K., Yi J., Ryu D. Y. (2009) Oxidative stress-dependent toxicity of silver nanoparticles in human hepatoma cells. Toxicol. In Vitro 23, 1076–1084 [DOI] [PubMed] [Google Scholar]
- 76. Arora S., Jain J., Rajwade J. M., Paknikar K. M. (2008) Cellular responses induced by silver nanoparticles: In vitro studies. Toxicol. Lett. 179, 93–100 [DOI] [PubMed] [Google Scholar]
- 77. Chen Y., Shertzer H. G., Schneider S. N., Nebert D. W., Dalton T. P. (2005) Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 280, 33766–33774 [DOI] [PubMed] [Google Scholar]
- 78. Piao M. J., Kang K. A., Lee I. K., Kim H. S., Kim S., Choi J. Y., Choi J., Hyun J. W. (2011) Silver nanoparticles induce oxidative cell damage in human liver cells through inhibition of reduced glutathione and induction of mitochondria-involved apoptosis. Toxicol. Lett. 201, 92–100 [DOI] [PubMed] [Google Scholar]
- 79. Freedman J. H., Ciriolo M. R., Peisach J. (1989) The role of glutathione in copper metabolism and toxicity. J. Biol. Chem. 264, 5598–5605 [PubMed] [Google Scholar]
- 80. Karlsson H. L., Gustafsson J., Cronholm P., Moller L. (2009) Size-dependent toxicity of metal oxide particles—A comparison between nano- and micrometer size. Toxicol. Lett. 188, 112–118 [DOI] [PubMed] [Google Scholar]
- 81. Zhang H., Ji Z., Xia T., Meng H., Low-Kam C., Liu R., Pokhrel S., Lin S., Wang X., Liao Y. P., Wang M., Li L., Rallo R., Damoiseaux R., Telesca D., Madler L., Cohen Y., Zink J. I., Nel A. E. (2012) Use of metal oxide nanoparticle band gap to develop a predictive paradigm for oxidative stress and acute pulmonary inflammation. ACS Nano 6, 4349–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. O'Reilly P. J., Hickman-Davis J. M., Davis I. C., Matalon S. (2003) Hyperoxia impairs antibacterial function of macrophages through effects on actin. Am. J. Respir. Cell Mol. Biol. 28, 443–450 [DOI] [PubMed] [Google Scholar]
- 83. Chiang P. K. (1998) Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol. Ther. 77, 115–134 [DOI] [PubMed] [Google Scholar]
- 84. Min H. W., Moochhala S., Eng K. H. (2000) Adenosine and its receptor agonists regulate nitric oxide production and RAW 264.7 macrophages via both receptor binding and its downstream metabolites-inosine. Life Sci. 66, 1781–1793 [DOI] [PubMed] [Google Scholar]
- 85. Midander K., Cronholm P., Karlsson H. L., Elihn K., Moller L., Leygraf C., Wallinder I. O. (2009) Surface characteristics, copper release, and toxicity of nano- and micrometer-sized copper and copper(II) oxide particles: a cross-disciplinary study. Small 5, 389–399 [DOI] [PubMed] [Google Scholar]
- 86. Kim B. E., Nevitt T., Thiele D. J. (2008) Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 4, 176–185 [DOI] [PubMed] [Google Scholar]
- 87. Hoogland C., Mostaguir K., Sanchez J. C., Hochstrasser D. F., Appel R. D. (2004) SWISS-2DPAGE, ten years later. Proteomics 4, 2352–2356 [DOI] [PubMed] [Google Scholar]
- 88. Park M., Lynch I., Ramirez-Garcia S., Dawson K. A., de la Fonteyne L., Gremmer E., Slob W., Briede J. J., Elsaesser A., Howard C. V., van Loveren H., de Jong W. H. (2011) In vitro evaluation of cytotoxic and inflammatory properties of silica nanoparticles of different sizes in murine RAW 264.7 macrophages. J. Nanopart. Res. 13, 6775–6787 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.