

Abstract
Genome wide association studies (GWAS) identify susceptibility loci for complex traits, but do not identify particular genes of interest. Integration of functional and network information may help in overcoming this limitation and identifying new susceptibility loci. Using GWAS and comorbidity data, we present a network-based approach to predict candidate genes for lipid and lipoprotein traits. We apply a prediction pipeline incorporating interactome, co-expression, and comorbidity data to Global Lipids Genetics Consortium (GLGC) GWAS for four traits of interest, identifying phenotypically coherent modules. These modules provide insights regarding gene involvement in complex phenotypes with multiple susceptibility alleles and low effect sizes. To experimentally test our predictions, we selected four candidate genes and genotyped representative SNPs in the Malmö Diet and Cancer Cardiovascular Cohort. We found significant associations with LDL-C and total-cholesterol levels for a synonymous SNP (rs234706) in the cystathionine beta-synthase (CBS) gene (p = 1 × 10−5 and adjusted-p = 0.013, respectively). Further, liver samples taken from 206 patients revealed that patients with the minor allele of rs234706 had significant dysregulation of CBS (p = 0.04). Despite the known biological role of CBS in lipid metabolism, SNPs within the locus have not yet been identified in GWAS of lipoprotein traits. Thus, the GWAS-based Comorbidity Module (GCM) approach identifies candidate genes missed by GWAS studies, serving as a broadly applicable tool for the investigation of other complex disease phenotypes.
Genome wide association studies (GWAS)1 meta-analyses have pinpointed a number of new gene regions contributing to multifactorial diseases. GWAS typically find limited numbers of loci that contribute modestly to complex phenotypes (1), and GLGC meta-analysis of GWAS data has reached the limit of what can be expected (2) without the use of alternative strategies. Given that susceptibility loci for complex traits are unlikely to be randomly distributed in the genome (3), we might expect that the genes associated with a disease will be more likely to be present within the same pathways or functional groupings. In published cases, pathway based GWAS analysis provides an alternative approach to the dissection of complex disease traits (4, 5). In addition, nominal GWAS p values superimposed upon the human molecular network have been used to identify genes associated with multiple sclerosis (6), and the disease association protein–protein link evaluator (DAPPLE) has been used to find significant interactions among proteins encoded by genes in loci associated with other particular diseases (7). Other approaches incorporate heterogeneous molecular data such as linkage studies, cross species conservation measures, gene expression data and protein–protein interactions to better understand GWAS results (8, 9). Integrating molecular network information, pathway analyses, and GWAS data thus holds promise for identifying new susceptibility loci and improving the identification of relevant candidate genes.
If a gene is involved in a specific functional process or disease, its molecular network neighbors might also be suspected to have some role (3). In line with this “local” hypothesis, proteins involved in the same disease show a high propensity to interact (10) or cluster together (11) with each other. Interactions between variations in multiple genes, each with strong or modest effects, perturbing the same pathways or modules, may govern complex traits (3, 6). The molecular triangulation (MT) algorithm can be applied to rank seed genes according to their common disease associated neighbors, assigning closer and more connected neighbors higher values (12). Interactions between modestly associated MT genes may be indicative of coherent disease pathways or of genes conferring susceptibility to disease in a coordinated manner. The jActiveModule method (13) combines seed gene scores with biologically relevant interactions to identify network modules where perturbations causative of disease are more likely to reside. Lastly, although not yet implemented at the module level, phenotypic coherence between interacting pairs of genes has been quantified using the combination of molecular level gene to disease relationships and Medicare comorbidity data (14, 15).
We believe that GWAS significant SNPs and variants representing potential candidate genes can use the above strategies to reveal more about the missing heritability of complex phenotypes. The most important risk factors for coronary artery disease (CAD) include serum concentrations of total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and triglycerides (TG). We present a GWAS-based meta-analysis Comorbid Module (GCM) approach that uses significant (p < 5 × 10−8) GWAS signals for these four traits in the context of molecular networks to prioritize modules of disease-associated candidate genes. We evaluate our approach experimentally through allelic association and genotyping within the Malmö Diet and Cancer Cardiovascular Cohort (MDC-CC) for SNPs representing top candidate genes.
MATERIALS AND METHODS
The GWAS Comorbid Module (GCM) approach to predict lipid/lipoprotein trait candidate genes involves the following steps:
Mapping of GLGC GWAS meta-analysis SNPs to genes.
Construction of a human interactome, pooling protein interaction data from different sources.
Identification of candidate genes associated with lipid/lipoprotein traits using molecular triangulation (MT).
Identification of modules of seed and neighboring genes using the jActiveModule method (jAM).
Selection of phenotypically coherent (GCM) modules of seed and candidate genes using comorbidity analyses.
Validation of pipeline outputs (MT, jAM, and GCM) and comparison to other methods (CANDID and MetaRanker).
Selection of SNPs representing GCM candidate genes for genotyping in the MDC-CC.
In summary, we curate GWAS-based seed genes (p < 5 × 10−8), constructed an interactome, implement the MT method, filter MT candidate genes by jActiveModule results, select phenotypically coherent modules, validate the outputs of every step, and genotype SNPs representing GCM candidate genes of interest for lipid and lipoprotein traits.
(1) Mapping of GLGC GWAS Meta-analysis SNPs to Genes
The GLGC GWAS meta-analyses data is based on 46 lipid/lipoprotein GWAS involving over 100,000 individuals of European descent as ascertained in the United States, Europe, or Australia (16). The GLGC consortium contributed genome wide analysis data for analyses, including ∼2.6 million genotyped or imputed SNPs associated with four traits (TC, LDL-C, HDL-C and TG). The entire set of HapMap phase III SNPs and pairwise LD estimates (Release 27) for the CEU population was downloaded, and LD pruning and SNP to gene mapping was performed as described previously (3). If a SNP could be mapped to more than one gene, all genes were included, and SNPs located in gene desert regions were excluded from our analysis. To be sure of the robustness of our results, we also annotated SNPs using the ProxyGeneLD tool (17) and found similar SNP to gene annotations. Genes representing SNPs with GWAS significant p values of less than 5 × 10−8 were used as “seed” genes.
(2) Construction of a Human Interactome, Pooling Interaction Data From Different Sources
We created a human interactome consisting of proteomic, transcriptional, and metabolic interactions. Protein-protein interactions from three high-throughput yeast-two-hybrid datasets were combined with the binary subset of interactions reported in the IntAct and MINT databases (18–22). Together, these data sets describe 15,315 interactions between 6101 gene-coding proteins. For regulatory interactions, we used the TRANSFAC database version 2008.2, which included 1340 links between 271 human transcription factors and their 564 targets (23). Metabolic coupling interactions were derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the Biochemically, Genetically, and Genomically structured genome scale metabolic network reconstruction (BiGG) database as described in (15); 10,642 such metabolic links between 921 enzymes were included. The union of these sets of interactions yielded an interactome of 7117 (N) proteins and 21,964 (M) links, with an average shortest path length <l> of 4.52.
(3) Identification of Candidate Genes Associated With Lipid/Lipoprotein Traits Using Molecular Triangulation (MT)
MT begins with sets of seed (disease) genes known to be associated with phenotypes and suggests additional disease genes, typically network neighbors of multiple seed genes (12, 24). Here, we used the strengths of GWAS signals for lipoprotein traits and SNP to gene mappings to assign primary evidence scores to seed genes. MT used these primary evidence scores and the position of these seed genes within the interactome to calculate secondary evidence scores for neighbors (12). To calculate the significance of the secondary evidence scores, we performed 1000 degree-preserving network randomizations. The significance of the scores was then calculated as described by Iossifov et al. (supplementary material) (24). We applied Benjamini and Hochberg false discovery rate (FDR) corrections with a 0.05 FDR threshold to our predictions; this meant that we expected less than 5% of our predictions to be false positives.
(4) Identification of Modules of Seed and Neighboring Genes Using the jActiveModule Method (jAM)
The MT method results in a large number of statistically significant predictions, but some of the predictions may be artifacts of low or excessive connectivity (24). To address this concern, we independently implemented the jActiveModule method to determine modules with maximal proportions of the lowest p value genes. Later we pruned the MT gene sets to only include genes that were within these modules.
The jActiveModule method uses GWAS association p values of seed genes and interactome context to produce aggregated module scores. The method compares real network modules to those derived from to 10,000 matched randomized network Monte-Carlo simulations (13). To examine the effect of the GWAS signal strength distribution by itself, we compared the real module scores to distributions based on randomized gene to trait association p values. Matched numbers of seed genes were chosen from the set annotated by NCBI (Ver. 36) and from the set described by the Online Mendelian Inheritance in Man database (OMIM, December 2009 release). The differences between outputs after either randomization strategy are described in supplementary material.
As additional controls for the jActiveModule results, we implemented the Molecular COmplex DEtection (MCODE) algorithm, the Markov Clustering algorithm (MCL), and the Klein-Ravi Steiner tree algorithm with submodule detection using MCL (GenRev package (25)). Parameters for the MCL and MCODE algorithms were adopted from a previous study (26). To compare the results from different approaches, we used the Jaccard similarity (J) between the sets of seed genes (S) and putative module genes (T) determined using each method:
where, |S∩T| is the intersection of sets S and T and |S∪T| is the union).
(5) Selection of Phenotypically Coherent (GCM) Modules of Seed and Candidate Genes Using Comorbidity Analyses
To further rank the modules, we used OMIM gene-disease associations to perform analyses of comorbidity based on the co-occurrence of ICD-9 codes taken from a 13 million patient Medicare data set (14). OMIM diseases were manually mapped to ICD9 codes so that interactions between genes could be supported by comorbidity between their associated diseases. To quantify comorbidity, Relative Risk (RR) scores were calculated for every pairwise combination of diseases associated with at least one of the genes in the module:
 |
C12 = number of patients who had both disease 1 and 2
P1 = number of patients who had disease 1
P2 = number of patients who had disease 2
np = 13,039,018 (total number of patients)
Lower and upper bounds (lb and ub) of 99% confidence intervals were calculated according to the Katz et al. method (24):
where σ is given by:
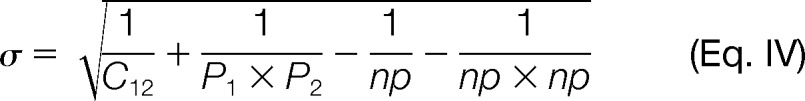 |
The relative risk was taken to be significant when the 99% confidence interval did not include the expected value of one, which would indicate findings of no consequence. To summarize the pairwise comorbidities for each module and rank them, we averaged the pairwise RR scores between associated ICD9 disease codes, creating a module relative risk (mRR) score. A Mann-Whitney U test was used to compare the observed mRR scores to those of 100 randomly constructed modules.
(6) Validation of Pipeline Outputs (MT, jActiveModule and GCM) and Comparison to Other Methods (CANDID and MetaRanker)
We validated the MT, jActiveModule and GCM steps using data from Teslovich et al. (1) as a benchmarking set. Several measures of predictive power were used: (1) precision [TP/(TP+FP)], (2) specificity [TN/(TN+FP)], and (3) accuracy [(TP+TN)/(TP+FP+FN+TN), where TP is number of true positives or candidate genes correctly identified as disease genes, TN is number of true negatives or correctly identified nondisease genes, FP is number of false positives or nondisease genes identified as candidate genes, and FN is number of false negatives of disease associated genes that were not identified as candidates. We evaluated the functional coherence of candidate genes relative to seed genes by comparing their enrichment, as a set, for functional annotations. These allowed us to evaluate the consistency of candidate gene sets with respect to phenotypically similar diseases (27). After determining the GO biological process terms enriched within the sets of seed genes, we tested the enrichment of these terms in candidate genes.
To perform a comprehensive comparison of GCM to other methods, we implemented MetaRanker (8) and CANDID (9). MetaRanker predicts candidate genes by integrating complementary layers of protein interaction, linkage, GWAS, differential expression and disease data. These five different data sources are integrated into a single meta-evidence rank, quantifying the likelihood of genes being involved in a disease of interest (8). CANDID is designed to rank candidate genes by eight evaluation criteria, considering associated publications, protein domains, conservation, expression, interactions, linkages, SNP associations, and custom data (9). We compared the top 200 MetaRanker and CANDID candidate genes to the outputs of the MT, jActiveModule and comorbidity analysis steps. We then benchmarked candidate gene sets sharing GO terms with the seed genes, computing precision, specificity and accuracy as described previously for the candidate genes from each step of the GCM method as well as the CANDID and MetaRanker outputs.
In addition, using GeneWanderer, we tested how parsimoniously candidate genes from the GCM approach were involved in obesity, which is known to be related to lipid/lipoprotein traits. GeneWanderer was provided with genomic locations 1Mb in either direction of SNPs representing GCM genes and ranked candidate genes within these windows according to their single shortest path through the STRING (28) network to obesity genes.
(8) Selection of SNPs representing GCM candidate genes for genotyping in the MDC-C
GCM genes with the strongest co-expression correlations to the seed genes were selected for genotyping. Genome wide mRNA expression data of 79 human tissues were obtained from the Gene Expression Atlas (29). Spearman's test was used to assess correlation between GCM gene and seed gene mRNA expression, with the criterion for significance set at Rho>0.5 and p < 0.05. The second criteria considered was sequence conservation of the regions in which SNPs were located, as alterations at conserved sites have more drastic functional effects when changed (30) The third criteria considered was the position of the SNPs relative to the candidate genes. Among conserved SNPs with GLGC GWAS p values <0.05, we used following hierarchy to rank importance for further genotyping: coding>intronic>5′UTR>3′UTR>5′upstream>3′upstream (31).
Study Population: The Malmö Diet and Cancer Cardiovascular cohort (MDC-CC)
The Malmö Diet and Cancer (MDC) study is a community-based prospective cohort of 28,449 persons originally recruited for baseline examination between 1991 and 1996 (32, 33). From this cohort, 6103 persons were randomly selected to participate in the Cardiovascular Cohort (MDC-CC), which seeks to investigate risk factors for cardiovascular disease. All participants underwent questioning regarding their medical history, a physical examination, and a laboratory assessment for cardiovascular risk factors. In fasting venous blood samples, TC, HDL-C, and triglyceride levels were measured according to standard procedures by the Department of Clinical Chemistry at University Hospital Malmö. Levels of LDL-C were calculated according to Friedewald's formula, with the assignment of missing values to subjects with a triglyceride level of more than 4.5 mmol per liter. DNA was available from 5763 individuals for genotyping, and of these individuals, lipid levels were available for 5056 individuals that were not on lipid lowering medication. The ethics committee of Lund University approved the MDC-CC study protocols, and all participants provided written informed consent.
Genotyping
Genotyping of the selected SNPs was performed using genomic DNA from 5763 individuals using the allelic discrimination method using an ABI 7900 instrument (Applied Biosystems, Foster City, CA). Samples that were successfully genotyped for at least 50% of the SNPs were included in further analyses (n = 5698). We confirmed that the genotypes were at Hardy-Weinberg equilibrium. The overall genotyping success rate was 98.2%. For this epistasis analysis, we created a variable indicating how many risk alleles (increasing total cholesterol, LDL-cholesterol or triglycerides and/or lowering HDL cholesterol) each individual in the population cohort was carrying, i.e. summing up the number of risk-alleles to a variable “risk-allele score.” For the four SNPs, the theoretical maximal number of risk-alleles was eight (for individuals homozygous for risk alleles of all four SNPs) and the minimum was zero. In MDC-CC cohort, individuals ranged from zero to six risk alleles, and the risk-allele score was used as a variable in a linear regression analysis adjusting for age and sex to analyze if the combined effect of the four SNPs resulted in an association with lipid levels.
Expression Quantitative Trait (eQTL) Analysis
RNA extracted from the livers of 206 patients patients undergoing aortic valve surgery and/or surgery for aortic aneurysms (34) was hybridized to Affymetrix ST 1.0 Exon arrays. DNA extracted from circulating blood cells was hybridized to Illumina 610w-Quad BeadArrays. The association was tested using a linear additive model with corrections for age and gender. The average age of patients was 63.9 ± 11.8 years, with average total cholesterol levels of 5.05 ± 1.09 mm and average LDL-C levels of 3.11 ± 0.93 mm. None of the patients were known to have liver disease.
Gene Set Enrichment for Biological Pathways
To find statistically over-represented Gene Ontology (GO) annotations for candidate genes at each of the analysis steps, we used the Biological Networks Gene Ontology tool (BiNGO) implemented in Cytoscape. Enrichment analyses were performed using a hyper-geometric test followed by a Benjamini and Hochberg multiple hypothesis correction with a 0.05 FDR threshold (35). Odds ratios to measure the magnitudes of the enrichment were calculated using raw BiNGO outputs.
RESULTS
Introducing a network-based integrative approach, we identified novel candidate genes for lipid and lipoprotein traits (Fig. 1).
Fig. 1.
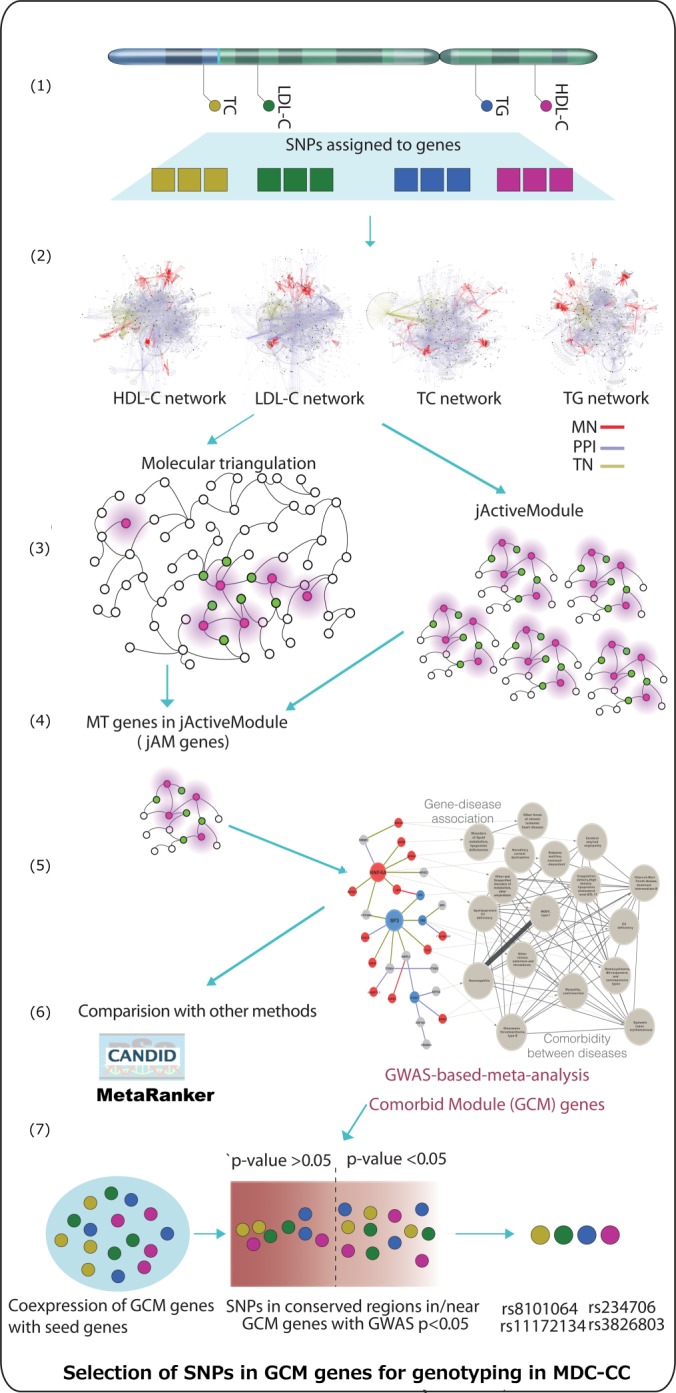
Schematic representation of GCM approach. (1): Mapping of GLGC GWAS meta-analysis SNPs to genes. (2): Construction of a human interactome by pooling protein interaction data from different sources and mapping seed genes within the network. (3): Identification of candidate genes associated with lipid/lipoprotein traits using molecular triangulation (MT). (4): Identification of seed and neighbouring gene modules using the jActiveModule (jAM) method, pruning of MT candidate gene sets. (5): Selection of phenotypically coherent (GCM) modules of seed and candidate genes using comorbidity analyses. (6): Validation of MT, jAM and GCM gene set outputs and comparison to CANDID and MetaRanker methods. (7): Selection of SNPs, representing GCM candidate genes, for genotyping in the MDC-CC. GCM genes were prioritized based on their co-expression with seed genes and hierarchical criteria including the genomic locations of SNPs, if the SNPs were synonymous variations, if the SNPs were in conserved regions of the genome, and GLGC GWAS-meta-analysis p-values (p < 0.05).
Prediction of Lipid/Lipoprotein Trait Candidate Genes Using Molecular Triangulation (MT)
We used the MT method to identify phenotypically related candidate genes (MT-genes) based on their proximity within the interactome to seed genes associated with HDL-C, LDL-C, TG and TC traits (supplemental Table S1). The MT method had an accuracy of 98% in classifying true positives and true negatives for the four traits, with 33% precision and 98% specificity (Fig. 2).
Fig. 2.
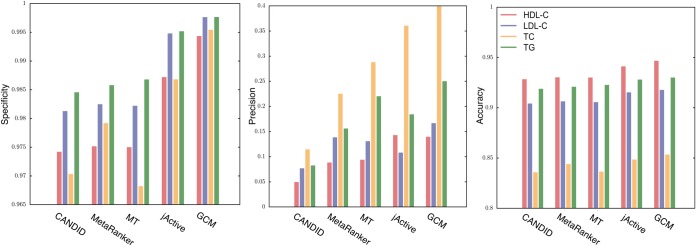
Performance of CANDID, MetaRanker, MT, jActiveModule and GCM with respect to benchmarking dataset. The histograms show an increase in specificity, precision and accuracy with each of the steps.
Refinement of Candidate Gene Sets Using the jActiveModules Method
Although direct interactions can be used to identify candidate genes, modules in biological networks represent connected components contributing to cellular functions in a coordinated manner. Disruptions of these modules, which include both identified and unidentified disease genes, result in disease phenotypes (36). To avoid method specific biases, we implemented the jActiveModule algorithm in parallel with MT and used the intersection of the two result sets to identify more cohesive modules of seed and MT genes. Comparision of MT and jAM genes, based on their degree of connectivity, indicated that only retaining genes within the intersection of the two groups removed a significant number of the nodes having only one or greater than one hundred connections. (p = 0.0017, odds ratio = 2.01, supplementry Fig. S1). Furthermore, after the jActiveModule filtering step, precision increased from 33% to 44%, with 99% specificity and accuracy (Fig. 2).
The jActiveModule method uses GWAS association p values of seed genes within the interactome context to produce aggregated module scores, and these scores determined the extent to which randomized inputs could create coherent modules. Confirming the usefulness of the network context using randomized controls, the top 20 jActiveModule subnetwork scores were significantly higher than those from 100 randomization controls for each of the four traits (p ≤ 0.001, supplemental Fig. S2).
Compared with the Steiner tree-MCL approach (supplemental Fig. S3), the jActiveModule algorithm identified modules with greater percentages of seed genes. Gene sets identified by jActivemodule were smaller and localized more tightly around seed genes within the interactome. This was likely because of jActivemodule's flexible search for multiple modules, as opposed to the Steiner tree based method's strategy of attempting to find a single module connecting all of the seed genes in the interactome (37). Comparison with conventional clustering methods such as MCL and MCODE suggest that these methods are more suited to the identification of individual protein complexes (37) as well, while the jActiveModule method is more suited to the identification of multiple modules of seed genes spread throughout the interactome.
Retention of Modules According to Comorbidity Analyses Using Medicare Data (GCM)
To determine those modules with the most phenotypically coherent associations, we quantified the strengths of comorbidities between diseases associated with their genes. We implemented mRR scores, as explained in the methods section, because we believed that co-occurring diseases might be driven by related molecular machinery. We found 48 comorbid modules with mRR scores higher than one for HDL-C, (mean mRR score of 1.8, average P∼0.002), 15 modules for LDL-C, (mean mRR score of 2.9, average P∼0.001), 15 modules for TC, (mean mRR score of 2.8, average P∼0.05 and 23 modules for TG, (mean mRR score of 1.8, average P∼0.001). (Table I). Filtering the modules to only include thoses with above average mRR scores, precision increased from 44% to 55%, with 99% specificity and accuracy (Fig. 2). ICD-9 codes associated with the retained GCM modules in all four of the primary traits included lipid metabolism, carbohydrate and transport metabolism, amino acid transport metabolism, being overweight, essential hypertension, cardiomyopathy, symptoms concerning nutrition, chronic ischemic heart disease, acute mycordial infraction, and diabetes mellitus (supplemental Fig. S4).
Table I. Mean relative risk (mRR) scores of the network modules. TC trait had the highest mRR score among the four. mRR-score is given as mean ± S.D.
| Trait | Number of modules | mRR-Score | *p value |
|---|---|---|---|
| HDL-C | 157 | 1.8 ± 1.0 | ∼0.002 |
| LDL-C | 64 | 2.9 ± 2.5 | ∼0.001 |
| TC | 89 | 2.8 ± 2.4 | 0.03 |
| TG | 85 | 1.8 ± 0.6 | ∼0.001 |
In addition to highlighting the phenotypic cohesiveness of the final GCM gene sets using ICD9 codes, the progressive benefit of the filtering steps was also quantified using GO term enrichment tests of the gene sets found at each step. We found that filtering MT genes by jActive module membership and only retaining modules with the most significant comorbidities yielded enrichments of progressively more specific GO terms annotating fewer genes (Fig. 3A). The cost of this was that the numbers of genes contributing to particular GO term enrichments decreased as genes were filtered away (Fig. 3B), and that except for the TG set of genes, fewer GO terms were detected (Fig. 3C) This filtered subset of more specific GO terms, however, displayed a trend of increasingly drastic effect size as measured by odds ratios of enrichment (Fig. 3D).
Fig. 3.
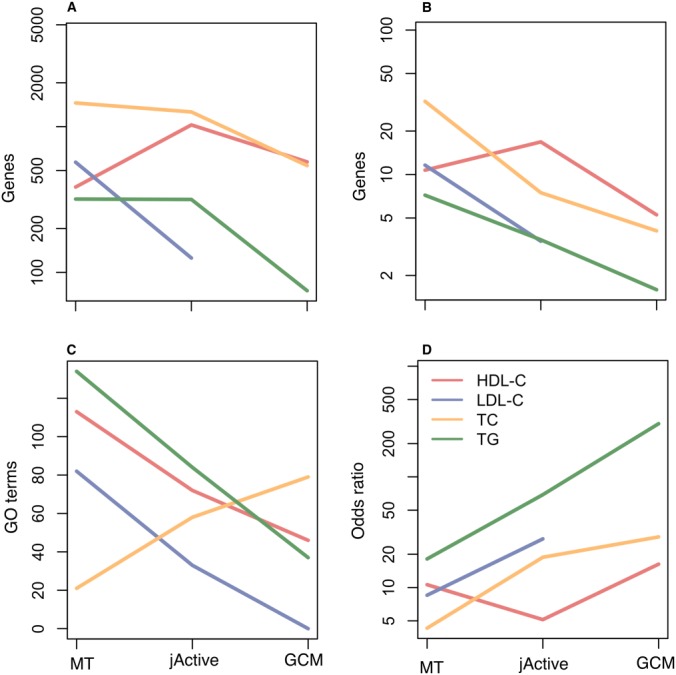
GO term enrichments for gene sets. A, Mean numbers of genes, anywhere in the genome, associated with GO terms for which significant enrichments were found. B, Mean numbers of genes, within the sets of genes being tested, found to be associated with GO terms for which the gene sets were enriched. C, Counts of GO terms for which sets of candidate genes were enriched. D, Median odds ratios of GO term enrichment as a measure of enrichment effect size.
Validation of Pipeline Outputs (MT, jActiveModule and GCM) and Comparison to Other Methods (CANDID and MetaRanker)
Enrichment for GO term effect sizes (supplemental Fig. S5) and functional coherence of candidate and seed gene sets (Fig. 2) for the MT, jActiveModule, and GCM methods was greater than that of MetaRanker (8) and CANDID (9). Comparing overlaps between the candidate gene sets (Fig. 4), we found that predictions from CANDID and MetRanker had 9% (p < 0.0001) overlap with each other. Each step in our method resulted in greater overlap with the consensus set of genes predicted by both CANDID and MetaRanker (supplemental Table S2). However, given that the maximum overlap between any pairwise combination of the gene sets was 15% (CANDID and MetaRanker versus TC-GCM), we were still assured of the complementarity of each of the gene sets.
Fig. 4.
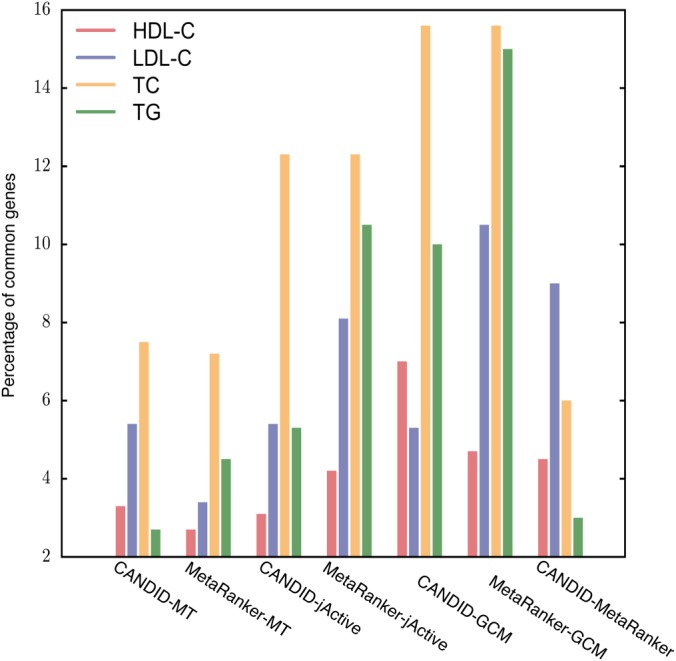
Percentage of overlapping candidate genes between CANDID or MetaRanker and each of the GCM steps.
We further evaluated our results using DAPPLE (7) and GRAIL (38). DAPPLE looks for significant protein-protein interaction connectivity among proteins encoded by genes in loci associated with disease (7). GRAIL describes the degree of functional connectivity between regions using literature based relationships between genes (38). Our method had 31% similarity to DAPPLE prioritized candidate genes when the same seed genes were used, and 18% similarity to GRAIL results.
Because of GeneWanderer's top rating among network based approaches for gene prioritization (39), we used it to rank GCM genes with respect to the polygenic trait of obesity. GeneWanderer identified 48 of the 51 GCM genes as highly ranked candidates in genomic locations related to obesity. In our interactome, these 48 genes were immediate neighbors of genes within loci identified by GLGC GWAS (p < 0.0001) (supplemental Fig. S6).
Relevance of GCM Genes to Lipid Related Diseases Based on Literature
Many of the GCM candidate genes associated with all four traits have been linked to lipid metabolism, cardiovascular disease and coronary artery disease (supplemental Table S3). Retinoid x-receptor alpha (RXRA) variant rs11185660 has been associated with low HDL-C and coronary heart disease (40, 41). TG and nonesterified fatty acid (NEFA) levels were increased in the livers and serum of cysthathionine-beta-synthase (CBS) knock out mice (41). Deletion of the four and a half LIM domains 2 (FHL2) gene attenuates the formation of atherosclerotic lesions normally present with a cholesterol-enriched diet (42).
Selection of SNPs for Genotyping in the MDC-CC
Because we expected elements of phenotype-specific modules to act cooperatively, we tested whether GCM genes were co-expressed with seed genes. Most of the GCM genes (90%) were significantly co-expressed with at least one of the seed genes (supplemental Table S1). Within the HDL-C, LDL-C, TC and TG GCM gene sets, 19, 9, 13 and 12 genes were co-expressed with seed genes having GWAS p values <0.05. As evolutionary conservation of genomic regions implies greater biological significance (30), we prioritized co-GCM genes represented by SNPs in evolutionarily conserved regions for further genotyping in MDC-CC cohort (Table III). The numbers of SNPs representing co-GCM genes in conserved regions for HDL-C, LDL-C, TC and TG traits were 11, 4, 4, and 6, respectively (Table II). The SNPs with the lowest GLGC GWAS p values in each of these gene groups were genotyped in the 5763 MDC-CC participants (Table III, Table IV).
Table III. Characteristics of the subjects in the Malmö Diet and Cancer Cardiovascular cohort.
| Clinical character | All (n = 5056) |
|---|---|
| Males/Females N (%) | 2054 (40.6)/3002 (59.4) |
| Age (years) | 57.5 ± 5.9 |
| Body mass index (kg/m2) | 25.8 ± 3.9 |
| HDL-cholesterol (mmol/l) | 1.4 ± 0.4 |
| LDL-cholesterol (mmol/l) | 4.2 ± 1.0 |
| Total cholesterol (mmol/l) | 6.2 ± 1.1 |
| Triglycerides (mmol/l) | 1.4 ± 0.8 |
| Diabetes N (%) | 416 (8.2) |
Table II. Prioritized co-GCM genes represented by SNPs in evolutionarily conserved regions for further genotyping in Malmö Diet and Cancer Cardiovascular cohort (MDC-CC) cohort.
| Trait | Gene | SNP | GLGC p-vlaue | Variant type/location | DAPPLE | Genewanderer ranking | GRAIL p value |
|---|---|---|---|---|---|---|---|
| HDL-C | INSR | rs8101064 | 2.03E-05 | INTRONIC | ✓ | 1 | 0.9 |
| ASCC2 | rs140147 | 0.0006 | SPLICE_SITE | 3 | 0.99 | ||
| CYP3A4 | rs12721617 | 0.002 | INTRONIC | 1 | 0.031 | ||
| APP | rs380713 | 0.003 | INTERGENIC | 1 | 0.3 | ||
| SMURF2 | rs6504248 | 0.006 | INTERGENIC | 1 | 0.96 | ||
| EHMT2 | rs9267659 | 0.008 | INTRONIC | 7 | 4.92E-11 | ||
| SKIL | rs6763533 | 0.009 | INTRONIC | 1 | 0.96 | ||
| PSMA1 | rs12362721 | 0.02 | INTRONIC | 3 | 0.126 | ||
| TERT | rs6554691 | 0.02 | INTRONIC | 1 | 0.636 | ||
| RALYL | rs6473532 | 0.03 | INTERGENIC | NA | 0.956 | ||
| FASN | rs6502051 | 0.04 | INTRONIC | 1 | 0.45 | ||
| LDL-C | NDUFA4L2 | rs11172134 | 0.0003 | UPSTREAM | ✓ | NA | 0.0006 |
| CDK5RAP2 | rs3739822 | 0.0008 | SYNONYMOUS_CODING | ✓ | 5 | 0.65 | |
| ITGB3BP | rs6588048 | 0.003 | INTRONIC | 3 | 0.98 | ||
| SH3GL3 | rs8025427 | 0.03 | INTRONIC | 1 | 0.48 | ||
| TC | CBS | rs234706 | 0.007 | SYNONYMOUS_CODING | 2 | 0.13 | |
| ASAP1 | rs7462286 | 0.02 | INTRONIC | ✓ | 1 | 0.42 | |
| ITSN1 | rs9984662 | 0.03 | 3PRIME_UTR | 13 | NA | ||
| EXOSC10 | rs11583740 | 0.03 | INTRONIC | 5 | 0.75 | ||
| TG | DNM2 | rs3826803 | 0.003 | INTRONIC | 5 | 0.85 | |
| HNF4A | rs3212198 | 0.003 | INTRONIC | 1 | 0.22 | ||
| PAFAH1B3 | rs3826706 | 0.02 | INTRONIC | ✓ | 5 | 0.7 | |
| COPS6 | rs2307345 | 0.02 | INTRONIC | 5 | 0.43 | ||
| ATP6V1E1 | rs3532 | 0.02 | 3PRIME_UTR | 3 | 0.86 | ||
| NR2F2 | rs4310804 | 0.04 | INTERGENIC | 2 | NA |
Table IV. Association of selected GCM gene SNPs with their respective traits in MDC-CC. A synonmyous SNP (rs234706) in CBS gene was significantly associated with the TC trait in MDC-CC.
| Trait | N | SNP (Chr) | Gene | Minor Allele (frequency, %) | Beta (S.E.) | p value |
|---|---|---|---|---|---|---|
| HDL-C | 4916 | rs8101064 (19) | INSR | T (3.9) | −0.001 (0.001) | 0.50 |
| LDL-C | 4822 | rs11172134 (12) | NDUFA4L2 | A (23.7) | 0.003 (0.02) | 0.91 |
| TC | 4962 | rs234706 (21) | CBS | A (45.9) | 0.06 (0.02) | 0.0032 |
| TG | 4933 | rs3826803 (19) | DNM2 | C (35.0) | 0.004 (0.01) | 0.68 |
Logistic regression analysis of the four SNPs revealed that the minor A-allele of the synonymous SNP rs234706 in CBS (Y233Y) was significantly associated with higher total cholesterol levels than the G-allele (p = 0.013 after Bonferroni correction, Table IV). The A-allele also associated significantly with higher LDL-C (p = 0.00001) and TG (p = 0.04) levels. The three other SNPs did not associate significantly with their respective traits. Despite this, we found that the combined effect of all four risk-alleles was nominally significant for an association with lower HDL-cholesterol and higher triglyceride levels (p = 0.041 and 0.026, respectively, Supplemental Table 4). No evidence for pairwise epistasis between the SNPs was found.
Expression of the CBS gene was associated with the directly measured rs234705 SNP, which served as a perfect proxy for rs234706 (HapMap CEU LD of R2 = 1). The rs234706 SNP was genotyped in the GLGC GWAS, and an association between CBS gene expression and the rs234706 genotype was determined (43). The minor allele of SNP rs234706 was significantly associated with mRNA levels of CBS in the 206 liver biopsy samples (p = 0.04). In the disease to gene mappings, homocystinuria, venous embolisms, and thrombosis were associated with the CBS gene (Fig. 5). Coagulation defects, Diabetes mellitus and Charcot-Marie-Tooth disease were associated with other genes in the CBS GCM module, and comorbidities were found between these diseases (with all disease pairs having RR >1, Fig. 5). We also found genes associated with homocystinuria and APOA1 associated amyloidosis within the CBS GCM module. These comorbid diseases have a RR score of 6.4, and the relationship between TC, CBS, homocystinuria, APOA1, and amyloidosis is supported by the observation that plasma cholesterol and APOA1 are significantly decreased in homozygous CBS-deficient mice (44).
Fig. 5.

GCM module with CBS gene and the associated diseases. Combination schema including protein-protein interactions (purple), metabolic interactions (red), and transcriptional interactions (yellow), gene-disease associations (dashed black), and relative risk associations between diseases greater with magnitude greater than 1 (black line). Seed genes (red ovals), CBS GCM genes (dark blue ovals) and diseases (gray) are linked within a highly interconnected module that includes Homocystinuria, venous embolism and thrombosis diseases associated with CBS gene in OMIM.
DISCUSSION
Although the human interactome is far from complete, merging network topological features with heterogeneous GWAS data provides experimentally verifiable insights into complex biological traits. Unlike approaches that test genes in GWAS identified loci for overrepresentation in pathways (3), our approach uses network context to prioritize specific candidate genes. The improvement in the precision of our predictions, related to the high coverage of seed genes by our modules, is supported by coherent gene-disease and comorbidity associations. This highlights how seemingly unrelated diseases may be the product of complex combinations of shared molecular mechanisms. We believe that this allows our three step procedure to compete with more established methods such as CANDID and MetaRanker, and to capture additional candidate genes missed by other methods (Table II).
The GCM approach prospectively allows us to use nominally significant GWAS p values in the hunt for missing heritability while minimizing spurious hits. Many of our prioritized candidates for lipid traits are related to cardiovascular and coronary artery disease in the literature (40, 42, 45) (supplemental Table S3), providing a common sense measure of the usefulness of our raw data and methodology.
The significant association of the synonymous SNP rs234706 within the CBS gene to the TC and LDL-C traits, together with the association between the synonymous SNP and variable CBS mRNA levels in the liver (likely because of linkage disequilibrium with the gene's transcriptional regulatory elements (46)) suggests that CBS expression levels are related to aberrant lipid profile traits in humans. It has been shown that CBS knockout mice have altered distributions of cholesterol and triglyceride lipoprotein fractions, and that mutations in the CBS gene cause altered lipoprotein metabolism as well as hyperhomocysteinemia (47). This finding demonstrates the usefulness of the GCM approach in selecting lipid and lipoprotein trait associated candidate genes.
Population level disease comorbidity between genes revealed interconnected complex phenotypes. Integrating lipid interactome data with patient medical records uncovered molecular associations for diseases unexpectedly comorbid with lipid related disorders. Despite the incompleteness of current protein-protein interactions and our incomplete knowledge of disease gene associations, the GCM method validated in one of the four SNPs tested. This 25% validation success rate surpasses that of other candidate gene prediction methods (8, 48, 49).
Although the GCM approach has been demonstrated using GWAS of lipid traits, it can be used to interpret GWAS of other traits as well. By capturing phenotypically coherent modules of candidate and seed genes, the GCM approach provides insights regarding involvement in complex phenotypes with multiple susceptibility alleles and low effect sizes. In this way, GCM as well as other network-based approaches may be of broad use in dissecting complex diseases in the coming era of systems medicine.
Supplementary Material
Acknowledgments
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Sekar Kathiresan for providing access to Global Lipids Genetics Consortium, GLGC- GWAS data. We are thankful to Iossifov for providing the program to run molecular traingulation algorithm.
Footnotes
* This study is supported by the Swedish Research Council, the Swedish Heart and Lung Foundation, the Region Skåne, the Skåne University Hospital, the Novo Nordic Foundation, the Albert Påhlsson Research Foundation, the Crafoord foundation equipment grant from the Knut and Alice Wallenberg Foundation and by Linnéus for the Lund University Diabetes Center (LUDC). MOM is a senior scientist at the Swedish Research Council. This work was supported by National Institutes of Health (NIH) grants P50-HG004233 CEGS.
 This article contains supplemental Figs S1 to S6 and Tables S1 to S4.
This article contains supplemental Figs S1 to S6 and Tables S1 to S4.
Conflict of interest: The authors declare that they have no conflict of interest.
1 The abbreviations used are:
- GWAS
- Genome wide association studies
- SNP
- single nucleotide polymorphism
- GO
- Gene Ontology
- GCM
- GWAS-based-meta analysis Comorbid Module
- GLGC
- Global Lipids Genetics Consortium
- MT
- molecular triangulation
- KEGG
- Kyoto Encyclopaedia of Genes and Genome
- BIGG
- biochemically, genetically, and genomically structured genome scale metabolic network reconstruction
- eQTL
- Expression quantitative trait loci.
REFERENCES
- 1. Manolio T. A., Collins F. S., Cox N. J., Goldstein D. B., Hindorff L. A., Hunter D. J., McCarthy M. I., Ramos E. M., Cardon L. R., Chakravarti A., Cho J. H., Guttmacher A. E., Kong A., Kruglyak L., Mardis E., Rotimi C. N., Slatkin M., Valle D., Whittemore A. S., Boehnke M., Clark A. G., Eichler E. E., Gibson G., Haines J. L., Mackay T. F., McCarroll S. A., Visscher P. M. (2009) Finding the missing heritability of complex diseases. Nature 461, 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hegele R. A. (2010) Genome-wide association studies of plasma lipids: have we reached the limit? Arterioscler. Thromb. Vasc. Biol. 30, 2084–2086 [DOI] [PubMed] [Google Scholar]
- 3. Holmans P., Green E. K., Pahwa J. S., Ferreira M. A., Purcell S. M., Sklar P., Owen M. J., O'Donovan M. C., Craddock N. (2009) Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am. J. Hum. Genet. 85, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang K., Zhang H., Kugathasan S., Annese V., Bradfield J. P., Russell R. K., Sleiman P. M., Imielinski M., Glessner J., Hou C., Wilson D. C., Walters T., Kim C., Frackelton E. C., Lionetti P., Barabino A., Van Limbergen J., Guthery S., Denson L., Piccoli D., Li M., Dubinsky M., Silverberg M., Griffiths A., Grant S. F., Satsangi J., Baldassano R., Hakonarson H. (2009) Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn Disease. Am. J. Hum. Genet. 84, 399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhong H., Yang X., Kaplan L. M., Molony C., Schadt E. E. (2010) Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am. J. Hum. Genet. 86, 581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baranzini S. E., Galwey N. W., Wang J., Khankhanian P., Lindberg R., Pelletier D., Wu W., Uitdehaag B. M., Kappos L., Polman C. H., Matthews P. M., Hauser S. L., Gibson R. A., Oksenberg J. R., Barnes M. R. (2009) Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 18, 2078–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rossin E. J., Lage K., Raychaudhuri S., Xavier R. J., Tatar D., Benita Y., Cotsapas C., Daly M. J. (2011) Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 7, e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pers T. H., Hansen N. T., Lage K., Koefoed P., Dworzynski P., Miller M. L., Flint T. J., Mellerup E., Dam H., Andreassen O. A., Djurovic S., Melle I., Børglum A. D., Werge T., Purcell S., Ferreira M. A., Kouskoumvekaki I., Workman C. T., Hansen T., Mors O., Brunak S. (2011) Meta-analysis of heterogeneous data sources for genome-scale identification of risk genes in complex phenotypes. Genet. Epidemiol. 35, 318–332 [DOI] [PubMed] [Google Scholar]
- 9. Hutz J. E., Kraja A. T., McLeod H. L., Province M. A. (2008) CANDID: a flexible method for prioritizing candidate genes for complex human traits. Genet. Epidemiol. 32, 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hartwell L. H., Hopfield J. J., Leibler S., Murray A. W. (1999) From molecular to modular cell biology. Nature 402, C47–52 [DOI] [PubMed] [Google Scholar]
- 11. Goh K. I., Cusick M. E., Valle D., Childs B., Vidal M., Barabási A. L. (2007) The human disease network. Proc. Natl. Acad. Sci. U. S. A. 104, 8685–8690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krauthammer M., Kaufmann C. A., Gilliam T. C., Rzhetsky A. (2004) Molecular triangulation: bridging linkage and molecular-network information for identifying candidate genes in Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A.101, 15148–15153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ideker T., Ozier O., Schwikowski B., Siegel A. F. (2002) Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18, S233–240 [DOI] [PubMed] [Google Scholar]
- 14. Hidalgo C. A., Blumm N., Barabási A. L., Christakis N. A. (2009) A dynamic network approach for the study of human phenotypes. PLoS Comput. Biol. 5, e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee D. S., Park J., Kay K. A., Christakis N. A., Oltvai Z. N., Barabási A. L. (2008) The implications of human metabolic network topology for disease comorbidity. Proc. Natl. Acad. Sci. U.S.A. 105, 9880–9885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Teslovich T. M., Musunuru K., Smith A. V., Edmondson A. C., Stylianou I. M., Koseki M., Pirruccello J. P., Ripatti S., Chasman D. I., Willer C. J., Johansen C. T., Fouchier S. W., Isaacs A., Peloso G. M., Barbalic M., Ricketts S. L., Bis J. C., Aulchenko Y. S., Thorleifsson G., Feitosa M. F., Chambers J., Orho-Melander M., Melander O., Johnson T., Li X., Guo X., Li M., Shin, Cho Y., Jin, Go M., Jin, Kim Y., Lee J. Y., Park T., Kim K., Sim X., Twee-Hee, Ong R., Croteau-Chonka D. C., Lange L. A., Smith J. D., Song K., Hua, Zhao J., Yuan X., Luan J., Lamina C., Ziegler A., Zhang W., Zee R. Y., Wright A. F., Witteman J. C., Wilson J. F., Willemsen G., Wichmann H. E., Whitfield J. B., Waterworth D. M., Wareham N. J., Waeber G., Vollenweider P., Voight B. F., Vitart V., Uitterlinden A. G., Uda M., Tuomilehto J., Thompson J. R., Tanaka T., Surakka I., Stringham H. M., Spector T. D., Soranzo N., Smit J. H., Sinisalo J., Silander K., Sijbrands E. J., Scuteri A., Scott J., Schlessinger D., Sanna S., Salomaa V., Saharinen J., Sabatti C., Ruokonen A., Rudan I., Rose LM., Roberts R., Rieder M., Psaty B. M., Pramstaller P. P., Pichler I., Perola M., Penninx B. W., Pedersen N. L., Pattaro C., Parker A. N., Pare G., Oostra B. A., O'Donnell C. J., Nieminen M. S., Nickerson D. A., Montgomery G. W., Meitinger T., McPherson R., McCarthy M. I., McArdle W., Masson D., Martin N. G., Marroni F., Mangino M., Magnusson P. K., Lucas G., Luben R., Loos R. J., Lokki M. L., Lettre G., Langenberg C., Launer L. J., Lakatta E. G., Laaksonen R., Kyvik K. O., Kronenberg F., König I. R., Khaw K. T., Kaprio J., Kaplan L. M., Johansson A., Jarvelin M. R., Janssens A. C., Ingelsson E., Igl W., Kees, Hovingh G., Hottenga J. J., Hofman A., Hicks A. A., Hengstenberg C., Heid I. M., Hayward C., Havulinna A. S., Hastie N. D., Harris T. B., Haritunians T., Hall A. S., Gyllensten U., Guiducci C., Groop L. C., Gonzalez E., Gieger C., Freimer N. B., Ferrucci L., Erdmann J., Elliott P., Ejebe K. G., Döring A., Dominiczak A. F., Demissie S., Deloukas P., de, Geus E. J., de, Faire U., Crawford G., Collins F. S., Chen Y. D., Caulfield M. J., Campbell H., Burtt N. P., Bonnycastle L. L., Boomsma D. I., Boekholdt S. M., Bergman R. N., Barroso I., Bandinelli S., Ballantyne C. M., Assimes T. L., Quertermous T., Altshuler D., Seielstad M., Wong T. Y., Tai E. S., Feranil A. B., Kuzawa C. W., Adair L. S., Taylor H. A., Jr, Borecki I. B., Gabriel S. B., Wilson J. G., Holm H., Thorsteinsdottir U., Gudnason V., Krauss R. M., Mohlke K. L., Ordovas J. M., Munroe P. B., Kooner J. S., Tall A. R., Hegele R. A., Kastelein J. J., Schadt E. E., Rotter J. I., Boerwinkle E., Strachan D. P., Mooser V., Stefansson K., Reilly M. P., Samani N. J., Schunkert H., Cupples L. A., Sandhu M. S., Ridker P. M., Rader D. J., van, Duijn C. M., Peltonen L., Abecasis G. R., Boehnke M., Kathiresan S.). (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hong M. G., Pawitan Y., Magnusson P. K., Prince J. A. (2009) Strategies and issues in the detection of pathway enrichment in genome-wide association studies. Hum. Genet. 126, 289–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rual J. F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D. S., Zhang L. V., Wong S. L., Franklin G., Li S., Albala J. S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R. S., Vandenhaute J., Zoghbi H. Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M. E., Hill D. E., Roth F. P., Vidal M. (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 19. Stelzl U., Worm U., Lalowski M., Haenig C., Brembeck F. H., Goehler H., Stroedicke M., Zenkner M., Schoenherr A., Koeppen S., Timm J., Mintzlaff S., Abraham C., Bock N., Kietzmann S., Goedde A., Toksoz E., Droege A., Krobitsch S., Korn B., Birchmeier W., Lehrach H., Wanker E. E. (2005) A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968 [DOI] [PubMed] [Google Scholar]
- 20. Venkatesan K., Rual J. F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T., Hao T., Zenkner M., Xin X., Goh K. I., Yildirim M. A., Simonis N., Heinzmann K., Gebreab F., Sahalie J. M., Cevik S., Simon C., de Smet A. S., Dann E., Smolyar A., Vinayagam A., Yu H., Szeto D., Borick H., Dricot A., Klitgord N., Murray R. R., Lin C., Lalowski M., Timm J., Rau K., Boone C., Braun P., Cusick M. E., Roth F. P., Hill D. E., Tavernier J., Wanker E. E., Barabási A. L., Vidal M. (2009) An empirical framework for binary interactome mapping. Nat. Methods 6, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ceol A., Chatr Aryamontri A., Licata L., Peluso D., Briganti L., Perfetto L., Castagnoli L., Cesareni G. (2010) MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 38, D532–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aranda B., Achuthan P., Alam-Faruque Y., Armean I., Bridge A., Derow C., Feuermann M., Ghanbarian A. T., Kerrien S., Khadake J., Kerssemakers J., Leroy C., Menden M., Michaut M., Montecchi-Palazzi L., Neuhauser S. N., Orchard S., Perreau V., Roechert B., van Eijk K., Hermjakob H. (2010) The IntAct molecular interaction database in 2010. Nucleic Acids Res. 38, D525–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matys V., Kel-Margoulis O. V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K., Voss N., Stegmaier P., Lewicki-Potapov B., Saxel H., Kel A. E., Wingender E. (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iossifov I., Rodriguez-Esteban R., Mayzus I., Millen K. J., Rzhetsky A. (2009) Looking at cerebellar malformations through text-mined interactomes of mice and humans. PLoS Comput. Biol. 5, e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng S., Zhao Z. (2012) GenRev: Exploring functional relevance of genes in molecular networks. Genomics 99, 183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun P. G., Gao L., Han S. (2011) Prediction of human disease-related gene clusters by clustering analysis. Int. J. Biol. Sci. 7, 61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schlicker A., Lengauer T., Albrecht M. (2010) Improving disease gene prioritization using the semantic similarity of Gene Ontology terms. Bioinformatics 26, i561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Köhler S., Bauer S., Horn D., Robinson P. N. (2008) Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 82, 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Su A. I., Wiltshire T., Batalov S., Lapp H., Ching K. A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Cooke M. P., Walker J. R., Hogenesch J. B. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reumers J., Conde L., Medina I., Maurer-Stroh S., Van Durme J., Dopazo J., Rousseau F., Schymkowitz J. (2008) Joint annotation of coding and non-coding single nucleotide polymorphisms and mutations in the SNPeffect and PupaSuite databases. Nucleic Acids Res. 36, D825–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berglund G., Elmstähl S., Janzon L., Larsson S. A. (1993) The Malmo Diet and Cancer Study. Design and feasibility. J. Intern Med. 233, 45–51 [DOI] [PubMed] [Google Scholar]
- 33. Jerntorp P., Berglund G. (1992) Stroke registry in Malmo, Sweden. Stroke 23, 357–361 [DOI] [PubMed] [Google Scholar]
- 34. Folkersen L., Wagsater D., Paloschi V., Jackson V., Petrini J., Kurtovic S., Maleki S., Eriksson M. J., Caidahl K., Hamsten A., Michel J. B., Liska J., Gabrielsen A., Franco-Cereceda A., Eriksson P. (2011) Unraveling the divergent gene expression profiles in bicuspid and tricuspid aortic valve patients with thoracic aortic dilatation - the ASAP study. Mol. Med. 17, 1365–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maere S., Heymans K., Kuiper M. (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 [DOI] [PubMed] [Google Scholar]
- 36. Barabási A. L., Gulbahce N., Loscalzo J. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scott M. S., Perkins T., Bunnell S., Pepin F., Thomas D. Y., Hallett M. (2005) Identifying regulatory subnetworks for a set of genes. Mol. Cell. Proteomics 4, 683–692 [DOI] [PubMed] [Google Scholar]
- 38. Raychaudhuri S., Plenge R. M., Rossin E. J., Ng A. C., Purcell S. M., Sklar P., Scolnick E. M., Xavier R. J., Altshuler D., Daly M. J. (2009) Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 5, e1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Navlakha S., Kingsford C. (2010) The power of protein interaction networks for associating genes with diseases. Bioinformatics 26, 1057–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peloso G. M., Demissie S., Collins D., Mirel D. B., Gabriel S. B., Cupples L. A., Robins S. J., Schaefer E. J., Brousseau M. E. (2010) Common genetic variation in multiple metabolic pathways influences susceptibility to low HDL-cholesterol and coronary heart disease. J. Lipid Res. 51, 3524–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Namekata K., Enokido Y., Ishii I., Nagai Y., Harada T., Kimura H. (2004) Abnormal lipid metabolism in cystathionine beta-synthase-deficient mice, an animal model for hyperhomocysteinemia. J. Biol. Chem. 279, 52961–52969 [DOI] [PubMed] [Google Scholar]
- 42. Chu P. H., Yeh H. I., Wu H. H., Hong R. C., Shiu T. F., Yang C. M. (2010) Deletion of the FHL2 gene attenuates the formation of atherosclerotic lesions after a cholesterol-enriched diet. Life Sci. 86, 365–371 [DOI] [PubMed] [Google Scholar]
- 43. Folkersen L., van't Hooft F., Chernogubova E., Agardh H. E., Hansson G. K., Hedin U., Liska J., Syvanen A. C., Paulsson-Berne G., Franco-Cereceda A., Hamsten A., Gabrielsen A., Eriksson P. (2010) Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circ. Cardiovasc Genet. 3, 365–373 [DOI] [PubMed] [Google Scholar]
- 44. Nuño-Ayala M., Guillen N., Navarro M. A., Lou-Bonafonte J. M., Arnal C., Gascon S., Barranquero C., Godino J., Royo-Canas M., Sarria A. J., Guzman M. A., Hernandez E., Bregante M. A., Garcia-Gimeno M. A., Osada J. (2010) Cysteinemia, rather than homocysteinemia, is associated with plasma apolipoprotein A-I levels in hyperhomocysteinemia: lipid metabolism in cystathionine beta-synthase deficiency. Atherosclerosis 212, 268–273 [DOI] [PubMed] [Google Scholar]
- 45. Palanker L., Tennessen J. M., Lam G., Thummel C. S. (2009) Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab. 9, 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aras O., Hanson N. Q., Yang F., Tsai M. Y. (2000) Influence of 699C–>T and 1080C–>T polymorphisms of the cystathionine beta-synthase gene on plasma homocysteine levels. Clin. Genet. 58, 455–459 [DOI] [PubMed] [Google Scholar]
- 47. Liao D., Tan H., Hui R., Li Z., Jiang X., Gaubatz J., Yang F., Durante W., Chan L., Schafer A. I., Pownall H. J., Yang X., Wang H. (2006) Hyperhomocysteinemia decreases circulating high-density lipoprotein by inhibiting apolipoprotein A-I Protein synthesis and enhancing HDL cholesterol clearance. Circ. Res. 99, 598–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tremblay K., Lemire M., Potvin C., Tremblay A., Hunninghake G. M., Raby B. A., Hudson T. J., Perez-Iratxeta C., Andrade-Navarro M. A., Laprise C. (2008) Genes to diseases (G2D) computational method to identify asthma candidate genes. PLoS One 3, e2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erlich Y., Edvardson S., Hodges E., Zenvirt S., Thekkat P., Shaag A., Dor T., Hannon G. J., Elpeleg O. (2011) Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 21, 658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.