

Abstract
Several recent reports suggest that inflammatory signals play a decisive role in the self-renewal, migration and differentiation of multipotent neural stem cells (NSCs). NSCs are believed to be able to ameliorate the symptoms of several brain pathologies through proliferation, migration into the area of the lesion and either differentiation into the appropriate cell type or secretion of anti-inflammatory cytokines. Although NSCs have beneficial roles, current evidence indicates that brain tumours, such as astrogliomas or ependymomas are also caused by tumour-initiating cells with stem-like properties. However, little is known about the cellular and molecular processes potentially generating tumours from NSCs. Most pro-inflammatory conditions are considered to activate the transcription factor NF-κB in various cell types. Strong inductive effects of NF-κB on proliferation and migration of NSCs have been described. Moreover, NF-κB is constitutively active in most tumour cells described so far. Chronic inflammation is also known to initiate cancer. Thus, NF-κB might provide a novel mechanistic link between chronic inflammation, stem cells and cancer. This review discusses the apparently ambivalent role of NF-κB: physiological maintenance and repair of the brain via NSCs, and a potential role in tumour initiation. Furthermore, it reveals a possible mechanism of brain tumour formation based on inflammation and NF-κB activity in NSCs.
Keywords: neural stem cells, brain tumours, inflammation, NF-kappaB
Neural stem cells
During mammalian central nervous system (CNS) development, multipotent cells undergo division, cell fate specification and maturation. These neural stem cells (NSCs) are commonly defined as undifferentiated cells with the ability to proliferate, exhibit self-renewal, generate a large number of progeny including the principal phenotypes of nervous tissue, and retain their differentiation potential over time. Within the adult brain NSCs are mainly located in the subventricular zone (SVZ) and the dentate gyrus of the hippocampus.
NSCs are believed to have the capacity to replace lost cells within the CNS, thus offering a potential starting point for therapy of neurodegenerative diseases such as Parkinson's and Alzheimer's disease [1, 2]. They also provide a promising method for treating brain cancers by delivering chemotherapeutic agents directly to the tumour cells (reviewed in [3]). On the other hand, multipotent subsets of NSCs such as radial glia are believed to generate all the phenotypically diverse cells that populate brain tumours (for review see [4]).
NF-κB
The transcription factor NF-κB plays a pivotal role in a variety of biological processes including innate and adaptive immunity (reviewed in [5]), neuroprotection and degeneration (reviewed in [6] and [7]), learning and memory formation and pathological tumour malignancies (reviewed in [8] and [9]). In particular NF-κB has distinct functions in multiple immune cell types via the regulation of target genes essential for cell proliferation, survival, effector functions and cell trafficking [10–12].
In the nervous system NF-κB is known to mediate either neuroprotection or apoptosis in a stimulus depending manner [13]. Concerning memory formation NF-κB regulates spatial memory formation, synaptic transmission and plasticity through protein kinase A (PKA) and cAMP responsive element binding protein (CREB) signalling as demonstrated in a forebrain neu-ronal conditional NF-κB-deficient mouse model [14].
The NF-κB protein family comprises p50, p52, p65, RelB and c-Rel, which form different heterodimeric complexes (reviewed in [15]). The most common NF-κB dimer within the CNS, p50-p65, exists as an inactive cytoplasmic complex bound to inhibitory proteins of the IκB family (see [6] for review). The trimeric NF-κB-IκB complex can be activated by several stimuli, such as inflammatory cytokines, neurotransmitters, mitogens and growth factors (reviewed in [16] and [6]).
TNF-α
Tumour necrosis factor-alpha (TNF-α) is one of the best-characterized mediators of inflammation. Among the cells of the haematopoietic system, TNF-α is mainly secreted by macrophages, mono-cytes, neutrophils, T cells and NK-cells after stimulation with bacterial lipopolysaccharides (LPS). In the event of pathological alterations within the CNS, TNF-α is secreted by astrocytes and microglial cells [17, 18]. TNF-α synthesis is induced by several different stimuli including interferons (IFN), interleukin 2 (IL2) and GM-CSF, whereas it is inhibited by IL6, TGF-α2 and dexamethasone [19].
In eukaryotes, members of the TNF receptor superfamily play pivotal roles in several biological processes like haematopoiesis, protection from bacterial infection and immune surveillance [19–24].
On the other hand, dysregulation of TNF and its superfamily members leads to various pathological symptoms like diabetes (Type II), heart failure, artheriosclerosis, tumourigenesis and tumour metastasis [25–28].
Activation of this family of receptors leads to activation of multiple signal transduction pathways including NF-κB signalling [29].
NF-κB activation via TNF-α
NF-κB can be activated by a wide range of stimuli including endotoxins (e.g. LPS), hypoxia, cytokines or bacterial and viral infection (see [16] for review). During inflammation, the activation of NF-κB is mediated mainly by TNF-α. In this canonical NF-κB activation pathway, the TNF ligand binds to the receptor, thereby transducing the signal to the IκB kinase (IKK) complex. This complex in turn leads to phosphorylation, ubiquitinylation and finally pro-teasomal degradation of the inhibitory IκB (reviewed in [6]). NF-κB is thereby released, enabling it to be translocated to the nucleus where it binds to specific promoter regions on the DNA, and finally the target genes are transcribed (see NF-κB target genes affecting NSCs and tumour formation).
Inflammation, NF-κB and neural stem cells
Brain inflammation is a complex phenomenon. In addition to the well-described neurodegenerative effect of inflammation, several studies suggest that inflammatory signals influence NSCs in respect of proliferation, migration and differentiation leading either to functional integration and improvement of the symptoms of inflammation or to secretion of antiinflammatory cytokines by the NSCs [15, 30–39]).
As described above, much inflammatory signal transduction can be considered an innate immune response triggered by TNF [40, 41]. The transcriptional profile of TNF-treated astroglioma cells has been investigated by Schwamborn et al. as a model for brain inflammation [42]. In this study, more than 800 TNF-regulated genes have been found and analysed by microarray analysis. Macrophage Chemoattractant Protein 1 (MCP-1) gene expression was demonstrated to be strongly up-regulated and secreted into the medium as shown by immunocyto-chemistry and ELISA.
It is well known that NSCs express various chemokine receptors as a result of brain pathology (for review see [43] and [15]). In addition to MCP-1, expression of stromal derived factor 1α (SDF1-α) [34], stem cell factor (SCF) [33] and vascular endothelial growth factor has been reported [44]. Subsequent experiments provided strong evidence that MCP-1 induces NSC migration [35].
In view of the well-characterized TNF secretion in the course of inflammatory diseases and the very potent induction of NSC migration by MCP-1, it has been proposed that in pathological situations like neuroinflammation these cells migrate from the SVZ to the area of the lesion. Belmadani et al. showed that in hippocampal slice cultures enhanced green-fluorescent protein (eGFP)-labelled neural progenitors migrate toward injected inflammatory stimuli and that chemokines are the major regulators of this process [38].
Functional chemokine signalling strongly depends on expression of the relevant receptors.
Robust in vivo expression of chemokine receptors in neurogenic regions of the brain has been recently demonstrated [45]. In this report, Tran and colleagues clearly showed the expression of CCR1, CCR2 (the cognate MCP-1 receptor), CCR5, CXCR3 and CXCR4 on NSCs in the dentate gyrus of the hip-pocampus, SVZ and olfactory bulb. These findings accord with a model proposed by Muller et al. stating that NSCs are attracted by inflammation, reactive astrocytosis and angiogenesis [43].
Thus, NSCs are exposed after migration to TNF at the area of inflammation. In this context, it is noteworthy that TNF-induced NF-κB activity results in increased proliferation of NSCs ([31, 32]). After migration and proliferation, NSCs may participate in the repair process. In accordance with this hypothesis, Pluchino et al. showed that NSCs are able to promote neuroprotection during CNS inflammation [30]. This phenomenon might be explained either by secretion of neuroprotective cytokines or by functional integration of the proliferated NSCs. In addition, the transplanted NSCs exerted immune-like functions by inducing apoptosis of blood-borne CNS-infiltrating encephalitogenic T cells [30].
In summary, the function of NF-κB in NSCs during acute inflammation consists in the increase of proliferation and in migration.
Inflammation, NF-κB and cancer
Inflammation is prerequisite for wound healing, elimination of infections and regeneration after pathological situations via, inter alia, the migration, proliferation and differentiation of stem cells (see above). On the other hand, cancer is associated with maladap-tive chronic inflammation.
In the 19th century, Rudolf Virchov hypothesized that chronic inflammation may cause several malignancies including cancer [46–48]. Interestingly, one of the most prominent inflammatory cytokines – TNF-α (see above) – is known to activate NF-κB strongly in several experimental cancer models and to act as a potent tumour promoter (reviewed in [9]). Brain tumours were demonstrated to express TNF receptors like TNFRI, TNFRII, DR6, Fas or Fn14 and the TNF receptor associated signalling molecules consisting of TRAIL-R1, TRAIL-R2, TRAIL, TRAF1, TRAF2 and TANK/I-TRAF [49–56]. In addition brain tumours express the chemokine receptors CXCR4, CCR1, CCR3, CCR5 and CCR2 [57–59].
The role of NF-κB in cancer development and progression has been extensively discussed ([8, 16, 60–67]). In particular, the activation of NF-κB blocks apoptosis via modulation of anti-apoptotic target genes, such as c-IAP, bcl-2 and bcl-xL and mediates tumour cell proliferation via up-regulation of targets like cyclin-D1 and c-myc [51, 68–76].
It also induces resistance to chemotherapeutic agents. In fact, numerous genes involved in tumour initiation, promotion and metastasis are regulated by the NF-κB pathway (for review see [16, 63]). NF-κB is constitutively active in most tumour specimens described to date (reviewed in [16] and [9]).
NSCs and brain cancer
Cancer is defined as a progressive disease, typically requiring initial mutations in proliferating cells. Physiologically, proliferation is tightly controlled and restricted to only a few cell types, including stem cells. In this context it is noteworthy that the ‘immortal strand hypothesis’ postulates that during the process of self-renewing primitive stem cells retain DNA strands with the fewest mutations acquired during DNA replication [77]. These stem cells remain primitive and divide slowly in asymmetric manner. The second one, more differentiated daughter cell, for example NSC divides fast. Thus, amplifying NSCs potentially have time to accumulate genetic mutations leading to tumour formation [78].
Recent studies on brain tumours have revealed stem-cell-like tumour cell populations. Uchida et al. described Nestin and Musashi-1 expressing cells within an infant brain tumour; this tumour was also positive for several immature neuronal and astrocytic markers [79]. Nestin – an intermediate filament [80], and Musashi, a RNA binding protein [81, 82] are very well-described markers for NSCs.
Hemmati et al.[83] isolated tumourigenic cells with stem cell properties from paediatric brain tumours. These neurosphere-forming, multipotent and self-renewing cells could differentiate into neural and glial lineages. Similarly, Tunici et al.[84] reported neu-rosphere-forming tumour cells with neural stem/precursor cell properties. Recently, Lee and colleagues found that TSCs derived directly from glioblastomas harbour extensive similarities to normal stem cells if they are cultured in basic fibroblast growth factor (bFGF)- and epidermal growth factor (EGF)-containing media [85–87].
These similarities include the formation of neu-rosphere-like structures in vitro, self-renewal, terminal differentiation into glial and neuronal lineage and of gene expression profile similar to NSCs.
TSCs have also been detected in brain tumours, such as ependymomas, glioblastomas and medul-loblastomas (see [88] for review). Some reports provide evidence that neuroblastomas – embryonic cancers of the neural crest – contain stem cell populations as well (see [89] for review). Moreover, Taylor et al.[90] discussed radial glia as potential stem cells for ependymomas in a recent review.
In summary, similarities between stem cells and cancer stem cells have been demonstrated in cell signalling pathways, differentiation and drug resistance [91–93].
During inflammation, stem cells are believed to switch from asymmetrical divisions that give rise to differentiated progeny to rapid symmetrical divisions resulting in an increased number of undifferentiated stem cells (reviewed in [94]). Several reports show that the molecular pathways regulating asymmetrical division in stem cells control the orientation of the mitotic spindle [95, 96]. The switch from asymmetrical to symmetrical cell division may increase the likelihood of aneuploidy – a frequently observed phenomenon in tumour cells.
Interestingly, Diamandis et al.[97] demonstrated in a recent report that small molecules known to affect neurotransmission pathways inhibit the proliferation of NSC also have inhibitory effects on brain cancer stem cell proliferation.
NF-κB target genes affecting NSCs and tumour formation
In this review, we focus on NF-κB targets regulating cell–cycle, anti-apoptosis, cellular ageing and multi-drug-resistance.
Several tumour specimens like malignant astrocytomas, especially glioblastomas show elevated levels of the c-myc proto-oncogene [51]. c-myc is a well-described NF-κB target with functional κB-binding site in its promoter region [98, 99]. In addition, in their study Bouragel-Rey and colleagues demonstrated that activated NF-κB strongly induces c-myc [100]. Recently, Faria and colleagues showed a positive correlation between c-Myc expression and the histopathologial grade and the proliferative status of astrocytic tumours [101].
Also, many human medulloblastomas express significantly elevated levels of myc oncogenes correlated with worse clinical outcome [102–104]. The c-Myc oncoprotein is well described to be a potent mitogen for neural precursors in vitro and in vivo[105].
In a recent study Xiaohua et al. showed that in NSCs, an elevated expression of c-Myc and neural restricted silencer factor (NRSF), a transcriptional repressor of neuronal differentiation causes cerebel-lar tumours [106]. Additionally, c-Myc enhances sonic-hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors [107]. Interestingly, TNF-α stimulated rat NSCs showed highly elevated c-Myc expression compared to untreated control (Widera et al. unpublished data).
All these data suggest c-myc to be one of the NF-κB target genes responsible for induction of tumours from NSCs in inflammatory situations.
A further NF-κB target gene known to trigger proliferation of tumour cells is cyclin D1. Cyclin D1 has two NF-κB binding sites in the promoter region. The stimulation of the transcription of cyclin D1 by NF-κB results in increased proliferation of several cell types (reviewed in [108]). Similar to c-myc, Cyclin D1 is widely up-regulated in several brain tumours like menin-giomas, olfactory neuroblastomas and gliomas and is closely related to oncogenesis, proliferation of tumour cells and worse clinical prognosis [76, 109, 110].
In contrast loss of cyclin D1 suppresses medul-loblastoma formation [111]. The NF-κB induced up-regulation of Cyclin D1 has been identified as one of the crucial events in cell cycle progression of tumour cells [75, 112].
In NSCs, Cyclin D1 seems to act in a similar man-ner. TNF-α treated rat NSCs show highly up-regulated Cyclin D1 expression and significantly increased proliferation compared to control cells [31]. This finding is in accordance with the hypothesis that inflammation activates the transcription factor NF-κB resulting in transcription of target genes that induce proliferation. In contrast, dexamethasone induced ubiquitination of Cyclin D1 or down-regulation of Cyclin D1 by GATA2 led to decreased proliferation of NSCs [113, 114]. Thus, cyclin D1 seems to be another NF-κB target gene potentially responsible for tumour formation and progression.
In addition to the deregulated cell cycle control, several pathways regulating apoptosis are also disrupted in brain tumours. Thus, malignant tumours often show intense resistance to apoptosis. NF-κB is known to directly activate the apoptosis inhibitors Bcl-xL and Bcl-2. In tumours a positive correlation between high expression of NF-κB and up-regulated levels of the target genes Bcl-xL and Bcl-2 has been demonstrated [69]. This phenomenon might partially explain the apoptosis resistance of tumours through NF-κB driven induction of anti-apoptotic genes like Bcl-xL and Bcl-2.
One problem of many brain malignant tumours is their resistance to chemotherapy [115]. This observable fact can be explained by the expression of ATP-binding cassette (ABC) drug efflux transporters (reviewed in [116] and [117]). Mutch and colleagues clearly demonstrated that the ABC transporter ABCB2 contains a functional NF-κB binding site in its promoter region [118]. Cells expressing ABCG2 are known to exclude Hoechst in flow cytometry, have been called side population (SP) cells [119]. Furthermore, it has been demonstrated that putative stem cells from solid tissues may also possess this SP phenotype [120].
A robust expression of ABC transporters ABCA2, ABCA3, ABCB1 and ABCG2 in NSCs has been described (reviewed in [121]). This fact suggests a potential role of these NF-κB targets in tumour biology.
Conclusion
Inflammatory signals and conditions have been described as inducing NSC proliferation via activation of the NF-κB pathway [31, 122–124]. Physiologically, NSCs proliferate slowly and asymmetrically or rest in the G0-Phase of the cell cycle. In an inflammatory environment, they start to proliferate rapidly and symmetrically [94]. This may be attributable to the release of pro-inflammatory cytokines, such as TNF-α from the injured tissue. In addition, the expression of mitogens like FGF-2 is increased in inflammation [125]. The expression of FGF and FGF receptors seems to be crucial for symmetrical division of embryonic and NSCs (self-renewal) [60–62]. Recently, evidence was provided that FGF-2 activates NF-κB [63]. In addition, FGF-2 acts in an anti-apoptotic and pro-proliferative manner through activated NF-κB [64, 65].
After attenuation of the acute phase of the inflammation and a decrease in local concentration of pro-inflammatory signals NSCs decrease their rate of proliferation and proceed either to slow proliferation or to the resting state (G0-Phase) (see Fig. 1).
1.
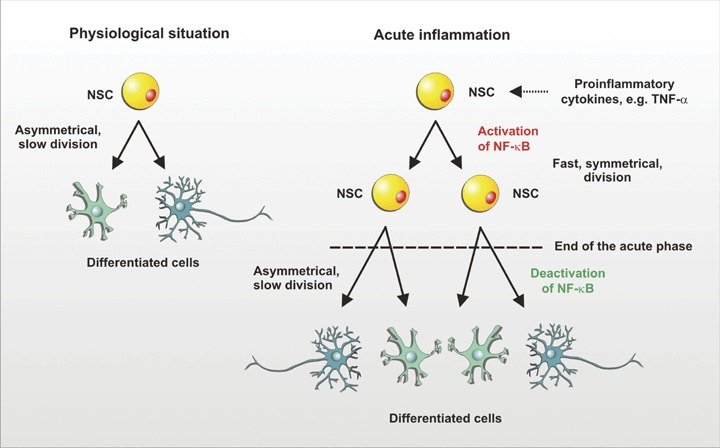
Neural stem cells (NSC) division in physiological situation and in acute inflammation. Physiologically, NSCs proliferate slowly and asymmetrically generating more committed precursors or differentiated cells. In an inflammatory environment, NSCs might start to proliferate rapidly and symmetrically in response to pro-inflammatory cytokines such as TNF-α. The pro-inflammatory stimuli activate the transcription factor NF-κB. After acute inflammation is attenuated, NF-κB becomes deactivated and NSCs switch back to the asymmetrical mode of division.
In contrast, during chronic inflammation, NSCs are permanently exposed to proliferation signals. Fast proliferation holds a much higher risk of mutation than slow cell division. These fast symmetric divisions may promote the expansion of NSCs and lead to aneuploidy (see Fig. 2). In rapidly proliferating NSCs, an initial mutation may be followed by additional mutations that ultimately lead to transformation. If the mutations occur in proto-oncogenes coding, for example for signalling molecules activating NF-κB, constitutive activity may lead to growth factor-independent proliferation of the transformed NSCs.
2.
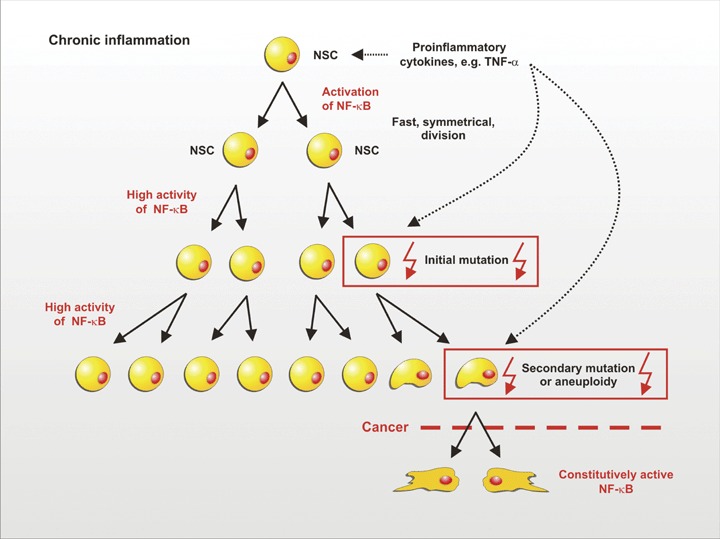
Model for the correlation between chronic inflammation, symmetric division of NSCs and cancer. In chronic inflammation, NSCs are permanently exposed to proliferation-inducing stimuli such as TNF-α. The ensuing rapid proliferation entails a much higher risk of mutation than slow cell division. Initial mutations could easily be propagated as a result of the fast symmetric division. Secondary mutations then may lead to constitutive NF-κB activity, aneuploidy and finally to cancer.
In fact, subsets of adult NSCs isolated from a long-term culture might become independent of exogenous growth factors. Such growth factor independent cells still expressed typical stemness markers as well as migratory activity, identifying them as stem cells. Moreover, these cells showed a constitutively high NF-κB activity and an aberrant, polyploid DNA content (Kaus et al., unpublished data).
From our point of view, NF-κB may be one of the most important regulators of brain tumour development via NSCs and later via TSCs. A primary physiological function of NF-κB may be to regulate stem cell proliferation via transcriptional regulation target genes like c-myc or cyclin D1 and their migration and differentiation. On the other hand, pathologically high activity of NF-κB during chronic inflammation may cause tumour initiation, progression and metastasis.
References
- 1.Gage FH. Mammalian neural stem cells. Science New York, NY. 2000;287:1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 2.Lie DC, Song H, Colamarino SA, Ming GL, Gage FH. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu Rev Pharmacol Toxicol. 2004;44:399–421. doi: 10.1146/annurev.pharmtox.44.101802.121631. [DOI] [PubMed] [Google Scholar]
- 3.Shah K, Hsich G, Breakefield XO. Neural precursor cells and their role in neuro-oncology. Dev Neurosci. 2004;26:118–30. doi: 10.1159/000082132. [DOI] [PubMed] [Google Scholar]
- 4.Poppleton H, Gilbertson RJ. Stem cells of ependy-moma. Br J Cancer. 2007;96:6–10. doi: 10.1038/sj.bjc.6603519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82:434–48. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 6.Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta. 2005;1745:287–99. doi: 10.1016/j.bbamcr.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–47. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- 9.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 10.Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, Schmidt-Supprian M. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–39. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki Y, Schmidt-Supprian M, Derudder E, Rajewsky K. Role of NFkappaB signaling in normal and malignant B cell development. Adv Exp Med Biol. 2007;596:149–54. doi: 10.1007/0-387-46530-8_13. [DOI] [PubMed] [Google Scholar]
- 12.Nenci A, Becker C, Wullaert A, Gareus R, Van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, Gumucio D, Neurath MF, Pasparakis M. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–61. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 13.Kaltschmidt B, Heinrich M, Kaltschmidt C. Stimulus-dependent activation of NF-kappaB specifies apoptosis or neuroprotection in cerebellar granule cells. Neuromolecular Med. 2002;2:299–309. doi: 10.1385/NMM:2:3:299. [DOI] [PubMed] [Google Scholar]
- 14.Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prullage M, Pfeiffer J, Lindecke A, Staiger V, Israel A, Kaltschmidt C, Memet S. NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol Cell Biol. 2006;26:2936–46. doi: 10.1128/MCB.26.8.2936-2946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Widera D, Mikenberg I, Kaltschmidt B, Kaltschmidt C. Potential role of NF-kappaB in adult neural stem cells: the underrated steersman? Int J Dev Neurosci. 2006;24:91–102. doi: 10.1016/j.ijdevneu.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–8. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J Immunol. 1990;144:2999–3007. [PubMed] [Google Scholar]
- 18.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excito-toxic and ischemic brain injury in mice lacking TNF receptors. Nature Medicine. 1996;2:788–94. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 19.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 20.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi T, Tanaka M, Ogasawara J, Suda T, Murakami H, Nagata S. Swapping between Fas and granulocyte colony-stimulating factor receptor. J Biol Chem. 1996;271:17555–60. doi: 10.1074/jbc.271.29.17555. [DOI] [PubMed] [Google Scholar]
- 22.Nagata S, Golstein P. The Fas death factor. Science New York, NY. 1995;267:1449–56. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 23.Kishimoto H, Surh CD, Sprent J. A role for Fas in negative selection of thymocytes in vivo. J Exp Med. 1998;187:1427–38. doi: 10.1084/jem.187.9.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song K, Chen Y, Goke R, Wilmen A, Seidel C, Goke A, Hilliard B, Chen Y. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J Exp Med. 2000;191:1095–104. doi: 10.1084/jem.191.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes. 1994;43:1271–8. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 26.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. 1994;91:4854–8. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bright JJ. Curcumin and autoimmune disease. Adv Exp Med Biol. 2007;595:425–51. doi: 10.1007/978-0-387-46401-5_19. [DOI] [PubMed] [Google Scholar]
- 28.Pacifico F, Leonardi A. NF-kappaB in solid tumors. Biochem Pharmacol. 2006;72:1142–52. doi: 10.1016/j.bcp.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 29.Baker SJ, Reddy EP. Modulation of life and death by the TNF receptor superfamily. Oncogene. 1998;17:3261–70. doi: 10.1038/sj.onc.1202568. [DOI] [PubMed] [Google Scholar]
- 30.Pluchino S, Zanotti L, Rossi B, Brambilla E, Ottoboni L, Salani G, Martinello M, Cattalini A, Bergami A, Furlan R, Comi G, Constantin G, Martino G. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodula-tory mechanism. Nature. 2005;436:266–71. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- 31.Widera D, Mikenberg I, Elvers M, Kaltschmidt C, Kaltschmidt B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006;7:64. doi: 10.1186/1471-2202-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu JP, Kuo JS, Liu YL, Tzeng SF. Tumor necrosis factor-alpha modulates the proliferation of neural progenitors in the subventricular/ventricular zone of adult rat brain. Neurosci Lett. 2000;292:203–6. doi: 10.1016/s0304-3940(00)01472-5. [DOI] [PubMed] [Google Scholar]
- 33.Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest. 2004;113:1364–74. doi: 10.1172/JCI20001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA. 2004;101:18117–22. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Widera D, Holtkamp W, Entschladen F, Niggemann B, Zanker K, Kaltschmidt B, Kaltschmidt C. MCP-1 induces migration of adult neural stem cells. Eur J Cell Biol. 2004;83:381–7. doi: 10.1078/0171-9335-00403. [DOI] [PubMed] [Google Scholar]
- 36.Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur J Neurosci. 2007;25:649–58. doi: 10.1111/j.1460-9568.2007.05309.x. [DOI] [PubMed] [Google Scholar]
- 37.Barkho BZ, Song H, Aimone JB, Smrt RD, Kuwabara T, Nakashima K, Gage FH, Zhao X. Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev. 2006;15:407–21. doi: 10.1089/scd.2006.15.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belmadani A, Tran PB, Ren D, Miller RJ. Chemokines regulate the migration of neural progenitors to sites of neuroinflammation. J Neurosci. 2006;26:3182–91. doi: 10.1523/JNEUROSCI.0156-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-Hur T. Immunomodulation by neural stem cells. J Neurol Sci. 2008;265:102–4. doi: 10.1016/j.jns.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegen-eration? Nat Rev Neurosci. 2002;3:216–27. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- 41.Turrin NP, Rivest S. Tumor necrosis factor alpha but not interleukin 1beta mediates neuroprotection in response to acute nitric oxide excitotoxicity. J Neurosci. 2006;26:143–51. doi: 10.1523/JNEUROSCI.4032-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwamborn J, Lindecke A, Elvers M, Horejschi V, Kerick M, Rafigh M, Pfeiffer J, Prullage M, Kaltschmidt B, Kaltschmidt C. Microarray analysis of tumor necrosis factor alpha induced gene expression in U373 human glioblastoma cells. BMC Genomics. 2003;4:46. doi: 10.1186/1471-2164-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller FJ, Snyder EY, Loring JF. Gene therapy: can neural stem cells deliver? Nat Rev Neurosci. 2006;7:75–84. doi: 10.1038/nrn1829. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H, Vutskits L, Pepper MS, Kiss JZ. VEGF is a chemoattractant for FGF-2-stimulated neural progenitors. J Cell Biol. 2003;163:1375–84. doi: 10.1083/jcb.200308040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tran PB, Banisadr G, Ren D, Chenn A, Miller RJ. Chemokine receptor expression by neural progenitor cells in neurogenic regions of mouse brain. J Comp Neurol. 2007;500:1007–33. doi: 10.1002/cne.21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Virchow R. Die Cellularpathologie in ihrer Begründung und in ihrer Auswirkung auf die physiol-ogische und pathologische Gewebelehre. Berlin: Verlag A. Hirschwald; 1871. [Google Scholar]
- 47.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt A, Weber OF. In memoriam of Rudolf vir-chow: a historical retrospective including aspects of inflammation, infection and neoplasia. Contrib Microbiol. 2006;13:1–15. doi: 10.1159/000092961. [DOI] [PubMed] [Google Scholar]
- 49.Kasof GM, Lu JJ, Liu D, Speer B, Mongan KN, Gomes BC, Lorenzi MV. Tumor necrosis factor-alpha induces the expression of DR6, a member of the TNF receptor family, through activation of NF-kappaB. Oncogene. 2001;20:7965–75. doi: 10.1038/sj.onc.1204985. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka S, Nagashima T, Hori T. In vitro inhibition of binding of tumor necrosis factor (TNF)-alpha by monoclonal antibody to TNF receptor on glioma cell and monocyte. Neurol Med Chir. 1998;38:812–7. doi: 10.2176/nmc.38.812. [DOI] [PubMed] [Google Scholar]
- 51.Hayashi S, Yamamoto M, Ueno Y, Ikeda K, Ohshima K, Soma G, Fukushima T. Expression of nuclear factor-kappa B, tumor necrosis factor receptor type 1, and c-Myc in human astrocytomas. Neurol Med Chir. 2001;41:187–95. doi: 10.2176/nmc.41.187. [DOI] [PubMed] [Google Scholar]
- 52.Konstantinidou AE, Korkolopoulou P, Patsouris E. Apoptotic markers for primary brain tumor prognosis. J Neurooncol. 2005;72:151–6. doi: 10.1007/s11060-004-3345-z. [DOI] [PubMed] [Google Scholar]
- 53.Kuijlen JM, Mooij JJ, Platteel I, Hoving EW, Van Der Graaf WT, Span MM, Hollema H, Den Dunnen WF. TRAIL-receptor expression is an independent prognostic factor for survival in patients with a primary glioblas-toma multiforme. J Neurooncol. 2006;78:161–71. doi: 10.1007/s11060-005-9081-1. [DOI] [PubMed] [Google Scholar]
- 54.Conti A, Ageunnouz M, La Torre D, Cardali S, Angileri FF, Buemi C, Tomasello C, Iacopino DG, D'Avella D, Vita G, Tomasello F. Expression of the tumor necrosis factor receptor-associated factors 1 and 2 and regulation of the nuclear factor-kappaB antiapoptotic activity in human gliomas. J Neurosurg. 2005;103:873–81. doi: 10.3171/jns.2005.103.5.0873. [DOI] [PubMed] [Google Scholar]
- 55.Tran NL, McDonough WS, Savitch BA, Sawyer TF, Winkles JA, Berens ME. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J Biol Chem. 2005;280:3483–92. doi: 10.1074/jbc.M409906200. [DOI] [PubMed] [Google Scholar]
- 56.Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, Nakada M, Cunliffe HE, Hostetter G, Hoelzinger DB, Rennert JL, Michaelson JS, Burkly LC, Lipinski CA, Loftus JC, Mariani L, Berens ME. Increased Fibroblast Growth Factor-Inducible 14 expression levels promote glioma cell Invasion via Rac1 and Nuclear Factor-{kappa}B and correlate with poor patient outcome. Cancer Res. 2006;66:9535–42. doi: 10.1158/0008-5472.CAN-06-0418. [DOI] [PubMed] [Google Scholar]
- 57.Bajetto A, Barbieri F, Dorcaratto A, Barbero S, Daga A, Porcile C, Ravetti JL, Zona G, Spaziante R, Corte G, Schettini G, Florio T. Expression of CXC chemokine receptors 1–5 and their ligands in human glioma tissues: role of CXCR4 and SDF1 in glioma cell proliferation and migration. Neurochem Int. 2006;49:423–32. doi: 10.1016/j.neuint.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Kouno J, Nagai H, Nagahata T, Onda M, Yamaguchi H, Adachi K, Takahashi H, Teramoto A, Emi M. Up-regulation of CC chemokine, CCL3L1, and receptors, CCR3, CCR5 in human glioblastoma that promotes cell growth. J Neurooncol. 2004;70:301–7. doi: 10.1007/s11060-004-9165-3. [DOI] [PubMed] [Google Scholar]
- 59.Galasso JM, Stegman LD, Blaivas M, Harrison JK, Ross BD, Silverstein FS. Experimental gliosarcoma induces chemokine receptor expression in rat brain. Exp Neurol. 2000;161:85–95. doi: 10.1006/exnr.1999.7249. [DOI] [PubMed] [Google Scholar]
- 60.Schwartz SA, Hernandez A, Mark Evers B. The role of NF-kappaB/IkappaB proteins in cancer: implications for novel treatment strategies. Surg Oncol. 1999;8:143–53. doi: 10.1016/s0960-7404(00)00012-8. [DOI] [PubMed] [Google Scholar]
- 61.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in onco-genic initiation and progression. Oncogene. 2006;25:6817–30. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 62.Hu MC, Hung MC. Role of IkappaB kinase in tumori-genesis. Future Oncol. 2005;1:67–78. doi: 10.1517/14796694.1.1.67. [DOI] [PubMed] [Google Scholar]
- 63.Shishodia S, Aggarwal BB. Nuclear factor-kappaB: a friend or a foe in cancer? Biochem Pharmacol. 2004;68:1071–80. doi: 10.1016/j.bcp.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 64.Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 65.Bharti AC, Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Bioch Pharmacol. 2002;64:883–8. doi: 10.1016/s0006-2952(02)01154-1. [DOI] [PubMed] [Google Scholar]
- 66.Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446:475–82. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 67.Escarcega RO, Fuentes-Alexandro S, Garcia-Carrasco M, Gatica A, Zamora A. The transcription factor nuclear factor-kappa B and cancer. Clin Oncol. 2007;19:154–61. doi: 10.1016/j.clon.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 68.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c- IAP2 to suppress caspase-8 activation. Science New York, NY. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 69.Yu HG, Yu LL, Yang Y, Luo HS, Yu JP, Meier JJ, Schrader H, Bastian A, Schmidt WE, Schmitz F. Increased expression of RelA/nuclear factor-kappa B protein correlates with colorectal tumorigenesis. Oncology. 2003;65:37–45. doi: 10.1159/000071203. [DOI] [PubMed] [Google Scholar]
- 70.Notarbartolo M, Poma P, Perri D, Dusonchet L, Cervello M, D'Alessandro N. Antitumor effects of curcumin, alone or in combination with cisplatin or doxorubicin, on human hepatic cancer cells. Analysis of their possible relationship to changes in NF-kB activation levels and in IAP gene expression. Cancer Lett. 2005;224:53–65. doi: 10.1016/j.canlet.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 71.Notarbartolo M, Cervello M, Poma P, Dusonchet L, Meli M, D'Alessandro N. Expression of the IAPs in multidrug resistant tumor cells. Oncol Rep. 2004;11:133–6. [PubMed] [Google Scholar]
- 72.Rishi L, Dhiman R, Raje M, Majumdar S. Nitric oxide induces apoptosis in cutaneous T cell lym-phoma (HuT-78) by downregulating constitutive NF-kappaB. Biochim Biophys Acta. 2007;1770:1230–9. doi: 10.1016/j.bbagen.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 73.Yang CC, Lin HP, Chen CS, Yang YT, Tseng PH, Rangnekar VM. Bcl-xL mediates a survival mechanism independent of the phosphoinositide 3-kinase/Akt pathway in prostate cancer cells. J Biol Chem. 2003;278:25872–8. doi: 10.1074/jbc.M301744200. [DOI] [PubMed] [Google Scholar]
- 74.Kaltschmidt B, Kaltschmidt C, Hehner SP, Droge W, Schmitz ML. Repression of NF-kappaB impairs HeLa cell proliferation by functional interference with cell cycle checkpoint regulators. Oncogene. 1999;18:3213–25. doi: 10.1038/sj.onc.1202657. [DOI] [PubMed] [Google Scholar]
- 75.Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol. 1999;19:2690–8. doi: 10.1128/mcb.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang X, Zhao M, Huang AY, Fei Z, Zhang W, Wang XL. The effect of cyclin D expression on cell proliferation in human gliomas. J Clin Neurosci. 2005;12:166–8. doi: 10.1016/j.jocn.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 77.Rando TA. The immortal strand hypothesis: segregation and reconstruction. Cell. 2007;129:1239–43. doi: 10.1016/j.cell.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 78.Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004;23:7267–73. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 79.Uchida K, Mukai M, Okano H, Kawase T. Possible oncogenicity of subventricular zone neural stem cells: case report. Neurosurgery. 2004;55:977–8. doi: 10.1227/01.neu.0000137891.99542.43. [DOI] [PubMed] [Google Scholar]
- 80.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–95. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 81.Sakakibara S, Imai T, Hamaguchi K, Okabe M, Aruga J, Nakajima K, Yasutomi D, Nagata T, Kurihara Y, Uesugi S, Miyata T, Ogawa M, Mikoshiba K, Okano H. Mouse-Musashi-1, a neural RNA-binding protein highly enriched in the mammalian CNS stem cell. Dev Biol. 1996;176:230–42. doi: 10.1006/dbio.1996.0130. [DOI] [PubMed] [Google Scholar]
- 82.Sakakibara S, Nakamura Y, Yoshida T, Shibata S, Koike M, Takano H, Ueda S, Uchiyama Y, Noda T, Okano H. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci USA. 2002;99:15194–9. doi: 10.1073/pnas.232087499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tunici P, Bissola L, Lualdi E, Pollo B, Cajola L, Broggi G, Sozzi G, Finocchiaro G. Genetic alterations and in vivo tumorigenicity of neurospheres derived from an adult glioblastoma. Mol Cancer. 2004;3:25. doi: 10.1186/1476-4598-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 86.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci USA. 2004;101:781–6. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–19. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 88.Nicolis SK. Cancer stem cells and “stemness” genes in neuro-oncology. Neurobiol Dis. 2007;25:217–29. doi: 10.1016/j.nbd.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 89.Ross RA, Spengler BA. Human neuroblastoma stem cells. Semin Cancer Biol. 2007;17:241–7. doi: 10.1016/j.semcancer.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 90.Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, Macdonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–35. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 91.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 92.Jogi A, Ora I, Nilsson H, Lindeheim A, Makino Y, Poellinger L, Axelson H, Pahlman S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci USA. 2002;99:7021–6. doi: 10.1073/pnas.102660199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 95.Yamashita YM, Jones DL, Fuller MT. Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science New York, NY. 2003;301:1547–50. doi: 10.1126/science.1087795. [DOI] [PubMed] [Google Scholar]
- 96.Kaltschmidt JA, Davidson CM, Brown NH, Brand AH. Rotation and asymmetry of the mitotic spindle direct asymmetric cell division in the developing central nervous system. Nat Cell Biol. 2000;2:7–12. doi: 10.1038/71323. [DOI] [PubMed] [Google Scholar]
- 97.Diamandis P, Wildenhain J, Clarke ID, Sacher AG, Graham J, Bellows DS, Ling EK, Ward RJ, Jamieson LG, Tyers M, Dirks PB. Chemical genetics reveals a complex functional ground state of neural stem cells. Nat Chem Biol. 2007;3:268–73. doi: 10.1038/nchembio873. [DOI] [PubMed] [Google Scholar]
- 98.La Rosa FA, Pierce JW, Sonenshein GE. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol Cell Biol. 1994;14:1039–44. doi: 10.1128/mcb.14.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee H, Wu M, La Rosa FA, Duyao MP, Buckler AJ, Sonenshein GE. Role of the Rel-family of transcription factors in the regulation of c-myc gene transcription and apoptosis of WEHI 231 murine B-cells. Curr Top Microbiol Immunol. 1995;194:247–55. doi: 10.1007/978-3-642-79275-5_29. [DOI] [PubMed] [Google Scholar]
- 100.Bourgarel-Rey V, Vallee S, Rimet O, Champion S, Braguer D, Desobry A, Briand C, Barra Y. Involvement of nuclear factor kappaB in c-Myc induction by tubulin polymerization inhibitors. Mol Pharmacol. 2001;59:1165–70. doi: 10.1124/mol.59.5.1165. [DOI] [PubMed] [Google Scholar]
- 101.Faria MH, Goncalves BP, Do Patrocinio RM, De Moraes-Filho MO, Rabenhorst SH. Expression of Ki-67, topoisomerase IIalpha and c-MYC in astrocyt-ic tumors: correlation with the histopathological grade and proliferative status. Neuropathology. 2006;26:519–27. doi: 10.1111/j.1440-1789.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 102.Eberhart CG, Kratz JE, Schuster A, Goldthwaite P, Cohen KJ, Perlman EJ, Burger PC. Comparative genomic hybridization detects an increased number of chromosomal alterations in large cell/anaplastic medulloblastomas. Brain Pathol. 2002;12:36–44. doi: 10.1111/j.1750-3639.2002.tb00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leonard JR, Cai DX, Rivet DJ, Kaufman BA, Park TS, Levy BK, Perry A. Large cell/anaplastic medul-loblastomas and medullomyoblastomas: clinicopathological and genetic features. J Neurosurg. 2001;95:82–8. doi: 10.3171/jns.2001.95.1.0082. [DOI] [PubMed] [Google Scholar]
- 104.Eberhart CG, Kratz J, Wang Y, Summers K, Stearns D, Cohen K, Dang CV, Burger PC. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anapla-sia. J Neuropathol Exp Neurol. 2004;63:441–9. doi: 10.1093/jnen/63.5.441. [DOI] [PubMed] [Google Scholar]
- 105.Fults D, Pedone C, Dai C, Holland EC. MYC expression promotes the proliferation of neural progenitor cells in culture and in vivo. Neoplasia. 2002;4:32–9. doi: 10.1038/sj.neo.7900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Su X, Gopalakrishnan V, Stearns D, Aldape K, Lang FF, Fuller G, Snyder E, Eberhart CG, Majumder S. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cere-bellar tumors by blocking neuronal differentiation. Mol Cell Biol. 2006;26:1666–78. doi: 10.1128/MCB.26.5.1666-1678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rao G, Pedone CA, Coffin CM, Holland EC, Fults DW. c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia. 2003;5:198–204. doi: 10.1016/S1476-5586(03)80052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15:158–63. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- 109.Alama A, Barbieri F, Spaziante R, Bruzzo C, Dadati P, Dorcaratto A, Ravetti JL. Significance of cyclin D1 expression in meningiomas: a preliminary study. J Clin Neurosci. 2007;14:355–8. doi: 10.1016/j.jocn.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 110.Wang SL, Chen WT, Li SH, Li SW, Yang SF, Chai CY. Expression of human telomerase reverse tran-scriptase and cyclin-D1 in olfactory neuroblastoma. Apmis. 2007;115:17–21. doi: 10.1111/j.1600-0463.2007.apm_446.x. [DOI] [PubMed] [Google Scholar]
- 111.Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development. 2006;133:3929–37. doi: 10.1242/dev.02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lin JK, Tsai SH. Chemoprevention of cancer and cardiovascular disease by resveratrol. Proc Natl Sci Counc Repub China B. 1999;23:99–106. [PubMed] [Google Scholar]
- 113.Sundberg M, Savola S, Hienola A, Korhonen L, Lindholm D. Glucocorticoid hormones decrease proliferation of embryonic neural stem cells through ubiquitin-mediated degradation of cyclin D1. J Neurosci. 2006;26:5402–10. doi: 10.1523/JNEUROSCI.4906-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.El Wakil A, Francius C, Wolff A, Pleau-Varet J, Nardelli J. The GATA2 transcription factor negatively regulates the proliferation of neuronal progenitors. Development. 2006;133:2155–65. doi: 10.1242/dev.02377. [DOI] [PubMed] [Google Scholar]
- 115.Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–41. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 116.Decleves X, Amiel A, Delattre JY, Scherrmann JM. Role of ABC transporters in the chemoresistance of human gliomas. Curr Cancer Drug Targets. 2006;6:433–45. doi: 10.2174/156800906777723930. [DOI] [PubMed] [Google Scholar]
- 117.Lepper ER, Nooter K, Verweij J, Acharya MR, Figg WD, Sparreboom A. Mechanisms of resistance to anticancer drugs: the role of the polymorphic ABC transporters ABCB1 and ABCG2. Pharmacogenomics. 2005;6:115–38. doi: 10.1517/14622416.6.2.115. [DOI] [PubMed] [Google Scholar]
- 118.Mutch DM, Anderle P, Fiaux M, Mansourian R, Vidal K, Wahli W, Williamson G, Roberts MA. Regional variations in ABC transporter expression along the mouse intestinal tract. Physiol Genomics. 2004;17:11–20. doi: 10.1152/physiolgenomics.00150.2003. [DOI] [PubMed] [Google Scholar]
- 119.Challen GA, Little MH. A side order of stem cells: the SP phenotype. Stem cells (Dayton, Ohio) 2006;24:3–12. doi: 10.1634/stemcells.2005-0116. [DOI] [PubMed] [Google Scholar]
- 120.Hadnagy A, Gaboury L, Beaulieu R, Balicki D. SP analysis may be used to identify cancer stem cell populations. Exp Cell Res. 2006;312:3701–10. doi: 10.1016/j.yexcr.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 121.Lin T, Islam O, Heese K. ABC transporters, neural stem cells and neurogenesis–a different perspective. Cell Res. 2006;16:857–71. doi: 10.1038/sj.cr.7310107. [DOI] [PubMed] [Google Scholar]
- 122.Zhu LL, Wu LY, Yew DT, Fan M. Effects of hypoxia on the proliferation and differentiation of NSCs. Mol Neurobiol. 2005;31:231–42. doi: 10.1385/MN:31:1-3:231. [DOI] [PubMed] [Google Scholar]
- 123.Zhang JM, Hoffmann R, Sieber-Blum M. Mitogenic and anti-proliferative signals for neural crest cells and the neurogenic action of TGF-beta1. Dev Dyn. 1997;208:375–86. doi: 10.1002/(SICI)1097-0177(199703)208:3<375::AID-AJA8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 124.Rice AC, Khaldi A, Harvey HB, Salman NJ, White F, Fillmore H, Bullock MR. Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp Neurol. 2003;183:406–17. doi: 10.1016/s0014-4886(03)00241-3. [DOI] [PubMed] [Google Scholar]
- 125.Zittermann SI, Issekutz AC. Basic fibroblast growth factor (bFGF, FGF-2) potentiates leukocyte recruitment to inflammation by enhancing endothelial adhesion molecule expression. Am J Pathol. 2006;168:835–46. doi: 10.2353/ajpath.2006.050479. [DOI] [PMC free article] [PubMed] [Google Scholar]