

Abstract
Cyclooxygenase-2 (COX-2)-dependent prostaglandin (PG) E2 synthesis in the spinal cord plays a major role in the development of inflammatory hyperalgesia and allodynia. Microsomal PGE2 synthase-1 (mPGES-1) isomerizes COX-2-derived PGH2 to PGE2. Here, we evaluated the effect of mPGES-1-deficiency on the noci-ceptive behavior in various models of nociception that depend on PGE2 synthesis. Surprisingly, in the COX-2-dependent zymosan-evoked hyperalgesia model, the nociceptive behavior was not reduced in mPGES-1-deficient mice despite a marked decrease of the spinal PGE2 synthesis. Similarly, the nociceptive behavior was unaltered in mPGES-1-deficient mice in the formalin test. Importantly, spinal cords and primary spinal cord cells derived from mPGES-1-deficient mice showed a redirection of the PGE2 synthesis to PGD2, PGF2α and 6-keto-PGF1α (stable metabolite of PGI2). Since the latter prostaglandins serve also as mediators of noci-ception they may compensate the loss of PGE2 synthesis in mPGES-1-deficient mice.
Keywords: pain, spinal cord, cyclooxygenase, prostaglandin, mPGES-1, hyperalgesia
Introduction
Prostaglanding E2 (PGE2) is an important mediator of inflammatory pain and fever [1–3]. It is synthesized by two cyclooxygenase (COX)-isoforms, COX-1 and COX-2, through conversion of arachidonic acid to the intermediate PGH2[4] which is subsequently isomer-ized by three specific terminal PGE2-synthases to PGE2. Due to its preferential coupling with COX-2, selective inhibition of the microsomal PGE-synthase-1 (mPGES-1) is regarded as a promising new target in order to a more selective suppression of the PGE2 production during inflammation and nociceptive processing [4]. Accordingly, several studies employing mPGES-1-deficient mice demonstrated a major role of mPGES-1 in PGE2-dependent processes such as the inflammatory response to lipopolysaccharide (LPS) [5], models of arthritis [3,5], and immune-induced pyresis [1] whereas its role in nociceptive processing has not been studied in detail.
The endoperoxide intermediate PGH2 serves as a common substrate for several prostaglandin synthas-es. Therefore the fate of PGH2 in the absence of mPGES-1 is of great importance for the evaluation of the effects of mPGES-1 inhibition [6]. Recently in peritoneal macrophages, the gastrointestinal system and the vascular system of mPGES-1-deficient mice, a redirection of eicosanoid metabolism was demonstrated [5,7–9]. Likewise, mice lacking prostacyclin synthase showed a marked increase in PGE2 and TxA2 levels in plasma, kidney, and lung [10], suggesting that the redirection of eicosanoid synthesis is not a specific phenomenon for inhibition of mPGES-1. Most importantly, previous studies employing prostaglandin receptor knockout mice or using intrathecal application of different prostaglandins demonstrated that not only PGE2 but also PGI2, PGD2, and PGF2α induce hyper-algesia [11–14]. Here, we investigated the effect of mPGES-1-deficiency on the nociceptive behavior in PGE2-dependent pain models. Surprisingly, we found during zymosan-induced thermal hyperalgesia, an animal model that depends on COX-2-derived PGE2, that the nociceptive behavior in mPGES-1-deficient mice was unaltered as compared to wild-type animals. In spinal cord cells of these knockout mice we observed a redirection of the eicosanoid synthesis to other prono-ciceptive prostaglandins, which might compensate the loss of PGE2 synthesis in these knockout mice.
Material and methods
Animals
Crl:CFW(Sw) mPGES-1-deficient mice were kindly provided by Aventis (Bridgewater, USA). In these animals the mPGES-1 expression was disturbed by the insertion of a neomycin cassette into exon 1 of the mPGES-1 gene [15]. In all experiments the ethics guidelines for investigations in conscious animals were obeyed and the procedures were approved by the local Ethics Committee. In all experiments, transgenic mice were compared with strain-, age-, and sex-matched controls.
Peritoneal macrophages
For cell culture, all substances were purchased from Gibco (Karlsruhe, Germany) unless otherwise specified. 0.5 ml 10% thioglycollate medium (Sigma, St. Louis, MO) was given intraperitoneally Peritoneal macrophages were prepared 4 days later as described previously [7].
Preparation of microsomal tissue fractions
Tissues were homogenized in phosphate-buffered saline (PBS) supplemented with 0.25 M sucrose and 1 M phenyl-methansulfonylfluoride (PMSF) (Sigma, St. Louis, MO) with a potter and by sonification. Then the homogenate was centrifuged for 10 min at 1000 ×g, for 15 min at 10,000 ×g and for 1 hr at 170,000 ×g. The pellet from the last centrifuga-tion step was re-suspended in 50 μl PBS, 0.25 M sucrose and 1 M PMSF. 30 μg protein was used for western blot analysis. COX-2, mPGES-2 and c-PGES were detected with antibodies from Cayman (Ann Arbor, MI), mPGES-1 antibody was from AgriSera (Vännäs, Sweden). Antibodies against extracellular regulated kinase-1/2 (ERK-1/2) (Promega, Madison, WI) or HSP-70 (BD Biosciences, Heidelberg, Germany) were used to control for equal protein loading.
Spinal cord cell culture
Primary embryonic spinal cord cell cultures were prepared as described previously [16]. Briefly, whole spinal cords were prepared from mice at embryonic day 16–18 and brought into culture. Cells were then incubated for 3 days in neurobasal-medium containing 10% foetal calf serum (FCS) and nerve growth factor followed by additional 3 day incubation in serum free neu-robasal-medium with mitosis inhibitors. Stimulation with LPS was performed in serum free medium without mitosis inhibitors.
LC-MS/MS analysis
The following eicosanoids were monitored by LC-MS/MS:PGF2α, PGE2, PGD2, TxB2, 11-dh-TxB2, 6-keto-PGF1α, Δ12-PGJ2, 15d-PGJ2, 13,14-dh-15-keto-PGD2, 13,14-dh-15-keto-PGE2, PGA2, PGB2, 15d-PGD2, and LTB4.
Preparation of spinal tissue
Lumbal spinal cords were prepared and directly frozen with liquid nitrogen. Then 200 μl PBS was added and the tissue quickly homogenized with a potter.100 μl of the homogenate were mixed with 50 μl water, 20 μl methanol and 20 μl internal standard solution (25ng/ml of [2H4]-PGE2, [2H4]-PGD2 and [2H4]-TXB2 and 10ng/ml of [2H4]-PGF2α and [2H4]-6-keto-PGF1α in methanol) and extracted twice with 800 μl ethyl acetate. The organic phase was removed at a temperature of 45°C under a gentle stream of nitrogen. The residues were reconstituted with 50 μl of acetonitrile/water/formic acid (20:80:0.0025, v/v, pH 4.0), and injected into the liquide chromatography-tandem mass spectrometry (LC-MS/MS). The prostanoids were normalized with protein concentrations in the sample.
Preparation of cell media
500 μl cell media were mixed with 500 μl 35 mM H3PO4, 20 μl methanol and 20 μl internal standard solution. Prostaglandins were extracted with solid-phase-extraction. Therefore, 1 ml Nexus cartridges (Varian, Darmstadt, Germany) were washed with 2 ml of hexane/ethylacetate/-isopropanol (35:60:5, v/v), dried for 20 sec and conditioned with 1 ml of methanol and 1 ml of water. 1040 μl of the prepared cell media (500 μl cell media, 500 μl 35 mM Hv3PO4, 20 μl prostaglandin standard/methanol and 20 μl internal standard solution) was loaded onto the column and washed with 1ml of water and 1ml of methanol/water (40:60, v/v). The cartridges were then dried for 7 min and eluted with 1 ml of hexane/ethylacetate/isopropanol (30:65:5, v/v).The organic phase was removed at a temperature of 45°C under a gentle stream of nitrogen. The residues were reconstituted with 50 μl of acetonitrile/water/formic acid (20:80:0.0025, v/v, pH 4.0), and injected into the LC-MS/MS. LC-MS/MS instrumentation and conditions are described in detail in the (see supplemental methods).
Behavioural tests
The investigator was unaware of the treatments or the genotypes during all behavioral experiments. Acidic acid writhing test: The writhing test was performed as described previously [3]. Briefly, 16 μl/g body weight of 0.7% acidic acid in PBS was injected intraperitoneally. Then writhes, defined as stretching of the torso accompanied by hyperextensions of the hind limbs, were monitored over 20 min. The formalin assay was performed as described previously [17]. Mechanical hyperalgesia: Mice were kept in the test cages for 4 hrs during the first day to allow accommoda-tion. During this time each mouse was tested several times to gain the baseline paw withdrawal thresholds. Animals were placed on an elevated wire grid and the plantar surface of the paw was stimulated using a Dynamic Plantar Aesthesiometer (Ugo Basile, Comerio VA, Italy). The device applies a constantly increasing force and stops on the withdrawal of the paw, recording the corresponding force. On day two 20 μl of a Zymosan A (Sigma, Deisenhofen, Germany) suspension (12.5 mg/ml in phosphate buffered saline) was injected subcutaneously into the plantar side of left hind paw. 10mg/kg etoricoxib was given i.p. 4hrs and 6hrs after zymosan injection. Control animals received dimethylsulfoxide (DMSO) alone at the same time points. Mechanical allodynia was assessed from 4 to 10 hrs after zymosan injection. Experiments were performed in air-conditioned rooms (22°C) between 8 a.m. and 6 p.m. Right (non-injected) and left (injected) paws were measured alternately in intervals of 5–10 min. At 2 hrs intervals, paw withdrawal thresholds were averaged. The measurement of paw withdrawal thresholds is reported in gram with an upper limit of 5 g and the data are presented as mean ± SEM. For statistical evaluation, the (injected) left paw of treated and untreated controls was compared. Data were analyzed by Student's t-test and P <0.05 was considered statistically significant.
Immunohistochemistry
Spinal (L4–5) tissues were cryostat sectioned at 10 μm and stored at 4°C until use. The staining was done as described previously [18] with the following exceptions. Sections were boiled in 0.1 M citric acid for 5 min with 240 watt (microwave) and then washed twice with PBS. Blocking was done in 3% bovine serum albumin (BSA) (Sigma, Deisenhofen, Germany) and 10% goat serum (Chemicon, Temecula, CA, USA) in PBS.
Multi-epitope ligand cartography (MELC) is an automated imaging technology using fluorescein isothiocyanate (FITC)-labelled antibodies [19].Spinal cords were removed from BL/6 wild-type mice, cryostat-sectioned at a thickness of 5 μm and fixed in acetone at –20°C for 10 min. Before use in the MELC system (MelTec, Magedburg, Germany) sections were re-hydrated in PBS at 20°C, incubated with normal goat serum for 30 min and washed again in PBS. Slides were placed on the stage of an inverted wide-field fluorescence microscope. Antibodies and wash solutions were added and removed robotically. After antibody incubation for 15 min, phase contrast and fluorescence images were acquired. Sections were then bleached using the excitation wavelength. After acquisition of post-bleaching images, the next cycle was started.
Primary polyclonal antibodies for PGD2-receptor (DP), PGF2α-receptor (FP) and prostacycline-receptor (IP) (1:50 dilution in PBS containing 3% BSA) were generated by immunization with receptor specific peptides. The peptides used were RNLTYVRGSVGPAT for the IP receptor, RYRSRC-SNSTNMESSL for the DP receptor and SPAAALLSNTTC-QTEN for the FP receptor. Immunization of rabbits was performed by Eurogentec (Seraing, Belgium) according to their standard protocol. Other antibodies used were against CGRP (Sigma, Deisenhofen, Germany), NF-200 (N 52) (Sigma, Deisenhofen, Germany) and NeuN (Chemicon, Temecula, CA, USA). As marker for lamina 2, the FITC-labelled lectin IB-4 (Sigma, Deisenhofen, Germany) was used.
Results
Lack of mPGES-1 expression in mPGES-1 knockout mice was confirmed by western blot analysis of kidney and lung tissue as well as in thioglycollate-elicited LPS-stimulated peritoneal macrophages (Fig. 1A and B). As described previously, the mPGES-1 deficiency did not alter the expression levels of the other PGE2 synthases, namely mPGES-2 and cPGES (Fig. 1B), but caused the re-direction of the eicosanoid synthesis to TxA2 (measured as the stable metabolite TxB2), PGI2 (measured by the stable metabolite 6-keto-PGF1α), PGF2α, and PGD2 (Fig. 1C) [5,7,8]. Importantly, mPGES-1-deficient mice exhibited normal motor function in the pole and the hanging wire test (Supplementary data 1: 1A, B) and normal basal pain thresholds in the tail flick and hot plate tests (supplementary data 1: 1C, D) as compared to wild-type mice.
1.
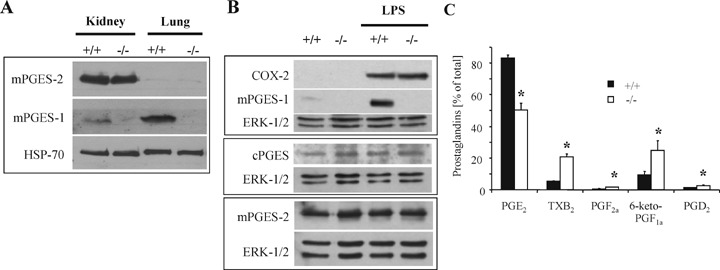
mPGES-1 expression and re-direction of eicosanoid metabolism in mPGES-1-deficient peritoneal macrophages. (A) Microsomal fractions (see Supplementary methods for details) of kidney and lung tissues from wild-type and mPGES-1-deficient mice were subjected to western blot analysis to detect relative protein amounts.(B) Peritoneal macrophages were stimulated with 5 μg/ml LPS for 15 hrs and then subjected to western blot analysis to detect relative protein amounts (see Supplementary methods for details). (C) Peritoneal macrophages were stimulated with 5 μg/ml LPS for 15hrs and prostaglandin levels determined from medium by LC-MS/MS. Data are shown as percent of all prostaglandins and are expressed as the mean ± S.E.M.(n= 9).*P <0.05.
Then we tested the mPGES-1-deficient mice in the writhing test, a commonly used model for visceral and acute pain where unselective COX inhibitors have a proven anti-nociceptive effect. As already described by others [3,5] a marked decrease in the number of writhes in mPGES-1-deficient mice as compared to wild-type animals was observed (Fig. 2A). However, in the writhing test nociceptive transmission depends on visceral chemoreceptors and although COX inhibitors decrease nociceptive behavior in this pain model the mode of action of PGE2 in this nociceptive response is still unclear [20]. In contrast, in the formalin test, a pain model that depends on the generation of PGE2 in the spinal cord, the mPGES-1-deficient mice showed only a slight, non-significant decrease in their nociceptive behavior as compared to wild-type mice (Fig. 2B) although the PGE2 concentrations in the spinal cord were significantly decreased (Fig. 2C).
2.
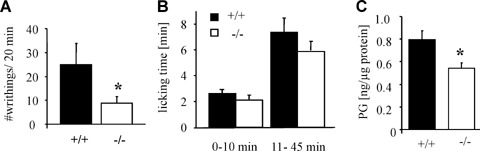
Nociceptive behavior of mPGES-1-deficient mice in the writhing and the formalin assay. (A) Responses in the acetic acid writhing assay. Total number of responses during 20 min after acid administration is shown as the mean±S.E.M.(n= 7–9).One-tailed Student's t-test:*P <0.05.(B) Formalin assay with mPGES-1−/– and wild-type ani-mals. Licking time between indicated time periods was measured. Data shown are the mean ±S.E.M.(n= 9).(C) PGE2 was extracted from spinal cord segments L4–L5 1hr after formalin injection. Data are shown as the mean ± S.E.M.(n= 6–7).
To characterize the effect of mPGES-1-deficiency in a model of inflammatory hyperalgesia that is known to depend on COX-2-mediated PGE2 synthesis, we determined the nociceptive behavior of mPGES-1-deficient mice in zymosan-evoked mechanical hyperalgesia. Zymosan injection into the left hind paw of wild-type animals resulted in a reduction of the mechanical threshold over 10 hrs. As expected administration of the selective COX-2 inhibitor etoricoxib (positive control) caused a marked decrease in the nociceptive behavior (antihy-peralgesia) of these animals reaching significance after 10 hrs (Fig. 3A). However, surprisingly the reduction of the mechanical threshold following zymosan injection was similar in wild-type and mPGES-1-deficient mice (Fig. 3B). In order to study the prostanoid synthetic pathways regarding a possible redirection of the prostanoid synthesis that may lead to a functional compensation of the loss of PGE2, we determined the concentrations of various eicosanoids in extracts from L4–L5 segments from spinal cords of zymosan-treated mice. As compared to wild-type mice the PGE2 levels were significantly lower in the mPGES-1-deficient mice (Fig. 3C). Since the difference in the total amount of PGE2 between both animal groups was relative small, it was not surprising that despite a tendency towards an increased synthesis of other prostaglandins (i.e. PGD2), it only reach significance for two eicosanoids, TxB2 and PGF2α (Fig. 3C). Most importantly, studies employing knockout mice or using intrathecal administration of different prostaglandins demonstrated that not only PGE2 but also PGI2, PGD2, and PGF2α induce hyper-algesia or sensitize nociceptive neurons [11–14].
3.
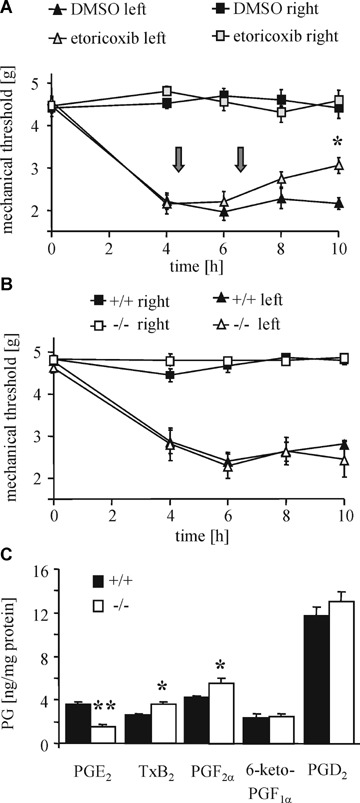
Nociceptive behavior of mPGES-1-deficient mice in zymosan-induced hyperalgesia.(A) Effect of etoricoxib on zymosan-evoked mechanical hyperalgesia. Zymosan was injected at time zero. Etoricoxib was given 4 and 6 hrs later (arrows). Data shown are the mean ± S.E.M. (n= 8–9).One-tailed Student's t-test:*P<0.05.(B) Same as panel A except wild-type and mPGES-1−/– mice were tested. Data are shown as the mean ± S.E.M. (n= 11). (C) PGE2 was extracted from spinal cord segments L4–L5 10 hrs after zymosan injection. Data are presented as mean ± S.E.M. (n= 3–8). Student's t-test: *P < 0.02, **P < 0.01.
Next, we employed primary cultures from embryonic spinal cords to study the effect of mPGES-1 deficiency on the prostanoid synthesis in more detail. LPS-stimulation of primary spinal cord neurons resulted in an up-regulation of the COX-2 expression in mPGES-1 as well as wild-type animals (Fig. 4A), while mPGES-1 was, as expected, only induced in cultures from wild-type mice. The expression of cPGES and mPGES-2 protein was unaltered, demonstrating that, like in LPS-stimulated macrophages (Fig. 1B), a compensatory up-regulation does not occur. In the spinal cord cultures only PGE2, PGD2, 6-keto-PGF1α, PGF2α, and TxB2 were produced at concentrations above the detection level of the LC-MS/MS-system used. Notably, the synthesis of 6-keto-PGF1α and PGF2α was already higher in unstimulated mPGES-1-deficient cells as compared to wild-type cells (Fig. 4B). After LPS-stimulation a 10-fold increase in the PGE2 synthesis occurred in wild-type cells (Fig. 4C). Cells from mPGES-1-deficient mice produced only 20% of the PGE2 levels seen in wild-type cells while the synthesis of 6-keto-PGF1α, PGF2α and PGD2 was significantly increased. A different pattern of an altered eicosanoid synthesis was seen in cells that were stimulated with TNF_ instead of LPS (Supplementary data 2). This finding indicates that the shift in the prostaglandin synthesis depends on the stimulus used and is probably due to differences in the induced expression of the terminal, synthases. Finally, in accordance with this observation the nonspecific mPGES-1 inhibitor MK-886 inhibited the PGE2 production in wild-type spinal cord cultures, while the 6-keto-PGF1α synthesis increased in an inverse manner (Fig. 4D). These data indicate that mPGES-1-inhibition either through genetic deletion or pharmacological inhibition induces a similar redirection of the eicosanoid synthesis.
4.
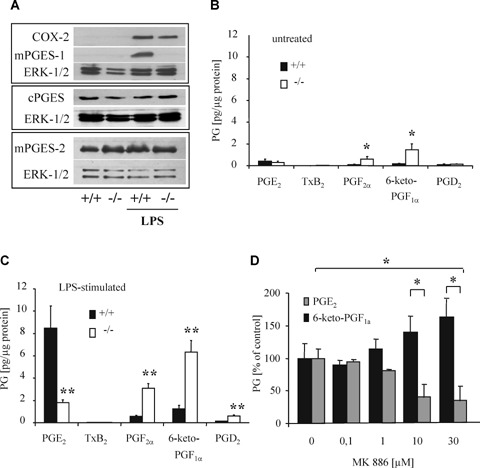
Eicosanoid re-direction in mPGES-1-deficient primary spinal cord cultures.(A) Primary spinal cord cultures were stimulated with 5 μg/ml LPS for 15 hrs and then subjected to western blot analysis. (B–C) Spinal cord cultures were incubated in serum-free medium for 15 hrs in absence (B) or presence (C) of 5 μg/ml LPS. Data are shown as the mean ±S.E.M.(n= 5, panel B; n= 14, panel C).Two-tailed Student's t-test:*P <0.05, **P <0.01.(D) Spinal cord cultures from wild-type mice were incubated with 5 μg/ml LPS and the indicated MK-886 concentrations for 15 hrs. Data are shown as percent of control and are the mean ± S.E.M.(n= 3). Student's t-test: *P <0.05.
To investigate whether or not the prostaglandins, which are compensatorily synthesized in absence of mPGES-1, might be involved in nociceptive processing in the outer laminae of the dorsal horn, we analysed the expression pattern of their corresponding receptors. Immunohistochemical staining of spinal cord sections showed that FP is expressed in neuronal and non-neuronal cells in the outer laminae of the dorsal horn (Fig. 5A). A similar expression pattern was observed for the DP and IP (Fig. 5B), which also co-localize in part with NeuN-immunostaining throughout the dorsal horn in spinal cords of adult mice.
5.
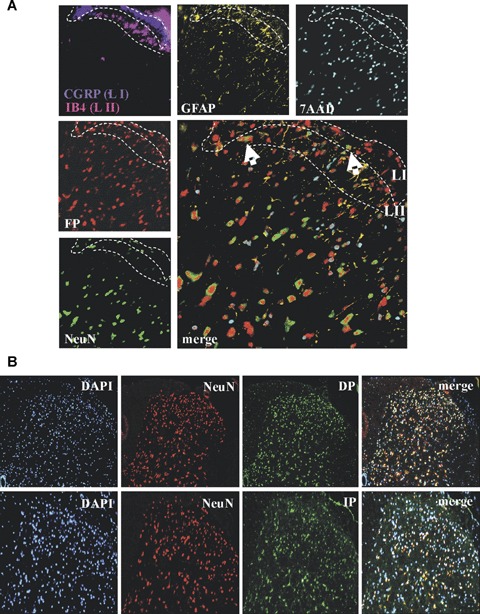
Prostaglandin receptors DP, FP and IP are expressed in dorsal horn neurons.(A) Immunohistochemical analysis of a L4–L5 spinal cord section from wild-type mice using the MELC technology. Signals are shown in false colors. CGRP (blue) was used as marker for lamina 1 (LI) and IB4 (pink) as marker for lamina 2 (LII). Arrows indicate FP-expressing neurons (white), or FP-expressing non-neuronal cells (yellow).(B) Immunohistochemical analysis of PGD2 (DP) and prostacyclin (IP) receptors in the spinal cord. Sections are from spinal cords (L4–5) of zymosan-treated (8 hrs) wild-type mice. DAPI was used for nuclear staining (blue), NeuN to stain neurons (red) and specific antibodies for the indicated prostaglandin receptors (green). The ipsilateral dorsal horn is shown.
Discussion
Here we show that mPGES-1-deficient mice, although the induced spinal PGE2 synthesis is significantly decreased, do not exhibit a reduced nociceptive behaviour in PGE2-dependent inflammatory pain models. Previously it has been demonstrated for several systems, such as peritoneal macrophages, the gastrointestinal system and the vascular system that in mPGES-1-deficient mice a redirection of eicosanoid metabolism occurs [5,7–9].We found that also in the spinal cord and in primary spinal cord cells, a redirection of the eicosanoid synthesis from PGE2 to other prostaglandins with pro-nociceptive properties (PGD2, PGF2α and prostacyclin) occurs. For these prostaglandins, it has been described that intrathecal injection of PGD2[14], PGI2[21] or PGF2α[11,13] induce allodynia or hyperalgesia [22] and it has been speculated that PGD2 might even mediate PGE2-evoked pain states, since PGDS-deficient mice do not develop PGE2-induced allodynia, [14, 23]. Furthermore, IP receptor-deficient mice as well as animals that received IP receptor antagonists exhibit a significantly reduced nociceptive behavior [12,24–26].
Taken together, these findings suggest that the redirection of the pro-staglandin synthesis in the spinal cord interferes with the anti-nociceptive effect of the decreased PGE2 synthesis. Since the PGE2 synthesis produces its pro-nociceptive actions mainly in neurons of lamina I and II in the dorsal horn, the expression of the receptors for PGD2, PGF2α and PGI2 in this region supports a possible compensation of the decreased PGE2 synthesis by one or more of these prostaglandins. Notably, the mechanisms by which FP, IP and DP receptors increase spinal noci-ceptive processing may be different from the PGE2 pathway. For example, it was recently shown that the re-direction of the prostaglandin synthesis in absence of mPGES-1 promotes iNOS expression and nitric oxide synthesis in embryonic fibroblasts [27].Thus, the ‘re-directed’prostanoids may increase spinal nociceptive processing through nitric oxide or other pro-nociceptive signaling elements.
Finally, and at a first view confusing, mPGES-1-deficient mice had fewer signs of inflammation in animal models of rheumatoid arthritis [3,5]. These data suggest that in these models, the re-direction of eicosanoid synthesis does not result in the synthesis of prostanoids that can fully compensate for the loss of PGE2 production. Since some prostanoids such as PGI2 and PGF2α, have anti-inflammatory effects and others, such as TXA2 and PGD2, exhibit pro- or antiinflammatory properties depending on the specific inflammatory setting [28], it can be speculated that in mPGES-1-deficient mice the redirected prostanoid synthesis may even help to enhance the desired antiinflammatory effect. Indeed, recently it was shown that in mPGES-1-deficient mice the re-direction of eicosanoid synthesis from PGE2 to PGI2 decreases the number of lesional macrophages and foam cells in atheriosclerotic plaques, thereby limiting atherio-genesis [29]. Whether or not mPGES-1 inhibitors may qualify as useful anti-inflammatory drugs needs further research. Additionally, results obtained in the writhing assay suggest some potency in the treatment of visceral pain. However, an anti-nociceptive effect of a reduced PGE2 production by inhibition of mPGES-1 seems to be compensated by the re-direction of the eicosanoid synthesis towards other prono-ciceptive prostaglandins, since not only PGE2 but also most other prostaglandins result in peripheral and central sensitization in the nociceptive system [23].However, due to the variability in the eicosanoid redirection patterns that even differ between the various mice strains [6], the effect of mPGES-1 inhibition on human pain perception can only be evaluated after the appropriate studies in humans employing selective mPGES-1-inhibitors have been performed.
Acknowledgments
The work was supported by the DFG grant GE695/2-2.The mPGES-1 knockout mice were a generous gift from Aventis (Bridgewater, USA).
Supporting Information
Legend to be supplied.
Table S1 Legend to be supplied.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Engblom D, Saha S, Engstrom L, Westman M, Audoly LP, J akobsson PJ, Blomqvist A. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat Neurosci. 2003;6:1137–8. doi: 10.1038/nn1137. [DOI] [PubMed] [Google Scholar]
- 2.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–5. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 3.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA. 2003;100:9044–9. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68–69:383–99. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- 5.Kamei D, Yamakawa K, Takegoshi Y, Mikami-Nakanishi M, Nakatani Y, Oh-Ishi S, Yasui H, Azuma Y, Hirasawa N, Ohuchi K, Kawaguchi H, Ishikawa Y, Ishii T, Uematsu S, Akira S, Murakami M, Kudo I. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004;279:33684–95. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- 6.Scholich K, Geisslinger G. Is mPGES-1 a promising target for pain therapy? Trends Pharmacol Sci. 2006;27:399–401. doi: 10.1016/j.tips.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Trebino CE, Eskra JD, Wachtmann TS, Perez JR, Carty TJ, Audoly LP. Redirection of eicosanoid metabolism in mPGES-1-deficient macrophages. J Biol Chem. 2005;280:16579–85. doi: 10.1074/jbc.M412075200. [DOI] [PubMed] [Google Scholar]
- 8.Boulet L, Ouellet M, Bateman KP, Ethier D, Percival MD, Riendeau D, Mancini JA, Methot N. Deletion of microsomal prostaglandin E2 (PGE2) synthase-1 reduces inducible and basal PGE2 production and alters the gastric prostanoid profile. J Biol Chem. 2004;279:23229–37. doi: 10.1074/jbc.M400443200. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and Cardiovascular function. J Clin Invest. 2006;116:1391–9. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokoyama C, Yabuki T, Shimonishi M, Wada M, Hatae T, Ohkawara S, Takeda J, Kinoshita T, Okabe M, Tanabe T. Prostacyclin-deficient mice develop ischemic renal disorders, including nephrosclerosis and renal infarction. Circulation. 2002;106:2397–403. doi: 10.1161/01.cir.0000034733.93020.bc. [DOI] [PubMed] [Google Scholar]
- 11.Muratani T, Nishizawa M, Matsumura S, Mabuchi T, Abe K, Shimamoto K, Minami T, Ito S. Functional characterization of prostaglandin F2alpha receptor in the spinal cord for tactile pain (allodynia) J Neurochem. 2003;86:374–82. doi: 10.1046/j.1471-4159.2003.01840.x. [DOI] [PubMed] [Google Scholar]
- 12.Murata T, Ushikubi F, Matsuoka T, Hirata M, Yamasaki A, Sugimoto Y, Ichikawa A, Aze Y, Tanaka T, Yoshida N, Ueno A, Oh-ishi S, Narumiya S. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–82. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 13.Turnbach ME, Spraggins DS, Randich A. Spinal administration of prostaglandin E(2) or prostaglandin F(2alpha) primarily produces mechanical hyperalge-sia that is mediated by nociceptive specific spinal dorsal horn neurons. Pain. 2002;97:33–45. doi: 10.1016/s0304-3959(01)00487-0. [DOI] [PubMed] [Google Scholar]
- 14.Eguchi N, Minami T, Shirafuji N, Kanaoka Y, Tanaka T, Nagata A, Yoshida N, Urade Y, Ito S, Hayaishi O. Lack of tactile pain (allodynia) in lipocalin-type prostaglandin D synthase-deficient mice. Proc Natl Acad Sci USA. 1999;96:726–30. doi: 10.1073/pnas.96.2.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeVito J, Devlin E, Schroeder K, Engle S, Swinton P, Sinha K, Bruno J, Souness J, Guehring H. Evaluation of inflammation and pain responses in mPGES-1 deficient mice. Inflamm Res. 2004;53:228. [Google Scholar]
- 16.Brenneis C, Maier TJ, Schmidt R, Hofacker A, Zulauf L, Jakobsson PJ, Scholich K, Geisslinger G. Inhibition of prostaglandin E2 synthesis by SC-560 is independent of cyclooxygenase 1 inhibition. FASEB J. 2006;20:1352–60. doi: 10.1096/fj.05-5346com. [DOI] [PubMed] [Google Scholar]
- 17.Hofacker A, Coste O, Nguyen HV, Marian C, Scholich K, Geisslinger G. Downregulation of cytosolic prostaglandin E2 synthase results in decreased nociceptive behavior in rats. J Neurosci. 2005;25:9005–9. doi: 10.1523/JNEUROSCI.2190-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehnert C, Tegeder I, Pierre S, Birod K, Nguyen HV, Schmidtko A, Geisslinger G, Scholich K. Protein associated with Myc (PAM) is involved in spinal noci-ceptive processing. J Neurochem. 2004;88:948–57. doi: 10.1046/j.1471-4159.2003.02229.x. [DOI] [PubMed] [Google Scholar]
- 19.Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Bockelmann R, Malykh Y, Gollnick H, Friedenberger M, Bode M, Dress AW. Analyzing pro-teome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 2006;24:1270–8. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- 20.Jett MF, Ramesha CS, Brown CD, Chiu S, Emmett C, Voronin T, Sun T, O’Yang C, Hunter JC, Eglen RM, Johnson RM. Characterization of the analgesic and anti-inflammatory activities of ketorolac and its enantiomers in the rat. J Pharmacol Exp Ther. 1999;288:1288–97. [PubMed] [Google Scholar]
- 21.Doi Y, Minami T, Nishizawa M, Mabuchi T, Mori H, Ito S. Central nociceptive role of prostacyclin (IP) receptor induced by peripheral inflammation. Neuroreport. 2002;13:93–6. doi: 10.1097/00001756-200201210-00022. [DOI] [PubMed] [Google Scholar]
- 22.Svensson CI, Yaksh TL. The spinal phospholi-pase–cyclooxygenase-prostanoid cascade in noci-ceptive processing. Annu Rev Pharmacol Toxicol. 2002;42:553–83. doi: 10.1146/annurev.pharmtox.42.092401.143905. [DOI] [PubMed] [Google Scholar]
- 23.Vanegas H, Schaible HG. Prostaglandins and cyclooxygenases in the spinal cord. Prog Neurobiol. 2001;64:327–63. doi: 10.1016/s0301-0082(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 24.Bley KR, Bhattacharya A, Daniels DV, Gever J, Jahangir A, O’Yang C, Smith S, Srinivasan D, Ford AP, Jett MF. RO1138452 and RO3244794: characterization of structurally distinct, potent and selective IP (prostacyclin) receptor antagonists. Br J Pharmacol. 2006;147:335–45. doi: 10.1038/sj.bjp.0706554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bley KR, Hunter JC, Eglen RM, Smith JA. The role of IP prostanoid receptors in inflammatory pain. Trends Pharmacol Sci. 1998;19:141–7. doi: 10.1016/s0165-6147(98)01185-7. [DOI] [PubMed] [Google Scholar]
- 26.Pulichino AM, Rowland S, Wu T, Clark P, Xu D, Mathieu MC, Riendeau D, Audoly LP. Prostacyclin antagonism reduces pain and inflammation in rodent models of hyperalgesia and chronic arthritis. J Pharmacol Exp Ther. 2006;319:1043–50. doi: 10.1124/jpet.106.110387. [DOI] [PubMed] [Google Scholar]
- 27.Kapoor M, Kojima F, Qian M, Yang L, Crofford LJ. Shunting of prostanoid biosynthesis in microsomal prostaglandin E synthase-1 null embryo fibroblasts: regulatory effects on inducible nitric oxide synthase expression and nitrite synthesis. FASEB J. 2006;20:2387–9. doi: 10.1096/fj.06-6366fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–66. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci USA. 2006;103:14507–12. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legend to be supplied.
Table S1 Legend to be supplied.