

Abstract
Novel 6,7-methylenedioxy-4-substituted phenylquinolin-2-one derivatives 12a–n were designed and prepared through an intramolecular cyclization reaction and evaluated for in vitro anticancer activity. Among the synthesized compounds, 6,7-methylenedioxy-4-(2,4-dimethoxyphenyl)quinolin-2(1H)-one (12e) displayed potent cytotoxicity against several different tumor cell lines at a sub-micromolar level. Furthermore, results of fluorescence-activated cell sorting (FACS) analysis suggested that 12e induced cell cycle arrest in the G2/M phase accompanied by apoptosis in HL-60 and H460 cells. This action was confirmed by Hoechst staining and caspase-3 activation. Due to their easy synthesis and remarkable biological activities, 4-phenylquinolin-2(1H)-one analogs (4-PQs) are promising new anticancer leads based on the quinoline scaffold. Accordingly, compound 12e was identified as a new lead compound that merits further optimization and development as an anticancer candidate.
Keywords: 4-Phenylquinolin-2(1H)-one (4-PQ), Anticancer agent, Podophyllotoxin, Apoptosis, Structure-activity relationships (SAR)
1. Introduction
Cancer is the leading cause of death in economically developed countries and the second leading cause of death in developing countries.1 Development of novel antitumor agents with cytotoxicity against cancer cell lines is an important focus of oncology. Natural products are often referred to as an inexhaustible source of lead compounds that are likely to exhibit multiple biological activities, including anticancer.2,3 Because of their intrinsic biorelevance, natural products represent a significant source of inspiration for the design of structural analogues with new pharmaceutical profiles.
The microtubule network is an essential component of the cytoskeleton of eukaryotic cells. Podophyllotoxin (PPT, 1) is a aryltetralin lignan isolated from the roots of Podophyllum peltatum L., P. emodi W., or P. pleianthum H..4 PPT inhibits tubulin assembly and polymerization through interaction with the protein at the colchicine binding site, thus preventing the formation of the spindle, and arresting cell division in metaphase (G2/M stage).5−7 Numerous efforts have been made to improve PPT’s safety profile while maintaining its potency. Thus, its use as a lead agent in the development of new anticancer drugs has led to the discovery of semisynthetic derivatives, such as etoposide (2) and teniposide (3), and a more soluble prodrug of etoposide, named etopophos (4) (Fig. 1). Semisynthetic derivatives of PPT are currently in clinical use for the treatment of various malignancies.
Figure 1.

Structures of podophyllotoxin and related compounds: podophyllotoxin (1) and its semi-synthetic derivatives in clinical use, etoposide (2), teniposide (3) and etopophos (4).
PPT’s structure is complex due to the presence of four chiral carbons in ring C (Fig. 1), which creates challenges for SAR (structure-activity relationship) studies.Research efforts to develop new analogues with simpler structures are ongoing ot identify the critical substructures for desired biological activity, and almost all rings (ring A to E) and positions on the cyclolignan skeleton have been modified.8 Importantly, elimination of the stereogenic centers at C-2 and C-3 removes the possibility of epimerization at C-2. Epimerization of the natural lignan has plagued its clinical development, because the rapidly formed in vivo cis-lactone metabolite is inactive.9 Numerous attempts have been made to obtain potent synthetically feasible analogues of PPT with a heteroatom incorporated in ring C to prevent epimerization. 4-Aza-podophyllotoxin (5) (Fig. 2) and related analogues were identified as structurally simpler derivatives of PPT with reported antiproliferative activity.10−12 In addition, podophyllic aldehyde (6) was found to be a highly selective antitumor agent against HT-29 colon and A-549 lung carcinomas. Several aldehydes related to this compound but with different configurations have been synthesized and evaluated for cytotoxic activities in neoplastic cell lines.13,14 These podophyllic aldehyde-related compounds lacked the lactone ring, but maintained cytotoxicity at, or under, micromolar level.
Figure 2.

Stuctures of 4-aza-PPT analogues (5), podophyllic aldehyde (6), combretastatin A-4 (7), 2-PQ derivatives and the target compounds (12a–n).
Another naturally occurring cis-stilbene, combretastatin A-4 (CA-4, 7) (Fig. 2) was isolated from the bark of the south African tree Combretum caffrum (Combretaceae).15 CA-4 is a potent tubulin assembly inhibitor and vascular disrupting agent at low concentrations.16 Its relative molecular simplicity suggests numerous practical approaches to the design of new antineoplastic agents, and several active stilbene-based compounds have been identified.17 The main disadvantage of CA-4 is the ready isomerization of its cis-double bond to an inactive trans-form during storage and administration.18 Consequently, this double bond in the cis-stilbene motif has often been used as a modification site and replaced with various heterocyclic or carbonyl functional groups.19−21
According to previous SAR studies on PPT and related analogues, structurally modified compounds that lack the trans-γ-lactone ring (ring D) can show potent and selective cytotoxicity against cancer cells. Thus, the γ-lactone ring is not an essential feature for cytotoxicity of PPT analogues. These results encouraged us to select 4-aza-podophyllotoxin (5) as the template and design much simpler structures without a γ-lactone ring. In our previous investigation, 2-phenylquinolin-4(1H)-one analogs (2-PQs) (Fig. 2) were identified as antimitotic agents.22 Recently, we shifted the phenyl ring on the quinolinone from the C-2 to C-4 position. Hence, a series of 4-phenylquinolin-2(1H)-one analogs (4-PQs) was designed that mimics the structures of 5–7 (Fig. 3). In addition, the two aromatic rings (B and E) of 4-phenylquinolin-2(1H)-one derivatives (4-PQs) adopt a conformation in which they are not coplanar, and the structural similarity between CA-4 and appropriately substituted 4-PQs might lead to interaction at the colchicine site to affect tubulin polymerization. Moreover, 4-PQ compounds would not be inactivated by cis to trans isomerization as with CA-4 derivatives.
Figure 3.

Rationally designed 4-PQ analogs mimic structures of PPT analogues and CA-4.
In this article, we describe the synthesis and cytotoxicity evaluation of 6,7-methylenedioxy-4-substituted phenylquinolin-2(1H)-one derivatives 12a–n (Fig. 3). Also, additional biological studies have been performed to analyze how novel compounds of this class affect the cell cycle.
2. Chemistry
The synthetic route to 6,7-methylenedioxy-4-substituted phenylquinolin −2(1H)-one derivatives is illustrated in Scheme 1. The 4-phenylquinolin-2(1H)-one> derivatives 12a–n were prepared using the general Knorr quinoline synthesis (acid-catalysis).23,24 The synthesis was initiated with alkoxycarbonylation of substituted acetophenones 8a–n with a diethyl carbonate (9) to provide the corresponding benzoylacetates 10a–n,25–27 which were condensed with 3,4-methylenedioxy aniline to give benzoylacetanilide intermediates 11a–n. The 4-phenylquinolin-2(1H)-one derivatives 12a–n were obtained by cyclization of key intermediates 11a–n with excess polyphosphoric acid (PPA) at 100–110 °C. The synthesized compounds 12a–n are reported for the first time. All synthesized compounds were characterized by IR, 1H NMR, 13C NMR and mass spectrometry. As an example, the target compound 12a possessed a characteristic peak at 6.02 ppm, representative of the methylenedioxy protons. Three singlet peaks at 6.18, 6.63 and 6.87 ppm were assignable to C(3)H, C(5)H and C(8)H protons in the 2-quinolone core, and one broad singlet (11.78 ppm) to an exchangeable NH group. The protons of the 4-phenyl ring appeared in the region 7.36–7.51 ppm. In the 13C NMR spectrum, compound 12a possessed a characteristic absorption at 5 161.70 ppm for the 2-quinolone amide carbon (NHC=O). The IR spectrum of 12a showed an absorption at 1653.07 cm−1 for the amide (NHC=O) carbonyl group.
Scheme 1.

Reagents and conditions: (a) NaH, toluene, reflux; (b) 3,4-methylenedioxy aniline, toluene, reflux; (c) PPA, 100–110 °C.
3. Biological evaluation
3.1.Invitro cytotoxicity assay
The newly synthesized analogues 12a–n were evaluated for cytotoxicity against Detroit 551 normal human cells and eight cancer cell lines: MCF-7 (breast adenocarcinoma), 2774 (ovarian carcinoma), SKOV-3 (ovarian carcinoma), HL-60 (leukemia), Hep 3B (hepatoma), H460 (non-small-cell-lung carcinoma), COLO205 (colorectal adenocarcinoma) and A498 (renal cell carcinoma). Etoposide and 5-FU was used as a positive control. The screening procedure was based on the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliun bromide (MTT) growth inhibition assay, and the results are summarized in Table 1.
Table 1.
Cytotoxicity of compounds 12a–n.
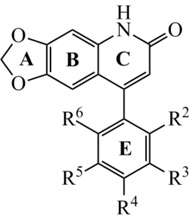 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | R2 | R3 | R4 | R5 | R6 | IC50(µM)a |
|||||||||
| MCF-7 | 2774 | SKOV-3 | HL-60 | Hep 3B | H460 | COLO 205 | A498 | Detroit 551 | |||||||
| Un-substituted | |||||||||||||||
| 12a | H | H | H | H | H | 18.5 | >30 | >30 | >50 | >50 | >50 | >50 | 42.7 | >50 | |
| Mono-substituted | |||||||||||||||
| 12b | OCH3 | H | H | H | H | 27.1 | 18.8 | 26.6 | 10 | 23.3 | >25 | 18.1 | 9.8 | >25 | |
| 12c | H | OCH3 | H | H | H | 14.9 | 15.6 | 24.6 | >10 | >10 | >10 | 39.6 | >50 | >10 | |
| 12d | H | H | OCH3 | H | H | >30 | >30 | 26.8 | >20 | >20 | >20 | >50 | >25 | >20 | |
| Di-substituted | |||||||||||||||
| 12e | OCH3 | H | OCH3 | H | H | 6.0 | 0.4 | 0.4 | 0.4 | 1.0 | 0.9 | 7.4 | 48 | >25 | |
| 12f | OCH3 | H | H | OCH3 | H | 3.7 | 6.5 | 20 | 3.5 | 10 | 10 | 6.1 | 28.9 | >100 | |
| 12g | OCH3 | H | H | H | OCH3 | 29.8 | >30 | >30 | >100 | >100 | >100 | >50 | >50 | >100 | |
| 12h | H | OCH3 | OCH3 | H | H | 14.3 | 6.6 | 6.4 | 4.1 | 14.6 | 14.4 | 16.8 | 25.6 | 23.6 | |
| 12i | H | OCH3 | H | OCH3 | >30 | 8.3 | >30 | 2.9 | >10 | >10 | >50 | 11.7 | >10 | ||
| Tri-substituted | |||||||||||||||
| 12j | OCH3 | H | OCH3 | H | OCH3 | 21 | >30 | >30 | 75 | >100 | 67.6 | 37.4 | >50 | >100 | |
| 12k | H | OCH3 | OCH3 | OCH3 | H | 4.5 | 2.1 | 0.93 | 2.3 | 5.0 | 7.6 | >50 | >50 | >25 | |
| Fluoro-substituted | |||||||||||||||
| 121 | F | H | H | H | H | >30 | 26.4 | >30 | >100 | >100 | >100 | >50 | >50 | >100 | |
| 12m | H | H | H | H | >30 | 24.5 | >30 | >20 | >20 | >20 | >50 | >50 | >20 | ||
| 12n | H | H | F | H | H | >30 | 17.6 | >30 | >60 | >20 | >20 | >50 | >50 | >20 | |
| Etoposide | 5.48 | 1.0 | |||||||||||||
| 5-FU | 22.3 | 26.7 | |||||||||||||
Human tumor cells were treated with different concentrations of samples for 48 h (n = 3 independent experiments).
Data are presented as IC50 (µM, the concentration of 50% proliferation-inhibitory effect).
Compound 12a, which contains an unsubstituted 4-phenyl E-ring, displayed poor cytotoxicity with IC50 values from 18.5 to greater than 50 µM. The steric parameters of hydrogen and fluorine are extremely similar (van der Waals radii of 1.2 Å for hydrogen vs. 1.35 Å for fluorine), and fluoro-substituted 12l, 12m, and 12n also displayed weak cytotoxicity against the cancer cell lines.
However, 12k, whose ring E bears the same 3,4,5-trimethoxy substitution found in PPT, exhibited significant antiproliferative activity with sub-micromolar IC50 (0.93–7.6 µM) against several cancer cell lines. However, potency dropped when the 3,4,5-trimethoxy group (12k) was changed to a 2,4,6-trimethoxy group (12j). In addition, the effects of dimethoxy substitution on the phenyl E-ring is noteworthy. Generally, dimethoxy substitution on the 4-phenyl ring (12e, 12f, 12h, 12i) led to more potent activity than monomethoxy substitution (12b–d). However, compound 12g with 2,6-dimethoxy substitution was less potent than the other four dimethoxy substituted analogues. Among the four more active dimethoxy compounds, analogue 12e with 2,4-dimethoxy substitution exhibited the most potent anticancer activity with IC50 less than or equal to 1 µM against five cell lines, 2774 (0.4 µM), SKOV-3 (0.4 µM), HL-60 (0.4 µM), Hep 3B (1.0 µM) and H460 (0.9 µM). Compound 12e was also generally more potent than the trimethoxy substituted 12k. Modification of the E-ring with different numbers and positions of methoxy substituents may affect its spatial rotation and the molecular orientation, which could increase binding to the biological target and anticancer activity of the optimized compounds. Finally, 12e showed only marginal toxicity against Detroit 551 normal human cells (IC50 > 25 µM). It is also noteworthy that the cell lines more resistant to 12e, including MCF-7, COLO205, and A498, harbor wild-type p53.
It is generally accepted that PPT’s cytotoxicity is attributable to inhibition of tubulin polymerization, while the target of etoposide is topoisomerase II. Recently, an alternative non-tubulin and non-antitopoisomerase mode of action has been proposed for some PPT derivatives. This mechanism is associated with cell death and sub-G1 apoptotic cell accumulation without any significant cell cycle arrest.13 Thus, mechanistic studies on 12e are underway in our laboratories.
3.2. Morphological changes and apoptosis
Apoptosis is one of the major pathways that lead to the process of cell death. Chromatin condensation, nuclear shrinking, and fragmented nuclei are known as classic characteristics of apoptosis. To examine the effect of 12e on apoptotic induction, the morphology changes of HL-60 and H460 cells were studied using Hoechst 33258 stain, which confirmed apoptosis as the cause of reduced cell viability. Compound 12e decreased cell viability and increased morphological changes in HL-60 and H460 cells. As shown in Figure 4, control cells with 0.1% DMSO treatment exhibited uniformly dispersed chromatin. Cells treated with compound 12e for 24 h demonstrated typical apoptotic characteristics, including condensation of chromatin and appearance of nuclear fragmentation and apoptotic bodies (the arrowhead indicates an apoptotic nucleus). These results clearly demonstrated that the compound 12e is effective in inducing cellular apoptosis.
Figure 4.

Fluorescent images of Hoechst staining showing compound 12e induced cell death. (A) Treatment of HL-60 cells with 12e for 24 h. The arrows indicate the formation of apoptotic bodies. (B) Treatment of H460 cells with 12e for 24 h. The arrows indicate the formation of apoptotic bodies. Scale bar = 50 µm.
3.3. Cell cycle analysis
Previous studies have indicated that PPT analogues induced DNA damage and caused G2/M arrest.28–30 To evaluate the effect of 12e on the cell cycle andgain further insight into the mode of action, we examined cell cycle accumulation at 24 h by propidium iodide staining and flow cytometry quantification in HL-60 and H460 cells treated with 12e. In HL-60 cells treated with 0.25 µM, 0.5 µM, and 1.0 µM concentrations of 12e, 11.87%, 25.02% and 40.54%, respectively, of cells were in G2/M phase, 10.51% of control (untreated) cells were in this phase (Fig. 5A). The percentage of G2/M cells also increased in H460 cells treated with 12e (Fig. 5B). As shown in Figure 5, the DNA cell cycle analysis revealed typical G2/M arrest and apoptosis (a sub-G1 peak appeared) in response to treatment with 12e.
Figure 5.

Compound 12e affected the cell cycle distribution. (A) HL-60 cells treated with 12e for 24 h. (B) H460 cells treated with 12e for 24 h.
3.4. Mechanism of action
Cyclin B1 and CDK1 are intricately involved in cell cycle progression through the G2/M phase transition.31 In our previous studies, the exposure of HL-60 cells to 2-PQ analogues led to G2/M phase arrest. In the Western blot analysis, a marked decrease in the expressions of cyclin B1 and CDK1 were detected in HL-60 cells.32,33 As shown in Figure 6, compound 12e induced a transient increase followed by a decrease in cyclin B1 protein expression, whereas CDK1 protein expression level decreased in a concentration-dependent manner in HL-60 cells (Fig. 6A). However, CDK1 protein expression level increased in a concentration-dependent manner in H460 cells (Fig. 6B). It was reported that treatment with deoxypodophyllotoxin (DPPT) in Hela cells induced G2/M phase arrest. In the Western blot analysis, cyclin B1 expression was observed to have a rapid increase within 3 h of DPPT treatment that remained elevated for up to 24 h. After 48 h, the expression of cyclin B1 returned to near control levels.34 Another study was reported that treatment of NTUB1 cells with higher concentration of 1-hydroxy-3-(3-alkylaminopropoxy)-9,10-anthraquinone (MHA) derivative for 24 h induced up-regulation of cyclinB1 and p21 expressions in NTUB1 cells while treatment with lower concentration of MHA for 24 h induced down-regulation of cyclinB1 and up-regulation of p21 expressions in NTUB1 cells.35 These results suggest that cell cycle progression is well regulated by the timing of the expression of cell-specific cyclins and different concentrations of the compounds.
Figure 6.

Regulation of mitotic phase- and apoptosis-associated proteins expression by compound 12e. (A) HL-60 cells treated with the indicated concentration of 12e for 24 h. (B) H460 cells treated with the indicated concentration of 12e for 24 h. After treatment, the cells were harvested and subjected to Western blot. The relative amounts of cyclin Bl, CDK1, and caspase 3 proteins were quantified and normalized to the corresponding β-actin protein amount. The quantitative data are shown under each protein, respectively.
Apoptosis is associated with activation of caspases, an expanding family of cysteine proteases that play important roles in the execution phase of apoptosis triggered by various stimuli.36 Among the capsases, caspase 3 is the best understood and mediates several apoptotic pathways. As shown in Figure 6, compound 12e induced caspase 3 cleavage (17 kDa) in HL-60 and H460 cells. The above preliminary investigation of the mechanism of action of compound 12e suggested that the anticancer effects of 12e against HL-60 and H460 cells are mediated via G2/M arrest and caspase-dependent apoptosis.
4. Conclusion
4-Phenylquinolin-2(1H)-one derivatives 12a–n were synthesized and tested for antiproliferative activity against several cancer cell lines. Compound 12e, containing 2,4-dimethoxy substitution on the 4-phenyl ring, demonstrated potent antiproliferative activities with IC50 values of 0.4, 0.4, 0.4 and 0.9 µM against 2774, SKOV-3, HL-60 and H460, respectively. The significant anticancer activity shown by 12e prompted us to evaluate cell viability of HL-60 and H460 cells, with the observation of morphological features of apoptotic cells in these cell lines. Furthermore, 12e could induce cell cycle arrest in the G2/M phase and apoptosis by activation of caspase-3 in both HL-60 and H460 cells. In conclusion, among the newly synthesized 4-PQ derivatives, compound 12e was identified as a promising candidate; due to its excellent antiproliferative potency, together with remarkable apoptosis-inducing activity. Further structure optimization of 12e is ongoing.
5. Experimental section
5.1. Materials and physical measurements
All solvents and reagents were obtained commercially and used without further purification. The progress of all reactions was monitored by TLC on 2 × 6 cm pre-coated silica gel 60 F254 plates of thickness 0.25 mm (Merck). The chromatograms were visualized under UV light at 254–366 nm. The following adsorbent was used for column chromatography: silica gel 60 (Merck, particle size 0.063–0.200 mm). Melting points (mp) were determined with a Yanaco MP-500D melting point apparatus and are uncorrected. IR spectra were recorded on Shimadzu IR-Prestige-21 spectrophotometers as KBr pellets. The NMR spectra were obtained on a Bruker Avance DPX-200 FT-NMR spectrometer at room temperature, and chemical shifts are reported in ppm (δ). The following abbreviations are used: s, singlet; d, doublet; t, triplet; dd, double doublet; and m, multiplet. Low-resolution mass spectra were performed by Finnigan/Thermo Qust MAT95XL at National Chung Hsing University, Taichung, Taiwan.
5.2. Chemistry
5.2.1. General procedure for the synthesis of benzoylacetates 10a–n
The substrate β-ketoesters 10a–n were either purchased or synthesized following published procedures. Some benzoylacetates were commercially available. Ethyl 3-oxo-3-phenyl propanoate (10a) was purchased. The reaction of benzoylacetates 10b–n was prepared as described in previous reports.25–27 A solution of a substituted acetophenone 8a–n (0.05 mol) dissolved in toluene (50 mL) was added dropwise to a solution containing diethyl carbonate (9) (0.10 mol) and sodium hydride (0.15 mol 60% dispersion in mineral oil). The mixture was stirred at room temperature, and then refluxed for 30 min. The mixture was poured into ice water, acidified with glacial acetic acid, and extracted with EtOAc (3 × 100 mL). The EtOAc extract was then dried over anhydrous MgSO4. After removal of the solvent in vacuo, the crude products were purified by silica gel column chromatography eluting with dichloromethane to afford benzoylacetates 10b–n. All synthetic compounds were in agreement with 1H NMR, 13C NMR, IR and mass spectroscopic data.
5.2.1.1. Ethyl 3-(2-methoxyphenyl)-3-oxopropanoate (10b)
Yield 86% from 8b as a yellow liquid; IR (KBr) ν (cm−1): 1735.93, 1674.21 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.14 (t, J = 7.2 Hz, 3H, -O-CH2CH3), 3.83 (s, 3H, -OCH3), 3.91 (s, 2H, -CO-CH2-CO-), 4.07 (q, J = 7.2 Hz, 2H, -O-CH2CH3), 7.03 (t, J = 7.6 Hz, 1H, Ar-H), 7.14 (d, J = 8.2 Hz, 1H, Ar-H), 7.56 (t, J = 7.6 Hz, 1H, Ar-H), 7.70 (d, J = 7.8 Hz, 1H, Ar-H); 13C NMR (50 MHz, DMSO-d6): δ 14.37, 50.48, 56.03, 60.80, 112.95, 120.99, 126.11, 130.50, 135.37, 159.37, 168.20, 193.25; MS (EI, 70 eV) m/z: 222.1 (M+); HRMS (EI) m/z: calculated for C12H14O4: 222.0892; found: 222.0884.
5.2.1.2. Ethyl 3-(3-methoxyphenyl)-3-oxopropanoate (10c)
Yield 80% from 8c as a yellow liquid; IR (KBr) ν (cm−1): 1743.65, 1689.64 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.16 (t, J = 7.2 Hz, 3H, -O-CH2CH3), 3.80 (s, 3H, -OCH3), 4.06–4.20 (m, 4H, -CO-CH2-CO- and -O-CH2CH3), 7.19–7.25 (m, 1H, ArH), 7.40–7.57 (m, 3H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.36, 46.12, 55.73, 61.05, 113.31, 120.18, 121.34, 130.38, 137.64, 159.91, 168.08, 193.67; MS (EI, 70 eV) m/z: 222.1 (M+); HRMS (EI) m/z: calculated for C12H14O4: 222.0892; found: 222.0898.
5.2.1.3. Ethyl 3-(4-methoxyphenyl)-3-oxopropanoate (10d)
Yield 87% from 8d as a yellow liquid; IR (KBr) ν (cm−1): 1735.93, 1674.21 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.16 (t, J = 7.2 Hz, 3H, -O-CH2CH3), 3.82 (s, 3H, -OCH3), 4.05–4.15 (m, 4H, -CO-CH2-CO- and -O-CH2CH3), 7.03 (d, J = 8.9 Hz, 2H, ArH), 7.93 (d, J = 8.9 Hz, 2H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.36, 45.71, 55.95, 60.97, 114.40 (2C), 129.24, 131.24 (2C), 164.04, 168.25, 192.06; MS (EI, 70 eV) m/z: 222.1 (M+); HRMS (EI) m/z: calculated for C12H14O4: 222.0892; found: 222.0899.
5.2.1.4. Ethyl 3-(2,4-dimethoxyphenyl)-3-oxopropanoate (10e)
Yield 70% from 8e as a yellow liquid; IR (KBr) ν (cm−1): 1735.93, 1666.50 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.16 (t, J = 7.2 Hz, 3H, -O-CH2CH3), 3.83–3.84 (m, 8H, 2 × -OCH3 and -CO-CH2-CO-), 4.08 (q, J = 7.2 Hz, 2H, -O-CH2CH3), 6.58–6.62 (m, 2H, ArH), 7.75 (d, J = 9.4 Hz, 1H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.39, 50.42, 56.03 (2C), 60.64, 98.59, 106.83, 119.11, 132.61, 161.57, 165.47, 168.46, 190.91; MS (EI, 70 eV) m/z: 252.1 (M+); HRMS (EI) m/z: calculated for C13H16O5: 252.0998; found: 252.0991.
5.2.1.5. Ethyl 3-(2,5-dimethoxyphenyl)-3-oxopropanoate (10f)
Yield 70% from 8f as a yellow liquid; IR (KBr) ν (cm−1): 1735.93, 1674.21 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.16 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 3.73 (s, 3H, -OCH3), 3.80 (s, 3H, -OCH3), 3.91 (s, 2H, -CO-CH2-CO-), 4.08 (q, J = 7.1 Hz, 2H, -O-CH2CH3), 7.12–7.15 (m, 2H, ArH), 7.22–7.24 (m, 1H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.38, 50.38, 55.90, 56.42, 60.75, 113.94, 114.51, 121.47, 126.35, 153.40, 153.81, 168.12, 192.75; MS (EI, 70 eV) m/z : 252.2 (M+); HRMS (EI) m/z: calculated for C13H16O5: 252.0998; found: 252.0996.
5.2.1.6. Ethyl 3-(2,6-dimethoxyphenyl)-3-oxopropanoate (10g)
Yield 78% from 8g as a colorless liquid; IR (KBr) ν (cm−1): 1743.65, 1705.07 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.14 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 3.75 (s, 8H, 2 × -OCH3 and -CO-CH2-CO-), 4.06 (q, J = 7.1 Hz, 2H, -O-CH2CH3), 6.70 (d, J = 8.4 Hz, 2H, ArH), 7.35 (t, J = 8.4 Hz, 1H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.30, 51.47, 56.26 (2C), 60.84, 104.71 (2C), 118.63, 132.07, 156.98 (2C), 166.82, 195.95; MS (EI, 70 eV) m/z: 252.2 (M+); HRMS (EI) m/z: calculated for C13H16O5: 252.0998; found: 252.1006.
5.2.1.7. Ethyl 3-(3,4-dimethoxyphenyl)-3-oxopropanoate (10h)
Yield 76% from 8h as a yellow liquid; IR (KBr) ν (cm−1): 1735.93, 1674.21 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.16 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 3.80 (s, 3H, -OCH3), 3.83 (s, 3H, -OCH3), 4.04–4.15 (m, 4H, -CO-CH2-CO- and -O-CH2CH3), 7.02 (d, J = 8.4 Hz, 1H, ArH), 7.43 (d, J = 1.4 Hz, 1H, ArH), 7.59 (dd, J = 8.4, 1.2 Hz, 1H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.34, 45.62, 55.85, 56.10, 60.98, 110.78, 111.20, 123.85, 129.19, 149.11, 154.03, 168.29, 192.09; MS (EI, 70 eV) m/z: 252.2 (M+); HRMS (EI) m/z: calculated for C13H16O5: 252.0998; found: 252.1003.
5.2.1.8. Ethyl 3-(3,5-dimethoxyphenyl)-3-oxopropanoate (10i)
Yield 81% from 8i as a yellow liquid; IR (KBr) ν (cm−1): 1735.93, 1689.64 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.17 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 3.79 (s, 6H, 2 × -OCH3), 4.06–4.15 (m, 4H, -CO-CH2-CO- and -O-CH2CH3), 6.77 (s, 1H, ArH), 7.07–7.08 (m, 2H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.35, 46.16, 55.87 (2C), 61.04, 105.95, 106.57 (2C), 138.21, 161.07 (2C), 168.05, 193.47; MS (EI, 70 eV) m/z: 252.2 (M+); HRMS (EI) m/z: calculated for C13H16O5: 252.0998; found: 252.0994.
5.2.1.9. Ethyl 3-oxo-3-(2,4,6-trimethoxyphenyl)propanoate (10j)
Yield 60% from 8j as a white solid; mp: 69–71 °C; IR (KBr) ν (cm−1): 1728.22, 1689.64 (C=O); 1H NMR (200 Mhz, DMSO-d6): δ 1.13 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 3.70 (s, 2H, -CO-CH2-CO-), 3.74 (s, 6H, 2 × -OCH3), 3.80 (s, 3H, -OCH3), 4.05 (q, J = 7.1 Hz, 2H, -O-CH2CH3), 6.25 (s, 2H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.41, 51.47, 55.95, 56.32 (2C), 60.72, 91.37 (2C), 111.65, 158.81 (2C), 163.17, 167.26, 194.47; MS (EI, 70 eV) m/z: 282.2 (M+); HRMS (EI) m/z: calculated for C14H18O6: 282.1103; found: 282.1106.
5.2.1.10. Ethyl 3-oxo-3-(3,4,5-trimethoxyphenyl)propanoate (10k)
Yield 69% from 8k as a colorless liquid; IR (KBr) ν (cm−1): 1743.65, 1681.93 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.18 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 3.75 (s, 3H, -OCH3), 3.84 (s, 6H, 2 × -OCH3), 4.07–4.19 (m, 4H, -CO-CH2-CO- and -O-CH2CH3), 7.25 (s, 2H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.36, 45.88, 56.41 (2C), 60.52, 61.01, 106.46 (2C), 131.51, 142.82, 153.24 (2C), 168.18, 192.68; MS (EI, 70 eV) m/z: 282.2 (M+); HRMS (EI) m/z: calculated for C14H18O6: 282.1103; found: 282.1102.
5.2.1.11. Ethyl 3-(2-fluorophenyl)-3-oxopropanoate (10l)
Yield 73% from 8l as a yellow liquid; IR (KBr) ν (cm−1): 1743.65, 1689.64 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.15 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 4.04–4.15 (m, 4H, -CO-CH2-CO- and -O-CH2CH3), 7.29–7.39 (m, 2H, ArH), 7.64–7.72 (m, 1H, ArH), 7.82–7.87 (m, 1H, ArH); 13C NMR (50 MHz, DMSO -d6): δ 14.36, 49.52 (d, 4JCF = 6.0 Hz, 1C), 61.09, 117.33 (d, 2 JCF = 23.0 Hz, 1C), 124.77 (d, 2JCF = 11.5 Hz, 1C), 125.33, 130.89, 136.27 (d, 3JCF = 9.0 Hz, 1C), 161.74 (d, 1 Jcf = 253.0 Hz, 1C), 167.76, 191.09; MS (EI, 70 eV) m/z: 210.1 (M+); HRMS (EI) m/z: calculated for C11H11FO3: 210.0692; found: 210.0700.
5.2.1.12. Ethyl 3-(3-fluorophenyl)-3-oxopropanoate (10m)
Yield 69% from 8m as a yellow liquid; IR (KBr) ν (cm−1): 1743.65, 1689.64 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.16 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 4.11 (q, J = 7.1 Hz, 2H, -O-CH2CH3), 4.20 (s, 2H, -CO-CH2-CO-), 7.46–7.64 (m, 2H, ArH), 7.68–7.83 (m, 2H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.36, 46.08, 61.13, 115.35 (d, 2 JCF = 22.5 Hz, 1C), 121.12 (d, 2 JCF = 21.5 Hz, 1C), 125.06, 131.45 (d, 3 JCF = 8.0 Hz, 1C), 138.45 (d, 3 JCF = 6.5 Hz, 1C), 162.64 (d, 1 JCF = 244.0 Hz, 1C), 167.90, 193.00; MS (EI, 70 eV) m/z: 210.1 (M+); HRMS (EI) m/z: calculated for C11H11FO3: 1210.0692; found: 210.0695.
5.2.1.13. Ethyl 3-(4-fluorophenyl)-3-oxopropanoate (10n)
Yield 77% from 8n as a yellow liquid; IR (KBr) ν (cm−1): 1743.65, 1689.64 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 1.16 (t, J = 7.1 Hz, 3H, -O-CH2CH3), 4.10 (q, J = 7.1 Hz, 2H, -O-CH2CH3), 4.17 (s, 2H, -CO-CH2-CO-), 7.30–7.39 (m, 2H, ArH), 8.00–8.07 (m, 2H, ArH); 13C NMR (50 MHz, DMSO-d6): δ 14.36, 45.94, 61.08, 116.28 (d, 2 JCF = 21.9 Hz, 2C), 131.43 (d, 3JCF = 9.6 Hz, 2C), 133.05, 165.80 (d, 1JcF = 251.0 Hz, 1C), 168.01, 192.53; MS (EI, 70 eV) m/z: 210.1 (M+); HRMS (EI) m/z: calculated for C11H11FO3: 210.0692; found: 210.0701.
5.2.2. General procedure for the synthesis of benzoylacetanilides 11a–n
A mixture of the substituted benzoyl acetate (10a–n, 1 equiv) and 3,4-methylenedioxy aniline (1 equiv) was stirred in 150 mL toluene and then heated at reflux for 1–2 h. The mixture was cooled to the room temperature and partitioned with 10% NaOH (3 × 50 mL). The aqueous layer was acidified to pH 4–5 with dropwise addition of glacial acetic acid. The resulting precipitate was isolated by suction filtration, washed with water and EtOH, and then air-dried air to give the desired benzoylacetanilide (11a–n) of sufficient purity for the next reaction.
5.2.2.1. N-(Benzo[d][1,3]dioxol-5-yl)-3-oxo-3-phenylpropanamide (11a)
Yield 45% from 10a as a light-yellow solid; mp: 149–151 °C; IR (KBr) ν (cm−1): 1654.03, 1684.89 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 4.09 (s, 2H, -CH2-), 5.97 (s, 2H, -O-CH2-O-), 6.84 (d, J = 8.4 Hz, 1H, ArH), 6.94 (dd, J = 8.4, 2.0 Hz, 1H, ArH), 7.28 (d, J = 2.0 Hz, 1H, ArH), 7.51–7.58 (m, 2H, ArH), 7.63–7.74 (m, 1H, ArH), 8.00 (d, J = 7.2 Hz, 1H, ArH), 10.11 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.41, 101.43, 101.67, 108.53, 112.33, 128.81 (2C), 129.25 (2C), 133.83, 134.03, 136.72, 143.45, 147.53, 165.39, 195.04; MS (EI, 70 eV) m/z: 283.2 (M+); HRMS (EI) m/z : calculated for C16H13NO4: 283.0845; found: 283.0836.
5.2.2.2. N-(Benzo[d][1,3]dioxol-5-yl)-3-(2-methoxyphenyl)-3-oxopropanamide (11b)
Yield 38% from 10b as a white solid; mp: 143–144 °C; IR (KBr) ν (cm−1): 1656.92, 1672.36 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.85 (s, 3H, -OCH3), 3.97 (s, 2H, -CH2-), 5.96 (s, 2H, -O-CH2-O-), 6.83 (d, J = 8.4 Hz, 1H, ArH), 6.94 (dd, J = 8.4, 1.6 Hz, 1H, ArH), 7.04 (t, J = 7.4 Hz, 1H, ArH), 7.15 (d, J = 8.2 Hz, 1H, ArH), 7.28 (d, J = 1.6 Hz, 1H, ArH), 7.56 (t, J = 7.8 Hz, 1H, ArH), 7.69 (d, J = 7.7 Hz, 1H, ArH), 10.01 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 52.96, 56.23, 101.39, 101.65, 108.51, 112.23, 113.01, 120.94, 126.91, 130.45, 133.99, 134.96, 143.29, 147.49, 159.23, 165.82, 195.04; MS (EI, 70 eV) m/z: 313.2 (M+); HRMS (EI) m/z: calculated for C17H15NO5: 313.0950; found: 313.0959.
5.2.2.3. N-(Benzo[d][1,3]dioxol-5-yl)-3-(3-methoxyphenyl)-3-oxopropanam ide (11c)
Yield 37% from 10c as a pale gray solid; mp: 141–142 °C; IR (KBr) ν (cm−1): 1656.92, 1685.86 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.81 (s, 3H, -OCH3), 4.08 (s, 2H, -CH2-), 5.97 (s, 2H, -O-CH2-O-), 6.84 (d, J = 8.4 Hz, 1H, ArH), 6.93 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.21–7.50 (m, 4H, ArH), 7.59 (d, J = 7.6 Hz, 1H, ArH), 10.12 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.57, 55.84, 101.44, 101.68, 108.53, 112.34, 113.36, 119.98, 121.34, 130.43, 133.81, 138.11, 143.46, 147.54, 159.91, 165.35, 194.79; MS (EI, 70 eV) m/z: 313.2 (M+); HRMS (EI) m/z: calculated for C17H15NO5: 313.0950; found: 313.0958.
5.2.2.4. N-(Benzo[d][1,3]dioxol-5-yl)-3-(4-methoxyphenyl)-3-oxopropanamide (11d)
Yield 38% from 10d as a light-brown solid; mp: 144–145 °C; IR (KBr) ν (cm−1): 1655.96, 1674.28 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.84 (s, 3H, -OCH3), 4.02(s, 2H, -CH2-), 5.97 (s, 2H, -O-CH2-O-), 6.84 (d, J = 8.4 Hz, 1H, ArH), 6.93 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.06 (d, J = 8.8 Hz, 2H, ArH), 7.27 (d, J = 1.6 Hz, 1H, ArH), 7.97 (d, J = 8.8 Hz, 2H, ArH), 10.09 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.15, 56.06, 101.42, 101.64, 108.52, 112.30, 114.45 (2C), 129.70, 131.23 (2C), 133.87, 143.40, 147.50, 163.89, 165.57, 193.34; MS (EI, 70 eV) m/z: 313.1 (M+); HRMS (EI) m/z: calculated for C17H15NO5: 313.0950; found: 313.0944.
5.2.2.5. N-(Benzo[d][1,3]dioxol-5-yl)-3-(2,4-dimethoxyphenyl)-3-oxopropanami de (11e)
Yield 44 % from 10e as a white solid; mp: 164–166 °C; IR (KBr) ν (cm−1): 1652.10, 1676.21 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.83 (s, 3H, -OCH3), 3.85 (s, 3H, -OCH3), 3.89 (s, 2H, -CH2-), 5.96 (s, 2H, -O-CH2-O-), 6.61–6.64 (m, 2H, ArH), 6.83 (d, J = 8.4 Hz, 1H, ArH), 6.93 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.27 (d, J = 1.6 Hz, 1H, ArH), 7.73 (d, J = 9.4 Hz, 1H, ArH), 9.96 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 52.90, 56.13, 56.29, 98.76, 101.36, 101.60, 106.78, 108.50, 112.16, 119.73, 132.62, 134.09, 143.21, 147.48, 161.52, 165.20, 166.19, 192.68; MS (EI, 70 eV) m/z: 343.2 (M+); HRMS (EI) m/z: calculated for C18H17NO6: 343.1056; found: 343.1049.
5.2.2.6. N-(Benzo[d][1,3]dioxol-5-yl)-3-(2,5-dimethoxyphenyl)-3-oxopropanami de (11f)
Yield 48% from 10f as a light-brown solid; mp: 144–146 °C; IR (KBr) ν (cm−1): 1646.32, 1668.50 (C=O); 1H NMR (200 MHz, CDCl3-d1): δ 3.73 (s, 3H, -OCH3), 3.80 (s, 3H, -OCH3), 3.95(s, 2H, -CH2-), 5.96 (s, 2H, -O-CH2-O-), 6.83 (d, J = 8.6 Hz, 1H, ArH), 6.89–6.95 (m, 1H, ArH), 7.08–7.31 (m, 4H, ArH), 10.00 (s, 1H, NH); 13C NMR (50 MHz, CDCl3-d1): δ 52.88, 56.00, 56.68, 101.38, 101.61, 108.53, 112.19, 114.04, 114.61, 121.01, 127.11, 133.98, 143.27, 147.49, 153.34, 153.65, 165.80, 194.58; MS (EI, 70 eV) m/z: 343.2 (M+); HRMS (EI) m/z: calculated for C18H17NO6: 343.1056; found: 343.1051.
5.2.2.7. N-(Benzo[d][1,3]dioxol-5-yl)-3-(2,6-dimethoxyphenyl)-3-oxopropanami de (11g)
Yield 59% from 10g as a white solid; mp: 131–133 °C; IR (KBr) ν (cm−1): 1654.03, 1709.97 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.72 (s, 6H, -OCH3), 3.74 (s, 2H, -CH2-), 5.96 (s, 2H, -O-CH2-O-), 6.69 (d, J = 8.4 Hz, 2H, ArH), 6.82 (d, J = 8.4 Hz, 1H, ArH), 6.92 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.25 (d, J = 1.8 Hz, 1H, ArH), 7.33 (t, J = 8.4 Hz, 1H, ArH), 9.94 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 53.89, 56.36 (2C), 101.40,101.76, 104.81 (2C), 108.46, 112.43, 119.26, 131.84, 133.89, 143.37, 147.44, 156.95 (2C), 164.02, 197.56; MS (EI, 70 eV) m/z: 342.9 (M+); HRMS (EI) m/z: calculated for C18H17NO6: 343.1056; found: 343.1062.
5.2.2.8. N-(Benzo[d][1,3]dioxol-5-yl)-3-(3,4-dimethoxyphenyl)-3-oxopropanami de (11h)
Yield 39% from 10h as a pale gray solid; mp: 191–192 °C; IR (KBr) ν (cm−1): 1653.07, 1676.21 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.80 (s, 3H, -OCH3), 3.84 (s, 3H, -OCH3), 4.03(s, 2H, -CH2-), 5.96 (s, 2H, -O-CH2-O-), 6.84 (d, J = 8.4 Hz, 1H, ArH), 6.94 (dd, J = 8.3, 1.8 Hz, 1H, ArH), 7.08 (d, J = 8.4 Hz, 1H, ArH), 7.27 (d, J = 1.6 Hz, 1H, ArH), 7.47 (d, J = 1.6 Hz, 1H, ArH), 7.67 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 10.12 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.15, 56.01, 56.25, 101.43, 101.65, 108.52, 110.90, 111.39, 112.30, 123.86, 129.67, 133.88, 143.42, 147.52, 149.08, 153.86, 165.57, 193.34; MS (EI, 70 eV) m/z: 343.1 (M+); HRMS (EI) m/z: calculated for C18H17NO6: 343.1056; found: 343.1050.
5.2.2.9. N-(Benzo[d][1,3]dioxol-5-yl)-3-(3,5-dimethoxyphenyl)-3-oxopropanami de (11i)
Yield 61% from 10i as a light-brown solid; mp: 158–160 °C; IR (KBr) ν (cm−1): 1653.07, 1695.50 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.79 (s, 6H, -OCH3), 4.06 (s, 2H, -CH2-), 5.97 (s, 2H, -O-CH2-O-), 6.77–6.86 (m, 2H, ArH), 6.92 (dd, J = 8.4, 2.0 Hz, 1H, ArH), 7.10 (d, J = 2.2 Hz, 2H, ArH), 7.26 (d, J = 2.0 Hz, 1H, ArH), 10.12 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.65, 56.03 (2C), 101.44, 101.64, 105.80, 106.60 (2C), 108.56, 112.31, 133.78, 138.67, 143.46, 147.53, 161.08 (2C), 165.30, 194.59; MS (EI, 70 eV) m/z: 342.9 (M+); HRMS (EI) m/z: calculated for C18H17NO6: 343.1056; found: 343.1059.
5.2.2.10. N-(Benzo[d][1,3]dioxol-5-yl)-3-oxo-3-(2,4,6-trimethoxyphenyl)propane mide (11j)
Yield 51% from 10j as a white solid; mp: 124–126 °C; IR (KBr) ν (cm−1): 1652.10, 1706.11 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.69 (s, 2H, -CH2 -), 3.72 (s, 6H, -OCH3), 3.79 (s, 3H, -OCH3), 5.96 (s, 2H, -O-CH2-O-), 6.24 (s, 2H, ArH), 6.82 (d, J = 8.4 Hz, 1H, ArH), 6.92 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.25 (d, J = 1.8 Hz, 1H, ArH), 9.90 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 53.90, 55.95, 56.36 (2C), 91.41 (2C), 101.38, 101.69, 108.46, 112.33 (2C), 133.96, 143.29, 147.42, 158.72 (2C), 162.91, 164.57, 197.17; MS (EI, 70 eV) m/z: 372.6 (M+); HRMS (EI) m/z: calculated for C19H19NO7: 373.1162; found: 373.1166.
5.2.2.11. N-(Benzo[d][1,3]dioxol-5-yl)-3-oxo-3-(3,4,5-trimethoxyphenyl)propane mide (11k)
Yield 27% from 10k as a white solid; mp: 186–188 °C; IR (KBr) ν (cm−1): 1649.21, 1672.36 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.74 (s, 3H, -OCH3), 3.83 (s, 6H, -OCH3), 4.08 (s, 2H, -CH2-), 5.97 (s, 2H, -O-CH2-O-), 6.84 (d, J = 8.4 Hz, 1H, ArH), 6.93 (dd, J = 8.4, 2.0 Hz, 1H, ArH), 7.26–7.29 (m, 3H, ArH), 10.16 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.42, 56.59 (2C), 60.65, 101.45, 101.67, 106.48 (2C), 108.56, 112.36, 131.98, 133.76, 142.65, 143.47, 147.53, 153.25 (2C), 165.46, 193.82; MS (EI, 70 eV) m/z: 373.0 (M+); HRMS (EI) m/z: calculated for C19H19NO7: 373.1162; found: 373.1170.
5.2.2.12. N-(Benzo[d][1,3]dioxol-5-yl)-3-(2-fluorophenyl)-3-oxopropanamide (11l)
Yield 61% from 10l as a white solid; mp: 147–148 °C; IR (KBr) ν (cm−1): 1653.07, 1691.64 (C=O); 1H NMR (200 MHz, CDCl3-d1): δ 4.10 (d, J = 2.4 Hz, 2H, -CH2-), 5.93 (s, 2H, -O-CH2-O-), 6.73 (d, J = 8.2 Hz, 1H, ArH), 6.86 (dd, J = 8.2, 2.2 Hz, 1H, ArH), 7.11–7.22 (m, 2H, ArH), 7.26 (d, J = 2.2 Hz, 1H, ArH), 7.53–7.60 (m, 1H, ArH), 7.84–7.92 (m, 1H, ArH), 9.00 (s, 1H, NH); 13C NMR (50 MHz, CDCl3-d1): δ 49.45 (d, 4JCF = 8.0 Hz, 1C), 101.27, 103.11, 108.05, 113.51, 117.01 (d, 2JCF = 23.0 Hz, 1C), 124.56 (d, 2 JCF = 21.5 Hz, 1C), 124.77, 130.75, 131.79, 135.84 (d, 3JCF = 9.5 Hz, 1C), 144.46, 147.79, 161.98 (d, 1Jcf = 254.5 Hz, 1C), 163 62, 194.75; MS (EI, 70 eV) m/z: 301.0 (M+); HRMS (EI) m/z: calculated for C16H12FNO4: 301.0750; found: 301.0760.
5.2.2.13. N-(Benzo[d][1,3]dioxol-5-yl)-3-(3-fluorophenyl)-3-oxopropanamide (11m)
Yield 62% from 10m as a pale gray solid; mp: 130–132 °C; IR (KBr) ν (cm−1): 1655.96, 1695.50 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 4.10 (s, 2H, -CH2-), 5.96 (s, 2H, -O-CH2-O-), 6.84 (d, J = 8.4 Hz, 1H, ArH), 6.92 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.25 (d, J = 1.8 Hz, 1H, ArH), 7.51–7.86 (m, 4H, ArH), 10.13 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.51, 101.41, 101.71, 108.56, 112.35, 115.31 (d, 2Jcf = 22.5 Hz, 1C), 120.94 (d, 2 Jcf = 21.0 Hz, 1C), 125.09, 131.39, 133.75, 138.88, 143.48, 147.55, 162.69 (d, 1JCF = 244.0 Hz, 1C), 165.13, 194.09; MS (EI, 70 eV) m/z: 300.9 (M+); HRMS (EI) m/z: calculated for C16H12FNO4: 301.0750; found: 301.0747.
5.2.2.14. N-(Benzo[d][1,3]dioxol-5-yl)-3-(4-fluorophenyl)-3-oxopropanamide (11n)
Yield 59% from 10n as a white solid; mp: 164–165 °C; IR (KBr) ν (cm−1): 1633.78, 1690.68 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 4.09 (s, 2H, -CH2-), 5.96 (s, 2H, -O-CH2-O-), 6.83 (d, J = 8.4 Hz, 1H, ArH), 6.93 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.27 (d, J = 1.8 Hz, 1H, ArH), 7.32–7.41 (m, 2H, ArH), 8.04–8.11 (m, 2H, ArH), 10.11 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 48.36, 101.44, 101.68, 108.53, 112.35, 116.29 (d, 2Jcf = 21.5 Hz, 2C), 131.90 (d, 3JCF = 9.5 Hz, 2C), 133.47, 133.77, 143.47, 147.53, 165.30, 165.69 (d, 1JCF = 250.5 Hz, 1C), 193.71; MS (EI, 70 eV) m/z: 300.9 (M+); HRMS (EI) m/z: calculated for C16H12FNO4: 301.0750; found: 301.0755.
5.2.3. General procedure for the synthesis of 6,7-methylenedioxy-4-substituted phenylquinolin-2-one derivatives 12a–n
A mixture of the benzoylacetanilide (11a–n) and PPA (10 g) was heated at 100–110 °C for 0.5 h to 1 h. The mixture was cooled after TLC monitoring indicated that the reaction was completed, and then the reaction mixture diluted with ice water, and extracted with CHCl3 (3 × 50 mL). The combined organic layers were dried over anhydrous MgSO4, and the solvent was removed in vacuo. The crude products were purified by column chromatography (silica gel, chloroform/ethyl acetate = 2/1) to give the corresponding pure 4-phenylquinolin-2-ones 12a–n.
5.2.3.1. 6,7-Methylenedioxy-4-phenylquinolin-2(1H)-one (12a)
Compound 12a (0.16 g, 0.60 mmol) was obtained by cyclization of 11a (0.27 g, 0.95 mmol) with PPA (10 g); Yield: 63%; white solid; mp: 274–275 °C; IR (KBr) ν (cm−1): 1653.07 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 6.02 (s, 2H, -O-CH2-O-), 6.18 (s, 1H, Ar-H), 6.63 (s, 1H, Ar-H), 6.87 (s, 1H, Ar-H), 7.36–7.51 (m, 5H, Ar-H), 11.78 (br. s, 1H, NH); 13C NMR (50 MHz, CDCl3-d1): δ 45.27, 101.25, 103.00, 108.04, 113.43, 128.57 (2C), 128.95 (2C), 131.81, 134.34, 136.05, 144.42, 147.76, 163.61, 196.51; MS (EI, 70 eV) m/z: 265.1 (M+); HRMS (EI) m/z: calculated for C16H11NO3: 265.0739; found: 265.0733.
5.2.3.2. 6,7-Methylenedioxy-4-(2-methoxyphenyl)quinolin-2(1H)-one (12b)
Compound 12b (0.19 g, 0.64 mmol) was obtained by cyclization of 11b (0.29 g, 0.93 mmol) with PPA (10 g); Yield: 68%; white solid; mp: 269–271 °C; IR (KBr) ν (cm−1): 1663.07 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.65 (s, 3H, -OCH3), 5.99, 6.01 (s, each 1H, -O-CH2-O-), 6.13 (s, 1H, Ar-H), 6.30 (s, 1H, Ar-H), 6.84 (s, 1H, Ar-H), 7.02 (t, J = 7.4 Hz, 1H, Ar-H), 7.10–7.17 (m, 2H, Ar-H), 7.43 (t, J = 7.4 Hz, 1H, Ar-H), 11.71 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 55.89, 95.79, 102.26, 103.90, 112.00, 113.69, 119.44, 121.17, 126.27, 130.37, 130.77, 136.09, 143.71, 149.69, 150.35, 156.41, 161.85; MS (EI, 70 eV) m/z: 295.1 (M+); HRMS (EI) m/z: calculated for C17H13NO4: 295.0845; found: 295.0835.
5.2.3.3. 6,7-Methylenedioxy-4-(3-methoxyphenyl)quinolin-2(1H)-one (12c)
Compound 12c (0.17 g, 0.58 mmol) was obtained by cyclization of 11c (0.30 g, 0.96 mmol) with PPA (10 g); Yield: 60%; white solid; mp: 278–279 °C; IR (KBr) ν (cm−1): 1653.07 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.75 (s, 3H, -OCH3), 6.02 (s, 2H, -O-CH2-O-), 6.19 (s, 1H, Ar-H), 6.66 (s, 1H, Ar-H), 6.85 (s, 1H, Ar-H), 6.90–6.93 (m, 2H, Ar-H), 7.00 (dd, J = 8.2, 2.2 Hz, 1H, Ar-H), 7.38 (t, J = 8.2 Hz, 1H, Ar-H), 11.73 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 55.65, 96.01, 102.38, 103.75, 112.87, 114.37, 114.75 118.75, 121.09, 130.23, 136.79, 138.98, 143.78, 150.49, 151.57, 159.80, 161.62; MS (EI, 70 eV) m/z: 295.2 (M+); HRMS (EI) m/z: calculated for C17H13NO4: 295.0845; found: 295.0851.
5.2.3.4. 6,7-Methylenedioxy-4-(4-methoxyphenyl)quinolin-2(1H)-one (12d)
Compound 12d (0.19 g, 0.64 mmol) was obtained by cyclization of 11d (0.29 g, 0.93 mmol) with PPA (10 g); Yield: 68%; white solid; mp: 269–271 °C; IR (KBr) ν (cm−1): 1663.07 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.82 (s, 3H, -OCH3), 6.06 (s, 2H, -O-CH2-O-), 6.19 (s, 1H, Ar-H), 6.75 (s, 1H, Ar-H), 6.90 (s, 1H, Ar-H), 7.07 (d, J = 8.6 Hz, 1H, Ar-H), 7.37 (d, J = 8.6 Hz, 1H, Ar-H), 11.69 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 55.70, 96.06, 102.37, 103.86, 113.13, 114.64 (2C), 118.60, 129.76, 130.32 (2C), 136.81, 143.82, 150,46, 151.53, 160.04, 161.73; MS (EI, 70 eV) m/z: 295.2 (M+); HRMS (EI) m/z: calculated for C17H13NO4: 295.0845; found: 295.0851.
5.2.3.5. 6,7-Methylenedioxy-4-(2,4-dimethoxyphenyl)quinolin-2(1H)-one (12e)
Compound 12e (0.11 g, 0.34 mmol) was obtained by cyclization of 11e (0.19 g, 0.55 mmol) with PPA (10 g); Yield: 61%; white solid; mp: 251–252 °C; IR (KBr) ν (cm−1): 1654.03 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.67 (s, 3H, -OCH3), 3.81 (s, 3H, -OCH3), 6.02, 6.04 (s, each 1H, -O-CH2-O-), 6.12 (s, 1H, Ar-H), 6.39 (s, 1H, Ar-H), 6.61–6.69 (m, 2H, Ar-H), 6.85 (s, 1H, Ar-H), 7.09 (d, J = 8.2 Hz, 1H, Ar-H), 11.70 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 55.79, 55.95, 95.76, 99.16, 102.24, 104.07, 105.74, 114.07, 118.73, 119.56, 131.07, 135.98, 143.69, 149.72, 150.28, 157.53, 161.57, 161.99; MS (EI, 70 eV) m/z: 325.1 (M+); HRMS (EI) m/z: calculated for C18H15NO5: 325.0950; found: 325.0954.
5.2.3.6. 6,7-Methylenedioxy-4-(2,5-dimethoxyphenyl)quinolin-2(1H)-one (12f)
Compound 12f (0.10 g, 0.31 mmol) was obtained by cyclization of 11f (0.17 g, 0.50 mmol) with PPA (10 g); Yield: 62%; white solid; mp: 240–241 °C; IR (KBr) ν (cm−1): 1669.46 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.59 (s, 3H, -OCH3), 3.69 (s, 3H, -OCH3), 5.99, 6.02 (s, each 1H, -O-CH2-O-), 6.14 (s, 1H, Ar-H), 6.33 (s, 1H, Ar-H), 6.74 (d, J = 2.8 Hz, 1H, Ar-H), 6.84 (s, 1H, Ar-H), 6.98 (dd, J = 8.9, 2.8 Hz, 1H, Ar-H), 7.06 (d, J = 9.2 Hz, 1H, Ar-H), 11.71 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 55.94, 56.38, 95.77, 102.27, 103.96, 113.22, 113.60, 115.31, 116.03, 119.38, 127.04, 136.06, 143.73, 149.50, 150.39 (2C), 153.61, 161.85; MS (EI, 70 eV) m/z: 325.2 (M+); HRMS (EI) m/z: calculated for C18H15NO5: 325.0950; found: 325.0959.
5.2.3.7. 6,7-Methylenedioxy-4-(2,6-dimethoxyphenyl)quinolin-2(1H)-one (12g)
Compound 12g (0.49 g, 1.51 mmol) was obtained by cyclization of 11g (0.75 g, 2.18 mmol) with PPA (10 g); Yield: 69%; white solid; mp: 280–281 °C; IR (KBr) ν (cm−1): 1654.03 (C=O); 1H NMR (200 MHz, CDCl3-d1): δ 3.68 (s, 6H, 2 × -OCH3), 5.93 (s, 2H, -O-CH2-O-), 6.48 (s, 2H, Ar-H), 6.65 (d, J = 8.4 Hz, 2H, Ar-H), 6.99 (s, 1H, Ar-H), 7.35 (t, J = 8.4 Hz, 1H, Ar-H), 12.99 (br. s, 1H, NH); 13C NMR (50 MHz, CDCl3-d1): δ 55.91 (2C), 96.48, 101.53, 103.72, 104.05 (2C), 114.71, 115.38, 119.94, 130.12, 135.68, 144.23, 146.92, 150.38, 157.62 (2C), 164.53; MS (EI, 70 eV) m/z: 325.3 (M+); HRMS (EI) m/z: calculated for C18H15NO5: 325.0950; found: 325.0954.
5.2.3.8. 6,7-Methylenedioxy-4-(3,4-dimethoxyphenyl)quinolin-2(1H)-one (12h)
Compound 12h (2.29 g, 7.04 mmol) was obtained by cyclization of 11h (3.00 g, 8.74 mmol) with PPA (10 g); Yield: 80%; white solid; mp: 272–273 °C; IR (KBr) ν (cm−1): 1647.28 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.77 (s, 3H, -OCH3), 3.80 (s, 3H, -OCH3), 6.05 (s, 2H, -O-CH2-O-), 6.23 (s, 1H, Ar-H), 6.81 (s, 1H, Ar-H), 6.88 (s, 1H, Ar-H), 6.93 (dd, J = 8.2, 1.8 Hz, 1H, Ar-H), 6.98 (d, J = 1.8 Hz, Ar-H), 7.06 (d, J = 8.2 Hz, 1H, Ar-H), 11.73 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 56.00 (2C), 96.00, 102.33, 103.94, 112.15, 112.61, 113.14, 118.59, 121.35, 129.97, 136.74, 143.78, 149.17, 149.56, 150.41, 151.71, 161.76; MS (EI, 70 eV) m/z: 325.1 (M+); HRMS (EI) m/z: calculated for C18H15NO5: 325.0950; found: 325.0954.
5.2.3.9. 6,7-Methylenedioxy-4-(3,5-dimethoxyphenyl)quinolin-2(1H)-one (12i)
Compound 12i (0.41 g, 1.26 mmol) was obtained by cyclization of 11i (0.75 g, 2.18 mmol) with PPA (10 g); Yield: 57%; white solid; mp: 283–284 °C; IR (KBr) ν (cm−1): 1654.03 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.74 (s, 6H, 2 × -OCH3), 6.03 (s, 2H, -O-CH2-O-), 6.20 (s, 1H, Ar-H), 6.49–6.57 (m, 3H, Ar-H), 6.71 (s, 1H, Ar-H), 6.86 (s, 1H, Ar-H), 11.73 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 55.83 (2C), 96.00, 100.87, 102.38, 103.79, 106.96 (2C), 112.87, 118.56, 136.73, 139.59, 143.82, 150.54, 151.70, 160.97 (2C), 161.64; MS (EI, 70 eV) m/z: 325.2 (M+); HRMS (EI) m/z: calculated for C18H15NO5: 325.0950; found: 325.0954.
5.2.3.10. 6,7-Methylenedioxy-4-(2,4,6-trimethoxyphenyl)quinolin-2(1H)-one (12j)
Compound 12j (0.12 g, 0.34 mmol) was obtained by cyclization of 11j (0.22 g, 0.59 mmol) with PPA (10 g); Yield: 57%; white solid; mp: 287–288 °C; IR (KBr) ν (cm−1): 1653.07 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.60 (s, 6H, 2 × - OCH3), 3.80 (s, 3H, -OCH3), 5.99, 6.03 (s, each 1H, -O-CH2-O-), 6.26 (s, 1H, Ar-H), 6.32 (s, 2H, Ar-H), 6.81 (s, 1H, Ar-H), 8.26 (s, 1H, Ar-H), 11.62 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 55.81, 56.17 (2C), 91.46 (2C), 95.73, 102.14, 103.52, 106.78, 114.72, 121.07, 136.01, 143.64, 146.02, 150.15, 158.19 (2C), 161.92, 162.10; MS (EI, 70 eV) m/z: 355.3 (M+); HRMS (EI) m/z: calculated for C19H17NO6: 355.1056; found: 355.1048.
5.2.3.11. 6,7-Methylenedioxy-4-(3,4,5-trimethoxyphenyl)quinolin-2(1H)-one (12k)
Compound 12k (0.17 g, 0.48 mmol) was obtained by cyclization of 11k (0.25 g, 0.67 mmol) with PPA (10 g); Yield: 71%; white solid; mp: 275–276 °C; IR (KBr) ν (cm−1): 1647.28 (C=O); 1H NMR (200 MHz, DMSO-d6): δ 3.68 (s, 3H, -OCH3), 3.75 (s, 6H, 2 × -OCH3), 6.03 (s, 2H, -O-CH2-O-), 6.23 (s, 1H, Ar-H), 6.65 (s, 2H, Ar-H), 6.79 (s, 1H, Ar-H), 6.86 (s, 1H, Ar-H), 11.72 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 56.48 (2C), 60.53, 96.00, 102.37, 103.97, 106.36 (2C), 113.07, 118.65, 133.12, 136.68, 138.03, 143.87, 150.52, 151.91, 153.39 (2C), 161.75; MS (EI, 70 eV) m/z: 355.2 (M+); HRMS (EI) m/z: calculated for C19H17NO6: 355.1056; found: 355.1050.
5.2.3.12. 4-(2-Fluorophenyl)-6,7-methylenedioxyquinolin-2(1H)-one (12l)
Compound 12l (0.11 g, 0.39 mmol) was obtained by cyclization of 11l (0.21 g, 0.70 mmol) with PPA (10 g); Yield: 55%; white solid; mp: 251–252 °C; IR (KBr) v (cm−1): 1653.07 (C=0); 1H NMR (200 MHz, DMSO-d6): 5 6.06 (s, 2H, -O-CH2-O-), 6.29 (s, 1H, Ar-H), 6.45 (d, J = 2.0 Hz, 1H, Ar-H), 6.91 (s, 1H, Ar-H), 7.33–7.47 (m, 3H, Ar-H), 7.51–7.62 (m, 1H, Ar-H), 11.87 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 95.99, 102.47, 103.42, 112.96, 116.38 (d, 2JCF = 21.5 Hz, 1C), 120.11, 124.89 (d, 2JcF = 15.8 Hz, 1C), 125.52, 131.39, 131.67 (d, 3JCF = 8.0 Hz, 1C), 136.46, 144.04, 146.18, 150.73, 159.02 (d, lJCF = 245.0 Hz, 1C), 161.47; MS (EI, 70 eV) m/z: 283.1 (M ); HRMS (EI) m/z: calculated for C16H10FNO3: 283.0645; found: 283.0641.
5.2.3.13. 4-(3-Fluorophenyl)-6,7-methylenedioxyquinolin-2(1H)-one (12m)
Compound 12m (0.13 g, 0.46 mmol) was obtained by cyclization of 11m (0.21 g, 0.70 mmol) with PPA (10 g); Yield: 65%; white solid; mp: 249–251 °C; IR (KBr) v (cm−1): 1654.03 (C=0); 1H NMR (200 MHz, DMSO-d6): 5 6.07 (s, 2H,-o-CH2-0-), 6.25 (s, 1H, Ar-H), 6.67 (s, 1H, Ar-H), 6.90 (s, 1H, Ar-H), 7.24–7.37 (m, 3H, Ar-H), 7.51–7.62 (m, 1H, Ar-H) 11.81 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 5 96.08, 102.44, 103.64, 112.62, 115.97 (d, 2Jcf = 23.0 Hz, 2C), 119.08, 125.22, 131.28 (d, 3JCF = 8.5 Hz, 1C), 136.83, 139.84 (d, 3JCF = 8.0 Hz, 1C), 143.94, 150.38, 150.64, 161.51, 162.54 (d, 1Jf = 243.5 Hz, 1C); MS (EI, 70 eV) m/z: 283.1 (M ); HRMS (EI) m/z: calculated for C16H10FNO3: 283.0645; found: 283.0648.
5.2.3.14. 4-(4-Fluorophenyl)-6,7-methylenedioxyquinolin-2(1H)-one (12n)
Compound 12b (0.17 g, 0.60 mmol) was obtained by cyclization of 11b (0.21 g, 0.70 mmol) with PPA (10 g); Yield: 85%; white solid; mp: 251–252 °C; IR (KBr) v (cm−1): 1668.50 (C=0); 1H NMR (200 MHz, DMSO-d6): 5 6.02 (s, 2H, -O-CH2-O-), 6.19 (s, 1H, Ar-H), 6.62 (s, 1H, Ar-H), 6.86 (s, 1H, Ar-H), 7.25–7.34 (m, 2H, Ar-H), 7.40–7.47 (m, 2H, Ar-H), 11.76 (br. s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): δ 96.06, 102.41, 103.67, 112.90, 116.11 (d, 2JCF = 22.0 Hz, 2C), 119.02, 131.15 (d, 3JCF = 8.5 Hz, 2C), 133.94, 136.79, 143.88, 150.56, 150.74, 161.58, 162.70 (d, lJCF = 244.5 Hz, 1C); MS (EI, 70 eV) m/z: 283.1 (M+); HRMS (EI) m/z: calculated for C16H10FNO3: 283.0645; found: 283.0636.
5.3. Biological evaluation
5.3.1. Antiproliferative assay
Human tumor cell lines of the cancer screening panel were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (GIBCO/BRL), penicillin (100 U/mL)/streptomycin (100 g/mL) (GIBCO/BRL) and 1% L-glutamine (GIBCO/BRL) at 37 °C in a humidified atmosphere containing 5% CO2. Human hepatoma Hep 3B and normal skin Detroit 551 cells were maintained in DMEM medium supplemented with 10% fetal bovine serum (GIBCO/BRL), penicillin (100 U/mL)/streptomycin (100 g/mL) (GIBCO/BRL) and 1% L-glutamine (GIBCO/BRL) at 37 °C in a humidified atmosphere containing 5% CO2. Logarithmically growing cancer cells were used for all experiments. The human tumor cell lines were treated with vehicle or test compounds for 48 h. Cell growth rate was determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliun bromide] reduction assay.37,38 After 48 h treatment, cell growth rate was measured by scanning with an ELISA reader with a 570 nm filter and the IC50 values of test compounds were calculated.
5.3.2. Hoechst 33258 staining
HL-60 cells were plated at a density of 1×105 cells per well in 24-well plates and then incubated with 0.25 µM, 0.5 µM, and 1.0 µM, of compound 12e for 24 h. H460 cells were plated at a density of 5×104 cells per well in 24-well plates and then incubated with 1.0 µM, 2.5 µM, and 5.0 µM, of compound 12e for 24 h. Cells were examined directly and photographed under a phase contrast microscope. Nuclei were stained with Hoechst 33258 (bis-benzimide, Sigma) to detect chromatin condensation or nuclear fragmentation, morphological characteristics of apoptosis. Compound 12e treated cells were stained with 5 µg/mL Hoechst 33258 for 10 min. After washing twice with PBS, cells were fixed with 4% paraformaldehyde (PFA) in PBS for 10 min at 25 °C. Fluorescence of the soluble DNA (apoptotic) fragments was measured in a Varian Fluorometer at an excitation wavelength of 365 nm and emission wavelength of 460 nm.39
5.3.3. Cell cycle distribution analysis
Cell cycle analysis by FACS® was performed as described in the previous paper. 40 HL-60 cells were co-treated with compound 12e (0.25 µM, 0.5 µM, and 1.0 µM) for 24 h, and H460 were also co-treated with compound 12e (1.0 µM, 2.5 µM, and 5.0 µM). After treatment, the cells were washed once with PBS and fixed with 70% ice-cold ethanol at −20 °C overnight. Then the cells were stained with a solution containing 0.1% Triton-X 100 (Sigma), 0.2 mg/mL RNase (Sigma) and 20 µg/mL propidium iodide (PI, Sigma) in the dark for 30 min. Cell cycle distribution were measured using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) and all histograms were analyzed by ModFit software.
5.3.4. Western blot assay
The treated cells were collected and washed with PBS. After centrifugation, cells were lysed in a lysis buffer. The lysates were incubated on ice for 30 min and centrifuged at 12,000 g for 20 min. Supernatants were collected, and protein concentrations were then determined using the Bradford Assay. After adding a 5× sample loading buffer containing 625 mM Tris–HCl, pH = 6.8, 500 mM dithiothreitol, 10% SDS, 0.06% bromophenol blue, and 50% glycerol, protein samples were electrophoresed on 10% SDS-polyacrylamide gels and transferred to a nitrocellulose membrane. Immunoreactivity was detected using the Western blot chemiluminescence reagent system (PerkinElmer, Boston, MA). β-Actin was used as a loading control.
Supplementary Material
Acknowledgments
This study was supported by research grant from the National Science Council (NSC) of the R.O.C. (NSC95–2320-B-039–011-MY3) awarded to L. J. H. Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities Center, Office of Research & Development, China Medical University, Taichung, Taiwan, R.O.C. This study was also supported in part by grant CA177584 from the National Cancer Institute, NIH awarded to K. H. L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. CA Cancer J. Clin. 2011;61:69. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Gordaliza M. Clin. Transl. Oncol. 2007;9:767. doi: 10.1007/s12094-007-0138-9. [DOI] [PubMed] [Google Scholar]
- 3.Man S, Gao W, Wei C, Liu C. Phytother. Res. 2012;26:1449. doi: 10.1002/ptr.4609. [DOI] [PubMed] [Google Scholar]
- 4.Lee KH, Xiao Z. Podophyllotoxin and Analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. Second Edition. Boca Raton, FL: CRC Press; 2012. pp. 95–122. and literature cited therein. [Google Scholar]
- 5.Kelleher JK. Mol. Pharmacol. 1911;13:232. [PubMed] [Google Scholar]
- 6.Cortese F, Bhattacharyya B, Wolff J. J. Biol. Chem. 1977;252:1134. [PubMed] [Google Scholar]
- 7.Hartley RM, Peng J, Fest GA, Dakshanamurthy S, Frantz DE, Brown ML, Mooberry SL. Mol. Pharmacol. 2012;81:431. doi: 10.1124/mol.111.075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castro MA, Miguel del Corral JM, Gordaliza M, Gómez-Zurita MA, García PA, San Feliciano A. Phytochem. Rev. 2003;2:219. [Google Scholar]
- 9.Gensler WJ, Murthy CD, Trammell MH. J. Med. Chem. 1977;20:635. doi: 10.1021/jm00215a004. [DOI] [PubMed] [Google Scholar]
- 10.Magedov IV, Manpadi M, Van Slambrouck S, Steelant WFA, Rozhkova E, Przheval’ skii NM, Rogelj S, Kornienko A. J. Med. Chem. 2007;50:5183. doi: 10.1021/jm070528f. [DOI] [PubMed] [Google Scholar]
- 11.Semenova MN, Kiselyov AS, Tsyganov DV, Konyushkin LD, Firgang SI, Semenov RV, Malyshev OR, Raihstat MM, Fuchs F, Stielow A, Lantow M, Philchenkov AA, Zavelevich MP, Zefirov NS, Kuznetsov SA, Semenov VV. J. Med. Chem. 2011;54:7138. doi: 10.1021/jm200737s. [DOI] [PubMed] [Google Scholar]
- 12.Shi C, Wang J, Chen H, Shi D. J. Comb. Chem. 2010;12:430. doi: 10.1021/cc100003c. [DOI] [PubMed] [Google Scholar]
- 13.Castro MA, Miguel del Corral JM, Garcia PA, Rojo MV, de la Iglesia-Vicente J, Mollinedo F, Cuevas C, San Feliciano A. J. Med. Chem. 2010;53:983. doi: 10.1021/jm901373w. [DOI] [PubMed] [Google Scholar]
- 14.Castro MA, Miguel del Corral JM, Gordaliza M, Garcia PA, Gomez-Zurita MA, Garcia-Gravalos MD, de la Iglesia-Vicente J, Gajate C, An F, Mollinedo F, San Feliciano A. J. Med. Chem. 2004;47:1214. doi: 10.1021/jm030978h. [DOI] [PubMed] [Google Scholar]
- 15.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experientia. 1989;45:209. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 16.Tozer GM, Kanthou C, Parkins CS, Hill SA. Int. J. Exp. Pathol. 2002:83. doi: 10.1046/j.1365-2613.2002.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. J. Med. Chem. 2006;49:3033. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 18.Ohsumi K, Hatanaka T, Fujita K, Nakagawa R, Fukuda Y, Nihei Y, Suga Y, Morinaga Y, Akiyama Y, Tsuji T. Bioorg. Med. Chem. Lett. 1998;8:3153. doi: 10.1016/s0960-894x(98)00579-4. [DOI] [PubMed] [Google Scholar]
- 19.Liou JP, Chang YL, Kuo FM, Chang CW, Tseng HY, Wang CC, Yang YN, Chang JY, Lee SJ, Hsieh HP. J. Med. Chem. 2004;47:4247. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]
- 20.Theeramunkong S, Caldarelli A, Massarotti A, Aprile S, Caprioglio D, Zaninetti R, Teruggi A, Pirali T, Grosa G, Tron GC, Genazzani AA. J. Med. Chem. 2011;54:4977. doi: 10.1021/jm200555r. [DOI] [PubMed] [Google Scholar]
- 21.Romagnoli R, Baraldi PG, Salvador MK, Preti D, Aghazadeh TM, Brancale A, Fu XH, Li J, Zhang SZ, Hamel E, Bortolozzi R, Basso G, Viola G. J. Med. Chem. 2011;55:475. doi: 10.1021/jm2013979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou LC, Tsai MT, Hsu MH, Wang SH, Way TD, Huang CH, Lin HY, Qian K, Dong Y, Lee KH, Huang LJ, Kuo SC. J. Med. Chem. 2010;53:8047. doi: 10.1021/jm100780c. [DOI] [PubMed] [Google Scholar]
- 23.Wlodarczyk N, Simenel C, Delepierre M, Barale JC, Janin YL. Synthesis. 2011;6:934. [Google Scholar]
- 24.Huang LJ, Hsieh MC, Teng CM, Lee KH, Kuo SC. Bioorg. Med. Chem. 1998;6:1657. doi: 10.1016/s0968-0896(98)00141-2. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence NJ, Patterson RP, Ooi LL, Cook D, Ducki S. Bioorg. Med. Chem. Lett. 2006;16:5844. doi: 10.1016/j.bmcl.2006.08.065. [DOI] [PubMed] [Google Scholar]
- 26.Vu AT, Cohn ST, Manas ES, Harris HA, Mewshaw RE. Bioorg. Med. Chem. Lett. 2005;15:4520. doi: 10.1016/j.bmcl.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Y, Zou X, Hu E, Yao C, Liu B, Yang H. J. Agric. Food Chem. 2005;53:9566. doi: 10.1021/jf051510l. [DOI] [PubMed] [Google Scholar]
- 28.Tseng CJ, Wang YJ, Liang YC, Jeng JH, Lee WS, Lin JK, Chen CH, Liu IC, Ho YS. Toxicology. 2002;175:123. doi: 10.1016/s0300-483x(02)00073-2. [DOI] [PubMed] [Google Scholar]
- 29.Shin SY, Yong Y, Kim CG, Lee YH, Lim Y. Cancer Lett. 2010;287:231. doi: 10.1016/j.canlet.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 30.Kao GD, McKenna WG, Yen TJ. Oncogene. 2001;20:3486. doi: 10.1038/sj.onc.1204445. [DOI] [PubMed] [Google Scholar]
- 31.Stewart ZA, Westfall MD, Pietenpol JA. Trends Pharmacol. Sci. 2003;24:139. doi: 10.1016/S0165-6147(03)00026-9. [DOI] [PubMed] [Google Scholar]
- 32.Chen CT, Hsu MH, Cheng YY, Liu CY, Chou LC, Huang LJ, Wu TS, Yang X, Lee KH, Kuo SC. Eur. J. Med. Chem. 2011;46:6046. doi: 10.1016/j.ejmech.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Hsu MH, Liu CY, Lin CM, Chen YJ, Chen CJ, Lin YF, Huang LJ, Lee KH, Kuo SC. Toxicol. Appl. Pharmacol. 2012;259:219. doi: 10.1016/j.taap.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 34.Yong Y, Shin SY, Lee YH, Lim Y. Bioorg. Med. Chem. Lett. 2009;19:4367. doi: 10.1016/j.bmcl.2009.05.093. [DOI] [PubMed] [Google Scholar]
- 35.Tu HY, Huang AM, Teng CH, Hour TC, Yang SC, Pu YS, Lin CN. Bioorg. Med. Chem. 2011;19:5670. doi: 10.1016/j.bmc.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 36.Herr I, Debatin KM. Blood. 2001;98:2603. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- 37.Hsu MH, Kuo SC, Chen CJ, Chung JG, Lai YY, Huang LJ. Leuk. Res. 2005;29:1399. doi: 10.1016/j.leukres.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 38.Hsu MH, Chen CJ, Kuo SC, Chung JG, Lai YY, Teng CM, Pan SL, Huang LJ. Eur. J. Pharmacol. 2007;559:14. doi: 10.1016/j.ejphar.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Lin YC, Chou LC, Chen SC, Kuo SC, Huang LJ, Gean PW. Bioorg. Med. Chem. Lett. 2009;19:3225. doi: 10.1016/j.bmcl.2009.04.101. [DOI] [PubMed] [Google Scholar]
- 40.Tsai JY, Lin YC, Hsu MH, Kuo SC, Huang LJ. Kaohsiung J. of Med. Sci. 2010;26:593. doi: 10.1016/S1607-551X(10)70091-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.