

Abstract
Mesenchymal stem cells (MSCs) are a potential novel delivery system for cell-based gene therapies. Although tumour necrosis factor (TNF)-α has been shown to have antitumour activity, its use in therapy is limited by its systemic toxicity. For the present study, we designed lentivirus-mediated signal peptide TNF-α-Tumstatin45–132-expressing mesenchymal stem cells (SPTT-MSCs) as a novel anti-cancer approach. We evaluated the effects of this approach on human prostate cancer cells (PC3 and LNCaP) by co-culturing them with either SPTT-MSCs or supernatants from their culture medium in vitro. The antitumour effects and possible mechanisms of action of SPTT-MSCs were then determined in PC3 cells in vivo. The results showed that efficient TNF-α-Tumstatin45–132-expressing MSCs had been established, and demonstrated that SPTT-MSCs inhibited the proliferation of and induced apoptosis in prostate cancer cells and xenograft tumours. As would be expected, given the properties of the individual proteins, the TNF-α-Tumstatin45–132 fusion exerted potent cytotoxic effects on human prostate cancer cells and tumours via the death receptor-dependent apoptotic pathway and via antiangiogenic effects. Our findings suggest that SPTT-MSCs have significant activity against prostate cancer cells, and that they may represent a promising new therapy for prostate cancer.
Keywords: mesenchymal stem cells, TNF-α, Tumstatin, prostate cancer, gene therapy
Introduction
Prostate cancer is the most commonly diagnosed cancer in men in the western world and the second leading cause of cancer death [1]. Despite recent advances in early detection and new therapeutic approaches, the occurrence of metastatic disease due to androgen independence and resistance to existing treatments is common [2–5]. New therapies that can limit the local advancement of primary tumours and distant metastases are urgently needed for prostate cancer.
The increase in knowledge about the molecular biology of prostate cancer and genetic engineering have made gene-based cancer therapy attractive. Unfortunately, the development of an efficient, safe, and reliable gene delivery system (a critical step) has so far limited the successful implementation of gene therapy. However, recent studies have shown the feasibility of using mesenchymal stem cells (MSCs) as a cellular vehicle for gene therapy [6–10]. MSCs have greater potential to contribute to the population of stromal cells than fully differentiated fibroblasts. This allows the development of therapeutic strategies that are based on the local production of tumoricidal biological agents by MSCs transduced to express specific genes of interest. Demonstrating this capacity, MSCs have previously been shown to exert antitumour effects when transduced with adenoviruses expressing a variety of transgenes [11–18]. Nevertheless, transduction of MSCs with adenoviral vectors is relatively inefficient compared with lentiviral gene transfer, as a result of the limited expression of the cellular coxsackie and adenovirus receptor on these cells [6, 11]. As a result of this inefficient transduction, the therapeutic transgene is diluted over time, limiting the use of adenoviral vectors to deliver gene therapy via MSCs. In contrast, lentivirus (LV)-based gene transfer compares favourably with other gene transfer methods for introducing genes into MSCs [19]. In particular, lentiviral vector systems have been shown to efficiently transduce both dividing and non-dividing cells, and are less prone to transcriptional silencing than retroviral systems, making lentiviral systems the ideal approach for gene delivery into MSCs [20, 21].
Tumour necrosis factor (TNF)-α, an inflammatory cytokine, is cytotoxic to tumour cells, and also disrupts tumour blood vessels. However, the clinical use of TNF-α as an antitumour drug is hampered by its severe systemic toxicity [22]. Another anticancer molecule with anti-angiogenic activity, Tumstatin, exerts its effects on the tumour vasculature via its binding to αVβ3 on endothelial cells and the tumour vascular endothelium, which shows increased expression of αVβ3. The region of Tumstatin spanning amino acids 54–132 is responsible for the observed in vitro and in vivo anti-angiogenic activity of the protein [23]. Luo et al. explored the possibility of fusing Tumstatin45–132 with human TNF-α in the hope of generating a specifically targeted, bi-functional protein for cancer therapy. In that study, the Tumstatin45–132-TNF-α fusion protein inhibited endothelial cell proliferation to a greater extent than TNF-α alone, and led to a similar level of apoptosis against L929 mouse fibroblast cells [24].
In the present study, we describe the potential of using genetically modified MSCs that constitutively express TNF-α-Tumstatin45–132 to inhibit the proliferation of prostate cancer cells in vitro and reduce xenograft tumour growth in vivo. We also examined the mechanisms of action of the approach.
Materials and methods
Cell culture
MSCs were isolated from bone marrow of 8-week-old Wistar rats and cultured as previously described [25, 26]. PC3, LNCaP and ECV304 cells were cultured in Dulbecco's modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA). Human umbilical vein endothelial cells (HUVEC) cells were generously provided by Dr. E. Tabengwa (University of Alabama at Birmingham, Birmingham, AL, USA) and cultured in MG199 containing 10% FBS.
Cloning and transfection
The signal peptide TNF-α-Tumstatin45–132 (SPTT) fragment was generated by PCR amplification using pBV220-TNF-α-Tumstatin45–132 (kindly provided by Dr. Y. Luo, The Affiliated Anhui Provincial Hospital of Anhui Medical University, Hefei, People's Republic of China) as a template. The primers used were SPTT-S (5′-GCGGATCCATGTGGCTGCAGAGCCTGCT-GCTCTTGGGCACTGTGGCCTGCAGCATCTCTGTCAGATCATCTTCTCGAACC-3′) and SPTT-A (5′-GCGAATTCTCAGGCGATCGCAGGACCTTCACA-3′). The signal peptide is underlined in the sense primer. A BamH I restriction site (codons in bold) was introduced into the 5’ end of the SPTT-S primer, and an EcoRI restriction site (codons in bold) was introduced into the 5′ end of the SPTT-A primer. The SPTT fragment was then subcloned into LV transfer vector FUGW (F, HIV-1 flap sequence; U, human polyubiquitin promoter; G, green fluorescent protein; W, woodchuck hepatitis virus post-transcriptional regulatory element) by BamHI and EcoRI double digestion. Recombinant lentiviral particles were produced in 293T cells by transient cotransfection involving a three-plasmid expression system [27], and MSCs were transfected as described previously [28]. For FACS analysis, transduced MSCs were trypsinized and analysed on a FACSCalibur instrument, and data were analysed using CellQuest software (Becton-Dickinson, Franklin Lakes, NJ, USA). Expression levels of SPTT were verified by ELISA and Western blotting analysis.
Cell growth curves, ex vivo differentiation and DNA contents of transduced MSCs
The growth curve analysis and DNA quantitation of transduced MSCs were performed as described previously [29]. In addition, cultured transduced MSCs were tested for the ability to differentiate into osteogenic and adipogenic lineages using a previously described protocol [29] with modifications. Briefly, the transduced and parental MSCs were plated at a density of 3 × 104 cells per well in either osteogenic media (supplemented with 100 mM (β-glycerophosphate, 50 mg/l ascorbic acid, 4 mg/l bFGF and 10−6 M dexamethasone; Sigma, St. Louis, MO, USA) or adipogenic media (supplemented with 10 μg/ml insulin, 10−6 M dexamethasone; Sigma) for up to 3 weeks. The conditioned media were changed every 3–4 days. Evidence of osteogenic differentiation was determined by alkaline phosphatase (ALP) staining, whereas adipogenic differentiation was monitored using oil red O staining for the appearance of intracellular lipid inclusion vacuoles.
Cell viability assay
Cell viability was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma) assay as described previously [30]. Briefly, PC3, LNCaP and ECV304 cells were seeded in 96-well plates at 3 × 103 cells per well, and were incubated at 37°C for 24 hrs. Supernatants from normal MSCs, and LV-EGFP- and LV-SPTT-transduced MSCs were added to the plates, which were then returned to the incubator for 2 days. Untreated cells served as a control. MTT (5 mg/ml) was added to each well for the last 4 hrs of treatment. The reaction was stopped by the addition of dimethyl sulphoxide, and the optical density was determined at 570 nm on a multiwell plate reader (Bio-TEK, Winooski, VT, USA). Background absorbance of the medium in the absence of cells was subtracted. All samples were assayed in triplicate, and the mean for each experiment was calculated. Results were expressed as a percentage of the control, which was considered to be 100%.
Cell proliferation assay
The effects of the infusion protein SPTT on cell proliferation were determined by BrdUrd incorporation assay (Oncogene, La Jolla, CA, USA), following the manufacturer's protocol. Cells were seeded in 96-well plates (6×10 cells per well) and incubated with supernatants from normal MSCs, LV-EGFP-transduced MSCs or LV-SPTT-transduced MSCs for 24 hrs. BrdUrd was added to the medium 8 hrs before termination of the experiment. The BrdUrd incorporated into cells was determined by anti-BrdUrd antibody and absorbance was measured at dual wavelengths of 450/540 nm with an OPTImax microplate reader (Molecular Devices, Sunnyvale, CA, USA). All experiments were performed in triplicate. Results were expressed as a percentage of the control, which was considered to be 100%.
Detection of apoptosis
Cells in early and late stages of apoptosis were detected using an Annexin V-FITC apoptosis detection kit from Bio-Vision (Mountain View, CA, USA) according to the manufacturer's protocol. In brief, 2–3 × 105 cells were exposed to the supernatants from normal MSCs, LV-EGFP- transduced and LV-SPTT-transduced MSCs and incubated for 48 hrs prior to analysis. The samples were analysed using a Becton-Dickinson FACS Calibur instrument (Ex = 488 nm; Em = 530 nm). Cells that were positive for Annexin V-FITC alone (early apoptosis) and Annexin V-FITC and PI (late apoptosis) were counted. All samples were assayed in triplicate. Results were expressed as a percentage of the control, which was considered to be 100%.
in vitro co-culture of prostate cancer cells with MSCs
PC3 cells (5 × 104 per well) were cultured either alone or with EGFP-MSCs or SPTT-MSCs in six-well plates (1 × 104 cells per well) for 72 hrs. Cells were then trypsinized, counted, and fixed with 70% ethanol overnight. The relative numbers of MSCs (diploid cells) and PC3 cells (aneuploid cells) were determined after labelling the cells with propidium iodide (Sigma) using ModFit software (Becton-Dickinson).
Endothelial tube formation assay
The endothelial tube formation assay was performed following the manufacturer's protocol (BD Biosciences, Franklin Lakes, NJ, USA). Matrigel was added (100 μl) to each well of a 24-well plate and allowed to polymerize. A suspension of 2 × 104 HUVEC cells was seeded into each well. The cells were then treated with conditioned medium from either EGFP-MSCs or SPTT-MSCs. Control cells were incubated with medium alone. Cells were incubated for 24–48 hrs at 37°C and viewed using a microscope. The cells were then photographed and the number of tubes was counted.
Animals and treatments
Six-week-old male Balb/c nu/nu mice (Laboratory Animal Center of Shanghai, Academy of Sciences, China) were injected with 2 × 106 PC3 cells in 200 μl PBS. After 2 weeks, subcutaneous tumours had reached approximately 50 mm3, and were directly injected with 2 × 106 MSCs that had been transduced with LV-EGFP (MOI 25) or LV-SPTT (MOI 25). The transduced MSCs were trypsinized and washed three times with PBS before their in vivo use. Two additional groups of mice bearing PC3 xenografts were injected with PBS or non-transduced MSCs as controls. Three animals per group were analysed for tumour growth. The MSCs were injected three times at weekly intervals. The growth of the tumours was then followed for one month. Tumour measurements in two dimensions were performed with calipers twice weekly and tumour volumes were calculated using the formula: volume = width2× length × 0.5. The animal studies were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee.
Western blotting
MSCs were transduced with LV-EGFP and LV-SPTT at a MOI of 25. After 72 hrs, the transduced cells were collected and lysed with RIPA buffer (Cell Signaling, Boston, MA, USA). PC3 cells co-cultured with MSCs, EGFP-MSCs and SPTT-MSCs at a 2:1 ratio for 48 hrs in a transwell system were also collected and lysed with RIPA buffer. After centrifugation at 13,000 rpm for 15 min. at 4°C, the supernatant was removed and kept for analysis. Total cellular protein concentrations were assessed using a Bio-Rad protein assay kit (Hercules, CA, USA). Aliquots containing identical amounts of protein were fractionated by SDS-PAGE, then transferred to methanol pre-activated PVDF membranes (Millipore, Termecula, CA, USA). Membranes were blocked and sequentially incubated with primary and secondary antibody, then bands of the proteins of interest were visualized using the ECL plus system from Amersham Pharmacia Biotech (Buckinghamshire, UK).
Immunohistochemistry
Animals were killed approximately 4 weeks after treatment, and the tumour tissues were harvested, and tumour weights were compared. Paraffin sections (5-mm thick) were made from the harvested tissue. Tissue sections were de-waxed in xylene and rehydrated in graded alcohol. Endogenous per-oxidase activity was blocked with 0.3% (v/v) hydrogen peroxide in methanol before washing the slides in water. Slides were incubated in citrate buffer (pH 6.0) for 20 min. in a steamer for antigen retrieval. Nonspecific binding was blocked by incubation in 5% (v/v) normal serum. The primary antibodies were used in the following dilutions: proliferating cell nuclear antigen (PCNA, 1:100; CD31, 1:200 and TNF-α, 1:2000. Following binding of the primary antibodies, slides were incubated with the respective secondary antibodies conjugated to horseradish peroxidase. The bound antibody was visualized using the peroxidase-based Vectastain Elite ABC Kit (Boster, Wuhan, China). The substrate reaction was stopped by washing the slides in running water. Finally, the slides were lightly counterstained in haematoxylin.
TUNEL staining
Apoptotic cells were visualized using a terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay. The TUNEL procedure was performed with an in situ cell death detection kit (Boster) according to the manufacturer's instructions. TUNEL+ apoptotic tumour cells were counted in 10 consecutive fields at 40× magnification in the tissue sections.
PCR and RT-PCR
Genomic DNA was isolated from the transduced MSCs by phenol-chloroform extraction and PCR was performed to detect the integration of the LV genome using primers specific for the ubiquitin promoter of the transfer vector. Total RNA was extracted from MSCs, and mock- and SPTT-transduced MSCs using Trizol (Invitrogen) according to the manufacturer's instructions. Equal quantities of RNA were subjected to reverse transcriptase followed by PCR using primers specific for SPTT and Oct4 as described in Table 1. β-actin was used as an internal control.
Table 1.
Primer sequences for the amplification of target genes and β-actin
| Gene | Primer sequence (5′-3′) | Product size | Annealing temp. |
|---|---|---|---|
| Ubiquitin | For: GTCTTGAGGCCTTCGCTAAT | 414 bp | 59°C |
| Rev: AACCGGAGCTTCAGCTACTT | |||
| SPTT | For: ATGTGGCTGCAGAGCCTGCTG | 813 bp | 63°C |
| Rev: TCAGGCGATCGCAGGACCTTC | |||
| Oct4 | For: ATACACAGGCCGATGTGG | 397 bp | 60°C |
| Rev: GTGCATAGTCGCTGCTTGA | |||
| β-actin | For: CACGAAACTACCTTCAACTCC | 600 bp | 60°C |
| Rev: CATACTCCTGCTTGCTGATC |
Statistical analysis
All data were expressed as means ± S.D. SPSS software was used to analyse the data using the Student's t-test. All reported P-values are two-tailed, and those <0.05 were considered statistically significant.
Results
Efficient lentiviral transduction of MSCs
The MSCs were transduced with recombinant LV at a multiplicity of infection (MOI) of 25, and were found to efficiently express either the reporter gene (EGFP) or the SPTT target gene. The MSCs transduced with LV-EGFP showed evidence of a green fluorescent signal in almost all cells (Fig. 1A-I, II), and FACS analysis revealed that 96.8% of all cells expressed the transgene (Fig. 1A-III). To confirm the expression of the transgene, MSCs transduced with LV-EGFP and LV-SPTT were harvested, and protein lysates were prepared 72 hrs after transduction. Western blotting indicated that the SPTT protein was detectable in the cells transduced with LV-SPTT (Fig. 1A-IV). To determine the transduction efficiency of LV-SPTT in MSCs, the cells were harvested three days after transduction, fixed on slides, and subjected to immunohistochemistry using a human TNF-α antibody. The results indicated that the LV-EGFP-trans-duced MSCs were negative for the transgenic SPTT TNF-α protein (Fig. 1A-V), whereas more than 90% of the MSCs transduced with LV-SPTT were positive for SPTT (Fig. 1A-VI). PCR results showed that the ubiquitin promoter gene was detectable in the genomic DNA of both LV-EGFP and LV-SPTT-transduced MSCs, that the SPTT gene was only detectable in the LV-SPTT-transduced MSCs, and that both the cells were still positive for Oct4, a stem cell marker (Fig. 1B).
Fig 1.
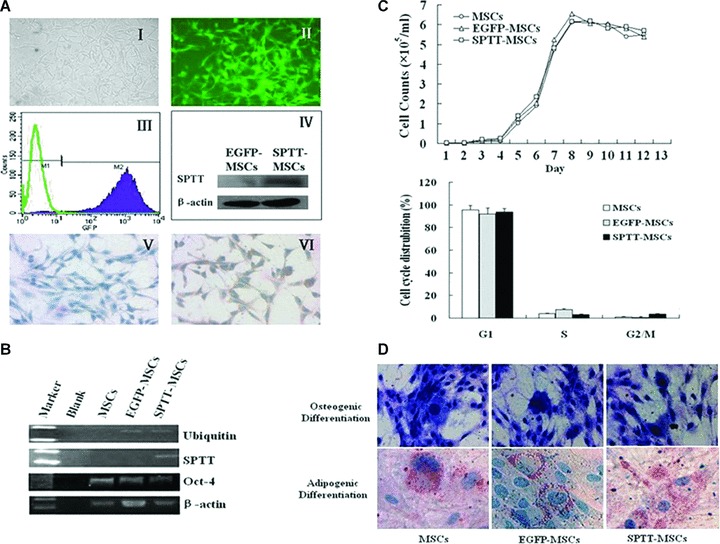
LV-transduced MSCs efficiently express human TNF-α-Tumstatin45–132 fusion protein and maintain their stem cell properties. (A) The morphology, transduction efficiency examination of LV-transduced MSCs. LV-EGFP-transduced MSCs (I, bright field; II, fluorescence field). FACS analysis showed that almost all of the MSCs expressed EGFP (III). The 66 kD SPTT protein was detectable by Western blot analysis in cell lysates from SPTT-MSCs (IV). Immunohistochemical staining revealed that whereas the mock-transduced MSCs were not stained for human TNF-α (V), but the SPTT-MSCs exhibited positive staining (VI). (B) RT-PCR showed that SPTT was only expressed in the tranduced SPTT-MSCs. Ubiquitin promoter gene was detectable in both LV-EGFP and LV-SPTT-transduced MSCs. Oct-4 was expressed in all of the cells (MSCs, EGFP-MSCs and SPTT-MSCs). (C) LV-transduced MSCs exhibited a virtually identical growth curve compared to that of untransduced parental cells (upper panel). Cell cycle analysis demonstrated that there were similar DNA contents between non-transduced and LV-transduced MSCs (lower panel). These results are representative of the average of three experiments performed in triplicates. (D) The transduced MSCs (LV-EGFP and LV-SPTT) could differentiate into either osteocyte or adipocyte lineages after induction, as indicated by ALP and oil red staining.
Stem cell properties of MSCs are not affected by LV transduction or transgene expression
As shown in Fig. 1C, EGFP-MSCs and SPTT-MSCs exhibited virtually identical growth curves (Fig. 1C, upper panel) and cell cycle profiles (Fig. 1C, lower panel) compared to those of untransduced parental cells. Analysis of DNA content showed that all of the MSCs had a high number of cells in the G1 phase, and a lower number in the S phase. The virally transduced MSCs retained their normal differentiation potential. Three weeks after initial lentiviral transduction, the EGFP-MSCs, SPTT-MSCs, and non-transduced parental MSCs were induced with adipogenic or osteogenic conditioned media. Cells that had differentiated into the adipogenic lineage were indicated by positive staining with oil Red O for intracellular fat droplets, whereas cells of osteogenic lineage stained positive for ALP (Fig. 1D).
SPTT inhibits the growth of prostate cancer cells and endothelial cells in vitro
ELISA experiments showed that up to 250 pg/ml of the SPTT protein could be detected in the supernatant of SPTT-MSCs between 3 and 4 days after transduction (Fig. 2A), which amounted to approximately 4 × 10−4 pg/cell. MTT assays demonstrated that the viability of human prostate cancer (PC3 and LNCaP) and endothelial (ECV304) cells was decreased by 51.6% (P< 0.004), 57.8% (P< 0.02) and 16.2% (P< 0.002), respectively (Fig. 2B-D). In contrast, the supernatants of normal MSCs and mocktransduced MSCs did not affect the growth of any of the cell lines. Demonstrating the activity of the fusion protein, only SPTT was able to decrease the viability of ECV304 endothelial cells, whereas TNF-α alone did not lead to any significant change (Fig. 2D).
Fig 2.
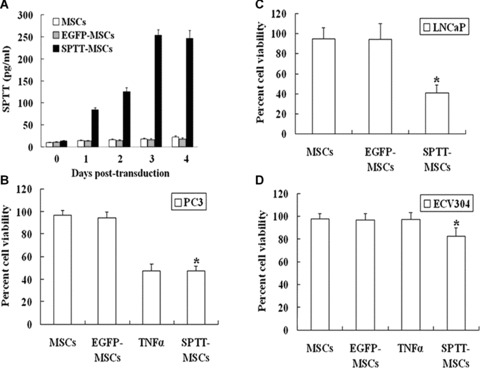
SPTT expression and its in vitro effects on the growth of prostate cancer and endothelial cells. (A) The level of SPTT protein in the culture super-natants of SPTT-MSCs increased with transduction time, reaching a peak at 3 to 4 days after transduction. (B) The SPTT protein from culture super-natants of SPTT-MSCs inhibited the growth of PC3 cells in vitro. Human TNF-α (5 μg/ml) was used as a positive control. (C) The SPTT protein from culture supernatants of SPTT-MSCs inhibited the growth of LNCaP cells in vitro. (D) The SPTT protein in culture supernatants from SPTT-MSCs inhibited the growth of ECV304 cells in vitro. Human TNF-α (5 μg/ml) was used as a negative control. The data are presented as means ± S.D. of triplicate experiments (*P< 0.05 compared to mock-transduced MSCs).
SPTT decreases the proliferation of prostate cancer cells in vitro
We then examined the effects of SPTT on prostate cancer cell proliferation. Treatment with the fusion resulted in anti-proliferative effects in both PC3 and LNCaP cells (Fig. 3A and B), with a decrease in the proliferation rate by 46.6% (P< 0.001) and 50.8% (P< 0.001) in PC3 and LNCaP cells, respectively. The supernatants of normal MSCs and mock-transduced MSCs did not affect the proliferation of either cell line.
Fig 3.
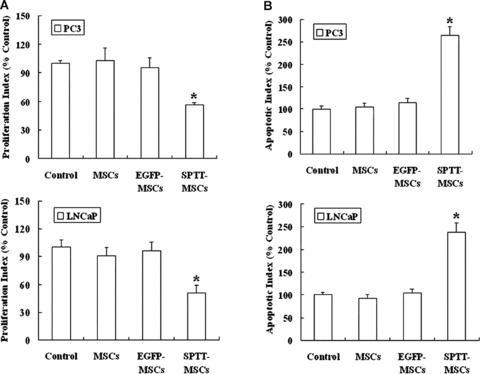
SPTT restrains proliferation and induces apoptosis of prostate cancer cells in vitro. (A) The results of BrdUrd assay showed that significant anti-proliferative effects were induced by SPTT in both PC3 and LNCaP cell lines, whereas the super-natants of normal MSCs and mock-transduced MSCs did not affect the proliferation of either cell line. (B) SPTT induced apoptosis of prostate cancer cells. Annexin V-FITC/PI staining demonstrated that the apoptosis rate of prostate cancer cells increased after treatment with conditioned media from SPTT-MSCs (*P< 0.05 compared to mock-transduced MSCs).
Induction of apoptosis by SPTT
We next determined whether SPTT induced apoptosis in inducing apoptosis of prostate cancer cells. PC3 and LNCaP cells were treated with conditioned media from SPTT-MSCs for 48 hrs, and apoptosis was analysed by Annexin V-FITC/PI staining. The apoptosis rate of prostate cancer cells significantly increased following exposure to conditioned media from SPTT-MSCs (PC3 15.2%; LNCaP 25.2%). In contrast, the conditioned media from MSCs (PC3 6.0%; LNCaP 9.2%) and EGFP-MSCs (PC3 6.2%; LNCaP 10.0%) did not cause any major increase in apoptosis, showing levels comparable to control cells (PC3 5.7%; LNCaP 9.6%) (Fig. 3C and D).
Co-culture with SPTT-MSCs inhibits the growth of PC3 cells in vitro
We next examined the therapeutic potential of MSCs as cellular vehicles for production of SPTT using a co-culture system with PC3 cells under in vitroconditions. When the two cell lines were co-cultured, the SPTT-MSCs directly inhibited the growth of the malignant prostate cells (Fig. 4A), whereas co-culture with mock-transduced MSCs did not have this inhibitory effect.
Fig 4.
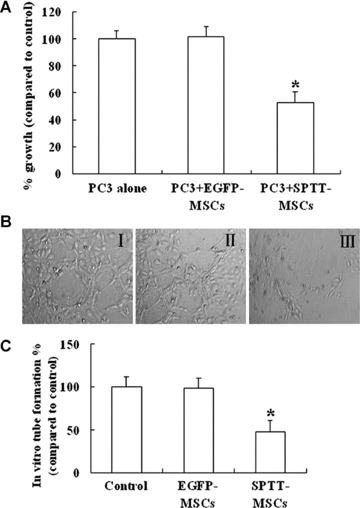
SPTT-MSCs directly inhibit the growth of PC3 cells in vitro, and decrease angiogenesis. (A) SPTT-MSCs and PC3 cells were mixed in vitro at a 1:5 ratio in a co-culture system for 72 hrs. The survival of PC3 cells was significantly decreased by co-culture with the SPTT-MSCs (*P< 0.05 compared to mock-transduced MSCs), whereas co-culture with EGFP-MSCs did not have any significant effect on their survival. (B) Compared with media from control MSCs (I) and EGFP-MSCs (II), the conditioned media from SPTT-MSCs (III), significantly inhibited endothe-lial tube formation. (C) The extent of tube formation after treatment with conditioned media from SPTT-MSCs was less than half of that observed in the control MSCs- and EGFP-MSCs-treated groups.
Inhibition of angiogenesis by SPTT in vitro
The ability of SPTT to block angiogenesis in vitro was evaluated using the endothelial tube formation assay. When HUVEC cells are cultured on matrigel, they rapidly align and form hollow tube-like structures. Compared to cells cultured with control media (Fig. 4B-I) and conditioned media from EGFP-MSCs (Fig. 4B-II), the conditioned media of SPTT-MSCs significantly inhibited endothelial tube formation (Fig. 4B-III). The extent of tube formation after treatment with conditioned media from SPTT-MSCs was less than half of that observed in the group treated with media from EGFP-MSCs (Fig. 4C).
LV-SPTT-transduced MSCs inhibit the growth of PC3 xenografts in mice
Given its potent in vitro activity, we wanted to know whether MSCs transduced with LV-SPTT would also have antitumour activity in vivo. Mice bearing PC3 xenograft tumours were injected peritumorally with parental (non-transduced) MSCs, EGFP-MSCs or SPTT-MSCs. Representative tumours harvested at the conclusion of the experiment are shown in Fig. 5A. Although the tumour mass was markedly smaller in the mice treated with SPTT-MSCs, the LV-EGFP-transduced MSCs did not lead to any significant difference in tumour size or weight compared to the mice treated with PBS or the control MSCs (Fig. 5A and B). The differential effects of the control MSCs and SPTT-MSCs were further illustrated by the tumour growth curves (Fig. 5C). All of the tumours in the animals treated with PBS, non-transduced MSCs, or LV-EGFP-transduced MSCs grew to about six times their original size. In contrast, the tumours treated with the LV-SPTT-transduced MSCs only grew by a factor of 2.0.
Fig 5.
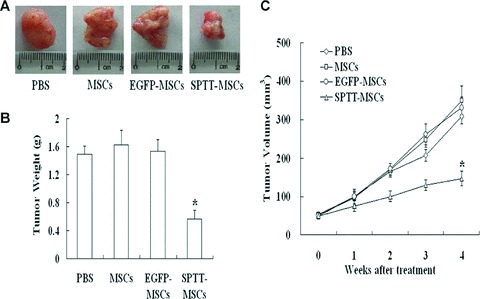
SPTT-MSCs suppress tumour growth in vivo. (A) Tumours from animals in the SPTT-MSCs-treated group were smaller than those treated with the controls. (B) The tumours from mice treated with SPTT-MSCs weighed much less than those from mice treated with the control MSCs or PBS. (C) Tumour growth curves showed that treatment with SPTT-MSCs suppresses tumour growth. Results reflect the average of three animals per group (*P< 0.05 compared to mock-transduced cells).
LV-SPTT-transduced MSCs decrease tumour cell proliferation, promote apoptosis and decrease tumour vascularity
Immunohistochemical examination of the expression of PCNA revealed that the number of proliferating tumour cells was significantly lower in the tumours from mice in the LV-SPTT-transduced MSCs group compared with the groups treated with PBS, MSCs or LV-EGFP-transduced MSCs (Fig. 6A, upper panels; 6B). Additionally, TUNEL staining demonstrated that there was an increase in apoptosis in the tumour cells in mice following treatment with LV-SPTT-transduced MSCs compared with the cells in mice treated with PBS, control MSCs, or LV-EGFP-transduced MSCs (Fig. 6A, middle panels; 6B). Immunohistochemical analysis of CD31, a marker for blood vessels, revealed that there were also significantly lower blood vessel counts in the tumours of mice treated with SPTT-MSCs compared with those treated with PBS, control MSCs, or LV-EGFP MSCs (Fig. 6A, lower panels; 6B). There were no significant differences in the apoptosis or vascularity of tumours in mice treated with MSCs expressing EGFP compared with those treated with PBS or control MSCs (Fig. 6A and B), demonstrating that the effects on apoptosis and angiogenesis were specific to the SPTT-transduced MSCs.
Fig 6.
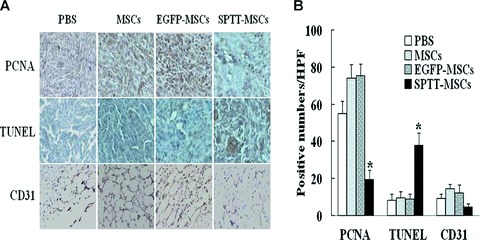
Immunohistochemical analysis of proteins related to cell proliferation, apoptosis, and the formation of neovasculature. For immunohistochemical analysis of tumour cell proliferation, apoptosis and microvessel density, tumours were paraffin embedded and sectioned. (A) Staining was performed with anti-PCNA antibody to assess proliferation, while TUNEL staining was performed to examine apoptosis, and a CD31 antibody was employed to detect endothelial cells, representing sites of vascularization. Slides were lightly counterstained with haematoxylin. (B) Quantitative analysis of proliferation (PCNA) and apoptotic (TUNEL) indices were calculated by counting positive cells in 10 random fields at 40× magnification. The number of proliferating tumour cells decreased, whereas the number of apoptotic cells increased in the group treated with SPTT-MSCs. Blood vessel counts were determined by counting the number of vessels in 10 randomly chosen areas of CD31 stained sections (40×). The number of CD31+ cells was decreased in the group treated with SPTT-MSCs (*P< 0.05 compared to mock-transduced MSCs).
Activation and induction of apoptosis-related proteins by SPTT treatment
To investigate the mechanisms responsible for the activity of LV-SPTT-transduced MSCs against prostate cancer, the prostate cancer cells and tumours were lysed and proteins were collected. Western blotting analysis of tumour cells and tissues showed that treatment with SPTT-MSCs led to increased cleavage of caspases-8 and –3 (Fig. 7A), In addition, we also found that the expression of Bax, a pro-apoptotic effector, increased in PC3 cells and tumours after treatment with SPTT-MSCs, whereas the expression of anti-apoptotic protein Bcl-2 decreased (Fig. 7A).
Fig 7.
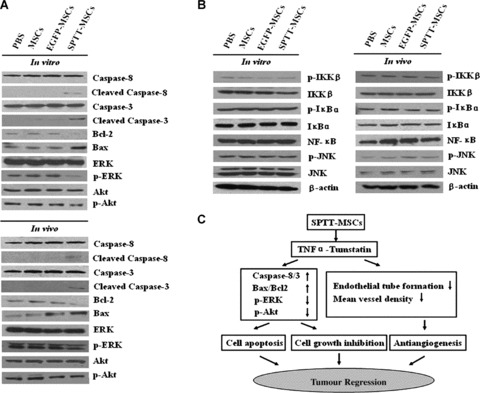
Putative mechanisms underlying the antitumour effects of SPTT-MSCs. (A) The proteins from PC3 cells and tumour tissues were acquired and analysed for different proteins. The SPTT-MSCs-treated group had increased cleavage of caspases-8 and -3, and an increase in the Bax/Bcl2 ratio, as well as decreased levels of phosphylated ERK-1/2 and Akt compared to the PBS group. (B) After the treatments with SPTT-MSCs, the expression of NF-κB protein and the phosphorylation status of IKK-β and Iκ-Bα in PC3 cells and tumour tissues did not change, nor was there any inhibition of JNK activity. (C) The possible mechanisms of anticancer action of SPTT-MSCs. In the death receptor-mediated caspase-dependent apoptotic pathway, the SPTT-MSCs can directly reduce prostate cancer cell viability by producing the active TNF-α peptide. In the anti-angiogenic pathway, the SPTT-MSCs directly inhibit the proliferation of vascular endothelial cells by producing the active Tumstatin45–132 peptide.
SPTT-MSCs reduce the phosphylation of ERK-1/2 and Akt, but do not affect NF-kB or JNK
The results of Western blotting analysis confirmed that the phosphorylation of ERK-1/2 and Akt was decreased in tumour cells and xenografts treated with SPTT-MSCs (Fig. 7A). Because TNF-α activates NF-κB signalling as well as apoptosis, it is possible that it may elicit a pro-survival response that might counteract its increase in cell death. To investigate this possibility, we analysed the response of PC3 cells to the SPTT fusion protein with regard to nuclear factor K-light-chain enhancer of activated B cells (NF-κB) and c-Jun N-terminal kinase (JNK) signalling. As shown in Fig. 7B, the expression of the NF-κB protein and the phosphorylation of inhibitor of nuclear factor k-B kinase (IKK)-β and κ-Bα did not change, nor was there any inhibition of JNK activity (Fig. 7B).
Discussion
The use of recombinant proteins for the treatment of cancer is often limited by their short half-life or excessive toxicity. More effective therapeutic strategies that can provide sustained antitumour effects, and that can preferentially deliver the therapeutic agent to the tumour will increase patient survival [31]. MSCs have been considered promising candidates for the expression of therapeutic proteins by gene transfer because their ability to be transduced with viral and non-viral vectors [32]. Studies have shown that MSCs efficiently transduced ex vivo with different viral vectors can migrate to and engraft at the site of gliomas, colorectal tumours, ovarian tumours and lung metastasis of breast cancer in animal models, suggesting that they can serve as cellular vehicles for anticancer agents, and can be used to specifically deliver them to malignant cells [17, 33–38].
Both highly efficient transduction of MSCs and efficient expression of the antitumour transgenic protein are needed in order to use the MSCs for therapeutic applications. Although many approaches have previously been tested on MSCs, including adenoviral vectors, adeno-associated viral vectors, and retroviral vector-based systems, most of these were unsuitable due to their relatively low transduction efficiency or unstable transgene expression [19]. We have recently observed highly efficient transduction of MSCs using a LV system. The transduction efficiency of MSCs reached up to 96.8%, and the transduced MSCs persistently expressed the transgene for more than 6 months while maintaining their capacity for self-renewal, differentiation, and survival after transplantation (data not shown). This efficient, safe, and reliable gene delivery system provides a platform for MSC-based gene therapy, and was the basis for the present study.
Prostate cancer is one of the most common malignant diseases and is the most lethal urological malignancy. The acquired drug resistance of tumour cells and cumulative side-effects of the cytotoxic agents used to treat advanced prostate cancer present serious clinical obstacles. It is necessary to explore new therapeutic strategies that can either enhance the effects of existing antitumour therapies or that exhibit potent activity against tumours and low cytotoxicity to normal tissues. MSC-based gene therapy may present just such an approach.
A large number of animal studies have shown that TNF-α has potent antitumour activity, however, the clinical use of TNF-α as an anticancer drug is hampered by its severe systemic toxicity. In addition, several malignant cell lines and most normal cells (including endothelial cells) are resistant to TNF-α. However, the combination of TNF-α with other molecules, and tumour-targeted delivery of TNF-α, still represent alternative approaches for its use as cancer therapy [24, 39–41]. Angiogenesis is a major pathogenic step in the process of tumour growth and metastasis, and tumour angiogenesis has become an important area in cancer treatment. Tumstatin, an endothelial cell-specific protein, has been shown to suppress tumour growth in human renal carcinoma (786O) and prostate carcinoma (PC3) in mouse xenograft models, and to induce endothelial cell-specific apoptosis [42–44]. Therefore, combining TNF-α with Tumstatin may provide a new therapeutic molecule for prostate cancer that is directly cytotoxic and can decrease angiogenesis.
In the present study, we established a LV-SPTT-transduced MSC line that efficiently expressed a human TNF-α-Tumstatin45–132 fusion protein. MTT assays indicated that the LV-SPTT-transduced MSCs inhibited the growth of prostate cancer cells (PC3 and LNCaP) and endothelial cells (ECV304) in vitro. Because recombinant human TNF-α alone was not able to suppress the proliferation of ECV304 cells, this suggests that the inhibitory effect of SPTT on ECV304 cells was due to the Tumstatin45–132 domain of the fusion protein. We found that the growth inhibitory effect was due to both suppression of proliferation and to induction of apoptosis by SPTT. Proliferation and cytotoxicity assays indicated that the TNF-α-tumstatin45–132 fusion protein retained both its TNF-α-like activities (demonstrated by its cytotoxicity to PC3 cells), as well as its Tumstatin45–132-like activity (demonstrated by its inhibition of ECV304 cell proliferation). This indicates that the fusion of TNF-α and Tumstatin45–132 by a linker amino acid does not interrupt the anticancer activities of the individual proteins. The activity of the SPTT-MSCs was further demonstrated in the PC3 xenograft prostate cancer model.
Based on our observations, we suggested that the inhibition of tumour growth by LV-SPTT-transduced MSCs occurred through both induction of apoptosis and inhibition of proliferation. To confirm the mechanisms of action of the LV-SPTT-transduced MSCs, we evaluated the expression of apoptotic effector caspases-8 and -3 in PC3 cells treated with the SPTT-MSCs or their culture medium. The cleavage of caspases-8 and -3 increased following exposure to the SPTT-MSCs, but not to any other MSCs, providing evidence to confirm our hypothesis. An up-regulation of the pro-apoptotic/ anti-apoptotic protein (Bax/Bcl2) ratio also supported our observed effects on apoptosis. Because ERK and Akt signalling plays a key role in the cell growth and survival of a variety of cell types, we then evaluated the changes in ERK-1/2, p-ERK-1/2, Akt and p-Akt in vitro and in vivo after treatment with SPTT-MSCs to further examine the mechanism(s) of action of the approach. Decreased phosphorylation of ERK-1/2 and Akt was observed both in vitro in cells co-cultured with the SPTT-MSCs and in vivoin xenograft tumours, suggesting that the antitumour effects may be at least partially mediated through the effects on the ERK and Akt pathways. In contrast, we did not observe any changes in the expression or phosphorylation status of NF-κB, IKK-β, Iκ-Bα or JNK proteins, indicating that these pathways are not affected by the TNF-α portion of the protein, and that the fusion protein does not have anti-apoptotic effects through these pathways (as TNF-α alone has been shown to do).
In addition to the effects on cell apoptosis and proliferation, the fusion protein has also been shown to decrease angiogenesis in both the in vitro endothelial tube formation assay and in the xenograft model, which contributes to the molecule's antitumour activity (Fig. 7C presents a cartoon illustrating the potential mechanisms of action of the SPTT-MSCs). Thus, the LV-transduced MSCs exert antitumour effects via several mechanisms of action: induction of apoptosis, inhibition of proliferation, and reduction in angiogenesis. It may also be possible to combine these transduced cells with conventional approaches to achieve an even greater decrease in tumour growth, angiogenesis and metastasis.
In conclusion, our results indicate that LV-SPTT-transduced MSCs can inhibit the proliferation and growth of prostate cancer cells (both androgen receptor-positive and negative cell lines) and tumours, as well as decrease their blood supply by inhibiting angiogenesis. Our findings provide a basis for future pre-clinical and clinical studies of MSC-based gene therapy for prostate cancer.
Acknowledgments
We thank Dr. Xiaosheng Wu (Mayo Clinic, Scottsdale, AZ, USA) and Jakob Reiser (LSU Health Sciences Center, New Orleans, LS, USA) for helpful guidance about lentiviral transduction. We thank Dr. Elizabeth R. Rayburn for assistance in editing this manuscript. We thank Dr. Edlue Tabengwa (University of Alabama at Birmingham) for her generous donation of HUVECs. This work was supported by the National Natural Science Foundation of China (Grant no. 30840053), the Natural Science Foundation of the Ministry of Public Health of China (Grant no. WKJ2005–2–024), Jiangsu Province's Outstanding Medical Academic Leader Program (Grant no. LJ200614), the Natural Science Foundation of Jiangsu Province (Grant no. BK2007705, BK2007092 and BK2008232) and the Sci-tech Innovation Team and Talents of Jiangsu University (Grant no. 2008–018–02).
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Naitoh J, Belldegrun A. Gene therapy for prostate cancer. Prostate Cancer Prostatic Dis. 1998;1:189–96. doi: 10.1038/sj.pcan.4500242. [DOI] [PubMed] [Google Scholar]
- 3.Shariat SF, Slawin KM. Gene therapy for prostate cancer. Rev Urol. 2000;2:81–7. [PMC free article] [PubMed] [Google Scholar]
- 4.Djavan B, Nasu Y. Prostate cancer gene therapy-what have we learned and where are we going? Rev Urol. 2001;3:179–86. [PMC free article] [PubMed] [Google Scholar]
- 5.Webster WS, Small EJ, Rini BI, Kwon ED. Prostate cancer immunology: biology, therapeutics, and challenges. J Clin Oncol. 2005;23:8262–9. doi: 10.1200/JCO.2005.03.4595. [DOI] [PubMed] [Google Scholar]
- 6.Pereboeva L, Komarova S, Mikheeva G, et al. Approaches to utilize mesenchymal progenitor cells as cellular vehicles. Stem Cells. 2003;21:389–404. doi: 10.1634/stemcells.21-4-389. [DOI] [PubMed] [Google Scholar]
- 7.Reiser J, Zhang XY, Hemenway CS, et al. Potential of mesenchymal stem cells in gene therapy approaches for inherited and acquired diseases. Expert Opin Biol Ther. 2005;5:1571–84. doi: 10.1517/14712598.5.12.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall B, Dembinski J, Sasser AK, et al. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. Int J Hematol. 2007;86:8–16. doi: 10.1532/IJH97.06230. [DOI] [PubMed] [Google Scholar]
- 9.Lee K, Majumdar MK, Buyaner D, et al. Human mesenchymal stem cells maintain transgene expression during expansion and differentiation. Mol Ther. 2001;3:857–66. doi: 10.1006/mthe.2001.0327. [DOI] [PubMed] [Google Scholar]
- 10.Aboody KS, Najbauer J, Danks MK. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 2008;15:739–52. doi: 10.1038/gt.2008.41. [DOI] [PubMed] [Google Scholar]
- 11.Conget PA, Minguell JJ. Adenoviral-mediated gene transfer into ex vivo expanded human bone marrow mesenchymal progenitor cells. Exp Hematol. 2000;28:382–90. doi: 10.1016/s0301-472x(00)00134-x. [DOI] [PubMed] [Google Scholar]
- 12.Komarova S, Kawakami Y, Stoff-Khalili MA, et al. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006;5:755–66. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- 13.Elzaouk L, Moelling K, Pavlovic J. Antitumor activity of mesenchymal stem cells producing IL-12 in a mouse melanoma model. Exp Dermatol. 2006;15:865–74. doi: 10.1111/j.1600-0625.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- 14.Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 15.Chen XC, Wang R, Zhao X, et al. Prophylaxis against carcinogenesis in three kinds of unestablished tumor models via IL12-gene-engineered MSCs. Carcinogenesis. 2006;27:2434–41. doi: 10.1093/carcin/bgl069. [DOI] [PubMed] [Google Scholar]
- 16.Studeny M, Marini FC, Champlin RE, et al. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603–8. [PubMed] [Google Scholar]
- 17.Nakamura K, Ito Y, Kawano Y, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11:1155–64. doi: 10.1038/sj.gt.3302276. [DOI] [PubMed] [Google Scholar]
- 18.Mohr A, Lyons M, Deedigan L, et al. Mesenchymal stem cells expressing TRAIL lead to tumour growth inhibition in an experimental lung cancer model. J Cell Mol Med. 2008;12:2628–43. doi: 10.1111/j.1582-4934.2008.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMahon JM, Conroy S, Lyons M, et al. Gene transfer into rat mesenchymal stem cells: a comparative study of viral and non-viral vectors. Stem Cells Dev. 2006;15:87–96. doi: 10.1089/scd.2006.15.87. [DOI] [PubMed] [Google Scholar]
- 20.Pfeifer A, Ikawa M, Dayn Y, et al. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc Natl Acad Sci USA. 2002;99:2140–5. doi: 10.1073/pnas.251682798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiser J, Harmison G, Kluepfel-Stahl S, et al. Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc Natl Acad Sci USA. 1996;93:15266–71. doi: 10.1073/pnas.93.26.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lejeune FJ, Ruegg C, Lienard D. Clinical applications of TNF-alpha in cancer. Curr Opin Immunol. 1998;10:573–80. doi: 10.1016/s0952-7915(98)80226-4. [DOI] [PubMed] [Google Scholar]
- 23.Maeshima Y, Sudhakar A, Lively JC, et al. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295:140–3. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- 24.Luo YQ, Wang LH, Ma XL, et al. Construction, expression, and characterization of a new targeted bifunctional fusion protein: tumstatin45–132-TNF. IUBMB Life. 2006;58:647–53. doi: 10.1080/15216540600981743. [DOI] [PubMed] [Google Scholar]
- 25.Breyer A, Estharabadi N, Oki M, et al. Multipotent adult progenitor cell isolation and culture procedures. Exp Hematol. 2006;34:1596–601. doi: 10.1016/j.exphem.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 26.Qian H, Yang H, Xu W, et al. Bone marrow mesenchymal stem cells ameliorate rat acute renal failure by differentiation into renal tubular epithelial-like cells. Int J Mol Med. 2008;22:325–32. [PubMed] [Google Scholar]
- 27.Lois C, Hong EJ, Pease S, et al. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 28.Zhang XY, La Russa VF, Reiser J. Transduction of bone-marrow-derived mesenchymal stem cells by using lentivirus vectors pseudotyped with modified RD114 envelope glycoproteins. J Virol. 2004;78:1219–29. doi: 10.1128/JVI.78.3.1219-1229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiao C, Xu W, Zhu W, et al. Human mesenchymal stem cells isolated from the umbilical cord. Cell Biol Int. 2008;32:8–15. doi: 10.1016/j.cellbi.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Li M, Wang H, et al. Antisense therapy targeting MDM2 oncogene in prostate cancer: Effects on proliferation apoptosis, multiple gene expression, and chemotherapy. Proc Natl Acad Sci USA. 2003;100:11636–41. doi: 10.1073/pnas.1934692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren C, Kumar S, Chanda D, et al. Therapeutic potential of mesenchymal stem cells producing interferon-alpha in a mouse melanoma lung metastasis model. Stem Cells. 2008;26:2332–8. doi: 10.1634/stemcells.2008-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nauta AJ, Kruisselbrink AB, Lurvink E, et al. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J Immunol. 2006;177:2080–7. doi: 10.4049/jimmunol.177.4.2080. [DOI] [PubMed] [Google Scholar]
- 33.Stoff-Khalili MA, Rivera AA, Mathis JM, et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat. 2007;105:157–67. doi: 10.1007/s10549-006-9449-8. [DOI] [PubMed] [Google Scholar]
- 34.Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96:1593–603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 35.Xin H, Kanehira M, Mizuguchi H, et al. Targeted delivery of CX3CL1 to multiple lung tumors by mesenchymal stem cells. Stem Cells. 2007;25:1618–26. doi: 10.1634/stemcells.2006-0461. [DOI] [PubMed] [Google Scholar]
- 36.Kucerova L, Altanerova V, Matuskova M, et al. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67:6304–13. doi: 10.1158/0008-5472.CAN-06-4024. [DOI] [PubMed] [Google Scholar]
- 37.Kanehira M, Xin H, Hoshino K, et al. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther. 2007;14:894–903. doi: 10.1038/sj.cgt.7701079. [DOI] [PubMed] [Google Scholar]
- 38.Chen X, Lin X, Zhao J, et al. A tumor-selective biotherapy with prolonged impact on established metastases based on cytokine gene-engineered MSCs. Mol Ther. 2008;16:749–56. doi: 10.1038/mt.2008.3. [DOI] [PubMed] [Google Scholar]
- 39.Tandle A, Hanna E, Lorang D, et al. Tumor vasculature-targeted delivery of tumor necrosis factor-alpha. Cancer. 2009;115:128–39. doi: 10.1002/cncr.24001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borsi L, Balza E, Carnemolla B, et al. Selective targeted delivery of TNFalpha to tumor blood vessels. Blood. 2003;102:4384–92. doi: 10.1182/blood-2003-04-1039. [DOI] [PubMed] [Google Scholar]
- 41.Mocellin S, Rossi CR, Pilati P, et al. Tumor necrosis factor, cancer and anti-cancer therapy. Cytokine Growth Factor Rev. 2005;16:35–53. doi: 10.1016/j.cytogfr.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Maeshima Y, Colorado PC, Torre A, et al. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J Biol Chem. 2000;275:21340–8. doi: 10.1074/jbc.M001956200. [DOI] [PubMed] [Google Scholar]
- 43.Maeshima Y, Colorado PC, Kalluri R. Tw o RGD-independent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J Biol Chem. 2000;275:23745–50. doi: 10.1074/jbc.C000186200. [DOI] [PubMed] [Google Scholar]
- 44.Maeshima Y, Manfredi M, Reimer C, et al. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J Biol Chem. 2001;276:15240–8. doi: 10.1074/jbc.M007764200. [DOI] [PubMed] [Google Scholar]