

Abstract
Hydrogen sulfide (H2S) has recently been proposed as an endogenous mediator of inflammation and is present in human synovial fluid. This study determined whether primary human articular chondrocytes (HACs) and mesenchymal progenitor cells (MPCs) could synthesize H2S in response to pro-inflammatory cytokines relevant to human arthropathies, and to determine the cellular responses to endogenous and pharmacological H2S. HACs and MPCs were exposed to IL-1β, IL-6, TNF-α and lipopolysaccharide (LPS). The expression and enzymatic activity of the H2S synthesizing enzymes cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE) were determined by Western blot and zinc-trap spectrophotometry, respectively. Cellular oxidative stress was induced by H2O2, the peroxynitrite donor SIN-1 and 4-hydroxynonenal (4-HNE). Cell death was assessed by 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and lactate dehydrogenase (LDH) assays. Mitochondrial membrane potential (DCm) was determined in situ by flow cytometry. Endogenous H2S synthesis was inhibited by siRNA-mediated knockdown of CSE and CBS and pharmacological inhibitors D,L-propargylglycine and aminoxyacetate, respectively. Exogenous H2S was generated using GYY4137. Under basal conditions HACs and MPCs expressed CBS and CSE and synthesized H2S in a CBS-dependent manner, whereas CSE expression and activity was induced by treatment of cells with IL-1β, TNF-α, IL-6 or LPS. Oxidative stress-induced cell death was significantly inhibited by GYY4137 treatment but increased by pharmacological inhibition of H2S synthesis or by CBS/CSE-siRNA treatment. These data suggest CSE is an inducible source of H2S in cultured HACs and MPCs. H2S may represent a novel endogenous mechanism of cytoprotection in the inflamed joint, suggesting a potential opportunity for therapeutic intervention.
Keywords: arthritis, cystathionine-γ-lyase, cystathionine-β-synthase, GYY4137, apoptosis, oxidative stress
Introduction
H2S is a pungent gas recently shown to be endogenously produced in a variety of mammalian tissues primarily from the amino acids cysteine and homocysteine by pyridoxal-5′-phosphate-dependent enzymes such as cystathionine-γ-lyase (CSE; E.C. 4.4.1.1) and cystathionine-β-synthetase (CBS; E.C. 4.2.1.22) (reviewed in [1,2]). Recent studies in animals have identified a role for H2S as a novel gaseous mediator in models of acute and chronic inflammation such as oedema, haemorrhagic and endotoxic shock, asthma, acute lung and burn injury and ischaemia-reperfusion injury (reviewed in [3]). In each of these disease models, tissue CSE expression was up-regulated leading to enhanced H2S biosynthesis and plasma H2S levels. Furthermore administration of sulfide salts such as sodium sulfide (Na2S) or sodium hydrosulfide (NaSH) as crude ‘H2S donors’ to animals has led to extensive tissue inflammation and oedema as well as increased expression of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 through NF-κB-mediated processes [4-7]. These studies have been interpreted as suggesting that H2S is a pro-inflammatory mediator.
Physiological levels of H2S in rodent and human serum and plasma have been reported in the range of 23–45 μmol/l (reviewed in [2]). Blood H2S levels are markedly increased in animals by pro-inflammatory mediators such as bacterial endotoxin and carrageenan, and are increased up to fourfold in intensive care patients with sepsis [8]. Recently, synovial fluid (SF) aspirates from patients with rheumatoid arthritis (RA) were shown to contain up to fourfold higher concentrations of H2S than in paired plasma samples (RA SF median concentration, 62.4 μmol/l) and more than twofold higher H2S levels than SF aspirates from patients with non-inflammatory arthritides (median, 25.1 μmol/l) [9]. However, increased synovial synthesis of H2S may not be specific to RA as other inflammatory joint diseases such as psoriatic and reactive arthritides also have significantly higher SF levels of H2S compared with matched plasma or patients with osteoarthritis (OA) [3].
Despite these observations, the precise role of H2S in inflammatory tissue destruction has not been elucidated. The effects of endogenous and pharmacological H2S on resident joint cells, such as articular chondrocytes, have not been studied in detail and the molecular signalling pathways leading to H2S synthesis in any cell type have not been examined. One recent study using synoviocytes isolated from RA patients [10] examined the effects of pharmacological H2S, generated by sodium hydrosulfide (NaSH). In this study NaSH transiently decreased and then increased IL-1β-induced synthesis of IL-6 via a mechanism independent of NF-κB activation but dependent upon the inactivation of the mean arterial pressure (MAP) extracellular regulated kinase (ERK). However, it should be noted that the pre-dominant effects were seen at NaSH concentrations equal to or greater than 125 μmol/l (often 1 mmol/l) which were considerably higher than the reported levels of H2S in SF [9]. Nevertheless, the same biphasic effect of H2S on inflammatory signalling have also been observed in LPS-treated murine macrophages, where low concentrations of H2S inhibited LPS-induced synthesis of PGE2,•NO, IL-1β and IL-6 and NF-κB activity, but higher concentrations of NaSH promoted the synthesis of pro-inflammatory mediators [11]. Furthermore, in an in vivo murine model of acute arthritis induced by kaolin/carrageenan [12], 30–50 μM of Na2S inhibited leucocyte adhesion in post-capillary venules in acutely inflamed mouse knees suggesting an anti-inflammatory role for H2S in this model of arthritis. However, the effects of H2S on cartilage-producing chondrocytes have not been investigated.
The loss of cartilage-producing chondrocytes is a hallmark on both OA and RA pathology. Chondrocytes are a differentiated cartilage-producing cell type derived from MPCs. The pluri-potent nature of MPCs results in the potential generation of several lineages including osteoblasts, adipocytes, myoblasts and tenocytes [13,14]. SF aspirates from OA patients contain immature mesenchymal cells, and normal adult cartilage contains MPCs capable of chondrogenic differentiation [15,16]. Furthermore, increased numbers of these cells are observed in cartilage from OA patients, strongly suggesting a role of MPCs in cartilage repair and pathological cartilage remodelling in various arthropathies. Cartilage-producing chondrocytes are known to undergo apoptotic-like cell death in OA [17,18] and RA ([19-22]), an event involving mitochondrial dysfunction [23-25], increased oxidative stress [26] and closely correlated to cartilage destruction [30-32].
Therefore with these observations in mind, we have investigated whether primary HACs and MPCs, differentiated into a chondrogenic lineage, are able to synthesize H2S in response to pro-inflammatory mediator stimulation. We have investigated the potential physiological and pathophysiological consequences of H2S production by these cells in regulating chondrocyte cell death and determined the effects of a novel slow releasing H2S donor (GYY4137) [33,34] on these cells. For the first time our study shows that endogenous H2S is inducible in chondrocytes and in MPCs, and that endogenous H2S or slowly released ‘pharmacological’ H2S from GYY4137 is cytoprotective. Our findings also suggest that during joint inflammation H2S may represent a novel endogenous mechanism for preserving joint integrity.
Materials and methods
Reagents
Tetramethylrhodamine methyl ester (TMRM) was obtained from Molecular Probes (Eugene, OR, USA). Triciribine, LY294002, wortmannin, PPM-18, SB203580, Ste-MEK113, FR180204 and human recombinant tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) were purchased from Calbiochem (San Diego, CA, USA). 1L-6-Hydroxymethyl-chiro-inositol-2-[(R)-2-O-methyl-3-O-octadecylcarbonate, AS601245, PD 169,316 and BAY 11–7085 were purchased from Enzo Life Sciences (Lausen, Switzerland). CSE and CBS siRNA were purchased from Abnova (Taipei, Taiwan). NF-κB p65 siRNA and non-coding siRNA negative controls (#4635 and #4611) and RNAi support reagents (Silencer® siRNA Transfection Kit) were purchased from Ambion (Carlsbad, CA, USA). Akt siRNA was purchased from Cell Signaling Technology (#6211; Beverly, MA, USA). Primary CBS and CSE antibodies were purchased from Abnova. All secondary horse radish peroxidase (HRP)-conjugated secondary antibodies for Western blotting were purchased from Cell Signaling Technology. Cytochrome c ELISA was purchased from Chemicon (Temecula, CA, USA; #APT200). All other reagents including bacterial LPS (Escherichia coli 0127:B8), SP600125, D,L-propargylglycine (PAG), aminooxyacetate (AOAA) and β-actin primary antibodies were purchased from Sigma-Aldrich (St. Louis, MO, USA). All cell culture flasks and micro-plates were obtained from Greiner Bio-One GmbH (Frickenhausen, Germany).
Isolation and characterization of human bone-marrow-derived MPCs
This study was approved by the ethics committee of the National University Hospital and National University of Singapore and informed written consent was obtained from each patient. MPCs were isolated from the trabecular bone chips of patients undergoing reconstructive surgery. After rinsing the sample with saline solution an explant culture system was performed. Colony forming units of spindle-shaped fibroblast-like cells were expanded in two-dimensional culture systems prior to using them for the experiments [30-32]. Cells were then differentiated into chondrocytic phenotype as described [30-32] in Ham’s F-12 media containing 2 mmol/l glutamine, 2 mmol/l dexamethasone, ascorbic acid 2 phosphate (50 μg/ml), 1 mmol/l sodium pyruvate, proline (40 μg/ml), transforming growth factor β-3 (10 ng/ml) and insulin, transferrin and selenium (ITS+3; Sigma-Aldrich) at a final concentration of 6.25 μg/ml [30-32]. Chondrocytic phenotype was confirmed by microscopic evaluation, staining for glycosaminoglycan production (Alcian Blue and Safranin O staining) and collagen type II expression [30-32]. We herein refer to these differentiated cells as MPCs. For comparative purposes primary HACs from a donor free from inflammatory or erosive joint disease were obtained from PromoCell (Singapore), expanded in monolayer culture up to passage 5 in chondrocyte growth medium (#C-27101; Promocell), supplemented with Supplement Mix (#C-39635; Promocell) and, unless otherwise stated, embedded in alginate beads as described in [32,35,36]. After treatment of chondrocytes with cytokines or oxidants, calcium alginate was dissolved in 100 μmol/l sodium citrate [32, 35, 36] and cellular viability determined as described later.
Synthesis of GYY4137
GYY4137 (morpholin-4-ium 4 methoxyphenyl(morpholino) phosphinodithioate) morpholine is a novel slow releasing H2S donor compound recently shown to exert potent anti-inflammatory and vasodilatory properties in vitro [11,34] and in vivo [33,34]. The effects of H2S or GYY4137 on human cells, including chondrocytes and MPCs, are not known. Unlike commonly used sulfide salt ‘H2S donors’ such as Na2S or NaSH, the release of H2S from GYY4137 is not instantaneous but sustained allowing for a more accurate comparison to enzymatic H2S production from CSE and CBS [11]. GYY4137 was synthesized from 2,4-bis(4-methoxyphenyl)-2,4-dithioxo-,3,2,4-dithiadiphosphetane as described [11,34] and purity (≥90%) was assessed by 1H nuclear magnetic resonance. To ensure any observed effects of GYY4137 were due to H2S and not to the GYY4137 parent molecule, experiments were performed using ‘decomposed’ (spent) GYY4137 [33]. Release of H2S was routinely confirmed by amperometry using a 2-mm H2S-selective micro-electrode (ISO-H2S-2; World Precision Instruments; WPI) attached to an TBR4100 Free Radical Analyzer (WPI) as described [11,34].
Induction of CSE and CBS expression and measurement of cellular H2S synthesis
Cells were exposed to the inflammatory mediators TNF-α; IL-1β and IL-6 and added to cell culture media at a final concentration of 5 ng/ml for up to 18 hrs. Initial pilot studies showed that treatment conditions did not induce significant cytotoxicity when assessed by MTT or LDH release assays as described later. Cells were lysed in RIPA buffer (#R0278; Sigma-Aldrich), pH 8.0 and protein expression of CSE, CBS and β-actin determined by standard Western blotting as described [37,38]. For antibody detection, an enhanced chemiluminescence detection kit (GE Healthcare, Amersham, Buckinghamshire, UK) was used followed by analysis using a Kodak Image Analyser (IS440CF; NEN Life Science, Boston, MA, USA). Images were captured and analysed using Kodak digital science one-dimensional image analysis software. To investigate potential pathways involved in induced CSE/CBS expression and activity, commercially available inhibitors were used and added to cells for 1 hr prior to cytokine or LPS stimulation. Inhibitors of NF-κB (PPM-18 and BAY 11-7085; 10 μmol/l), p38 (SB203580 and PD169316; 10 μmol/l), JNK (SP600125 and AS601245; 10 μmol/l) and ERK1/2 (Ste-MEK1(13) and FR180204; 10 μmol/l) were used [30].
Cellular biosynthesis of H2S was assessed by zinc-trap spectrophotometry as described previously [8,11,34]. Briefly, after cytokine treatment, cells were lysed by freeze-thawing and protein concentration determined using a commercial kit (Bradford assay; BioRad Hercules, CA, USA). The assay mixture contained cell lysate, L-cysteine (10 mmol/l), pyridoxal 5′-phosphate (2 mmol/l), in phosphate buffer, pH 7.4 (50 mmol/l). To inhibit CSE or CBS activity, PAG or AOAA were added, respectively, at final concentrations of 1.0 mmol/l. Incubation was carried out in tightly sealed amber glass vials (Chromacol #08-CRV) with polytetrafluoroethylene seals (Chromacol #2-SC-ST2) in the dark. After incubation (37°C, 30 min.), zinc acetate (1% w/v) was injected to trap the generated H2S followed by trichloroacetic acid (10% w/v) to precipitate protein and thus stop the reaction. Subsequently, N,N-dimethyl-p-phenylenediamine sulfate (20 μmol/l) in 7.2M HCl was added followed by FeCl3 (30 μmol/l) in 1.2M HCl, and absorbance (670 nm) determined using a SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA, USA) [8,11,34]. The H2S concentration of each sample was calculated against a calibration curve of Na2S and results are expressed as nanomoles H2S formed per milligram soluble protein.
Assessment of cell death and determination of mitochondrial membrane potential (DCm)
There is extensive evidence for a role of oxidative and nitrosative stress in inflammatory joint diseases (reviewed in [39]). Pro-inflammatory oxidants such as reactive nitrogen species [26,40] and hydrogen peroxide (H2O2) [41] as well as the products of biomolecule oxidation such as lipid-derived aldehyde 4-HNE [42] are well known to induce apoptotic cell death in human chondrocytes. Similarly, there is increasing evidence for a predominant role of mitochondrial dysfunction in inflammatory and degenerative joint diseases [23,25,43].
Cellular viability was assessed using standard laboratory techniques: trypan blue dye exclusion, reduction of MTT and LDH release assays [44]. MTT data are expressed as percentage of untreated cells, and leakage of LDH into the culture media was measured at 340 nm using a commercially available kit (CytoTox96, Promega) after 18 hrs [26]. Mitochondrial membrane potential (DCm) in whole cells [45] was assessed using the potentiometric dye tetramethylrhodamine methyl ester (TMRM) and flow cytometry [26,31,38]. Cells were loaded for 30 min. at 37°C with TMRM at a final concentration of 50 nmol/l [26,31,38]. Levels of adenosine triphosphate (ATP) were determined with luciferase [44,46] and chemiluminescence measured using an LMax micro-plate reader (Molecular Devices) [44]. ATP levels were normalized to protein content using the Bradford assay (Bio-Rad). Cytosolic and mitochondrial levels of cytochrome c were assessed by ELISA (#APT200; Chemicon, Temecula, CA, USA) according to the manufacturer’s instructions and absorbance at 450 nm read using a SpectraMax190 microplate reader (Molecular Devices) [38].
Effects of H2S on pro-cell survival signalling
To investigate whether H2S could activate pro-cell survival signalling, cells were treated with GYY4137 or L-cysteine in the presence and absence of CSE and CBS inhibitors. Phosphorylation of Akt (Thr308) and ERK1/2 was assessed by ELISA (Cell Signaling Technology) according to the manufacturer’s instructions using 30 μg protein. To determine whether activation of PI3K/Akt and ERK was required for protection against oxidative stress-induced cell death, MPC and chondrocytes were treated for 1 hr with inhibitors of Akt {1L-6-Hydroxymethyl-chiro-inositol-2-[(R)-2-O-methyl-3-O-octadecylcarbonate]; 6-HO} or triciribine; 5 μmol/l), ERK1/2 (Ste-MEK113 or FR180204) or PI3K (LY29402 or wortmannin; 5 μmol/l and 10 μmol/l, respectively). After this time GYY4137 (100–500 μmol/l) was added for a further 1 hr followed by SIN-1, H2O2 and 4-HNE. Cell death was then assessed by MTT and LDH release assays as described earlier.
siRNA-mediated protein knockdown
For siRNA-mediated protein knockdown [31], cells were cultured in monolayers and transfected for 48 hrs in Ham’s F-12 media containing CSE siRNA or CBS siRNA (10 nmol/l; Abnova), Akt siRNA, p65NF-κB siRNA (80 nmol; Cell Signaling Technology) and a Silencer® siRNA Transfection Kit (Ambion) according to the manufacturers’ instructions. These treatment conditions were identified from preliminary optimization experiments. Non-coding siRNA transfections were also performed as negative controls using two commercially available negative control siRNAs (#4635 and #4611; termed herein as non-coding controls, NCC-1 and NCC-2, respectively; Ambion). Each of these comprised of a 19 base-pair non-targeting sequence with 3′ dT overhangs and had no significant similarity to any known human gene sequence (Ambion). These transfection conditions were chosen based on preliminary optimization experiments to ensure that siRNA-mediated protein knockdown, siRNA support reagents or transfection conditions did not significantly reduce cell viability (MTT and LDH assays) as described earlier.
Statistical analysis
Data are expressed as mean ± standard deviation of the mean (S.D.) of separate experiments (n ≥ 6) performed on separate days using freshly prepared reagents. Where significance testing was performed, ANOVA was used (*P < 0.05, **P < 0.01, ***P < 0.001) and concentration-dependent effects investigated with post-hoc Dunnett’s test using SPSS v15.0 software.
Results
Inducible expression and activity of CSE in MPC and articular chondrocytes
Figure 1A–C shows that under basal conditions chondrogenically differentiated MPCs expressed CBS and had detectable CSE. Densitometric analysis of CSE and CBS expression is shown in Figure 1D–E, respectively. Basal levels of H2S synthesis were significantly inhibited by AOAA (a CBS inhibitor) (Fig. 1F). In contrast, a small statistically insignificant decrease in H2S synthesis was observed after treatment with the CSE inhibitor PAG, suggesting that CBS was the predominant source of H2S under basal conditions. In contrast, treatment of MPCs with the pro-inflammatory cytokines TNF-α, IL-1β and IL-6 significantly increased expression (Fig. 1A–E) and activity (Fig. 1F) of CSE but not CBS. Incubation of chondrocytes in alginate culture with TNF-α, IL-1β and IL-6 under the same experimental conditions as MPCs also resulted in significant increases in expression (Fig. 2A and B) and activity (Fig. 2C) of CSE but not CBS. In MPCs and chondrocytes PAG, but not AOAA, significantly inhibited cytokine-induced CSE activity, suggesting that CSE is an inducible source of H2S in these cells.
Fig 1.
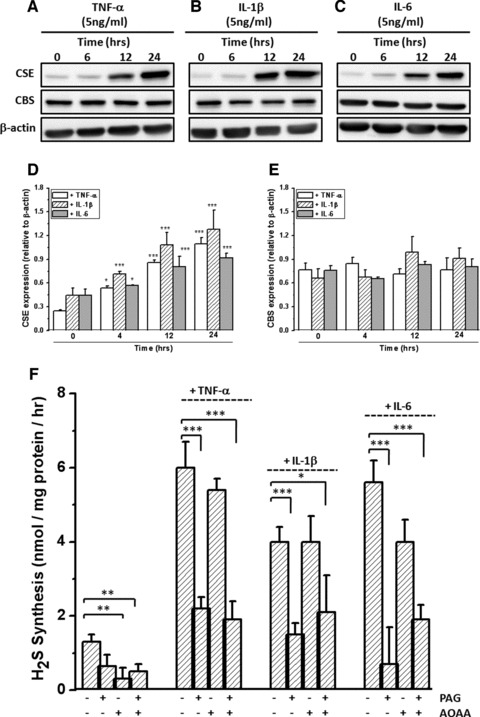
Inducible expression and activity of CSE but not CBS in human chondrogenically differentiated mesenchymal progenitor cells (MPCs). (A–C) CSE and CBS protein expression determined by Western blotting and (D, E) Western blot analysis by densitometry. (F) Cytokine induced H2S synthesis in MPC. Cells were treated with TNF-α, IL-1β or IL-6 at a final concentration of 5 ng/ml for 18 hrs. Cells were lysed with RIPA buffer and 20 μg protein analysed by Western blotting for CSE, CBS and β-actin expression. H2S synthesis was determined by zinc-trap specrophotometry in the presence and absence of D,L-propargylglycine (PAG; to inhibit CSE) or aminooxyacetate (AOAA; to inhibit CBS) added a final concentration of 1 mmol/l as described in Materials and Methods. Data are expressed as mean ± S.D. of six determinations. Figure 1(D) ***P < 0.001, cf. untreated cells; Figure 1(F) *P < 0.05, **P < 0.01, ***P < 0.001.
Fig 2.
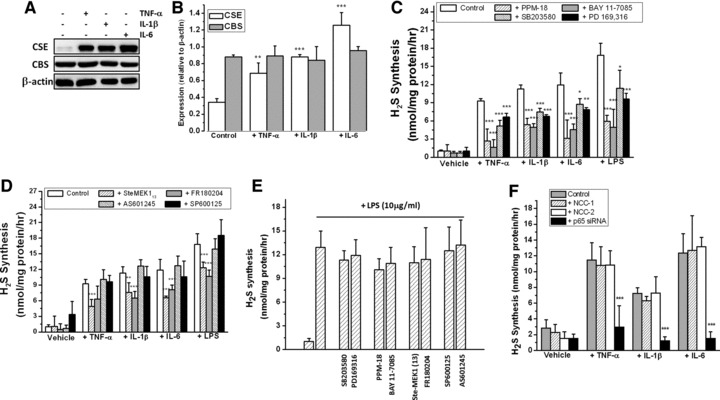
Inducible expression and activity of CSE is dependent on MAPK and NF-κB activation in human articular chondrocytes. (A, B) Inducible expression of CSE determined by Western blotting (A) and densitometric analysis of protein levels (B). (C, D) Cytokine and LPS-inducible synthesis of H2S in the presence of inhibitors of (C) NF-κB and p38 and (D) JNK and ERK. (E) Lack of inhibition of CSE activity by inhibitors of NF-κB, p38, ERK and JNK. (F) Effect of NF-κB p65siRNA treatment on inducible CSE activity. Chondrocytes were cultured in alginate and exposed to TNF-α, IL-1β, IL-6 (5 ng/ml) or LPS (10 μg/ml) for 18 hrs and CSE, CBS and β-actin expression determined by Western blotting. H2S synthesis was determined by zinc-trap spectrophotometry as described in Materials and Methods. Inhibitors of NF-κB (PPM-18 and BAY 117085), ERK (Ste-MEK113 and FR180204), p38 (SB203580 and PD169316) and JNK (SP600125 and AS601245) (C, D) were added at final concentrations of 10 μmol/l for one hr prior to cytokine or LPS treatment and H2S synthesis determined after 18 hrs. To determine the effects of these inhibitors on CSE activity (E), chondrocytes were treated with LPS for 18 hrs and inhibitors subsequently added (10 μmol/l) for one hr prior to H2S synthesis assay. For siRNA treatment, cells were transfected with siRNA for 48 hrs as described in Materials and Methods. Data are expressed as mean ± S.D. of six determinations. Figure 2(B) **P < 0.01, ***P < 0.001, cf. untreated cells; Figure 2(C), (D) and (F), *P < 0.05, **P < 0.01, ***P < 0.001, cf. cytokine or LPS-treated cells.
To identify potential pathways regulating cytokine-induced H2S synthesis, chondrocytes were incubated with TNF-α, IL-1β and IL-6 in the presence of various inhibitors of p38, ERK1/2, JNK and NF-κB; pathways strongly implicated in chronic inflammatory disease [47-50]. Pharmacological inhibition of p38, NF-κB (Fig. 2C) and ERK1/2 but not JNK (Fig. 2D) significantly inhibited cytokine- and LPS-induced H2S synthesis in chondrocytes. To investigate whether the compounds used to inhibit p38, ERK1/2, JNK and NF-κB could also inhibit the CSE activity, we incubated these compounds with cell lysates from LPS-treated chondrocytes. Figure 2E shows that the compounds used in our study did not significantly lower H2S synthesis, for example the compounds themselves did not inhibit CSE/CBS activity directly. To confirm the molecular requirement for NF-κB in cytokine-induced H2S synthesis, we performed additional experiments using NF-κB p65-siRNA. Figure 2F shows that in MPCs, p65 NF-κB siRNA treatment, but not non-coding controls (NCC-1 and NCC-2), significantly lowered cytokine-induced H2S synthesis, confirming the requirement for NF-κB activation in mediating cytokine-inducible synthesis of H2S.
Effects of endogenous and pharmacological H2S on oxidative/nitrosative stress-induced cell death
We initially investigated the effects of the commonly used sulfide salts, sodium sulfide (Na2S) and sodium hydrosulfide (NaSH), and the slow-release H2S donor compound GYY4137 on cell viability using MTT and LDH release assays. Na2S, and to a lesser extent NaSH, induced significant concentration-dependent cytotoxicity in MPCs (Fig. 3A and B) and in alginate cultured chondrocytes (Fig. 3C and D). These observations are consistent with the effects of Na2S and NaSH on other primary cell monolayers [51-54]. In contrast, no significant cytotoxicity was observed in MPCs and chondrocytes exposed to GYY4137 over the same concentration range (Fig. 3). This observation is also consistent with the findings of others [34,55]. Therefore future experiments using Na2S and NaSH were precluded due to overt cytotoxicity. Preliminary control experiments showed that in the cell culture media in the absence of cells, Na2S, NaSH or GYY4137 treatment alone (up to 500 μmol/l) did not significantly reduce MTT to formazan or inhibit LDH activity (data not shown), for example, the H2S donors alone did not interfere with the viability assays. Further control experiments showed that the addition of ‘decomposed’ GYY4137 did not significantly reduce cell viability (MTT assay; mean ± S.D. 98.5 ± 3.2%; cf. control, 102.1 ± 3.1%) or induce significant LDH release (mean ± S.D. 2.3 ± 1.2%; cf. control, 3.4 ± 2.1%).
Fig 3.
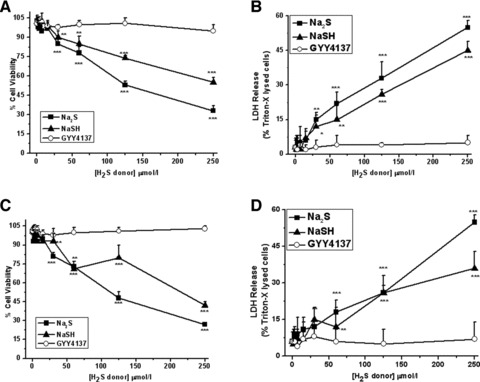
Induction of cell death by rapid release H2S donors Na2S and NaSH. MPC (A, B) and chondrocytes cultured in alginate (C, D) were exposed to Na2S, NaSH or the slow release H2S donor GYY4137 at the concentrations stated for 18 hrs. After this time cell viability was assessed by MTT (A, C) and LDH release (B, D) assays as described in Materials and Methods. MTT data are expressed as percentage of untreated cells. LDH data are expressed as percentage of Triton-X released LDH. Data are expressed as mean ± S.D. of six determinations. *P < 0.05, **P < 0.01, ***P < 0.001, cf. vehicle (PBS)-treated cells.
Figure 4 shows that, in agreement with previous studies using chondrocytes, exposure of MPC cells and chondrocytes to the peroxynitrite donor SIN-1 (100 and 500 μmol/l), H2O2 (100 and 500 μmol/l) and the 4-HNE (20 and 50 μmol/l) induced significant cell death as determined by MTT (Fig. 4A) and LDH release (Fig. 4B) assays. These concentrations and treatment conditions were identified from the literature and from preliminary investigations by us. These treatment conditions were then employed in subsequent experiments to determine whether endogenous and/or pharmacological H2S could be cytoprotective.
Fig 4.
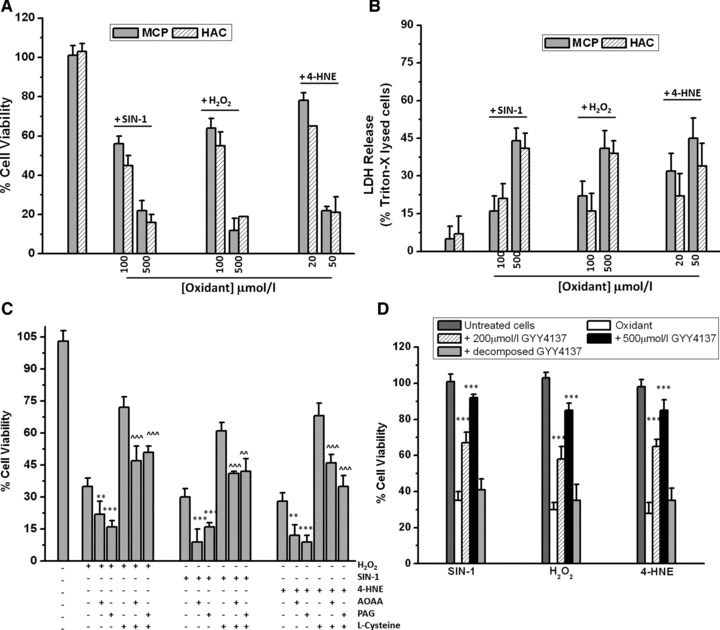
Inhibition of oxidative stress-induced cell death by endogenous and pharmacological H2S. MPCs and chondrocytes were exposed to the peroxynitrite donor SIN-1 and H2O2 (100 μmol/l and 500 μmol/l) and the lipid peroxidation product 4-HNE (20 μmol/l and 50 μmol/l) and cell death determined by MTT (A) and LDH release (B) assays. (C) Effect of endogenously generated H2S on oxidative stress-induced cell death. MPCs were exposed to H2O2 (200 μmol/l), SIN-1 (500 μmol/l) and 4-HNE (25 μmol/l) for 18 hrs in the presence and absence of CSE and CBS inhibitors PAG and AOAA (1 mmol/l), respectively. L-cysteine (1 mmol/l) was added as a substrate for CSE and CBS. Cell death was determined by MTT assay. (D) Effect of GYY4137 and ‘decomposed’ GYY4137 on oxidative stress-induced cell death. MPCs were exposed to oxidative insult in the presence and absence of the slow release H2S donor GYY4137 (200 μmol/l and 500 μmol/l) or decomposed GYY4137 and cell death determined by MTT assay as described in Materials and Methods. Data are expressed as mean ± S.D. of six determinations. *P < 0.05, **P < 0.01, ***P < 0.001, cf. oxidant treatment alone.
Oxidative stress-induced cell death in MPCs (Fig. 4C and D) was significantly inhibited by treatment of cells with L-cysteine. However, this protective effect was significantly removed in the presence of the CSE inhibitor PAG and by the CBS inhibitor AOAA. Furthermore, oxidative stress treatment in the presence of PAG and AOAA alone significantly increased cell death in H2O2-, SIN-1- and 4-HNE-treated cells collectively, suggesting that endogenous H2S could protect against cell injury. Because CSE- and CBS-generated H2S occur in a slow and steady manner [11,56,57], we next investigated whether the slow release H2S donor compound GYY4137 could prevent oxidative stress-induced cell death. Figure 4D shows GYY4137 preserved cellular viability against all three oxidative insults in a concentration-dependent manner. This effect was not observed with ‘decomposed’ GYY4137 added as a control.
To confirm the molecular requirement for CSE- and CBS-derived H2S in mediating cytoprotection, we next examined the effects of siRNA-mediated CSE and CBS protein knockdown in chondrocytes. In CSE- and CBS-siRNA-treated cells the cytoprotective effect of L-cysteine against oxidative injury was significantly reduced but preserved in cells transfected with two non-coding siRNA (Fig. 5A and B). Small but statistically significant increases in cell death induced by SIN-1 (Fig. 5A), H2O2 (Fig. 5B) and 4-HNE (Fig. 5C) were observed with CSE and CBS-siRNA treatment, in agreement with the effects of PAG and AOAA in MPCs (Fig. 4C and D). Pharmacological H2S, supplied via GYY4137, further significantly inhibited oxidative stress-induced cell death when added to CSE-treated and CBS-siRNA-treated cells, that is, the lack of enzymatically generated H2S could be overcome by pharmacological H2S.
Fig 5.
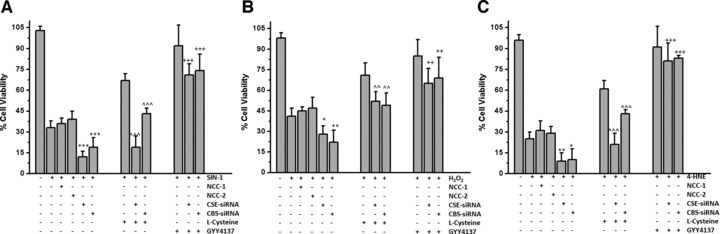
Effect of CSE and CBS protein knockdown on oxidative stress induced cell death. MPCs were treated with CSE-siRNA, CBS-siRNA or two non-coding controls (NCC-1 and NCC-2) as described in Materials and Methods. L-cysteine (1 mmol/l) was added as a substrate for CSE and CBS. GYY4137 (500 μmol/l) was added as a source of pharmacological (exogenous) H2S. Cell death was assessed by MTT assay after treatment with (A) SIN-1 (500 μmol/l), (B) H2O2 (200 μmol/l) and (C) 4-HNE (35 μmol/l). Data are expressed as mean ± S.D. of six determinations. ^^^P < 0.001, cf. L-cysteine only treatment, ++P < 0.01, +++P < 0.001, cf. siRNA + oxidant treatment.
Effects of pharmacological H2S on pro-cell survival signalling
To determine the potential pathways mediating H2S-mediated cytoprotection we next investigated the effects of GYY4137 on Akt and ERK1/2 activation. Figure 6A shows that GYY4137, but not the ‘decomposed’ donor, induced a time- and concentration-dependent increase in the phosphorylation (Thr308) of the pro-cell survival protein Akt in chondrocytes. Figure 6B shows that L-cysteine treatment of MPCs for 6 hrs significantly increased the phosphorylation of Akt whereas PAG and AOAA (added as inhibitors of CSE and CBS, respectively) significantly lowered L-cysteine-induced Akt phosphorylation. Figure 6C shows that incubation of cells in the presence of GYY4137 (0–500 μmol/l) and L-cysteine (1 mmol/l) for 6 hrs also significantly increased ERK1/2 phosphorylation.
Fig 6.
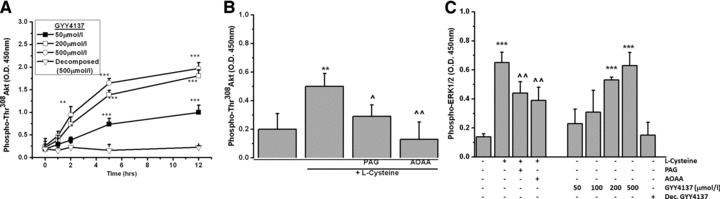
Activation of ERK/AKT signalling by GYY4137. Chondrocytes were cultured in alginate beads and treated (A) GYY4137 (50–500 μmol/l) and (B) L-cysteine (1 mmol/l) for the time stated in the presence and absence of PAG and AOAA and phosphorylation of Akt determined by commercial ELISA. (C) Phosphorylation of ERK induced by L-cysteine (1 mmol/l), GYY4137 (50–500 μmol/l) or 'decomposed' GYY4137 (500 μmol/l) added to chondrocytes for six hrs. Data are expressed as mean ± S.D. of six determinations. *P < 0.05, **P < 0.01, ***P < 0.001, cf. vehicle (PBS) treated cells; ^P < 0.05, ^^P < 0.01, cf. L-cysteine only treated cells.
To determine whether Atk was required for H2S-mediated cytoprotection, MPCs and chondrocytes were treated with pharmacological inhibitors of Akt signalling in the presence of GYY4137 and cell death induced by oxidant stress determined as described earlier. Figure 7 shows that pharmacological inhibition of Akt and upstream PI-3 kinase and to a lesser extent ERK1/2, significantly reduced the ability of GYY4137 (200 μM) to prevent cell death in MPCs induced by SIN-1 (200 μM; Fig. 7A), H2O2 (200 μM; Fig. 7B), and 4-HNE (25 μM; Fig. 7C) suggesting activation of Akt and ERK1/2 signalling pathways was required for H2S-mediated cytoprotection. Furthermore, siRNA-mediated Akt protein knockdown in chondrocytes showed that activation of Akt was required to confer cytoprotection by GYY4137 against oxidative stress–mediated cell death (Fig. 7D).
Fig 7.
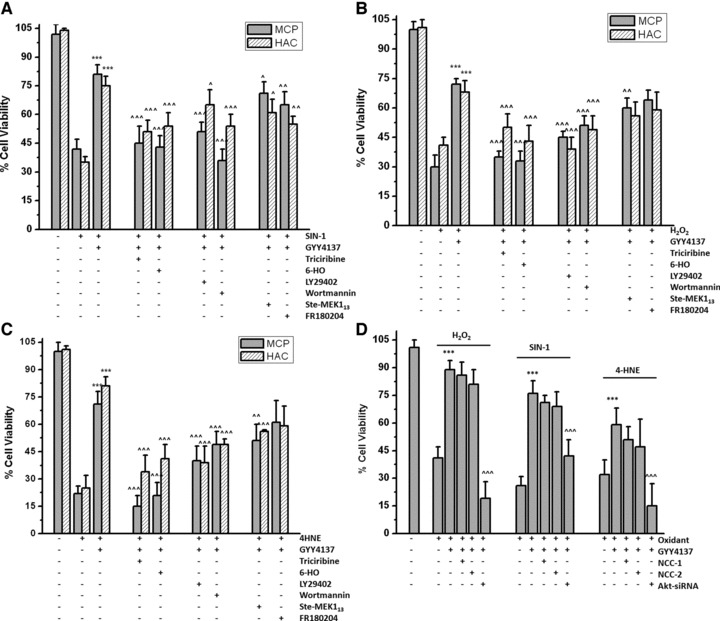
Role of ERK, Akt and PI3-kinase in mediating GYY4137-induced cytoprotection. (A–C) MPCs and chondrocytes were incubated with inhibitors of ERK (Ste-MEK113 and FR180204), Akt (triciribine and 6-HO) and PI3-kinase (LY29402 and wortmannin) at a final concentration of 5 μmol/l and GYY4137 (500 μmol/l) added. Cell death induced by (A) SIN-1 (500 μmol/l), (B) H2O2 (200 μmol/l) and (C) 4-HNE (25 μmol/l) was assessed after 18 hrs by MTT assay as described in Materials and Methods. (D) Effects of Akt siRNA-treatment on GYY4137-induced cytoprotection. Chondrocytes were transfected with Akt siRNA (80 nmol) or non-coding controls (NCC-1 and NCC-2; 80 nmol) for 48 hrs. Cell death induced by SIN-1 (500 μmol/), H2O2 (200 μmol/l) and 4-HNE (25 μmol/l) in the presence or absence of GYY4137 (500 μmol/l) after 18 hrs was determined by MTT assay. Data are expressed as mean ± S.D. of six determinations. ***P < 0.001, cf. oxidant-treated cells. ^^P < 0.01, ^^^P < 0.001, cf. GYY4137-treated cells.
Effects of H2S on oxidative stress-mediated mitochondrial dysfunction
There is increasing evidence for perturbed mitochondrial function in inflammatory and degenerative joint disease [29,43,58] and that mitochondrial dysfunction may potentiate inflammation [29]. Because SIN-1 [26], 4-HNE [42] and H2O2 [59] have previously been shown to collapse mitochondrial DCm in human chondrocytes, we next investigated whether pharmacological H2S could protect mitochondria in situ from oxidative injury by examining mitochondrial DCm, mitochondrial ATP and cytoplasmic cytochrome c. In MPCs, inhibition of CSE (with PAG) or CBS (with AOAA) significantly increased the loss of mitochondrial DCm induced by SIN-1 (Fig. 8A), H2O2 and 4-HNE (Fig. 8B), whereas GYY4137 added at a final concentration of 200 μmol/l or 500 μmol/l significantly inhibited mitochondrial toxicity. In agreement with these observations, treatment of chondrocytes with CSE-siRNA or CBS-siRNA but not non-coding controls (see figure legend) similarly increased oxidative stress-induced collapse of mitochondrial DCm. In these additional experiments, GYY4137 (500 μmol/l) also inhibited SIN-1-, H2O2- and 4-HNE-induced mitochondrial toxicity in CSE-siRNA and CBS-siRNA-treated chondrocytes (Fig. 8C).
Fig 8.
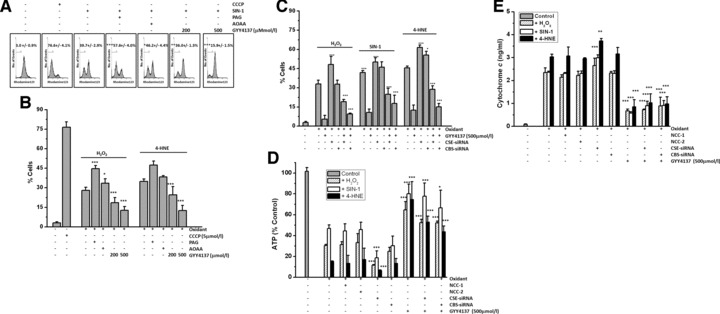
Effect of H2S on oxidative stress induced mitochondrial dysfunction. (A–C) Mitochondrial membrane potential (DCm) and assessment of (D) ATP and (E) cytochrome c levels. MPCs were exposed to (A) SIN-1 (500 μmol/l), (B) H2O2 (200 μmol/l) and 4-HNE (25 μmol/l) for 18 hrs and mitochondrial membrane potential (DCm) determined by flow cytometry using tetramethylrhodamine methyl ester (TMRM; 50 nmol/l) as described in Materials and Methods. PAG (1 mmol/l) and AOAA (1 mmol/l) were used to inhibit endogenous CSE and CBS activity, respectively. Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) was added as a positive control. (C) Effect of CSE and CBS-siRNA treatment on oxidant induced loss of mitochondrial DCm in MPCs. (D, E) Effect of CSE and CBS-siRNA treatment on oxidant induced loss of ATP (D) and cytoplasmic levels of cytochrome c (E). Cells were treated with CSE or CBS siRNA or non-coding controls (NCC-1 and NCC-2) as described in Materials and Methods. Data are expressed as mean ± S.D. of six determinations. GYY4137 *P < 0.05, **P < 0.01, ***P < 0.001, cf. oxidant-treated cells.
Because Figure 8A–C suggested H2S could preserve mitochondrial function, we next examined whether GYY137 could prevent oxidant-induced ATP depletion. Under basal conditions, MPCs contained 135.1 ± 2.4 nmol ATP/mg protein. Treatment with PAG (1 mM) or (AOAA) alone did not significantly decrease ATP levels (131.7 ± 7.5 nmol/mg protein and 122.8 ± 7.6 nmol/mg protein, respectively). Similarly treatment of MPC with GYY4137 or ‘decomposed’ GYY4137 alone at final concentrations, 500 μmol/l, did not significantly reduce cellular ATP content (138.7 ± 4.7 nmol/mg protein and 127.6 ± 10.6 nmol/mg protein, respectively). Figure 8D shows that treatment of MPCs with H2O2, SIN-1 or 4-HNE significantly reduced ATP levels, and ATP loss was significantly increased further with siRNA-mediated CSE and CBS protein knockdown but not by non-coding siRNA controls NCC-1 and NCC-2 (Fig. 8D). In contrast, GYY4137 (200 and 500 μmol/l) but not ‘decomposed’ GYY4137 (500 μmol/l; data not shown) significantly inhibited oxidant stress-induced cellular ATP depletion. Furthermore oxidative stress-induced cytoplasmic accumulation of cytochrome c (Fig. 8E), indicative of mitochondrial dysfunction, was significantly inhibited by GYY4137 (500 μmol/l) but attenuated by CSE siRNA treatment.
Discussion
Hydrogen sulfide is emerging as an important mediator in acute and chronic inflammation [3] as well as vascular pathologies such as hypertension, obesity, diabetes and kidney disease (reviewed in [60]). However, the role of H2S in human inflammatory joint disease, whether human joint cells synthesize H2S and how joint cells respond to H2S has not been investigated in detail. Previous indirect evidence has suggested perturbed H2S synthesis in RA. For example, serum levels of the CSE/CBS substrates L-homocysteine, L-cysteine and L-cystathionine are elevated [61,62], and erythrocyte levels of S-thiolmethyltransferase (TMT), an enzyme potentially capable of ‘detoxifying’ H2S in vivo are decreased in RA plasma compared to healthy controls [63]. More recently H2S has been shown to be present in knee-joint SF aspirates from RA and OA patients [9] and other arthritides [3]. Consistent with a role of H2S in mediating inflammatory signalling, significantly higher H2S levels were recently observed in RA compared to OA SF and levels significantly correlated with inflammatory scores, including neutrophil and total white cell count [9], but the precise source of H2S in SF has not been determined. Furthermore, in an in vivo murine model of acute arthritis induced by kaolin/carrageenan [12], 30–50 μM of Na2S inhibited leucocyte adhesion in post-capillary venules in acutely inflamed mouse knees. In sharp contrast to the effects of H2S on the systemic vasculature, Na2S induced vessel constriction rather than vasodilatation. Although the precise reasons for this observation are unclear, one possibility is that H2S induces vessel constriction to counteract local pro-inflammatory vasodilatory mediators such as PGE2, •NO and histamine. Activated rodent neutrophils, macrophages and vascular endothelial cells have been shown to synthesize H2S in response to pro-inflammatory stimuli and if equivalent human cells are present in the inflamed RA joint they could also generate H2S. This study also suggests that resident joint cells, when exposed to a strongly pro-inflammatory milieu, would also synthesize H2S.
We were unable to compare the effects of Na2S and NaSH with the previous studies on rodent cell lines or immortalized chondrocyte monolayers using these compounds as Na2S and NaSH induced significant cytotoxicity in MPCs and chondrocytes (Fig. 3). These findings are in agreement with studies on other primary cells such as acinar cells, pancreatic β-cells and vascular smooth muscle cells and could highlight important differences in responsiveness to H2S between primary cells and cell lines. Furthermore, NaSH (and Na2S) may not be ideal compounds for studying the physiology (or pathophysiology) of H2S because these sulfide salts generate an instantaneous and very short lived (<5 sec.) bolus of concentrated H2S (as well as Na+), whereas enzymatic CSE and CBS-derived H2S synthesis is considerably slower over a much longer time period [56,57,64]. The manner in which cells and tissues are exposed to H2S may influence the cellular or tissue response [11]. To this end, we used GYY4137 to generate ‘pharmacological’ H2S and L-cysteine to generate ‘endogenous’ H2S to more accurately study the role of physiologically generated H2S.
Our study also suggests that the control of H2S synthesis through the induced activity of CSE proceeds through well-defined pathways strongly associated with the regulation of inflammatory signalling, NF-κB, p38 and ERK1/2. The pathways controlling H2S synthesis have largely been overlooked and the published studies to date have only focused on the effects of added H2S (invariably as Na2S or NaSH) on cells and tissues. The H2S donor GYY4137 has recently been shown to generate H2S in a manner comparable to enzymatically synthesized H2S from CSE/CBS and does not induce significant cytotoxicity even at mmol/l concentrations [11,34]. GYY4137 has also been shown to inhibit the synthesis of pro-inflammatory mediators (e.g. TNF-α, IL-6, IL-1β, PGE2 and •NO) in LPS-stimulated murine macrophages [11] and IL-8 secretion by primary human pulmonary airway smooth muscle cells in vitro [65]. Furthermore, in a rat model of endotoxemia GYY4137 decreased the plasma levels of these inflammatory mediators as well as inhibiting their synthesis in isolated whole blood, limiting endotoxin-induced tissue damage, oedema and inflammation [33]. It is therefore possible that the up-regulation of H2S synthesis, previously observed in RA SF, may represent a novel endogenous mechanism for limiting inflammation in the joint.
There is strong evidence for a contribution of oxidative stress in mediating the various aspects of inflammatory and degenerative joint disease, for example, cartilage destruction, mitochondrial dysfunction, cell death of resident joint cells and pro-inflammatory signalling (reviewed in [39]). This study suggests an additional role for H2S in human joint cells: cytoprotection against oxidative injury. Is it therefore possible that elevated H2S synthesis in the inflamed joint counteracts increased oxidative/nitrosative stress? In MPCs and chondrocytes oxidative stress–induced mitochondrial dysfunction and cell death was significantly inhibited when cells were exposed to GYY4137 within the concentration range of 100–500 μmol/l (but not its decomposed control) or L-cysteine. The ability of L-cysteine to protect against cell death was inhibited by the treatment of cells with CSE and CBS inhibitors (PAG and AOAA, respectively) and by siRNA-mediated protein knockdown. PAG and AOAA have been used extensively to reduce cellular and tissue synthesis of H2S in a variety of cell and whole animal model systems, but these compounds target the PLP binding site of CSE and CBS and may also inhibit other PLP-dependent enzymes. There are currently no completely selective inhibitors of CSE- and CBS-derived H2S synthesis (reviewed in [3]) necessitating the requirement for complementary and confirmatory experiments using CSE and CBS siRNA.
Recently, in simple in vitro and cell free assays, Na2S and NaSH have been shown to ‘scavenge’ pro-inflammatory oxidants such as •NO [66], ONOO− [67], OCl− [68,69] H2O2 [70] and superoxide (O2•−) [71] as well as ‘destroy’ lipid peroxides [72,73] and inhibit atherogenic modification of low density lipoprotein [74]. However, more recent in vitro kinetic studies have suggested that the rate constants for the reaction of NaSH-generated H2S with the above oxidants are not sufficiently high enough for oxidant ‘scavenging’ to solely account for the cytoprotective effects of H2S on cells, suggesting additional cellular mechanisms must be involved [75]. In support of this, NaSH and Na2S have been shown to up-regulate glutathione synthesis via γ-glutamylcysteine synthetase, to increase cysteine uptake in neuronal cells [76,77] and to activate Nrf-2 signalling, thereby conferring cytoprotection in cardiac cells and tissues [78,79] and preserving mitochondrial integrity. However, the mechanisms for these observations are unclear. In this study, slow release of H2S by GYY4137 or stimulation of endogenous H2S production with L-cysteine induced significant time- and concentration-dependent phosphorylation of Akt and ERK1/2 and prevented the loss of mitochondrial DCm and cell death induced by oxidant species that have been well characterized to have detrimental effects on the extracellular matrix and cartilage producing cells. Furthermore, inhibitors of PI3K, Akt and ERK significantly reduced the protective effect of GYY437. The protective effects of L-cysteine were significantly reduced in the presence of PAG and AOAA and nearly absent in CSE- or CBS-siRNA-treated cells. This suggests that a potential role of endogenous H2S is to activate PI3K-Akt/ERK cytoprotective pathways and preserve mitochondrial and cellular integrity and function. Recently H2S, albeit derived from NaSH, was shown to decrease IL-6 and IL-8 synthesis in the immortalized human C29/I2 chondrocyte cell line [80] grown in monolayer culture suggesting the possibility that H2S could also regulate the inflammatory response in cartilage cells, in agreement with previously published studies in other cell types.
In summary, our study shows for the first time that (1) chondrogenically differentiated MPCs and HACs synthesize H2S via CSE and CBS, (2) pro-inflammatory cytokines induce CSE expression and activity via p38-ERK-NF-κB-dependent pathways, (3) endogenous and pharmacological H2S inhibit oxidant-induced mitochondrial dysfunction and cell death through pathways involving Akt/PI3K-dependent signalling and (4) induced H2S synthesis may represent a novel endogenous mechanism to limit cartilage destruction, cell death and inflammation in the joint. Therefore, the role of H2S in chronic joint inflammation deserves further attention. Controlling H2S synthesis may represent a novel opportunity for therapeutic intervention in human chronic inflammatory diseases such as rheumatoid arthritis.
Acknowledgments
This work was supported by the European Union Economic Development Fund (POC04/10–11) and the Devon Arthritis Appeal Research Trust (DAART-RR100196).
Conflict of interest
The authors have no conflict of interest.
References
- 1.Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–87. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 2.Whiteman M, Moore PK. Hydrogen sulfide and the vasculature: a novel vasculoprotective entity and regulator of nitric oxide bioavailability. J Cell Mol Med. 2009;13:488–507. doi: 10.1111/j.1582-4934.2009.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whiteman M, Winyard PG. Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev Clin Pharmacol. 2011;4:13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- 4.Zhi L, Ang AD, Zhang H, et al. Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappaB pathway. J Leukoc Biol. 2007;81:1322–32. doi: 10.1189/jlb.1006599. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Moochhala SM, Bhatia M. Endogenous hydrogen sulfide regulates inflammatory response by activating the ERK pathway in polymicrobial sepsis. J Immunol. 2008;181:4320–31. doi: 10.4049/jimmunol.181.6.4320. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Zhi L, Moore PK, et al. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1193–201. doi: 10.1152/ajplung.00489.2005. [DOI] [PubMed] [Google Scholar]
- 7.Bhatia M, Zhi L, Zhang H, et al. Role of substance P in hydrogen sulfide-induced pulmonary inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2006;291:L896–904. doi: 10.1152/ajplung.00053.2006. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Bhatia M, Zhu YZ, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19:1196–8. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- 9.Whiteman M, Haigh R, Tarr JM, et al. Detection of hydrogen sulfide in plasma and knee-joint synovial fluid from rheumatoid arthritis patients: relation to clinical and laboratory measures of inflammation. Ann N Y Acad Sci. 2010;1203:146–50. doi: 10.1111/j.1749-6632.2010.05556.x. [DOI] [PubMed] [Google Scholar]
- 10.Kloesch B, Liszt M, Broell J. H2S transiently blocks IL-6 expression in rheumatoid arthritic fibroblast-like synoviocytes and deactivates p44/42 mitogen-activated protein kinase. Cell Biol Int. 2010;34:477–84. doi: 10.1042/CBI20090436. [DOI] [PubMed] [Google Scholar]
- 11.Whiteman M, Li L, Rose P, et al. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal. 2010;12:1147–54. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andruski B, McCafferty DM, Ignacy T, et al. Leukocyte trafficking and pain behavioral responses to a hydrogen sulfide donor in acute monoarthritis. Am J Physiol Regul Integr Comp Physiol. 2008;295:R814–20. doi: 10.1152/ajpregu.90524.2008. [DOI] [PubMed] [Google Scholar]
- 13.Caplan AI, Elyaderani M, Mochizuki Y, et al. Principles of cartilage repair and regeneration. Clin Orthop Relat Res. 1997. pp. 254–69. [PubMed]
- 14.Yoo JU, Barthel TS, Nishimura K, et al. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J Bone Joint Surg Am. 1998;80:1745–57. doi: 10.2106/00004623-199812000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Alsalameh S, Amin R, Gemba T, et al. Identification of mesenchymal progenitor cells in normal and osteoarthritic human articular cartilage. Arthritis Rheum. 2004;50:1522–32. doi: 10.1002/art.20269. [DOI] [PubMed] [Google Scholar]
- 16.Marinova-Mutafchieva L, Taylor P, Funa K, et al. Mesenchymal cells expressing bone morphogenetic protein receptors are present in the rheumatoid arthritis joint. Arthritis Rheum. 2000;43:2046–55. doi: 10.1002/1529-0131(200009)43:9<2046::AID-ANR16>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Blanco FJ, Guitian R, Vazquez-Martul E, et al. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998;41:284–9. doi: 10.1002/1529-0131(199802)41:2<284::AID-ART12>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 18.Thomas CM, Whittles CE, Fuller CJ, et al. Variations in chondrocyte apoptosis may explain the increased prevalence of osteoarthritis in some joints. Rheumatol Int. 2010. doi: 10.1007/s00296-010-1471-9. [DOI] [PubMed]
- 19.Kim HA, Song YW. Apoptotic chondrocyte death in rheumatoid arthritis. Arthritis Rheum. 1999;42:1528–37. doi: 10.1002/1529-0131(199907)42:7<1528::AID-ANR28>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Saito S, Murakoshi K, Kotake S, et al. Granzyme B induces apoptosis of chondrocytes with natural killer cell-like cytotoxicity in rheumatoid arthritis. J Rheumatol. 2008;35:1932–43. [PubMed] [Google Scholar]
- 21.Polzer K, Schett G, Zwerina J. The lonely death: chondrocyte apoptosis in TNF-induced arthritis. Autoimmunity. 2007;40:333–6. doi: 10.1080/08916930701356721. [DOI] [PubMed] [Google Scholar]
- 22.Liu H, Pope RM. The role of apoptosis in rheumatoid arthritis. Curr Opin Pharmacol. 2003;3:317–22. doi: 10.1016/s1471-4892(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 23.Moodley D, Mody G, Patel N, et al. Mitochondrial depolarisation and oxidative stress in rheumatoid arthritis patients. Clin Biochem. 2010;41:1396–401. doi: 10.1016/j.clinbiochem.2008.08.072. [DOI] [PubMed] [Google Scholar]
- 24.Miesel R, Murphy MP, Kroger H. Enhanced mitochondrial radical production in patients which rheumatoid arthritis correlates with elevated levels of tumour necrosis factor alpha in plasma. Free Radic Res. 1996;25:161–9. doi: 10.3109/10715769609149921. [DOI] [PubMed] [Google Scholar]
- 25.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7:161–9. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 26.Whiteman M, Armstrong JS, Cheung NS, et al. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004;18:1395–7. doi: 10.1096/fj.03-1096fje. [DOI] [PubMed] [Google Scholar]
- 27.Thomas CM, Fuller CJ, Whittles CE, et al. Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthritis Cartilage. 2007;15:27–34. doi: 10.1016/j.joca.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 28.Yatsugi N, Tsukazaki T, Osaki M, et al. Apoptosis of articular chondrocytes in rheumatoid arthritis and osteoarthritis: correlation of apoptosis with degree of cartilage destruction and expression of apoptosis-related proteins of p53 and c-myc. J Orthop Sci. 2000;5:150–6. doi: 10.1007/s007760050142. [DOI] [PubMed] [Google Scholar]
- 29.Cillero-Pastor B, Caramés B, Lires-Deán M, et al. Mitochondrial dysfunction activates cyclooxygenase 2 expression in cultured normal human chondrocytes. Arthritis Rheum. 2008;58:2409–19. doi: 10.1002/art.23644. [DOI] [PubMed] [Google Scholar]
- 30.Whiteman M, Spencer JP, Zhu YZ, et al. Peroxynitrite-modified collagen-II induces p38/ERK and NF-kappaB-dependent synthesis of prostaglandin E2 and nitric oxide in chondrogenically differentiated mesenchymal progenitor cells. Osteoarthritis Cartilage. 2006;14:460–70. doi: 10.1016/j.joca.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Whiteman M, Chu SH, Siau JL, et al. The pro-inflammatory oxidant hypochlorous acid induces Bax-dependent mitochondrial permeabilisation and cell death through AIF-/EndoG-dependent pathways. Cell Signal. 2007;19:705–14. doi: 10.1016/j.cellsig.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 32.Schantz J-T, Ng KWA. Manual for primary human cell culture. Singapore: World Scientific; 2004. p. 23–46.
- 33.Li L, Salto-Tellez M, Tan CH, et al. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic Biol Med. 2009;47:103–13. doi: 10.1016/j.freeradbiomed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Li L, Whiteman M, Guan YY, et al. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation. 2008;117:2351–60. doi: 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez C, Mateus MM, Defresne MP, et al. Metabolism of human articular chondrocytes cultured in alginate beads. Longterm effects of interleukin 1beta and nonsteroidal antiinflammatory drugs. J Rheumatol. 2002;29:772–82. [PubMed] [Google Scholar]
- 36.Legendre F, Bauge C, Roche R, et al. Chondroitin sulfate modulation of matrix and inflammatory gene expression in IL-1beta-stimulated chondrocytes: study in hypoxic alginate bead cultures. Osteoarthritis Cartilage. 2008;16:105–14. doi: 10.1016/j.joca.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 37.Armstrong JS, Whiteman M, Rose P, et al. The coenzyme Q10 analog decylubiquinone inhibits the redox-activated mitochondrial permeability transition: role of mitcohondrial [correction mitochondrial] complex III. J Biol Chem. 2003;278:49079–84. doi: 10.1074/jbc.M307841200. [DOI] [PubMed] [Google Scholar]
- 38.Whiteman M, Rose P, Siau JL, et al. Hypochlorous acid-mediated mitochondrial dysfunction and apoptosis in human hepatoma HepG2 and human foetal liver cells: role of mitochondrial permeability transition. Free Radic Biol Med. 2005;38:1571–84. doi: 10.1016/j.freeradbiomed.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 39.Pattison DJ, Winyard PG. Dietary antioxidants in inflammatory arthritis: do they have any role in etiology or therapy. Nat Clin Pract Rheumatol. 2008;4:590–6. doi: 10.1038/ncprheum0920. [DOI] [PubMed] [Google Scholar]
- 40.Blanco FJ, Ochs RL, Schwarz H, et al. Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995;146:75–85. [PMC free article] [PubMed] [Google Scholar]
- 41.Asada S, Fukuda K, Nishisaka F, et al. Hydrogen peroxide induces apoptosis of chondrocytes: involvement of calcium ion and extracellular signal-regulated protein kinase. Inflamm Res. 2001;50:19–23. doi: 10.1007/s000110050719. [DOI] [PubMed] [Google Scholar]
- 42.Vaillancourt F, Fahmi H, Shi Q, et al. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: the protective role of glutathione-S-transferase. Arthritis Res Ther. 2008;10:R107. doi: 10.1186/ar2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanco FJ, Lopez-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. 2004;4:715–28. doi: 10.1016/j.mito.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 44.Whiteman M, Rose P, Siau JL, et al. Nitrite-mediated protection against hypochlorous acid-induced chondrocyte toxicity: a novel cytoprotective role of nitric oxide in the inflamed joint? Arthritis Rheum. 2003;48:3140–50. doi: 10.1002/art.11284. [DOI] [PubMed] [Google Scholar]
- 45.Maneiro E, Lopez-Armada MJ, de Andres MC, et al. Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. Ann Rheum Dis. 2005;64:388–95. doi: 10.1136/ard.2004.022152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whiteman M, Hooper DC, Scott GS, et al. Inhibition of hypochlorous acid-induced cellular toxicity by nitrite. Proc Natl Acad Sci U S A. 2002;99:12061–6. doi: 10.1073/pnas.152462399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bamborough P, Morse MA, Ray KP. Targeting IKKbeta for the treatment of rheumatoid arthritis. Drug News Perspect. 2010;23:483–90. doi: 10.1358/dnp.2010.23.8.1447844. [DOI] [PubMed] [Google Scholar]
- 48.Roman-Blas JA, Jimenez SA. Targeting NF-kappaB: a promising molecular therapy in inflammatory arthritis. Int Rev Immunol. 2008;27:351–74. doi: 10.1080/08830180802295740. [DOI] [PubMed] [Google Scholar]
- 49.Coulthard LR, White DE, Jones DL, et al. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15:369–79. doi: 10.1016/j.molmed.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47:409–14. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- 51.Yang G, Yang W, Wu L, et al. H2S, endoplasmic reticulum stress, and apoptosis of insulin-secreting beta cells. J Biol Chem. 2007;282:16567–76. doi: 10.1074/jbc.M700605200. [DOI] [PubMed] [Google Scholar]
- 52.Adhikari S, Bhatia M. H2S-induced pancreatic acinar cell apoptosis is mediated via JNK and p38 MAP kinase. J Cell Mol Med. 2008;12:1374–83. doi: 10.1111/j.1582-4934.2008.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li W, Jin HF, Liu D, et al. Hydrogen sulfide induces apoptosis of pulmonary artery smooth muscle cell in rats with pulmonary hypertension induced by high pulmonary blood flow. Chin Med J (Engl) 2009;122:3032–8. [PubMed] [Google Scholar]
- 54.Zhang JH, Dong Z, Chu L. Hydrogen sulfide induces apoptosis in human periodontium cells. J Periodontal Res. 2010;45:71–8. doi: 10.1111/j.1600-0765.2009.01202.x. [DOI] [PubMed] [Google Scholar]
- 55.Yu F, Zhao J, Tang CS, et al. Effect of synthesized GYY4137, a slowly releasing hydrogen sulfide donor, on cell viability and distribution of hydrogen sulfide in mice. Beijing Da Xue Xue Bao. 2010;42:493–7. [PubMed] [Google Scholar]
- 56.Chiku T, Padovani D, Zhu W, et al. H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J Biol Chem. 2009;284:11601–12. doi: 10.1074/jbc.M808026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh S, Padovani D, Leslie RA, et al. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–66. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maneiro E, Martin MA, de Andres MC, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48:700–8. doi: 10.1002/art.10837. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi T, Kitaoka K, Ogawa Y, et al. Lysosomal dysfunction on hydrogen peroxide-induced apoptosis of osteoarthritic chondrocytes. Int J Mol Med. 2004;14:197–200. [PubMed] [Google Scholar]
- 60.Beltowski J, Jamroz-Wisniewska A, Tokarzewska D. Hydrogen sulfide and its modulation in arterial hypertension and atherosclerosis. Cardiovasc Hematol Agents Med Chem. 2010;8:173–86. doi: 10.2174/187152510792481207. [DOI] [PubMed] [Google Scholar]
- 61.Lazzerini PE, Capecchi PL, Bisogno S, et al. Reduction in plasma homocysteine level in patients with rheumatoid arthritis given pulsed glucocorticoid treatment. Ann Rheum Dis. 2003;62:694–5. doi: 10.1136/ard.62.7.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giustarini D, Lorenzini S, Rossi R, et al. Altered thiol pattern in plasma of subjects affected by rheumatoid arthritis. Clin Exp Rheumatol. 2005;23:205–12. [PubMed] [Google Scholar]
- 63.Bradley H, Waring RH, Emery P. Reduced thiol methyl transferase activity in red blood cell membranes from patients with rheumatoid arthritis. J Rheumatol. 1991;18:1787–9. [PubMed] [Google Scholar]
- 64.Sun Q, Collins R, Huang S, et al. Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J Biol Chem. 2009;284:3076–85. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 65.Perry MM, Hui CK, Whiteman M, et al. Hydrogen sulfide inhibits proliferation and release of IL-8 from human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2011. doi: 10.1165/rcmb.2010-0304OC. [DOI] [PMC free article] [PubMed]
- 66.Whiteman M, Li L, Kostetski I, et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulfide. Biochem Biophys Res Commun. 2006;343:303–10. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 67.Whiteman M, Armstrong JS, Chu SH, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–8. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 68.Whiteman M, Cheung NS, Zhu YZ, et al. Hydrogen sulfide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain. Biochem Biophys Res Commun. 2005;326:794–8. doi: 10.1016/j.bbrc.2004.11.110. [DOI] [PubMed] [Google Scholar]
- 69.Nagy P, Winterbourn CC. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem Res Toxicol. 2010. doi: 10.1021/tx100266a. [DOI] [PubMed]
- 70.Tyagi N, Moshal KS, Sen U, et al. H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid Redox Signal. 2009;11:25–33. doi: 10.1089/ars.2008.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muzaffar S, Shukla N, Bond M, et al. Exogenous hydrogen sulfide inhibits superoxide formation, NOX-1 expression and Rac1 activity in human vascular smooth muscle cells. J Vasc Res. 2008;45:521–8. doi: 10.1159/000129686. [DOI] [PubMed] [Google Scholar]
- 72.Muellner MK, Schreier SM, Laggner H, et al. Hydrogen sulfide destroys lipid hydroperoxides in oxidized LDL. Biochem J. 2009;420:277–81. doi: 10.1042/BJ20082421. [DOI] [PubMed] [Google Scholar]
- 73.Schreier SM, Muellner MK, Steinkellner H, et al. Hydrogen sulfide scavenges the cytotoxic lipid oxidation product 4-HNE. Neurotox Res. 2010;17:249–56. doi: 10.1007/s12640-009-9099-9. [DOI] [PubMed] [Google Scholar]
- 74.Laggner H, Muellner MK, Schreier S, et al. Hydrogen sulfide: a novel physiological inhibitor of LDL atherogenic modification by HOCl. Free Radic Res. 2007;41:741–7. doi: 10.1080/10715760701263265. [DOI] [PubMed] [Google Scholar]
- 75.Carballal S, Trujillo M, Cuevasanta E, et al. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic Biol Med. 2011;50:196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- 76.Kimura Y, Dargusch R, Schubert D, et al. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid Redox Signal. 2006;8:661–70. doi: 10.1089/ars.2006.8.661. [DOI] [PubMed] [Google Scholar]
- 77.Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–7. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 78.Calvert JW, Elston M, Nicholson CK, et al. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–9. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calvert JW, Jha S, Gundewar S, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–74. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kloesch B, Liszt M, Steiner G, et al. Inhibitors of p38 and ERK1/2 MAPkinase and hydrogen sulfide block constitutive and IL-1beta-induced IL-6 and IL-8 expression in the human chondrocyte cell line C-28/I2. Rheumatol Int. 2010; doi: 101007/s00296–010-1682–0. [DOI] [PubMed]