

Abstract
Combining scanning force and fluorescent microscopy allows simultaneous identification of labeled biomolecules and analysis of their nm level architectural arrangement. Fluorescent polystyrene nano-spheres were used as reliable objects for alignment of optical and topographic images. This allowed the precise localization of different fluorescence particles within complex molecular assemblies whose structure was mapped in nanometer detail topography. Our experiments reveal the versatility of this method for analysis of proteins and protein-DNA complexes.
Keywords: Scanning force microscopy, Fluorescence microscopy, DNA-protein complex, Single-molecule studies, Fluorescent probes
1. Introduction
Imaging processes at the single-molecule level reveals information otherwise inaccessible by “bulk” experiments where individual features are usually hidden by ensemble averaging. Studying individual molecules allows observation of intermediate-transient states in dynamic reactions, extraction of precise information from asynchronous mixtures, and determination of the proportion of different heterogeneous forms within a population. Scanning force microscopy (SFM, also known as atomic force microscopy or AFM) is one such single molecule tool that produces images of biomolecules with nanometer resolution in the absence of external contrast agents. Three-dimensional information is collected from “soft” biological matter like protein complexes or nucleic acids likely without affecting relevant conformation and retaining overall native structure (for example as shown in ref.1). In the last two decades, a wealth of information about different proteins and their interactions with DNA has been produced using this technique [2-7]. Proteins can easily be distinguished from DNA. Their influence on DNA structure can be quantitatively determined from such images due to the distinct shape and the well described mechanical properties of DNA [8]. However in complex assemblies it is not often possible to distinguish proteins from each other based on shape and size. Several advances in SFM techniques are aimed at overcoming this limitation. Because the interaction force varies between different materials, the oscillations of the cantilever could be used to identify different molecules. For instance using intermittent contact mode imaging the phase signal can be used to produce surface hydrophobicity maps [9]. Another recently developed approach can identify different materials based on resonance frequencies of the cantilevers as in “bimodal atomic force microscopy”[10]. Molecular recognition with functionalized tips has been reported for the identification of specific proteins (for a review see ref.11) but involves a trade off with decreased spatial resolution as coated tips become larger and less well defined. Although, the above-mentioned methods have partially solved the molecular recognition problem, they all lack one of the most attractive features of conventional SFM: its simplicity.
In biological imaging specific molecules are often identified by attaching different florescent dyes or objects. Fluorescence detection methods thus allow molecular identification based on emission wavelength. Current technical advances in optical imaging allow detection down to the level of single fluorophores and can report on multiple fluorescent properties. Time resolution in the order of nanoseconds allows the study of dynamic process of individual molecules. However, due to Rayleigh criterion, spatial resolution is limited in the best case to half of the excitation wavelength. Super-resolution beyond the diffraction limit, down to nanometer precision, can be achieved by techniques such as stimulated emission depletion (STED) microscopy [12], photoactivated localization microscopy (PALM) [13] and stochastic optical reconstruction microscopy (STORM) [14]. Super-resolution of many fluorescent dyes is possible with STED although the best resolution requires depletion intensities as high as GW/cm2. PALM and STORM require photo-switchable fluorophores but only need intensities in the range of W/cm2. These advanced optical methods can localize fluorophores with nm precision however even at hypothetical atomic resolution structural details would still be limited to the labeled components.
A combination of SFM and fluorescence microscopy merges imaging capabilities to overcome limitations of the individual techniques. This also has the advantage of relatively simple SFM and standard optical imaging. Several reports have already described the development and use of such combined platforms. Identification of fluorescence markers on cell membranes or measuring adhesion forces constitute the main applications [15-17]. Other dedicated set-ups have probed their utility in combining fluorescence spectroscopy and SFM for identifying DNA and polystyrene beads [18], lipid organization [19] or light harvesting complexes in membranes [20]. However, imaging of biomolecules such as DNA-protein complexes remains technically challenging because the objects of interest are smaller than typical optical resolution. One of the main challenges of combining SFM and fluorescence imaging is the unambiguous assignment of a fluorescence signal (typically a few hundred of nanometers wide) to a globular structure of a few nanometers, like a protein complex. Although triangulation based on the center of fluorescence of quantum dots can be useful [21], blinking, photobleaching or heterogeneous excitation of isolated fluorophores make the correlation with topography difficult.
We show here nanometer localization of single particles using a combined scanning force and fluorescence microscope. The set up is based on a conventional wide-field fluorescence microscope using a mercury arc lamp combined with common excitation and emission filters. Intermittent contact mode scanning force microscopy in air was done with a commercially available scanner coupled to an optical microscope. Our experiments reveal the versatility of this method for recognition and simultaneous localization of different fluorescent-tagged objects, proteins and protein-DNA complexes. We describe the use of fluorescence polystyrene nano-spheres as reliable objects for alignment of optical and topographic images. This allowed us to precisely localize different fluorescent particles with nanometer resolution within complex molecular assemblies whose structure was mapped in nanometer detail topography.
2. Materials and Methods
2.1 Reagents
We used the following fluorescent dyes and objects: FluoSpheres® carboxylate-modified microspheres (0.04 μm diameter, yellow-green fluorescent (505/515), orange fluorescent (540/560) and red fluorescent (580/605) from Invitrogen), semiconductor nanocrystals streptavidin coupled Qdot525 and Qdot655 (“quantum dots”) (Invitrogen) and Alexa-488 (495/519) dye, AlexaFluor-633 (632/647) conjugated streptavidin (Invitrogen).
2.2. Sample preparation
Linear dsDNA (2054-bp) with a single 5′ biotin was generated by PCR amplification of pBluescript DNA using a modified biotinylated oligo essentially as described previously [22]. Nucleoprotein filaments, RAD51 coated dsDNA, were made by incubation of λ DNA (Roche) and Alexa-488 conjugated human RAD51 as previously described [23].
Unless otherwise stated, the indicated amount of product was diluted in 20 μL of deposition buffer (10mM HEPES-KCl, pH7.5, 10mM MgCl2). Samples were deposited over freshly cleaved mica (muscovite V-1 quality, from Electron Microscopy Science) mounted on glass cover slips (24 mm round from Menzel-glazer) with optical adhesive NOA88 (Norland products) cured with UV light (350 nm) for 3 min at room temperature. Before gluing, the mica disc was cleaved with tape until almost transparent. Then a minimum amount of optical adhesive was put on the cover slip. The mica disc was attached and, by pressing down on it, the low viscosity adhesive was homogeneously spread. After curing the adhesive and before using the mica-cover slip, the mica surface was made as thin as possible, by cleaving as many layers as possible off with tape. This produces a mica glued glass substrate that allows focusing on the sample, with the objective lens we have used here that has a 170 micron working distance the mica layer will be 30 microns or less. After one minute the sample drop was rinsed with MilliQ water and dried with filtered air. Before depositing the sample, autofluorescence on the mica surface was eliminated by reduction with a drop of sodium borohydride [24] (0.25% w/v) for 20 minutes at room temperature and rinsed with MilliQ water.
2.3 Fluorescence microscopy
Samples were observed with a Nikon TE 2000U microscopy using a Nikon 60× (NA 1.45) TIRF oil objective and collected by a Cascade II:512B EMCCD camera (Princeton Instruments) driven with MetaMorph software (Molecular Devices). Excitation of the fluorophores was performed with a mercury arc lamp and standard filter cubes (center wavelength/bandwidth, excitation-emission, in nm): FITC (480/30-535/40) and TxRed (560/40-630/60) from Nikon, and GFP-3035B (472/30-520/35) from Semrock.
2.4 Scanning force microscopy
Scanning force microscopy was done with a NanoWizard®II scanner (JPK instruments) mounted on the fluorescence microscope. Air dried samples were scanned in intermittent contact mode (air). Silicon Tapping/Non-Contact Mode tips 125 μm in length with a spring constant of 25–75 N/m were from Applied Nanostructures. Drive frequency of the cantilevers used was on average 300 kHz. Images were, in general, acquired at 2 Hz line rate. Correlation of fluorescence and topographic images was accomplished first by the JPK DirectOverlay™ software then refined to nanometer accuracy as describe in results making use of structures with distinct identifiable shape and fluorescence such as nanospheres.
3. Results
3.1 Identification and nanometer localization of individual fluorescent particles
In order to validate precision alignment of optical and topographic images to nm accuracy, we used commercially-available fluorescently-label polystyrene-beads, referred to as fluorospheres. With these we could distinguish three different fluorescence particles with a combined SFM/fluorescence microscope. Such objects are needed as “fiduciary markers” or reference points in these combined images. Yellow-green (505/515), orange (540/560) and red fluorescent (580/605) 40 nm diameter beads were mixed (60 pM each) and sonicated in a water bath for 30 min. A solution (3 pM each) was diluted and deposited on a glued-mica glass surface as described in the experimental section. Samples were first observed with the inverted epifluorescence microscope. Fluorescent emission signal from “green” and “red” beads were recorded after filtering the emission spectrum with a FITC (480/30-535/40) and TxRed (560/40-630/60) cubes respectively. A high numerical aperture objective (1.45) with a short working distance (0.17 mm at 23°C) was used. So, mica thickness was limited to 30 microns excluding the glass. We did not observe a reduction of fluorescence intensity due to the presence of the mica compare to glass in these conditions. Nor were birefringence effects observed (Supplementary Figure 1). Lenses and mirrors guiding the light produce a variable degree of optical aberrations reflected in the final image. The DirectOverlay™ software allows the automatic calibration of the optical images by mapping the actual position of the cantilever (determined by the piezo precision and not by the optical pathway) at 25 different positions. We calibrated regions of 40×40 μm, that are extrapolated to cover 50% of the 60×60 μm surface, in each experiment. This allowed us to precisely correlate SFM images with optical images of 512×512 pixels (~100nm/pixel using a 60X objective). However, because the initial position of the cantilever is unknown, there is an offset of a few microns in X and Y between the optical and topographic images, as is shown in Figure 1 A. When imaging structures at the single molecule level (nm), it becomes apparent that in the absence of a clear reference the offset will result in a serious disorientation and inability to identify specific objects. We used the pattern of randomly deposited fluorospheres evident in both topographic and optical images for alignment as shown in Figure 1. The red fluorescence pattern had an X,Y offset (-4.8 μm, -3.6 μm) with respect to the height image (Figure 1 B). After alignment of the red spot mass-centers with the height structure mass-centers both pictures correlate with nanometer accuracy (Figure 1 C). Overlapping red and green signals identified the orange beads as shown in Figure 1 A. The boxed area in Figure 1 A was selected for SFM scanning, shown in Figure 1 E. The separate red and green emission images are overlaid on the topography (Figures 1 C and 1 D) for correlation and identification of two red, two green and one orange sphere. Volume analyses of the particles (calculated as previously described [6]) indicated the presence of one or more sphere per fluorescence spot in this example (Figure 2 B). Intensity measurements from non-saturated images where plotted against their respective volume revealing a linear correlation (Figure 2 A). Different fluorescence objects were identified in a complex mixture based on emission wavelength and their intensity correlated with size or number of fluorescent particles.
Figure 1.

Unique identification of different fluorophores by combined optical and scanning force microscopy. A) Optical image of a mixture of three different fluorospheres: red, green and orange. Green and red channel were overlaid. Orange polystyrene beads are recognized by the colocalization of both signals. The indicated area (green frame) was chosen for SFM. B) Alignment procedure using fluorospheres. Red fluorescent signal emitted from the polystyrene beads shows an offset in X and Y coordinates after the image calibration procedure. C) Height features in topographic image (scale as in panel E) are easily aligned with the fluorescent pattern. Overlay of the red channel with the SFM image is shown. D) Green channel and the overlay with the SFM image. E) Height image.
Figure 2.

Linear correlation between fluorescence intensity and particle volume. A) Volume in arbitrary units (a.u.) [6] versus integrated fluorescence intensity (also in a.u.) [23] coming from the same regions were plotted and analyzed by regression fit. The red and green lines and squares represent data from individual particles red fluorospheres and green fluorospheres, respectively. Note that some particles are smaller than complete fluorospheres and some represent more than one sphere, in the range of particle sizes shown here. B) Fluorescence intensity distribution of the spheres. Intensity bin size equivalent to fluorescence from one fluorosphere (volume 1.9 ± 1.2×105 a.u, N=22), representing 3.5×105 a.u. (green) and 8×105 a.u. (red). Nred = 95, Ngreen = 99.
3.2 Topography reveals a large fraction of dark Quantum dots
We similarly characterized quantum dots by depositing a mixture of green Qdots and red fluorospheres or red Qdots and green fluorespheres (semiconductor nanocrystals streptavidin coupled Qdot525 and Qdot655, Invitrogen). To account for quantum dot blinking, consecutive images were collected and compiled into a single average composite (500 ms exposition time, 60 frames, 30 s total exposure time per image). Figure 3 A shows the final Qdot image overlaid with signal from the fluorospheres. The indicated areas were selected for topographic imaging (Figures 3 B and C). Fluorescence signal from the fluorospheres could be overlaid onto structures with the expected dimensions (~40 nm height). There was a homogenous population of smaller structures corresponding to the size of Qdots (between 5-10 nm height). Around 10% (17 fluorescent objects out of 160 countable structures in the best case like Figure 4 B) of these Qdot sized entities were detected in their corresponding fluorescence channel (green in Figures 3 A, B and D; and red in Figures 3 E and F). The presence of a majority of Qdot sized structures that were not fluorescence is similar to a previous report [21]. A detailed look at the height images revealed additional information difficult to obtain from fluorescence alone. For instance, intensity over time traces for the two red Qdots (1 and 2) highlighted in panel 3E show blinking behavior as expected (Figure 3 G). Although there was no clear signature that allowed us to ascribe this fluorescence behavior to more than one fluorophore, a zoomed scan of the selected area (Figure 3 F) revealed the presence of two Qdot sized structures very close together contributing to this one fluorescent spot.
Figure 3.

Fluorospheres as molecular fiduciary markers. A) Optical image showing red fluorospheres and green quantum dots. B) Green channel picture from the selected region in A and height images overlaid. C) Red channel picture from the same region and height images overlaid. D) Enlarged area from white rectangle in B. E) Green fluorospheres and red quantum dots: height image and red fluorescence channel overlaid. Qdots analyzed in panel G are labeled by numbers 1 and 2. F) Zoomed region from panel E; height image and fluorescence channels overlaid showing the double nature of one specific red fluorescent spot (labeled 2). G) Time traces of Qdot655 fluorescence. Total pixel intensity in regions around the indicated two red dots in panel E (1 and 2) were measured from 60 frames acquired for 500 ms each in a total time of 30 seconds. Scale bar 2μm.
Figure 4.

SFM topographic image of DNA at nm resolution and localization of attached fluorescent label. A) Optical image showing the fluorescence signal from red fluorospheres and B) green quantum dots associated with biotinylated DNA, overlaid with the height image. C) Zoomed overlay region marked in B. D) Height image, Z scale 0-2 nm, dark to bright. Scale bar 0.5 μm.
3.3 Fluorescent labeled DNA molecules
We produced nm resolution images of DNA identified by a fluorescent label coupled specifically to one end. For identification and localization of specifically labeled proteins in complex assemblies with DNA it will be impossible to correlate topography and fluorescence for structures smaller than optical resolution without a marker object. DNA can be fluorescently labeled and is often made visible by intercalating dyes [25; 26]. However, such dyes distort DNA structure and frequently interfere with protein DNA binding activity. Here, in order to identify DNA by a fluorescent marker we use a fluorescent label coupled specifically to the end of DNA molecules.
Streptavidin coupled quantum dots (Qdot525) were attached to linear 2054-bp DNA molecules that included one 5′-biotin. This DNA was deposited for imaging together with red fluorospheres (3 pM). Green and red signals, from the Qdots and the fluorospheres respectively, were recorded separately (Figure 4). The overlay of topography again indicated most Qdots were not fluorescent in the conditions used (Figure 4 C). However, DNA molecules can be clearly identified in topography by length (~0.6 μm) and width (~10 nm) most with Q-dots at one terminus (Figure 4 D). Note that more than one DNA could attach to a Qdot because streptavidin is multivalent (glowing or not).
3.4 Detection of fluorescent proteins
To demonstrate nm resolution imaging of individual proteins we deposited fluorescent streptavidin (red) together with fluorospheres (green) and first detected emission signals from both fluorophores (Figure 5 A). A region including both streptavidin and fluorosphere signals was selected for topography imaging. The 60-kD streptavidin protein is expected to be approximately a 5 nm diameter sphere, smaller than the 40 nm polystyrene fluorospheres. In addition, each streptavidin, (reported to have 4 dye molecules per tetramer, Molecular Probes-Certificate of analysis, or our measurement of 2.7), will have far fewer dye molecules than the fluorospheres (3.5×102 fluorescein equivalents in the yellow-green fluorospheres, Molecular Probes). In the zoomed images (Figure 5 B, C and D) the fluorospheres were distinctly identifiable by both fluorescence and topography. There were in addition many small objects visible in topography that corresponded to the size of streptavidin. Smaller objects were proven not to be scanning noise by shifting the SFM scan direction and observing the same constellation of objects (data not shown). With the simple optical imaging we applied here, most of the single proteins did not show obvious fluorescence above background. After using the fluorospheres to align the optical and topography images, the red fluorescence signal could be identified corresponding to a complex likely including several streptavidin proteins, larger than the predominant single protein molecules. Thus we are able to distinguish individual protein molecules and observe fluorescence for complexes corresponding to several proteins.
Figure 5.
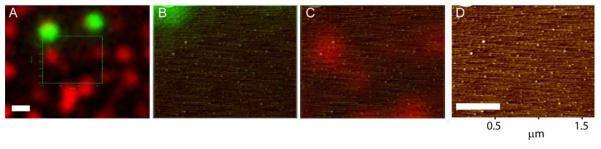
Localization of individual fluorescence streptavidin particles. A) Optical image showing the fluorescence signal from green fluorospheres and AlexaFluor-633 conjugated streptavidin (in red). B) and C) Selected area in A shown individual fluorescence channels overlaid with the height image. D) Height image alone. Scale bar 0.5 μm.
3.5 Visualizing DNA protein complexes
Our eventual aim is to identify proteins in complex structures based on different fluorescent labels. As an example of this type of analysis we used a version of the human RAD51 protein labeled with a single fluorophore in a defined position [23]. Alexa Fluor488 labeled RAD51 filaments covering double-stranded DNA, 48 kbp phage lambda genome, were mixed with red fluorospheres and deposited for imaging. Green and red channels were individually recorded and overlaid after acquisition (Figure 6). Green filamentous structures are DNA covered with RAD51. In this case we could detect the presence of the protein by its color and additionally define the structure of the filaments by the topography. The topography revealed structural detail, indistinguishable in the optical image, such as multiple filaments close together (Figures 6 B and C) and the filament end curled into a loop (Figures 6 D and E). Here the DNA filament is obvious in both optical and topographic images due to the fluorescent protein. However, it is clear that even non-fluorescent proteins and bare DNA would be visible for analysis in such topographic images, where the relative location of labeled proteins within complex assemblies could be accurately determined.
Figure 6.

Fluorescence human RAD51 protein filaments on DNA. A) Optical image showing green fluorescence filamentous structures originated by the association of the Alexa 488 conjugated RAD51 with the long DNA molecules of lambda phage (~48 kbp). Height image overlaid with the optical image from red fluorospheres (B) or green RAD51 (C) from the same region shown in A. D) Overlay of height and green fluorescence images of one filament end. E) Extra resolution in the height image shows a lariat at the end of this filamentous structure. Scale bar 2 μm.
4. Discussion
We describe here the use of a commercial instrument and simple sample preparation methods for simultaneous optical and topographic imaging of nano-objects, proteins and DNA. Biomolecules such as DNA, proteins and their complexes were deposited on mica as they are for many standard SFM imaging experiments. The methods described avoid chemical cross-linking of the complexes to be observed thus avoiding possible structural artifacts this can cause. In addition we use untreated surfaces to preserve DNA conformation and allow quantitative measurement of changes induced by proteins [8]. In contrast, a recent similar demonstration of combined SFM/optical imaging [25] relied on treated surfaces and DNA stretching methods that would preclude analysis of important protein-induced changes in DNA conformation that are often of interested. Topographic images of biomolecules such as DNA and proteins are also more reliable on atomically flat mica surfaces compared with much rougher glass.
We present here several improvements over the previously reported combined SFM-fluoresence imaging of protein-DNA complexes that make our set-up more efficient. We show labeled-RAD51 protein associated with unlabeled DNA (Figure 6). The fluorescence and height images are correlated using fluorospheres added for this purpose. Moreover, using the same image we show the position of fluorospheres of different color and their correlated height image (Figures 1 and 2). We addressed the suitability of Qdots for correlating topography and fluorescence by studying the dark fraction and observe a high dark fraction making them unacceptable for this purpose (Figure 3). DNA labeled at one end by Qdots was imaged by SFM showing the expected height, width, shape and resolution for DNA. The fluorescent end-bound Qdots could be identified. We show a small protein that is hard to unambiguously identify in topography, Alexa fluor 633 labeled-streptavidin but can be identified in clusters by fluorescence (Figure 5). In all of the presented cases, the correlation of topography and fluorescence relies on the use of fluorospheres and not on the fortuitous emergence of discernable shape in the sample objects themselves, and is therefore generally applicable to aligning similar images for any biomolecules. Another important advantage of our set up is the use of 25 pictures of the cantilever that are used by the manufacturer’s software to calibrate the optical image and determine the position of the tip in the topographical images, avoiding in this way, optical aberrations. We are currently using this approach to analyze complexes involving several proteins acting together on DNA in the DNA repair processes we study. Identification of the relative position of specific proteins will provide a wealth of information about their functional arrangements.
Many applications that we envision combining optical and SFM topographic imaging will involve objects that are smaller than the resolution limit of standard optical imaging. In order to accurately map optical signals and topography, both the available calibration of optical images by the JPK DirectOverlay™ system and fine tuning offset adjustment to align optical and topographic images to nm precision are needed to obtain useful information. We demonstrate here the use of fluorescent polystyrene nanospheres as convenient fiduciary objects for this purpose. The fluorospheres attach to mica together with the biomolecules during standard deposition and fall at random on the surface. Any image including at least 3 fluorospheres can be accurately corrected for offset by aligning the nm resolution topography images of the spheres with the center of the optical spots. Thus fluoropsheres are robust, convenient, economical and non-interfering markers for coordinated SFM and optical imaging. We used this alignment to identify clusters of fluorescent streptavidin among the multitude of monomeric streptavidin detected in the topographic image. Correlating topography and optical images also revealed that under the conditions we use a majority of quantum dots are not fluorescent, and thus cannot be used as robust fiduciary markers. Furthermore, it was possible to address unambiguously the proportion of dark particles in our semiconductor nanocrystals. Fluorescence correlation spectroscopy (FCS) studies [27], reported a quantum dot preparation as 44% dark, 36% blinking and the remaining fraction was assumed to be glowing. In our experimental set up individual particles could be observed indefinitely with an equivalent dwell time of 500 ms. We did not observe a glowing fraction but a non-dark fraction that was entirely blinking. However, likely due to limited freedom of the particles in ambient humidity conditions the proportion of the dark fraction was higher than expected from solution. Fluorescence properties of quantum dots are very sensitive to surface changes [28].Our data support the idea that the quantum yield reported for the particles is proportional to the non-dark fraction. Recently, the generation of non-blinking Qdots that would overcome this limitation has been reported [29]. Other fluorophores and defined molecular complexes such as Alexa dye coupled proteins with known stoichiometry will have to be analyzed to determine if the surface and imaging conditions in general influence the efficiency of detecting fluorescent molecules. For instance the Alexa labeled RAD51, already characterized to include one fluorophore per monomer [23; 30], may be useful for characterizing our sample preparation and imaging system with respect to this feature.
With a relatively simple optical set up and samples prepared on a mica-glue-glass substrate we have not yet demonstrated single fluorophore sensitivity. However this sensitivity is only limited by the sophistication of the optical set up and analysis. The number of different fluorophores that can be detected is limited by the filters installed and can easily be increased, for instance by exciting the Quantum dots with shorter wavelengths from the arc lamp spectrum or by using a dedicated laser. In addition, higher power laser excitation of specific fluorophores will increase detection sensitivity. Similarly, nm localization of single fluorophores should also be possible with sufficiently bright or accumulated emission signals. Using a higher magnification objective (100X) and the calibration of a smaller area of 30×30 μm will increase the localization accuracy of the fluorescence emitter. Expanding the capability of a combined SFM/FM set up to quantitative analysis of fluorescent signals is likely achievable based on characterizing calibration objects, such as defined identifiable fluorophores, to be included within the sample.
5. Conclusions
The combined SFM/fluorescent microscope will allow identification of specific proteins within complex assemblies on DNA and other biologically relevant molecular complexes. Appropriate marker objects are essential to achieve nm accuracy in alignment of optical and topographic images when the molecules of interest are smaller than optical resolution. We describe here the convenient use of Fluorospheres as robust fiduciary markers. In addition, using an optically suitable mica glued glass sample substrate, we take advantage of this atomically flat surface characterized in greater than 15 years of SFM publications and well established protocols for deposition and characterization of biological molecules like DNA-protein complexes. Our test sample, the RAD51 filament protein-DNA complex, reveals the additional structural information that can be obtained from nm resolution topography images of objects identified by the presence of fluorescent proteins.
Supplementary Material
Acknowledgments
This work was supported by a Marie Curie grant from the European Commission (to H.S.), a VICI (to C.W.) and a TOP (to R.K.) grant from the Netherlands Organization for Scientific Research (NWO), Chemical Sciences division, the Netherlands Genomics Initiative/NWO) and NCI (USA) program project (CA92584).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ristic D, Modesti M, van der Heijden T, van Noort J, Dekker C, Kanaar R, Wyman C. Nucleic Acids Res. 2005;33:3292–302. doi: 10.1093/nar/gki640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dame RT, Wyman C, Goosen N. J Microsc. 2003;212:244–53. doi: 10.1111/j.1365-2818.2003.01243.x. [DOI] [PubMed] [Google Scholar]
- [3].Hansma HG, Golan R, Hsieh W, Daubendiek SL, Kool ET. J Struct Biol. 1999;127:240–7. doi: 10.1006/jsbi.1999.4170. [DOI] [PubMed] [Google Scholar]
- [4].Janicijevic A, Ristic D, Wyman C. J Microsc. 2003;212:264–72. doi: 10.1111/j.1365-2818.2003.01254.x. [DOI] [PubMed] [Google Scholar]
- [5].Sanchez H, Cardenas PP, Yoshimura SH, Takeyasu K, Alonso JC. Nucleic Acids Res. 2008;36:110–20. doi: 10.1093/nar/gkm759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].van der Linden E, Sanchez H, Kinoshita E, Kanaar R, Wyman C. Nucleic Acids Res. 2009;37:1580–8. doi: 10.1093/nar/gkn1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rees WA, Keller RW, Vesenka JP, Yang G, Bustamante C. Science. 1993;260:1646–9. doi: 10.1126/science.8503010. [DOI] [PubMed] [Google Scholar]
- [8].Rivetti C, Guthold M, Bustamante C. J Mol Biol. 1996;264:919–32. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- [9].Sahagun E, Garcia-Mochales P, Sacha GM, Saenz JJ. Energy Dissipation due to Capillary Interactions: Hydrophobicity Maps in Force Microscopy. APS. 2007:176106. [Google Scholar]
- [10].Martinez NF, Patil S, Lozano JR, Garcia R. Applied Physics Letters. 2006;89 [Google Scholar]
- [11].Hinterdorfer P, Dufrene YF. Nature Methods. 2006;3:347–355. doi: 10.1038/nmeth871. [DOI] [PubMed] [Google Scholar]
- [12].Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Proc Natl Acad Sci U S A. 2000;97:8206–10. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642–5. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- [14].Rust MJ, Bates M, Zhuang X. Nat Methods. 2006;3:793–5. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Neagu C, van der Werf KO, Putman CA, Kraan YM, de Grooth BG, van Hulst NF, Greve J. J Struct Biol. 1994;112:32–40. doi: 10.1006/jsbi.1994.1004. [DOI] [PubMed] [Google Scholar]
- [16].Madl J, Rhode S, Stangl H, Stockinger H, Hinterdorfer P, Schutz GJ, Kada G. Ultramicroscopy. 2006;106:645–51. doi: 10.1016/j.ultramic.2005.12.020. [DOI] [PubMed] [Google Scholar]
- [17].Puech PH, Poole K, Knebel D, Muller DJ. Ultramicroscopy. 2006;106:637–44. doi: 10.1016/j.ultramic.2005.08.003. [DOI] [PubMed] [Google Scholar]
- [18].Kolodny LA, Willard DM, Carillo LL, Nelson MW, Van Orden A. Anal Chem. 2001;73:1959–66. doi: 10.1021/ac001472z. [DOI] [PubMed] [Google Scholar]
- [19].Chiantia S, Kahya N, Ries J, Schwille P. Biophys J. 2006;90:4500–8. doi: 10.1529/biophysj.106.081026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kassies R, van der Werf KO, Lenferink A, Hunter CN, Olsen JD, Subramaniam V, Otto C. J Microsc. 2005;217:109–16. doi: 10.1111/j.0022-2720.2005.01428.x. [DOI] [PubMed] [Google Scholar]
- [21].Owen RJ, Heyes CD, Knebel D, Rocker C, Nienhaus GU. Biopolymers. 2006;82:410–4. doi: 10.1002/bip.20414. [DOI] [PubMed] [Google Scholar]
- [22].Ristic D, Modesti M, Kanaar R, Wyman C. Nucleic Acids Res. 2003;31:5229–37. doi: 10.1093/nar/gkg729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Modesti M, Ristic D, van der Heijden T, Dekker C, van Mameren J, Peterman EJ, Wuite GJ, Kanaar R, Wyman C. Structure. 2007;15:599–609. doi: 10.1016/j.str.2007.04.003. [DOI] [PubMed] [Google Scholar]
- [24].Raghavachari N, Bao YP, Li G, Xie X, Muller UR. Anal Biochem. 2003;312:101–5. doi: 10.1016/s0003-2697(02)00440-2. [DOI] [PubMed] [Google Scholar]
- [25].Ebenstein Y, Gassman N, Kim S, Weiss S. J Mol Recognit. 2009 doi: 10.1002/jmr.956. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hilario J, Amitani I, Baskin RJ, Kowalczykowski SC. Proc Natl Acad Sci U S A. 2009;106:361–8. doi: 10.1073/pnas.0811965106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yao J, Larson DR, Vishwasrao HD, Zipfel WR, Webb WW. Proc Natl Acad Sci U S A. 2005;102:14284–9. doi: 10.1073/pnas.0506523102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Alivisatos AP. Science. 1996;271:933–937. [Google Scholar]
- [29].Spinicelli P, Mahler B, Buil S, Quelin X, Dubertret B, Hermier JP. Chemphyschem. 2009;10:879–82. doi: 10.1002/cphc.200800827. [DOI] [PubMed] [Google Scholar]
- [30].van Mameren J, Modesti M, Kanaar R, Wyman C, Peterman EJ, Wuite GJ. Nature. 2009;457:745–8. doi: 10.1038/nature07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.