

Abstract
According to epidemiological studies, type-2 diabetes increases the risk of Alzheimer’s disease. Here, we induced hyperglycaemia in mice overexpressing mutant amyloid precursor protein and presenilin-1 (APdE9) either by cross-breeding them with pancreatic insulin-like growth factor 2 (IGF-2) overexpressing mice or by feeding them with high-fat diet. Glucose and insulin tolerance tests revealed significant hyperglycaemia in mice overexpressing IGF-2, which was exacerbated by high-fat diet. However, sustained hyperinsulinaemia and insulin resistance were observed only in mice co-expressing IGF-2 and APdE9 without correlation to insulin levels in brain. In behavioural tests in aged mice, APdE9 was associated with poor spatial learning and the combination of IGF-2 and high-fat diet further impaired learning. Neither high-fat diet nor IGF-2 increased β-amyloid burden in the brain. In male mice, IGF-2 increased β-amyloid 42/40 ratio, which correlated with poor spatial learning. In contrast, inhibitory phosphorylation of glycogen synthase kinase 3β, which correlated with good spatial learning, was increased in APdE9 and IGF-2 female mice on standard diet, but not on high-fat diet. Interestingly, high-fat diet altered τ isoform expression and increased phosphorylation of τ at Ser202 site in female mice regardless of genotype. These findings provide evidence for new regulatory mechanisms that link type-2 diabetes and Alzheimer pathology.
Keywords: Alzheimer’s disease, β-amyloid, hyperinsulinaemia, insulin growth factor 2, insulin resistance, type-2 diabetes mellitus
Introduction
Alzheimer’s disease (AD) and type-2 diabetes mellitus (T2DM) are rapidly increasing disabling conditions among the aging population that reach a dimension of worldwide epidemic. Several population-based epidemiological studies have indicated that AD and T2DM often co-occur and that these diseases are risk factors for one another [1-7]. On the other hand, it is a well-established fact that T2DM is an integral part of the metabolic syndrome, which is the most important risk factor for cardiovascular diseases [8]. However, it is impossible to conclude whether T2DM, or more specifically hyperinsulinaemia and insulin resistance, are independent risk factors for AD solely on the basis of the epidemiological studies. Nevertheless, it was recently shown that insulin resistance increased the formation of neuritic plaques (NP), whereas no relationship was found between diabetes-related factors and neurofibrillary tangles (NFT) [9]. Although this study addressed the association of diabetic phenotype with AD pathology, other population-based neuropathological studies have not been able to establish the anticipated link between diabetes and amyloid-β (Aβ) accumulation [1, 7, 10]. Therefore, these population-based data collectively underscore the importance of using alternative approaches, such as experimental animal models, which display the key features of AD and T2DM, to study the common pathogenic mechanisms underlying these diseases. There are two important overlapping features in the metabolism of insulin and Aβ, a peptide generated from the amyloid precursor protein (APP) after β- and γ-secretase–mediated cleavages. The first common feature is that both insulin and Aβ are substrates of insulin-degrading enzyme (IDE) [11] and high extracellular levels of insulin compete with Aβ for binding to IDE [12]. Because peripheral Aβ is mainly degraded in the liver by IDE, it has been postulated that decreased IDE levels in the liver of T2DM patients lead to augmented Aβ levels both in the periphery and the brain [13]. Secondly, glycogen synthase kinase 3 (GSK3), a protein kinase, is intimately involved in the molecular mechanisms of insulin resistance and AD [14]. Insulin negatively regulates GSK3 activity, whereas GSK3 opposes the action of insulin. At the same time, GSK3β is the most important kinase responsible for the hyperphosphorylation of τ protein, which is the prime component of NFTs in the AD brain. Also, it should be noted that GSK3α, another GSK3 isoform, has been shown to regulate the production of Aβ both in vitro and in vivo [15]. So far, only a few animal studies have investigated the co-occurrence of AD pathology and metabolic features related to T2DM, such as hyperinsulinaemia and/or insulin resistance [16-18]. Results in these animal models have been controversial in terms of alterations in APP processing, Aβ accumulation, and insulin signalling. Therefore, we have cross-bred well-known AD model mice carrying human APPswe and PS1dE9 mutations (APdE9 mice) [19] with an established mouse model of T2DM, which overexpresses IGF-2 specifically in the pancreatic islet cells [20]. In the APdE9 χ IGF-2 (A+I+) mice, primary hyperinsulinaemia led to insulin resistance, which subsequently allowed us to study the effects of both genetic and diet-induced diabetic phenotype on Aβ pathology and neurological symptoms in aging mice. Consequently, we found that APdE9 genotype exacerbated hyperinsulinaemia and insulin resistance phenotype, whereas genetic or diet-induced insulin resistance did not augment beta-amyloid pathology in the brain of A+I+ mice.
Materials and methods
Animals
As a model for AD, we used transgenic mice expressing mutated human amyloid precursor protein (APPswe) and presenilin-1 (PS1dE9) genes [19]. Formation of amyloid plaques in the cortex and hippocampus begins at the age of 3 months and proceeds faster in female mice. As a model of insulin resistance we used transgenic mice overexpressing IGF-2 [20]. In these mice, β-islet hyperplasia leads to hyperinsulinaemia and development of insulin resistance [20]. Both mouse lines were back-crossed for more than 10 generations to the common C57B6 strain. The experiments were conducted according to the Council of Europe (Directive 86/609) and Finnish guidelines, and approved by the National Animal Ethics Committee.
Experimental setup
The study consisted of two different age groups. The effect of genetic manipulation on glucose metabolism was first assessed in 4-month-old female and male mice (acute group). These mice first underwent glucose tolerance test (GTT) and 2 weeks later insulin tolerance test (ITT), and were euthanized at the age of 18 weeks.
The combined effect of genetic and dietary manipulations was investigated in another group of female and male mice (diet group). Beginning at the age of 5 (female mice) or 8 (male mice) months, the mice were fed with either normal lab chow [containing 5% (w/w) fat and no cholesterol; referred to as standard diet, STD] or a chow mimicking typical Western diet [TWD; Adjusted Calories diet, TD 88137, Harlan Tecklad with 21% (w/w) fat and 0.15% cholesterol]. The mice were killed at the age of 9 (female mice) or 12 (male mice) months. The female mice underwent GTT 9 weeks and ITT 11 weeks after the onset of either diet. Both female and male mice were subjected to a neurological test battery during the last 2 weeks before killing.
Behavioural testing
Spontaneous exploratory activity was monitored in a transparent test cage (L 26 χ W 26 χ H 39 cm) with automated infrared photo detection (TruScan; Coulbourn Instruments, Allentown, PA, USA) for 10 min. in two separate sessions with a retention interval of 48 hrs.
Spatial memory was assessed with the Morris swim navigation task (water maze). The apparatus was a black plastic pool with a diameter of 120 cm. The experiment was performed for 5 consecutive days with five 60 sec. trials (T1–T5) per day and a single 40 sec. probe trial without the platform on day 6 to determine the search bias.
Insulin and glucose metabolism
Glucose tolerance test (GTT)
After an overnight (16 hrs) fasting, an i.p. injection of 2 mg/g D-glucose as a 20% solution (prepared in normal saline) was given. Blood samples for the determination of glucose and insulin levels (50–75 μl) were collected at 0, 15, 30 and 60 min. from the saphenous vein and the glucose values were determined immediately using a glucometer.
Insulin tolerance test (ITT)
After fasting for 4 hrs in the morning, an i.p. injection of insulin at 0.25 mU/g was given. Blood samples were collected at 0, 20, 40 and 80 min., and the glucose values were recorded using a glucometer.
Analysis of insulin levels
During GTT, blood samples were collected into fine glass capillary tubes lined with anti-coagulant. Plasma obtained by centrifugation (13,000 r.p.m. for 1 min.) was snap-frozen in liquid nitrogen and analysed with rat insulin enzyme-linked immunosorbent assay kit and mouse insulin standards (INSKR020 and INSSM021; Crystal Chem Inc., Chicago, IL, USA).
Insulin gene expression
A separate cohort of 4-month-old mice (n ∇ 10 of all four genotypes; four male mice + six female mice) was euthanized with cervical dislocation and pancreatic islets were isolated as described previously [21]. Total islet RNA was isolated using RNeasy Plus Micro kit (QIAGEN GmbH, Hilden, Germany) and liver RNA with TRIzol Reagent (Life Technologies, Rockville, MD, USA). Quantitative RT-PCR reactions were performed with the following TaqMan Gene Expression Assays: Ins1 (Mm01259683_g1), Ins2 (Mm00731595_gH) and Arbp (Mm00725448_s1) (Applied Biosystems, Foster City, CA, USA), G6Pc3 (Mm00440636-m1), pck1 (Mm00616234_m1) and β-actin (Assay by design from Applied Biosystems). Insulin gene expression was normalized to Arbp (36B4 gene) and liver G6Pc3 and pck1 gene expression to β-actin, and calculated as a percentage related to wild-type gene expression.
Western blot analysis
For Western blot analyses, total proteins were extracted from mechanically homogenized brain and liver samples using TPER extraction buffer (Pierce, Thermo Scientific, Rockford, IL, USA) containing EDTA-free phosphatase and protease inhibitor cocktail (Thermo Scientific, Rockford, IL, USA). After protein quantification using BCA Protein Assay Kit (Pierce), 30 μg of total protein lysates were subjected to 4–12% Bis-Tris-polyacrylamide gel electrophoresis (Invitrogen, Carlsbad, CA, USA) and subsequently blotted onto Immun-Blot polyvinylidene fluoride membranes (PVDF; Bio-Rad, Hercules, CA, USA). Primary antibodies against APP (A8717, 1:1000; Sigma-Aldrich, St. Louis, MO, USA), phospho-Akt (pAKT, Ser473, 1:1000; Cell Signaling Technology, Danvers, MA, USA), Akt (1:1000; Cell Signaling Technology), pGSK3α/β (pan antibody, 1:1000; Cell Signaling Technology), pGSK3β (Ser 9, 1:1000; Cell Signaling Technology), GSK3α (1:1000; Cell Signaling Technology), GSK3β (1:1000; Cell Signaling Technology), IDE (1:1000; Abcam, Cambridge, MA, USA), GAPDH (1:15,000; Abcam), PHF-τ (AT8, 1:1000; Thermo Scientific) and total τ (ab47579, 1:1000; Abcam) were used for immunoblotting. After incubation with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (GE Healthcare, Piscataway, NJ, USA), enhanced chemiluminescence substrate (Amersham Biosciences, GE Healthcare, Piscataway, NJ, USA) was applied to membranes and detection of protein bands was performed with ImageQuant RT ECL camera (GE Healthcare). Western blot images were quantified using Quantity One software (Bio-Rad).
ELISA analysis of liver and brain Aβ levels
Liver Aβ levels were analysed from female mice of the acute group at 4 months of age. The tissue was homogenized in PBS with phosphatase and protease inhibitor cocktail. Ventral cortex was homogenized into 5 M guanidine, pH 8.0 with EDTA-free phosphatase and protease inhibitor cocktail. The levels of transgenic human Aβ1–40 and Aβ1–42 were quantified using the Signal Select TM β Amyloid ELISA Kits (BioSource International Inc., Camarillo, CA, USA).
Insulin measurements from the ventral posterior cortex
Brain insulin levels were measured by using AlphaLISA Insulin Kit (#AL204C; PerkinElmer LAS, Inc., Boston, MA, USA) according to the manufacturer instructions with a minor change. Briefly, the total protein samples from the ventral posterior cortex were centrifuged (13,000 r.p.m. for 30 min.) and the supernatant was collected and stored at −70°C until the assay was performed. For the insulin assay, 5 μl of supernatant of each sample was added in triplicate to each well of 384-well plate. To this, 20 μl of a 2.5χ mixture of AlphaLISA Anti-Analyte Acceptor beads (10 μg/ml final concentration) and Biotinylated Antibody Anti-Analyte (1 nM final concentration) was added and incubated at 23°C for 60 min. Next, 25 μl of 2χ SA-Donor beads (40 μg/ml final concentration) were added to each well and the plate was incubated at 23°C for 60 min. in the dark. Results were obtained after correlating with the standard curve from human insulin standard dilutions by using the EnVision 2104 Multilabel Reader (Perkin Elmer, Waltham, MA, USA). Insulin concentrations were normalized to total protein levels.
Histological analysis
The pancreas was fixed in 4% paraformaldehyde (PFA), embedded in paraffin and processed into 4-μm sections. Three sections separated by 100 μm were stained with polyclonal guinea pig anti-swine insulin antibody (A0564, 1:500; DakoCytomation, Carpinteria, CA, USA) The sections were counterstained with haematoxylin to stain the total pancreatic area. The left hemi-brain from female mice in the diet group was immersion fixed in 4% PFA for 4 hrs, whereas all male mice in the diet group were transcardially perfused with 4% PFA. Coronal sections of 35 μm were stained with human anti-Aβ antibody WO-2 (1:20,000; Genetics, Schlieren, Switzerland) and CD45 antibody detecting microglia (1:1000; Serotec, Oxford, UK), counterstained with Congo Red to visualize amyloid plaques. The insulin-positive area as percentage of total pancreatic area and amyloid plaque area as percentage of total cortical or hippocampal area analysed were measured from digital micrographs using Adobe Photoshop CS3 extended (version 10).
Statistical analysis
Male and female groups were analysed separately. The acute groups were analysed with univariate ANOVA with APdE9 and IGF-2 genotypes as factors. The analysis of diet groups included diet (STD versus TWD) as a third factor. Metabolic and behavioural tests including repetitions were analysed with ANOVA for repeated measures (ANOVA-R) with time as within-subject, and APdE9 and IGF-2 genotypes and diet as between-subjects factors. Western blot data were analysed first separately for STD and TWD samples, and finally the effect of diet was analysed in a combined ANOVA model with the two genotypes and diet as factors. Spearman correlation coefficients were used to assess correlation between phosphoproteins of the insulin signalling pathway and brain Aβ levels, as well as between these two factors and spatial learning.
Results
Metabolic phenotypes of the transgenic mice
The effects of APdE9 (A+) and IGF-2 (I+) transgenes on glucose metabolism were investigated at two different time points. The first evaluation was performed in female and male mice at 4 months of age. The second experiment assessed the combined effect of genetic (A+ and I+) background and dietary manipulation using standard (STD) or typical Western diet (TWD) in 7-month-old female mice.
Body weight
At 4 months of age, mice carrying the APdE9 genotype, that is A+Iw and A+I+ mice, had a lower body mass as compared to the APdE9 non-transgenic mice with either AwIw or AwI+ genotypes [female mice: F(1,20) ∇ 8.0, P ∇ 0.01; male mice: F(1,12) ∇ 4.1, P ∇ 0.07] (Table 1). At 7 months of age, female mice on TWD, regardless of their genetic background, weighed significantly more than mice harbouring the cognate genotype but fed with the standard diet [F(1,68) ∇ 14.5, P < 0.001]. Furthermore, the weight of these mice continued to rise during the months preceding the metabolic tests [F(1,68) ∇ 83.4, P < 0.001] (Table 1). In contrast to the A+ genotype, the I+ genotype was associated with a slightly higher body mass [F(1,68) ∇ 9.1, P ∇ 0.004] in 4-month-old mice. During the two follow-up months, the body weight tended to further increase, as observed in female mice at 6 and 7 months of age [F(1,68) ∇ 4.0, P ∇ 0.05] (Table 1).
Table 1.
Body weights in the four different genotype groups with respect to age and diet
| Gender | Age (mo) | Diet | Weight in the four genotypes (g, mean + S.E.M.) | |||
|---|---|---|---|---|---|---|
| AwIw | AwI+ | A+Iw | A+I+ | |||
| Acute groups | ||||||
| Male | 4 | STD | 24.1 + 0.8 | 25.6 + 1.9 | 22.5 + 0.7 | 22.6 + 0.5 |
| Female | 4 | STD | 19.4 + 0.4 | 19.4 + 0.5 | 17.5 + 0.4* | 18.8 + 0.5 |
| Diet groups | ||||||
| Female | 6 | STD | 22.2 + 0.4 | 23.7 + 0.6 | 22.3 + 0.7 | 23.3 + 0.6 |
| Female | 6 | TWD | 23.4 + 0.7 | 25.5 + 1.3 | 21.7 + 0.9 | 23.7 + 1.0 |
| Female | 7 | STD | 22.8 + 0.3 | 24.5 + 0.7 | 22.9 + 0.5† | 24.2 + 0.6 |
| Female | 7 | TWD | 27.6+ 1.3† | 30.5 + 1.1‡ | 24.3 + 1.1† | 28.6 + 1.4‡ |
The bold fonts signify significant diet effect, P < 0.05 (t-test).
Significantly different from AwIw controls, P < 0.05 (Dunnett’s test).
Significant increase in follow-up, P < 0.05.
Significant increase in follow-up, P < 0.01 (paired t-test).
Glucose tolerance test (GTT)
At 4 months of age, the fasting blood glucose (B-gluc) levels in both genders of A+Iw or A+I+ mice were significantly lower than those in APdE9 non-transgenic mice (AwIw or AwI+) [female mice: F(1,20) ∇ 6.9, P ∇ 0.02; male mice: F(1,12) ∇ 12.3, P ∇ 0.004], suggesting that the APdE9 genotype associated with lower blood glucose levels. In contrast, the B-gluc levels in AwI+ or A+I+ female mice were significantly elevated as compared to AwIw or A+Iw female mice [F(1,20) ∇ 6.7, P ∇ 0.02]. This effect was not observed among male mice (data not shown). GTT in the 4-month-old mice revealed elevated B-gluc levels in both female and male mice carrying the IGF-2 transgene (AwI+ or A+I+) [female mice: F(1,19) ∇ 105.5, P < 0.001; male mice: F(1,12) ∇ 15.6, P ∇ 0.02]. In contrast, the A+ genotype (A+Iw or A+I+) was associated with lower B-gluc levels among male mice but not in female mice [F(1,12) ∇ 6.0, P ∇ 0.03] (Fig. S1A and B).
In the 7-month-old mice, the IGF-2 genotype (AwI+ or A+I+) [F(1,69) ∇ 26.0, P < 0.001] associated with elevated B-gluc levels when compared to AwIw or A+Iw mice (Fig. 1A and B). The B-gluc levels were also increased in all mice fed with TWD [F(1,69) ∇ 31.3, P < 0.001] as compared with mice on the STD (Fig. 1A and B). In addition, we observed a significant interaction with the transgenic genotype and diet. TWD did not affect B-gluc in A+ mice (A+Iw or A+I+; Fig. 1B), but it significantly elevated B-gluc in APdE9 non-transgenic (AwIw or AwI+) mice (Fig. 1A). Furthermore, the impact of the IGF-2 genotype on B-gluc levels was smaller in A+I+ than in AwI+ mice (Fig. 1A and B). GTT further revealed that both IGF-2 genotype [F(1,67) ∇ 141.0, P < 0.001] and TWD [F(1,67) ∇ 13.0, P ∇ 0.001] elevated B-gluc concentrations in Aw and A+ mice (Fig. 1A and B). The highest B-gluc levels were observed in IGF-2–positive mice on the TWD. Both factors also prolonged the elevation in B-gluc after a single i.p. injection of glucose so that at 60 min, the B-gluc levels remained the highest in IGF-2–positive mice fed with TWD when compared to the other mice [time χ IGF-2, F(3,65) ∇ 53.9, P < 0.001; time χ diet, F(3,65) ∇ 3.6, P ∇ 0.02] (Fig. 1A and B). In addition, there was a significant APdE9 χ IGF-2 [F(1,67) ∇ 6.0, P ∇ 0.02] and APdE9 χ TWD [F(1,67) ∇ 4.8, P ∇ 0.03] interaction, such that the presence of the APdE9 transgene attenuated the effects of both IGF-2 and TWD factors (Fig. 1B).
Fig 1.
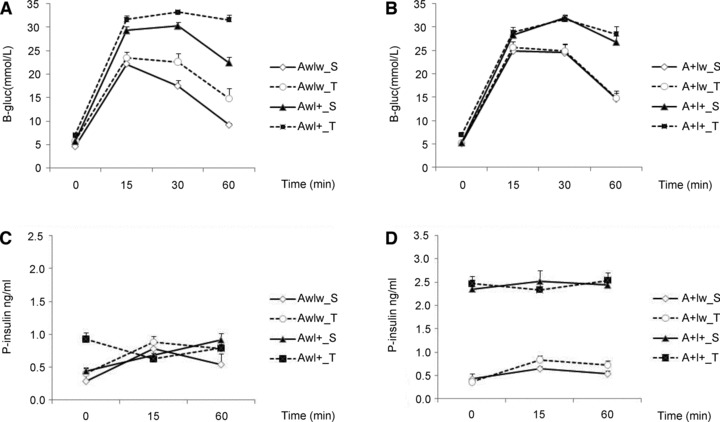
Glucose tolerance test. Time course of B-gluc changes in (A) wild-type littermates (Aw) and (B) APdE9 transgenic (A+) female mice at 7 months of age. Corresponding changes in plasma insulin levels during the test in (C) wild-type littermates (Aw) and (D) APdE9 transgenic (A+) female mice. S: standard diet; T: typical Western diet. Mean + S.E.M.s are shown; n ∇ 4–6 mice/group.
In the 4-month-old mice, fasting plasma insulin levels (P-ins) were significantly dependent on both APdE9 and IGF-2 genotypes and their interaction [female mice F(1,19) > 26.9, P < 0.001; male mice F(1,12) > 21.7, P ≤ 0.001 for all comparisons]. Mice with increased P-ins did not significantly further increase their insulin secretion in response to glucose challenge (Fig. S1C and D).
In the 7-month-old mice, P-ins levels (time 0 in GTT) varied significantly depending on the APdE9 [F(1,67) ∇ 111.1, P < 0.001] and IGF-2 genotype [F(1,67) ∇ 195.0, P < 0.001] and their interaction [F(1,67) ∇ 100.3, P < 0.001]. Mice with both APdE9 and IGF-2 transgenes (A+I+) had approximately threefold higher insulin levels than the other groups (Fig. 1C and D). TWD had only a marginal effect on insulin levels [F(1,67) ∇ 4.0, P ∇ 0.05]. In general, however, mice fed with TWD had higher insulin levels when compared to mice on STD. Mice with increased P-ins did not significantly further increase their insulin secretion in response to glucose challenge (Fig. 1C and D).
Insulin tolerance test (ITT)
ITT was performed only in the 7-month-old female mice. Because B-gluc levels were greatly influenced by the genotypes and the diet, and because gluconeogenesis was triggered already at 40 min. from the insulin injection, we considered the change from individual baseline to the 20 min. time point as a measure of insulin resistance. This analysis revealed significant time χ APdE9 [F(1,68) ∇ 4.1, P < 0.05] and time χ APdE9 χ IFG2 interactions [F(1,68) ∇ 6.4, P ∇ 0.01]. Among IGF-2 non-transgenic mice (Iw), neither APdE9 genotype nor TWD influenced B-gluc response to acute insulin injections (Fig. 2A). In contrast, among I+ mice, the APdE9 transgene (A+I+) induced insulin resistance (Fig. 2B). TWD did not further enhance insulin resistance in these mice (Fig. 2B).
Fig 2.
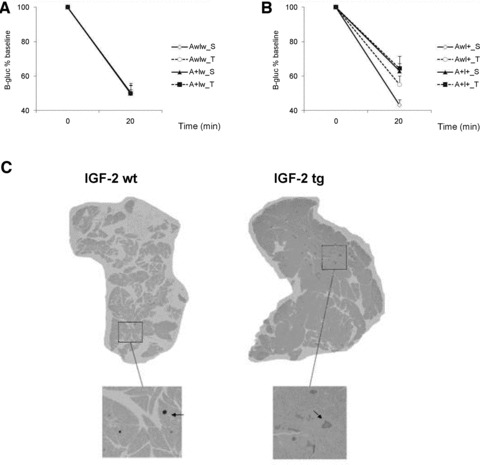
Insulin tolerance test. Changes in glucose levels from baseline in (A) IGF-2 wild-type (Iw) and (B) IGF-2 transgenic (I+) mice. (C) Representative examples of pancreatic β-islets visualized with anti-insulin antibody of IGF-2 wild-type (wt) and IGF-2 transgenic (tg) mice; n ∇ 4–6 mice/group.
Assessment of potential mechanisms of hyperinsulinaemia
To delve into the mechanisms of hyperinsulinaemia caused by the combined effect of APdE9 and IGF-2 transgenes, we first studied the morphology of pancreatic islets in 9-month-old female mice (at the age of perfusion) receiving STD. Immunohistochemistry revealed a significant hypertrophy of islet β cells in I+ mice independent of their APdE9 genotype [F(1,16) ∇ 11.4, P ∇ 0.004] (Figs. 2C and S2A). To further assess insulin production per β-cell, we analysed insulin-1 and insulin-2 mRNA levels in isolated pancreatic islets of 4-month-old female and male mice. This analysis revealed that IGF-2 overexpression in AwI+ or A+I+ mice was associated with lower insulin 1 [F(1,25) ∇ 6.3, P ∇ 0.02] and insulin 2 [F(1,25) ∇ 5.9, P ∇ 0.02] mRNA levels as compared to the IGF-2 non-transgenic AwIw or A+Iw mice. The presence of APdE9 transgene had an opposite effect [Ins1: F(1,25) ∇ 8.9, P ∇ 0.006; Ins2: F(1,25) ∇ 4.8, P ∇ 0.04], such that A+Iw or A+I+ mice displayed increased insulin 1 or insulin 2 mRNA levels as compared to the AwIw or AwI+ mice (Fig. S2B). Hyperinsulinaemia may also arise in response to increased gluconeogenesis. To test this possibility, we assessed the mRNA levels of two rate-limiting enzymes in glucose production, glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, from frozen liver samples of 4-month-old female mice. When normalized to β-actin levels, neither mRNA exhibited significant differences between the genotypes (data not shown).
Finally, hyperinsulinaemia may result from retarded insulin degradation in the liver by IDE. To this end, we determined IDE protein levels in the liver of 4-month-old female mice. There was on average 20% reduction in IDE levels in APdE9-positive mice independent of their IGF-2 genotype (A+Iw or A+I+ mice). However, this reduction was not statistically significant (Fig. S2C). In addition to insulin, IDE is also known to degrade Aβ. Aβ1–40 levels in the liver of 4-month-old APdE9-positive female mice (n ∇ 6) were 1.7 ± 0.2 ng/g. These levels are about six times higher than the plasma insulin concentration of wild-type mice (∼0.3 ng/ml). On the other hand, the affinity of insulin to IDE is about 20-fold higher than that of Aβ. Therefore, it seems unlikely that Aβ would slow down insulin degradation and in this way cause hyperinsulinaemia in these mice.
In conclusion, our data suggest that the extensive hyperinsulinaemia in A+I+ mice results from a combined effect of islet hyperplasia induced by IGF-2 overexpression and increased insulin production (at least at the mRNA level) per islet cell as a result of APdE9 transgene. Additional contributing factors present in A+I+ mice may be the decreased IDE proteins levels in the liver and direct substrate competition between Aβ and insulin for IDE in the liver.
Neurological phenotype of the transgenic mice
Behavioural manifestations of hyperglycaemia and hyperinsulinaemia
Female mice underwent neurological testing at the age of 8 months and male mice at the age of 12 months. Female APdE9-positive mice exhibited hyperactivity in a new environment [F(1,68) ∇ 18.4, P < 0.001]. However, this was dependent on the IGF-2 genotype (F ∇ 4.7, P ∇ 0.03), so that I+ mice were not hyperactive compared to IwAw mice on STD (Fig. 3A and B). Among male mice, the APdE9 genotype had little effect on exploratory activity (Fig. 3E and F). On the other hand, the effect of diet approached significance in male mice [F(1,66) ∇ 3.6, P ∇ 0.06], as all mice on TWD, except the A+I+ mice, moved less than the mice on STD (Fig. 3E and F). Spatial learning was assessed with the Morris swim navigation task. Among 8-month-old female mice, APdE9-positive mice, as expected, were inferior in the task than APdE9 non-transgenic mice (Fig. 3C and D). However, the effect of the APdE9 transgene on escape latency only approached significance [F(1,67) ∇ 3.1, P ∇ 0.08]. In contrast, there was a significant IGF-2 χ diet interaction (F ∇ 4.3, P ∇ 0.04). TWD did not affect learning in IwAw or IwA+ mice but it had a deleterious effect in I+Aw or I+A+ mice (Fig. 3C and D). The genotypes or the diet did not significantly modulate swimming speed, so escape latency can be taken as a valid measure of the task acquisition.
Fig 3.
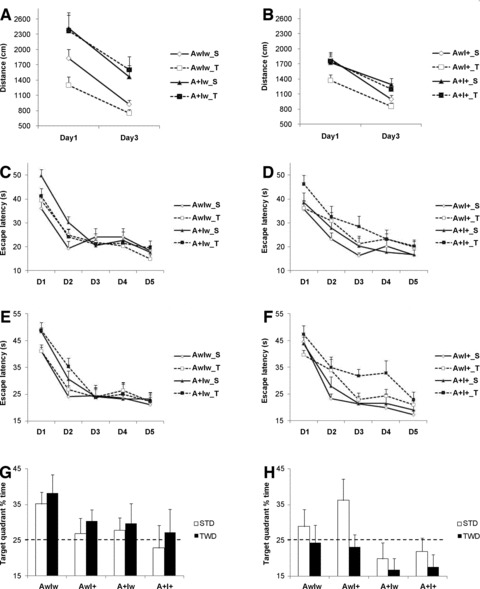
Summary of behavioural tests in mice on standard diet (STD) or high-fat diet (TWD). Ambulatory distance (gross horizontal locomotion) in a new test cage during 10 min. in (A) IGF-2 wild-type (Iw) and (B) IGF-2 transgenic (I+) female mice. Escape latency to reach the hidden platform in the Morris swim navigation task in (C) IGF-2 wild-type (Iw) and (D) IGF-2 transgenic (I+) female mice at 8 months of age. Corresponding data are shown for (E) IGF-2 wild-type (Iw) and (F) IGF-2 transgenic (I+) male mice at 12 months of age. Note the impairment in A+I+ mice on TWD groups in both genders. A probe trial on day 6 tests memory for the platform location in terms of time spent in the target quadrant of the pool. (G) Among female mice, only AwIw mice independent of diet show a significant search bias, whereas (H) AwIw and AwI+ male mice on STD search in the correct target quadrant. The dashed line indicates probability of a random search. S: standard diet; T: typical Western diet; n ∇ 4–6 mice/group.
Search bias was assessed in a separate probe test without the platform on day 6 as an indicator of memory retention. This analysis revealed a significant effect of the APdE9 genotype [F(1,67) ∇ 4.8, P ∇ 0.03] in female mice, such that especially the AwIw mice on STD or TWD showed a significant search bias over chance as compared to the mice with other genotypes (Fig. 3G). The IGF-2 transgene or TWD or their interaction did not affect the performance (Fig. 3G).
Among 12-month-old male mice, task acquisition as measured by escape latency was impaired by the APdE9 genotype in A+Iw or A+I+ mice [F(1,66) ∇ 5.0, P ∇ 0.03] and TWD (F ∇ 4.7, P ∇ 0.03) (Fig. 3E and F). However, the swimming speed was slower in mice on TWD (P ∇ 0.01), which may have contributed to this parameter. Therefore, task acquisition was also analysed by measuring the swim path length. This was significantly longer in A+Iw or A+I+ mice than in APdE9 non-transgenic mice (F ∇ 16.9, P < 0.001). TWD did not affect this parameter (P ∇ 0.40). An additional common measure of task acquisition is the tendency to swim near the wall. This is an ineffective strategy that may prevent or delay the mouse from finding the platform located half-way between the wall and the pool centre. Both the APdE9 genotype (A+Iw or A+I+ mice; F ∇ 11.2, P ∇ 0.001) and TWD (F ∇ 9.7, P ∇ 0.003) prolonged the time the mice were swimming near the pool wall. In fact, A+I+ mice on TWD continued this behaviour until the very last day of task acquisition. Search bias on day 6 was marginally impaired by the APdE9 genotype (A+Iw or A+I+ mice) [F(1,66) ∇ 3.7, P ∇ 0.06]. Only AwIw or AwI+ male mice on STD exhibited significant search bias over chance (Fig. 3H). In summary, whereas significant hyperactivity was only observed in female A+Iw mice independent of the diet, spatial learning was most robustly impaired in A+I+ mice on TWD in both genders.
Effects of genotype and diet on Aβ, τ, insulin and insulin signalling in the cerebral cortex
APP processing, Aβ pathology and IDE expression
Next, we assessed the effects of genotype and diet on APP processing, Aβ pathology and IDE expression in the brain, focusing on the ventral posterior (temporo-occipital) cortex. According to the Western blot analysis of total protein lysates, APdE9-positive mice showed an expected significant increase in APP695 isoform (∼2-fold) and APP C-terminal fragment (CTF) (∼3.5-fold) levels when compared to wild-type mice (AwIw; Fig. 4A). Apart from these differences, examination of female (Fig. 4B and D) and male (Fig. 4C and E) mice did not reveal significant changes in GAPDH-normalized total APP or APP CTF levels with respect to genotypes or diet.
Fig 4.
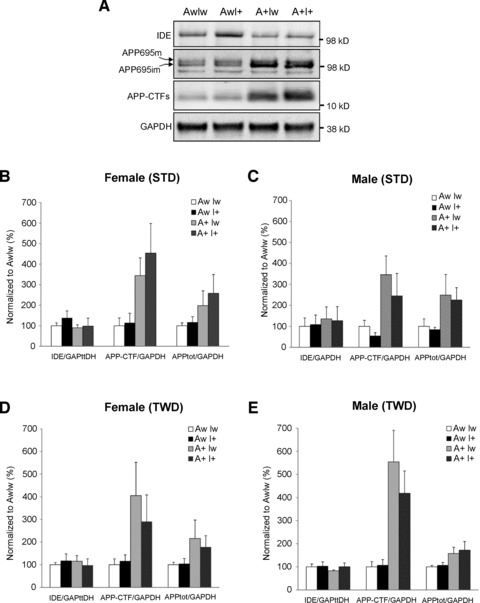
Effects of genotype and diet on APP processing and IDE expression in the ventral posterior cortex. (A) Total protein lysates of female mice on STD were analysed with Western blotting using the APP C-terminal–specific antibody. GAPDH-normalized levels of total APP (APPtot), APP C-terminal fragments (APP CTF), and IDE in (B) female mice on STD, (C) male mice on STD, (D) female mice on TWD and (E) male mice on TWD. Mean + S.D.s are shown; n ∇ 4–6 mice/group.
In addition, insoluble Aβ1–40 (Fig. 5A and B) and Aβ1–42 (Fig. 5C and D) levels were measured from the same protein lysates. Among female mice, Aβ levels did not differ between the genotypes or diet groups. However, IGF-2 χ diet interaction revealed a borderline significance for increased Aβ1–40 levels in A+I+ female mice on TWD [F(1,26) ∇ 4.0, P ∇ 0.06] (Fig. 5A). In contrast, male A+I+ mice on STD or TWD had lower Aβ1–40 [F(1,32) ∇ 5.6, P ∇ 0.03] levels as compared to A+Iw mice on STD or TWD (Fig. 5B). Insoluble Aβ1–42 levels did not differ between the genotypes or diet groups in either female or male mice (Fig. 5C and D). However, male A+I+ mice showed a higher Aβ 42/40 ratio than A+Iw mice (F ∇ 8.6, P ∇ 0.006; Fig. 5F), while neither the IGF-2 transgene nor the diet modulated this ratio in female mice (Fig. 5E).
Fig 5.
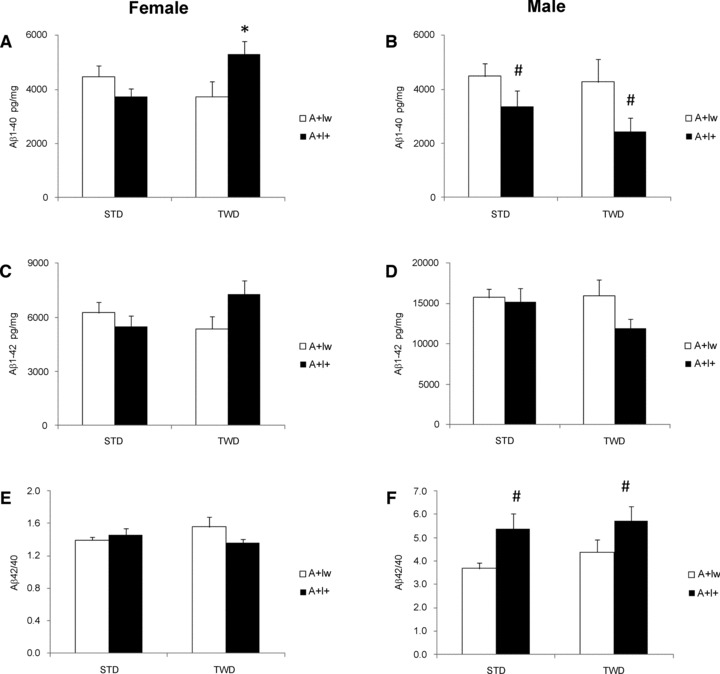
Aβ levels in the cortex of female and male APdE9 mice without (Iw) or with (I+) IFG2 transgene and on standard diet (STD) or typical Western diet (TWD). ELISA results (mean + S.E.M.) are shown separately for Aβ1-40 (A and B), Aβ1-42 (C and D) and the Aβ42/40 ratio (E and F). *Different from the Iw genotype mouse on the same diet (P < 0.05, t-test). #Main effect of IGF-2 (P < 0.05, ANOVA); n ∇ 4–6 mice/group.
Aβ load was assessed in histological sections in male A+I+ and A+Iw mice. Both hippocampus and the overlying parietal cortex were analysed, but no overall changes in Aβ load with respect to IGF-2 genotype or diet were found (P > 0.11; data not shown). In contrast, IGF-2 transgene had a significant effect on microglial activation as assessed by CD45-positive relative surface area (Fig. 6A). A+I+ mice had less microglial activation both in the cortex [F(1,32) ∇ 6.4, P ∇ 0.02; Fig. 6B] and the hippocampus (F ∇ 7.1, P ∇ 0.01) than other groups (data not shown). Finally, we assessed the effects of the genotype and diet on IDE protein levels in the ventral posterior cortex of female and male mice (Fig. 4A–E). A significant overall genotype effect by the IGF-2 transgene was observed in female mice on STD (F ∇ 4.0, P ∇ 0.02; Fig. 4B), but not in male mice on either STD or TWD (Fig. 4C and E). GAPDH-normalized IDE levels were increased ∼1.4-fold in female AwI+ mice on STD when compared to the other female mice on STD.
Fig 6.
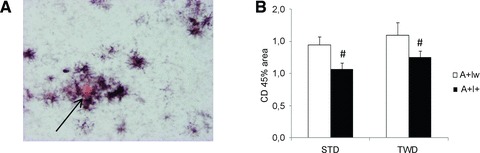
Effect of IGF-2 transgene on microglial activation in mice on standard diet (STD). (A) Representative section of the A+Iw mouse cortex double-stained with Congo Red (arrow) to show the amyloid plaque core and CD45 antibody to show activated microglia/macrophages around the plaques. (B) Mean (+S.E.M.) surface area as percentage of total analysed area that was positive for CD45 in A+Iw and A+I+ male mice on STD or typical Western diet (TWD). #Main effect of IGF-2 (P < 0.05, ANOVA); n ∇ 4–6 mice/group.
τIsoform expression and phosphorylation status
To elucidate the possible effects of the genotype and diet on τ isoform expression and Ser202 phosphorylation status in the brain tissue, Western blotting analysis was performed from the total protein lysates extracted from the ventral posterior cortex (Fig. 7). Using protein lysate from HEK293 cells transiently overexpressing human τ-0N4R isoform as a size control, three major τ isoforms were detected in both female (Fig. 7A) and male (Fig. 7D) mice on STD independently of the genotype. Based on the previously determined τ isoform profile in the adult mouse brain [22] as well as on the estimated molecular weights of τ protein bands, it was concluded that the three τ-specific bands most likely represent 0N4R, 1N4R and 2N4R τ isoforms. Comparison of the GAPDH-normalized total τ (all isoforms) levels or τ phosphorylation status, as assessed with a τ-phospho-Ser202-specific antibody (AT8), between the different genotypes revealed no differences in female or male mice on STD (Fig. 7B and E). Interestingly, all female mice on TWD independently of the genotype showed a prominent appearance of a τ isoform (∼62 kD) that was not observed in female mice on STD (Fig. 7A). Based on the molecular weight, this isoform likely represents τ-2N3R. Furthermore, in female mice on TWD, AT8 antibody staining revealed a consistent increase in the phosphorylation of a τ-specific band, which was approximately the same in size as the τ-0N4R (∼52 kD) isoform, when compared to female mice on STD (Fig. 7A). Comparison of total τ and phosphorylated τ levels between different genotypes did not reveal statistically significant changes in female mice on TWD (Fig. 7A and C). Male mice on STD showed a similar τ isoform expression pattern as the female mice on STD (Fig. 7A and D), and no statistically significant changes were observed in total τ levels between the genotypes (Fig. 7E). On the other hand, male mice on TWD showed similar kind of alterations in τ isoform expression profile as the female mice on TWD (Fig. 7A and D). However, due to fluctuation of the expression of specific τ isoforms, total τ levels did not significantly differ between the genotypes among male mice on TWD (Fig. 7F). Male mice on STD or TWD revealed a moderate, but statistically non-significant increase in the phosphorylation status of τ in the A+Iw and A+I+ mice when compared to AwIw mice (Fig. 7E and F). There were no significant correlations with measures of spatial memory and the levels of total τ or phosphorylated τ in mice on STD or TWD (data not shown).
Fig 7.
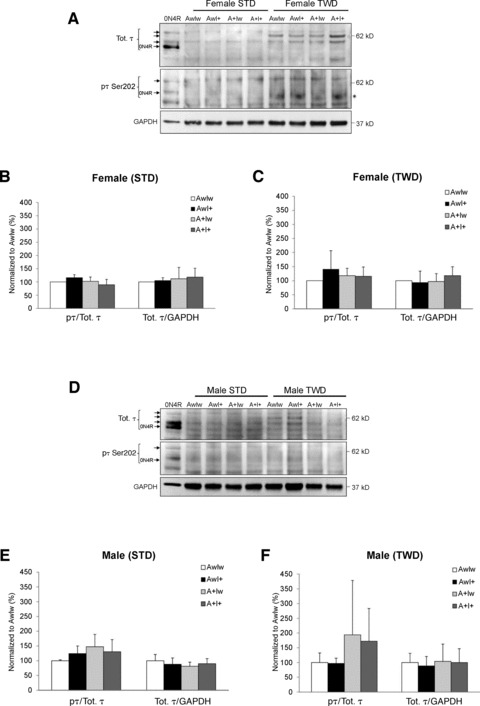
Effects of genotype and diet on τ isoform expression and τ Ser202 phosphorylation in the ventral posterior cortex. Total protein lysates of female (A) and male (D) mice on standard diet (STD) or typical Western diet (TWD) were analysed with Western blotting using the total τ (Tot. τ) and Ser202-phosphorylation-specific (pτ Ser202, AT8) antibodies. Quantification of GAPDH-normalized levels of the total τ and the τ phosphorylation status (pτ/Tot. τ) in female (B, C) or in male (E, F) mice on STD (B, E) and TWD (C, F). Phosphorylation status of ∼52-kD τ isoform in female mice on TWD was increased when compared to female mice on STD (A, indicated as*). τ levels in wild-type female mice (AwIw) were set to 100% in individual Western blot gels (B, C). Mean + S.D.s are shown; n ∇ 4–6 mice/group.
Insulin levels and insulin signalling
Because the A+I+ mice showed peripheral hyperinsulinaemia and insulin resistance, we next studied the effects of the genotype and diet on the levels of insulin and components of the insulin signalling pathway in the brain. Total protein-normalized insulin levels in the ventral posterior cortex were not significantly altered in the female (Fig. 8A) or male (Fig. 8B) mice with respect to the genotype or the diet. Although there were no statistically significant changes based on the ANOVA analysis with multiple comparisons, brain insulin levels in male A+I+ mice on TWD were significantly decreased an average 20% when compared to AwIw mice on TWD (P < 0.05). There were no correlations with fasting insulin levels in plasma and brain in mice on STD or TWD (data not shown).
Fig 8.
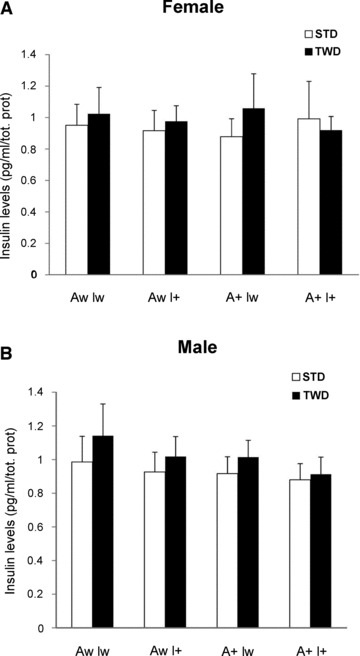
Effects of genotype and diet on insulin levels in the ventral posterior cortex. Total protein-normalized insulin levels were measured from female (A) or male (B) mice on standard diet (STD) or typical Western diet (TWD). Mean + S.D.s are shown; n ∇ 4–6 mice/group.
Western blot analysis of phosphorylation sites in GSK3α (pSer21), GSK3β (pSer9) and Akt (pSer473) revealed that the IGF-2-positive genotype significantly increased the inhibitory phosphorylation of GSK3β (pGSK3β/total GSK3β) in female AwI+ and A+I+ mice on STD [F(1,18) ∇ 7.9, P ∇ 0.01; Fig. 9A and C], but not in female (Fig. 9D) or male mice on TWD (Fig. S3B). On average 1.4-fold increase in pGSK3β/total GSK3β ratio was observed in female A+I+ mice on STD when compared to female A+Iw mice on STD. This difference was not due to alterations in total GSK3β levels (Fig. 9A and C). Inhibitory phosphorylation of GSK3α (pGSK3α/total GSK3α) was similarly increased in IGF-2 transgenic female mice on STD [F(1,18) ∇ 6.2, P ∇ 0.02; Fig. 9A and C]. An ANOVA model including both the genotype and the diet as factors revealed a significant diet χ IGF-2 interaction in both GSK3α [F(1,39) ∇ 11.2, P ∇ 0.002] and GSK3β (P ∇ 0.02) phosphorylation in female mice. The diet alone had a significant effect on GSK3α (P ∇ 0.002), but not GSK3β phosphorylation (P > 0.50). None of these effects was significant in male mice. In contrast, Ser473 phosforylation of Akt was not affected in female or male mice of any genotype on STD or TWD (Figs. 9B–D and S3A and B).
Fig 9.
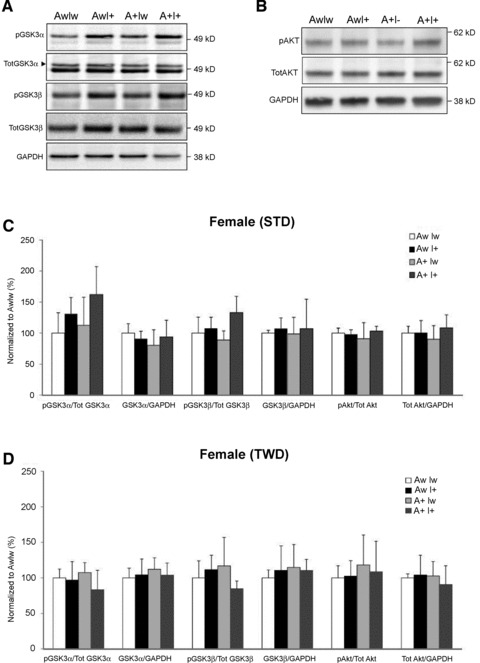
Effects of genotype and diet on insulin signalling in the ventral posterior cortex. (A) Total protein lysates of female mice on standard diet (STD) were analysed with Western blotting using antibodies specific for phosphorylated GSK3α (Ser 21) and GSK3β (Ser 9), as well as total GSK3α and GSK3β. (B) Western blots from the same mice analysed using antibodies specific for phosphorylated Akt (Ser473) and total Akt. Phosphorylated and total levels of GSK3α, GSK3β and Akt in (C) female mice on STD and (D) female mice on typical Western diet (TWD). Mean + S.D.s are shown; n ∇ 4–6 mice/group.
The influence of genetic and dietary factors on insulin signalling in the cerebral cortex is summarized in Table 2. Notably, APdE9 genotype itself did not significantly modulate insulin signalling pathways in female mice, but increased the ratio of pGSK3α to total GSK3α in male mice [STD: F(1,20) ∇ 5.8, P ∇ 0.03; Fig. S3A; TWD: [F(1,16) ∇ 7.8, P ∇ 0.01; Fig. S3B]. To test whether this change relates to the observation that IGF-2 transgene decreases Aβ1–40 levels and increases Aβ42/40 ratio in male APdE9 mice, we ran correlation analyses between these variables. Interestingly, in IGF-2 wild-type mice (AwIw or A+Iw), pGSK3β positively correlated with Aβ1–40 levels (r ∇ 0.76, P < 0.01) and negatively with Aβ42/40 ratio (r ∇ −0.82, P < 0.01). Total levels of GSK3α or GSK3β exhibited an opposite correlation with Aβ1–40 levels and Aβ42/40 ratio (Aβ1–40: r ∇ −0.69 and −0.64; Aβ42/40: r ∇ 0.68 and 0.64, all P < 0.05). In contrast, no significant correlation between Aβ and GSK3 variables was found among IGF-2 transgenic mice (AwI+ or A+I+). Furthermore, brain insulin levels positively correlated with increased inhibitory phosphorylation of GSK3β in female mice regardless of the genotype on STD (r ∇ 0.49, P < 0.05), but not on TWD (r ∇ −0.04, P ∇ 0.84).
Table 2.
Influence of APdE9 and IGF-2 genotypes on key components associated with insulin signalling pathway in mice on standard (STD) and typical Western (TWD) diet
| Females, STD* | Females, TWD* | |||
|---|---|---|---|---|
| APdE9 | IGF-2 | APdE9 | IGF-2 | |
| Akt | 0 | 0 | 0 | 0 |
| pAkt | 0 | 0 | 0 | 0 |
| GSK3α | 0 | 0 | 0 | 0 |
| pGSK3α | 0 | + | 0 | 0 |
| GSK3β | 0 | 0 | 0 | 0 |
| pGSK3β | 0 | + | 0 | 0 |
| IDE | (−) | (+) | 0 | 0 |
| Males, STD* | Males, TWD* | |||
| Akt | 0 | 0 | 0 | 0 |
| pAkt | 0 | 0 | 0 | 0 |
| GSK3α | 0 | 0 | + | 0 |
| pGSK3α | + | 0 | + | 0 |
| GSK3β | 0 | 0 | 0 | − |
| pGSK3β | 0 | 0 | 0 | 0 |
| IDE | 0 | 0 | 0 | 0 |
Parentheses show a trend with borderline significance, + indicates increased levels, − decreased levels and 0 no effect.
Finally, we wanted to study whether changes in Aβ levels and insulin signalling pathways contributed to the observed learning impairment, particularly in A+I+ mice on TWD. To this end, we correlated these factors with the main measure of spatial learning in the swim navigation task, the escape latency (Table 3). First, high Aβ42/40 ratio in general appeared to correlate with poor learning in male mice. An opposite correlation was found in female mice. Secondly, activation of Akt and consequent inhibitory phosphorylation at Ser9 of GSK3β correlated with improved learning in female mice, particularly in APdE9-positive mice. In this regard, it is intriguing that A+I+ mice on STD demonstrated increased pGSK3β levels compared to other groups (Fig. 9C), while this increase was blunted in A+I+ mice on TWD (Fig. 8D), which also showed impaired spatial learning. Third, despite the fact that most correlations were opposite in female and male mice, the correlation between good spatial learning and GSK3β-Ser9 phosphorylation in APdE9-positive mice was evident and consistent across both genders (Table 3).
Table 3.
Correlation of escape latency in Morris swim task to Aβ and to the key components in the insulin signalling pathway
| Females‡ | |||||
|---|---|---|---|---|---|
| All | Aw | A+ | Iw | I+ | |
| Aβ1–40 | 0.33 | N/A | 0.33 | 0.47 | 0.17 |
| Aβ1–42 | 0.28 | N/A | 0.28 | 0.40 | 0.23 |
| Aβ42/40 | −0.31 | N/A | −0.31 | −0.53* | 0.03 |
| pAkt | −0.32* | −0.42* | −0.32 | −0.33 | −0.31 |
| pGSK3α | -0.29* | −0.28 | −0.38 | −0.11 | −0.54* |
| pGSK3β | -0.32* | −0.14 | −0.57† | −0.37 | −0.07 |
| Males‡ | |||||
| Aβ1–40 | −0.28 | N/A | −0.28 | −0.49* | −0.25 |
| Aβ1–42 | −0.35* | N/A | −0.35* | −0.36 | −0.37 |
| Aβ42/40 | 0.32 | N/A | 0.32 | 0.58† | 0.17 |
| pAkt | 0.15 | 0.39 | −0.15 | 0.38 | 0.11 |
| pGSK3α | 0.02 | 0.25 | −0.25 | −0.11 | 0.05 |
| pGSK3β | -0.10 | 0.22 | −0.47* | −0.34 | 0.11 |
Negative correlation indicates good learning.
P < 0.01.
P < 0.05.
In summary, it is apparent that genotype and diet encompassed differential effects in terms of Aβ pathology, τ isoform expression, τ phosphorylation, as well as GSK3 activities between female and male mice. Importantly, high-fat diet or IGF-2 genotype did not increase Aβ plaque burden in the brain of female or male mice. In male mice, however, IGF-2 transgene increased Aβ42/40 ratio, which in turn correlated with poor spatial learning. Conversely, increased inhibitory phosphorylation of GSK3β correlated with good spatial learning and the inhibitory phosphorylation of both GSK3β and GSK3α were increased in A+I+ female mice on the standard diet, but not on the high-fat diet. Finally, high-fat diet, but not any of the genotypes, increased the expression of a specific τ isoform (∼62 kD) and the Ser202 phosphorylation of τ (∼52 kD) in female mice when compared to the female mice on standard diet.
Discussion
Although several epidemiological studies have shown an intimate link between AD and T2DM [1-7], the molecular mechanisms underlying this co-morbidity are not well-understood. Here we demonstrate that the APdE9 transgene enhanced hyperinsulinaemia and insulin resistance induced by genetic or dietary factors. Extensive hyperinsulinaemia and insulin resistance in APdE9 χ IGF-2 (A+I+) overexpressing mice likely resulted from increased insulin secretion due to the pancreatic islet hyperplasia and preserved insulin production by islet cells in combination. These may have been further exacerbated by the competition of insulin and peripheral Aβ for IDE-mediated degradation in the liver. This is an important finding because it suggests that increased circulating levels of Aβ can aggravate diabetic phenotype, at least in mouse models of AD. Similar exacerbation of diabetic phenotype was recently observed in APP+-ob/ob and APP+-NSY mice, which are well-established AD mouse models with hyperinsulinaemia and insulin resistance [18]. In our study, hyperglycaemia and insulin resistance did not exacerbate Aβ pathology, the expression of τ isoforms, or the phosphorylation status of τ in the brain of APdE9 transgenic mice, which is another widely used mouse model of AD [19]. These results with respect to Aβ pathology are in contrast to diet-induced hyperinsulinaemia and insulin resistance in APPswe mice [16], but fully consistent with observations in APP+-ob/ob and APP+-NSY mice [18]. In contrast to APP+-ob/ob and APP+-NSY mice, however, we did not observe increased cerebral inflammation or cerebral amyloid angiopathy in A+I+ mice. Quite the opposite, A+I+ mice showed less microglial activation both in the cortex and hippocampus. Furthermore, extensive peripheral hyperinsulinaemia in combination with insulin resistance in aged A+I+ mice resulted in impaired spatial learning only after dietary manipulation with a high-fat diet (21% fat, 0.15% cholesterol). This suggests that the cognitive phenotype of A+I+ mice was influenced by dietary lipids in conjunction with the diabetic genotype rather than the diabetic genotype itself. Consistent with this, we observed decreased GSK3α (Ser21) and GSK3β (Ser9) inhibitory phosphorylation in female A+I+ mice after dietary manipulation with the high-fat diet. Furthermore, brain insulin levels were moderately decreased in A+I+ mice on high-fat diet similarly to those seen previously in APP+-ob/ob mice on standard diet [18]. Because increased GSK3β-Ser9 inhibitory phosphorylation correlated with improved spatial learning in all genotypes, but especially in APdE9-positive mice, this mechanism likely contributes to the selective spatial learning impairment observed only in A+I+ mice on high-fat diet. Our findings are in line with the recent observations in triple transgenic 3xTg-AD mice, in which high-fat diet increased the activity of c-Jun N-terminal kinase and thereby augmented insulin receptor substrate-1 (IRS-1) phosphorylation at the Ser616 site [23]. Because Ser616 phosphorylation leads to the uncoupling of IRS-1 from PI3-kinase and Akt [24], it was suggested that the activity of GSK3α and GSK3β is affected due to the disrupted insulin signalling [23]. Collectively, our data suggest that hyperglycaemia and insulin resistance alone may not lead to cognitive impairment unless these metabolic changes are accompanied by high-fat and high-cholesterol diet. The results related to metabolic, pathological, behavioural and insulin signalling phenotypes with respect to genotype and diet are summarized in Table 4.
Table 4.
Summary of the metabolic, behavioural, pathological and insulin signalling changes in A+I+ mice on standard (STD) or typical Western (TWD) diet compared to corresponding A+I− mice
| A+I+ STD | A+I+ TWD | |
|---|---|---|
| Metabolic phenotype in periphery | ||
| Hyperglycaemia | - | - |
| Hyperinsulinaemia | - | - |
| Insulin resistance | - | - |
| Behavioural phenotype | ||
| Spatial learning | – | ↓ |
| Aβ pathology | ||
| APP expression and processing | – | – |
| Aβ levels† | – | – |
| IDE levels | – | – |
| τ Pathology | ||
| τ Expression (total τ levels)* | – | – |
| τ Phosphorylation | – | – |
| Inflammation | ||
| CD45+ microglia | ↓ | - |
| Insulin and insulin signalling in brain | ||
| Insulin levels | – | ↓ |
| Akt activity | - | – |
| GSK3α activity | ↓ | – |
| GSK3β activity | ↓ | – |
Female mice on TWD diet showed specific appearance of 62 kD τ isoform (2N3R) when compared to female mice on STD.
Male A+I+ mice on STD and TWD showed a decrease in Aβ1–40 levels, whereas the ratio of Aβ42/40 was increased.
Although the expression levels of total soluble τ isoforms and the Ser202 phosphorylation status of τ remained unchanged relative to the genotype in mice on standard or high-fat diet, there were extensive differences in the expression of specific τ isoforms in female mice on high-fat diet as compared to the standard diet. Importantly, τ isoform of ∼62 kD was strongly up-regulated in all female mice on high-fat diet regardless of the genotype. Based on the size and the previous findings in adult mouse brain [22], it was concluded that the ∼62 kD τ represents the τ-2N3R isoform. Similar kind of τ isoform expression profile was observed in male mice on high-fat diet, but the appearance of the ∼62 kD τ isoform varied from mouse to mouse with the same genotype. Similarly to our data, a recent study showed that high-fat diet in 3xTg-AD mice increased the soluble τ levels when compared to 3xTg-AD mice on standard diet [25], indicating that diet plays an important role in the regulation of τ expression. Apart from the altered τ isoform expression profile, a robust enhancement of Ser202 phosphorylation of ∼52 kD τ was detected in female mice on high-fat diet when compared to mice on standard diet. This is an important finding because GSK3β is the key kinase responsible for Ser202 phosphorylation in τ, providing a direct link between τ phosphorylation and insulin signalling pathway. The fact that we observed hyperphosphorylation of τ in mice on high-fat diet is consistent with a previous study reporting high-cholesterol diet-induced τ hyperphosphorylation in apolipoprotein E-deficient mice [26]. However, because male mice on high-fat diet did not show prominent changes in the phosphorylation status of τ, it is likely that in addition to diet, gender-specific factors contribute to the underlying τ phenotype.
To date, only a few animal studies have addressed the co-occurrence of diabetic phenotype (hyperglycaemia/glucose intolerance and/or hyperinsulinaemia/insulin resistance) and AD pathology [16-18]. These studies have been partially contradictory with respect to Aβ production and accumulation in the brain, similarly to the population-based neuropathological studies in human beings [1, 7, 9, 10]. Diet-induced insulin resistance in Tg2576 mice [16] promoted Aβ40 and Aβ42 generation and consequently exacerbated Aβ plaque burden in the brain, while no effect on Aβ load was observed in APP+-ob/ob or APP+-NSY mice [18]. It should be noted, however, that both of these studies demonstrated significant cognitive dysfunction and alterations in the activity of insulin signalling pathway components, such as Akt and/or GSK3, in the brain. Interestingly, IRS-2–deficient mice crossed with Tg2576 mice showed alterations in both insulin receptor and IGF1 receptor signalling (insulin and IGF-1 resistance), which coincided with decreased APP processing and Aβ40 levels in a gender-independent manner in the brain. Here we also observed slightly decreased Aβ40 levels in A+I+ mice on the standard diet in a gender-independent manner, while the Aβ42 levels particularly in male mice remained unchanged leading to increased Aβ42/40 ratio. Similar increase in Aβ42/40 ratio was also observed in male A+I+ mice after dietary manipulation with high-fat diet, suggesting that the IGF-2 genotype modulated γ-secretase cleavage at the γ-site of the APP C-terminal fragment. This idea is consistent with the fact that we observed no changes in APP synthesis or processing, such as APP holoprotein or APP C-terminal fragment levels, in these mice. In this context, it should be noted that intranasal administration of insulin in patients with early AD improved cognition and also decreased the ratio of Aβ42/40 in plasma [27], suggesting that alterations in insulin signalling may modulate γ-secretase activity also in human beings. Changes in γ-secretase activity have been previously linked to the diet-induced insulin resistance in the brain of Tg2576 mice [16]. Increased γ-secretase activity, which was assessed using the in vitro APP intracellular domain generation assay, displayed a positive correlation with decreased inhibitory phosphorylation status of both GSK3α (Ser 21) and GSK3β (Ser 9). This further suggests that alterations in brain insulin levels and insulin signalling can influence Aβ generation. In line with this idea, GSK3α has been shown to regulate Aβ generation both in vitro and in vivo [15]. However, it appears that the effects of glucose intolerance, hyperinsulinaemia and insulin resistance on Aβ generation and deposition are highly dependent on the AD mouse model used. For instance, cerebral amyloid angiopathy observed in APP+-ob/ob mice is likely related to the increased propensity of the aging APP+ (APP23) mice, which mainly produce Aβ1–40, to develop prominent deposition of cerebrovascular amyloid [28]. In contrast, cerebral amyloid angiopathy is uncommon in APdE9 mice that primarily produce Aβ1–42 (our unpublished observation).
The A+I+ mice of this study were generated by cross-breeding a well-known AD mouse model, APdE9 [19] with diabetic IGF-2 overexpressing mice. In these mice, IGF-2 overexpression was specifically targeted to pancreas under the control of rat insulin-1 promoter [20]. IGF-2-overexpressing mice developed T2DM phenotype, which was accompanied with islet hyperplasia, increased β-cell mass, hyperinsulinaemia and insulin resistance [20]. Similar kind of morphological changes in pancreas were also observed in leptin-deficient ob/ob mice [29], indicating comparable phenotypic features in these two diabetic mouse models used for cross-breeding with different AD mouse models. Because IGF-2 overexpression was targeted specifically to pancreas and because it was not detected in the liver or muscle [20], it is therefore unlikely that I+ mice had significant amounts of IGF-2 in the circulation. However, we cannot completely exclude the possibility that the pancreas-derived IGF-2 had entered from blood to the brain via a receptor-mediated transcytosis mechanism through the blood–brain barrier, as shown previously [30]. Recent evidence suggests that IGF-2 is a potent enhancer of memory consolidation [31]. It is thus possible that improved spatial memory in AwI+ mice (Fig. 3H) could have resulted from a direct IGF-2 action in the brain. On the other hand, gene expression profiling has demonstrated significantly increased IGF-2 expression in the brain of APPswe overexpressing mice (Tg2576) showing memory impairment [32]. It is unknown whether APP or its cleavage products affect IGF-2 expression in the peripheral organs, such as pancreas. However, such regulation could explain the exacerbated diabetic phenotype observed in the A+I+ mice. APP/PS1dE9 overexpression in APdE9 mice was driven by a neuron-specific prion promoter. Therefore, it is possible that APP cleavage products, such as circulating soluble APP (sAPP) and/or Aβ, rather than APP holoprotein itself could specifically be responsible for the regulation of peripheral IGF-2 expression. Accordingly, sAPPα can induce the expression of several neuroprotective genes, including IGF-2 [33]. Collectively, this body of evidence suggests that combination of the IGF-2 and APdE9 genotypes in A+I+ mice influences the phenotypic outcome (e.g. hyperinsulinaemia, insulin signalling and cognitive functions) via complex regulatory mechanisms.
In conclusion, this study provides experimental in vivo evidence that Aβ- producing APdE9 genotype may exacerbate hyperinsulinaemia and insulin resistance in mice. This finding agrees with epidemiological studies suggesting that AD can be a risk factor for T2DM [1]. We did not find evidence supporting the idea that T2DM would increase brain Aβ load or τ pathology. However, in accordance with previous studies [16-18], our study suggests that diabetic phenotype can interfere with the production of specific Aβ species, thus affecting especially the Aβ42/40 ratio and the inflammatory reaction around Aβ plaques. These factors may thus explain the increased AD pathology induced by T2DM. Also, consistent with the previous findings that insulin resistance did not influence NFT pathology in the human brain [9], our study emphasizes that genetically induced insulin resistance does not modulate τ isoform expression or the phosphorylation status of τ. Our study also confirms the robust difference between peripheral and brain insulin levels, reinforcing the concept that hyperinsulinaemia/insulin resistance may affect brain insulin uptake through the blood–brain barrier as previously suggested [13, 18]. Finally, comparison of genetic and diet-induced T2DM suggests that high-fat diet, which is the most common way to induce T2DM phenotype in rodents, has important effects of its own in addition to inducing hyperglycaemia and insulin tolerance. According to our study, only the combination of the strongest T2DM-associated genetic background (A+I+) and a high-fat diet led to cognitive impairment. In human beings, this would suggest that persons with a genetic background predisposing them to T2DM can significantly influence their risk of getting AD by active dietary choices. In addition to diet effects, the data from this study provide potential pathophysiological insights to the clinical settings of AD and T2DM. First, it is likely that the novel therapeutic strategies targeted to mitigate insulin signalling defects in early AD or its prodrome are feasible treatment options. In accordance with this idea, it was recently shown that intranasal administration of insulin improved cognition in patients with early AD or mild cognitive impairment [27]. Second, the aggravated diabetic phenotype in Aβ-producing mice (A+I+) implies that it would be reasonable to monitor the diabetic status of AD patients during the course of the disease, as all the intervention measures related to altered glucose metabolism in the periphery could be beneficial in preventing T2DM in AD patients.
Acknowledgments
The authors thank Saara Stavén and Elina Latonummi for technical assistance. Dr. David Borchelt and Joanna Jankowsky (Johns Hopkins University, Baltimore, MD, USA) for the APPswe/PS1dE9 breeder mice and Dr. Fatima Bosch (Universitat Autònoma de Barcelona, Bellaterra, Spain) for the IGF-2 breeder mice. This study was supported by Sigrid Juselius Foundation (H.T., M.H.), Finland and Centre for International Mobility (Finland) grant to V.K., the Kuopio University Hospital (EVO grant 5772708 to H.S.), the strategic funding of the University of Eastern Finland (M.H., H.T., A.H., H.S.) and the Health Research Council of the Academy of Finland (M.H., A.H., H.S., H.T.).
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Glucose tolerance test in the acute group mice at the age of 4 months. Time course of B-gluc changes in (A) female and (B) male mice of the four genotypes. Corresponding changes in plasma insulin levels during the test in (C) female and (D) male mice. Mean + SEMs are shown, n =4–6 mice/group.
Fig. S2. (A) Area of beta-islets as percent of total area of pancreatic crosssections of 7-month-old female mice of different genotypes on standard diet. ** I+ mice differed from Iw mice (p < 0.01, ANOVA). (B) Insulin 1 and insulin 2 mRNA levels in different genotypes isolated from 4-month-old female mice (all on standard diet). *A+ mice differed from Aw mice (p < 0.05, **p < 0.01, ANOVA); #I+ mice differed from Iw mice (p < 0.05, ANOVA). (C) Protein levels in the liver of insulin-degrading enzyme (IDE) in 4-month-old female mice. Mean + SEMs are shown, n ∇ 4–6 mice/group.
Fig. S3. Effects of genotype and diet on insulin signaling in the ventral posterior cortex. (A) Total protein lysates of male mice on standard diet (STD) were analyzed with Western blotting using antibodies specific for phosphorylated GSK3α (Ser 21), GSK3β (Ser 9), and Akt (Ser473) as well as total GSK3α, GSK3β, and Akt.
Quantifications of the protein levels are shown. (B) Quantification of the Western blots from male mice on typical Western diet (TWD) with the same antibodies. Mean + SDs are shown, n ∇ 4–6 mice/group.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Janson J, Laedtke T, Parisi JE, et al. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–81. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 2.Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63:1187–92. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 3.Vanhanen M, Soininen H. Glucose intolerance, cognitive impairment and Alzheimer’s disease. Curr Opin Neurol. 1998;11:673–7. doi: 10.1097/00019052-199812000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Irie F, Fitzpatrick AL, Lopez OL, et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch Neurol. 2008;65:89–93. doi: 10.1001/archneurol.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ronnemaa E, Zethelius B, Sundelof J, et al. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology. 2008;71:1065–71. doi: 10.1212/01.wnl.0000310646.32212.3a. [DOI] [PubMed] [Google Scholar]
- 6.Xu WL, von Strauss E, Qiu CX, et al. Uncontrolled diabetes increases the risk of Alzheimer’s disease: a population-based cohort study. Diabetologia. 2009;52:1031–9. doi: 10.1007/s00125-009-1323-x. [DOI] [PubMed] [Google Scholar]
- 7.Ahtiluoto S, Polvikoski T, Peltonen M, et al. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology. 2010;75:1195–202. doi: 10.1212/WNL.0b013e3181f4d7f8. [DOI] [PubMed] [Google Scholar]
- 8.Cannon CP. Mixed dyslipidemia, metabolic syndrome, diabetes mellitus, and cardiovascular disease: clinical implications. Am J Cardiol. 2008;102:5L–9L. doi: 10.1016/j.amjcard.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaki T, Sasaki K, Tanizaki Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–70. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- 10.Alafuzoff I, Aho L, Helisalmi S, et al. Beta-amyloid deposition in brains of subjects with diabetes. Neuropathol Appl Neurobiol. 2009;35:60–8. doi: 10.1111/j.1365-2990.2008.00948.x. [DOI] [PubMed] [Google Scholar]
- 11.Vekrellis K, Ye Z, Qiu WQ, et al. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000;20:1657–65. doi: 10.1523/JNEUROSCI.20-05-01657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasparini L, Xu H. Potential roles of insulin and IGF-1 in Alzheimer’s disease. Trends Neurosci. 2003;26:404–6. doi: 10.1016/S0166-2236(03)00163-2. [DOI] [PubMed] [Google Scholar]
- 13.Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–52. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- 14.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Phiel CJ, Wilson CA, Lee VM, et al. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–9. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 16.Ho L, Qin W, Pompl PN, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18:902–4. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 17.Freude S, Hettich MM, Schumann C, et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 2009;23:3315–24. doi: 10.1096/fj.09-132043. [DOI] [PubMed] [Google Scholar]
- 18.Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–41. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–70. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 20.Devedjian JC, George M, Casellas A, et al. Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. J Clin Invest. 2000;105:731–40. doi: 10.1172/JCI5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacy PE, Kostianovsky M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–9. doi: 10.2337/diab.16.1.35. [DOI] [PubMed] [Google Scholar]
- 22.Takuma H, Arawaka S, Mori H. Isoforms changes of tau protein during development in various species. Brain Res Dev Brain Res. 2003;142:121–7. doi: 10.1016/s0165-3806(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 23.Ma QL, Yang F, Rosario ER, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–89. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DAlessandris C, Lauro R, Presta I, et al. C-reactive protein induces phosphorylation of insulin receptor substrate-1 on Ser307 and Ser 612 in L6 myocytes, thereby impairing the insulin signalling pathway that promotes glucose transport. Diabetologia. 2007;50:840–9. doi: 10.1007/s00125-006-0522-y. [DOI] [PubMed] [Google Scholar]
- 25.Julien C, Tremblay C, Phivilay A, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–31. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 26.Rahman A, Akterin S, Flores-Morales A, et al. High cholesterol diet induces tau hyperphosphorylation in apolipoprotein E deficient mice. FEBS Lett. 2005;579:6411–6. doi: 10.1016/j.febslet.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 27.Reger MA, Watson GS, Green PS, et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70:440–8. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- 28.Calhoun ME, Burgermeister P, Phinney AL, et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96:14088–93. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shino A, Matsuo T, Iwatsuka H, et al. Structural changes of pancreatic islets in genetically obese rats. Diabetologia. 1973;9:413–21. doi: 10.1007/BF01239438. [DOI] [PubMed] [Google Scholar]
- 30.Duffy KR, Pardridge WM, Rosenfeld RG. Human blood-brain barrier insulin-like growth factor receptor. Metabolism. 1988;37:136–40. doi: 10.1016/s0026-0495(98)90007-5. [DOI] [PubMed] [Google Scholar]
- 31.Chen DY, Stern SA, Garcia-Osta A, et al. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–7. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–8. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stein TD, Anders NJ, DeCarli C, et al. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci. 2004;24:7707–17. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Glucose tolerance test in the acute group mice at the age of 4 months. Time course of B-gluc changes in (A) female and (B) male mice of the four genotypes. Corresponding changes in plasma insulin levels during the test in (C) female and (D) male mice. Mean + SEMs are shown, n =4–6 mice/group.
Fig. S2. (A) Area of beta-islets as percent of total area of pancreatic crosssections of 7-month-old female mice of different genotypes on standard diet. ** I+ mice differed from Iw mice (p < 0.01, ANOVA). (B) Insulin 1 and insulin 2 mRNA levels in different genotypes isolated from 4-month-old female mice (all on standard diet). *A+ mice differed from Aw mice (p < 0.05, **p < 0.01, ANOVA); #I+ mice differed from Iw mice (p < 0.05, ANOVA). (C) Protein levels in the liver of insulin-degrading enzyme (IDE) in 4-month-old female mice. Mean + SEMs are shown, n ∇ 4–6 mice/group.
Fig. S3. Effects of genotype and diet on insulin signaling in the ventral posterior cortex. (A) Total protein lysates of male mice on standard diet (STD) were analyzed with Western blotting using antibodies specific for phosphorylated GSK3α (Ser 21), GSK3β (Ser 9), and Akt (Ser473) as well as total GSK3α, GSK3β, and Akt.
Quantifications of the protein levels are shown. (B) Quantification of the Western blots from male mice on typical Western diet (TWD) with the same antibodies. Mean + SDs are shown, n ∇ 4–6 mice/group.