

Abstract
Platinum anticancer drugs have been used for three decades despite their serious side effects and the emerging of resistance phenomena. Recently, a phosphine copper(I) complex, [Cu(thp)4][PF6] (CP), gained special attention because of its strong antiproliferative effects. CP killed human colon cancer cells more efficiently than cisplatin and oxaliplatin and it overcame platinum drug resistance. CP preferentially reduced cancer cell viability whereas non-tumour cells were poorly affected. Colon cancer cells died via a programmed cell death whose transduction pathways were characterized by the absence of hallmarks of apoptosis. The inhibition of 26S proteasome activities induced by CP caused intracellular accumulation of polyubiquitinated proteins and the functional suppression of the ubiquitin–proteasome pathway thus triggering endoplasmic reticulum stress. These data, providing a mechanistic characterization of CP-induced cancer cell death, shed light on the signaling pathways involved in paraptosis thus offering a new tool to overcome apoptosis-resistance in colon cancer cells.
Keywords: copper(I) phosphine complexes, proteasome, colon cancer, endoplasmatic reticulum stress, non-apoptotic cell death
Introduction
Cisplatin(CisPt)-based combination regimens form the mainstay of current treatment of various solid malignancies [1]. Unfortunately, CisPt is characterized by some inherited setbacks, which significantly limit its usefulness. Indeed, CisPt has an unfavourable toxicological profile and some common cancers (e.g. colon adenocarcinoma) are intrinsically refractory, whereas in tumours initially sensitive, acquired resistance is often developed in the course of therapy [2]. To avoid these problems, thousands of platinum and other metal-based compounds have been tested for their potential antitumour properties in the last 40 years. A series of compounds showing encouraging perspectives were the phosphine complexes of group 11 metal ions [3]. Their biological properties were little explored until late 1970s when a thioglucose derivative of triethylphosphine gold(I) (auranofin) was found to have antitumour activity in murine models [4]. In an attempt to identify gold complexes with a wider spectrum of activity, Berners-Price demonstrated the antitumour activity of [Au(dppe)2][Cl] (dppe = 1,2-bis(diphenylphosphino)ethane) against a range of tumour models in mice [5]. Unfortunately, the presence of several phenyl groups appended to the phosphorus donors caused undesired side effects [6], thus precluding clinical trials. A rational extension of diphosphine gold(I) chemistry to the first row congener copper indicated that analogous copper(I) complexes of ‘CuP4’ stoichiometry could be efficiently designed [7, 8]. Among them, a series of hydrophilic copper(I) derivatives including the water soluble tris(hydroxymethyl)phosphine (thp) ligand, either alone [9] or in combination with hydrophilic scorpionates were studied [10]. Copper(I,II) complexes investigated for antitumour activity have been recently reviewed [11–13].
CP (Fig. 1), a monocationic copper(I) complex highly soluble and stable in water solution, distinguished itself for its remarkable cancer cell killing ability. Against human cancer cells, CP cytotoxic potency was over 40-fold that of CisPt and it was not cross-resistant with this platinum-based drug; moreover, it was able to overcome the multi-drug resistance phenomenon [9]. It has been proposed that the cytotoxicity of CP may be correlated to its ability to induce a programmed non-apoptotic death termed paraptosis or type-III cell death [9, 14]. Paraptosis lacks of apoptotic morphology such as pyknosis, plasma membrane blebbing, caspase-3 activation and DNA fragmentation [15], and it is characterized by a massive cytoplasmic vacuolization [15, 16]. Various studies have described paraptotic-like processes in various models, but the mechanisms underlying paraptosis, in particular the signals responsible for triggering cytoplasmatic vacuolisation, have not been fully determined yet. Vacuolization has been progressively more recognized as a morphological change indicative of a disruption of endoplasmic reticulum (ER)-proteasome functional link [17, 18]. Many studies pointed out that copper complexes inhibit proteasome activity in cancer cells [19, 20], indicating that they may work as antiproliferative agents throughout the accumulation of misfolded proteins and, consequently, by ER homeostasis disorder. In particular, paraptosis induced in human cancer cells by a thioxotriazole copper(II) complex was recently found to be associated with the inhibition of the ubiquitin–proteasome system (UPS) and unfolded protein response (UPR) induction [16]. Furthermore, it has been also demonstrated that some proteasome inhibitors induced in colon cancer cells a Bax- and caspase-independent paraptosis that could be suppressed by translational or transcriptional inhibitors [21]. These findings clearly contribute to enlarge the potential therapeutic utility of proteasome inhibitors as they could overcome drug resistance which is because of compromised apoptotic machinery, such as deficiency of Bax, in cancer cells [21].
Fig 1.
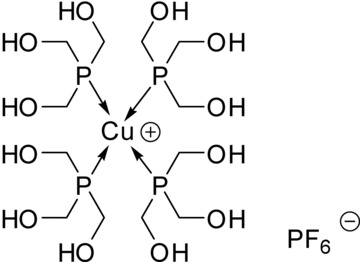
Chemical structure of CP.
Among neoplasias, colorectal cancer is one of the leading causes of cancer-related death in the world, largely because of the poor clinical response of colorectal tumours to conventional chemotherapeutics [22]. Moreover, colon cancer cells commonly develop resistance to chemotherapy. Resistance can emerge from failure to execute apoptosis caused by either predominance of antiapoptotic factors or defects in downstream effectors [23, 24].
In this study, we sought to investigate cell growth inhibition of CP in a panel of human colon carcinoma cell lines. CP provoked a strong effect on cell viability that was preferential for cancer cells as it did not harm normal colon fibroblasts. Against colon cancer cells, CP was several-fold more active than CisPt and, most interestingly, than oxaliplatin (OxPt), key drug in FOLFOX regimens in the treatment of colorectal cancers [25]. Moreover, treatment with CP efficiently overcame acquired resistance to OxPt. Colon cancer cells treated with CP died via a paraptotic cell death characterized by a massive formation of cytoplasmatic vacuoles, derived from ER swelling. We provided evidence that ER stress induced by CP was coherent with the inhibition of the proteasome 26S and UPR induction.
Materials and methods
Chemicals
CP was prepared as described elsewhere [9]. CisPt, OxPt, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), cell culture media (RPMI, DMEM, MEM and F-12 HAM'S), fluorogenic proteasome substrates [N-Suc-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (AMC), Boc-Gln-Ala-Arg-AMC and Z-Leu-Leu-Glu-AMC for chymotrypsin-like (CT-L), trypsin-like (T-L) and caspase-like (C-L) activities, respectively], rabbit polyclonal anti-calnexin antibody and caspase substrates (N-Acetyl-Asp-Glu-Val-Asp-AMC for caspase-3 and -7, N-Acetyl-Val-Glu-Ile-Asp-AMC for caspase-6, N-Acetyl-Ile-Glu-Thr-Asp-AMC for caspase-8, N-Acetyl-Leu-Glu-His-Asp-AMC for caspase-9) were from Sigma Chemical Co, St. Louis, MO, USA. Antibody for β-actin and ubiquitin were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phosphorylated-PERK and anti-phosphorylated-IRE1 were from Abcam (Cambridge, MA, USA). CP, CisPt and OxPt were dissolved in physiological solution to stock solutions of 1 mg/ml, which were diluted to the required concentration. Protein content was estimated with Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
Cell cultures
Human colon LoVo, DLD1, HCT-15, SW480 and Caco-2 cancer cell lines along with human lung MRC-5 and human foreskin HFF-1 fibroblasts were obtained from the American Tissue Culture Collection (ATCC, RocKville, MD, USA). Human colon CCD-18Co fibroblasts were obtained from European Collection of Cell Cultures (ECACC, Salisbury, UK). The LoVo–OxPt cells were derived using a standard protocol [26]. Cell lines were maintained using the following culture media containing 10% foetal calf serum (Euroclone, Milan, Italy), antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin) and 2 mM l-glutamine: (i) RPMI for HCT-15, cells, (ii) F-12 HAM'S for LoVo and LoVo–OxPt cells, (iii) DMEM medium for SW480 and HFF-1 cells and (iv) MEM for CaCo-2, MRC-5 and CCD-18Co cells.
Cytotoxicity assays
MTT
The growth inhibitory effect towards tumour cells was evaluated by means of MTT assay as previously described [9]. Mean absorbance for each drug dose was expressed as percentage of the control and plotted versus drug concentration. Dose–response curves were fitted and IC50 values were calculated with four parameter logistic model (4PL).
Clonogenic assay
LoVo and CCD-18Co cells (1.5 × 105) were seeded in growth medium. After 24 hrs, cells were incubated for 6 hrs with increasing concentrations of CP. Aliquots of 200 cells were seeded in fresh medium and incubated for 12 days. The colonies were then stained and counted, discarding colonies of fewer than 50 cells. The efficiency of clonal growth, that is the ratio between the number of colonies formed and the number of cells seeded, was then calculated. Plating efficiency was about 80% for LoVo cells and about 70% for CCD-18Co cells.
Cell death studies
Alkaline single-cell gel electrophoresis assay (Comet assay)
LoVo (105) cells were incubated for 6, 12 and 24 hrs with IC50 of tested compounds. Subsequently, cells were processed and comet assay was performed as previously described [27].
Nuclear DNA fragmentation
Mono- and oligo-nucleosome formation was detected with the ELISAplusCell-Death-Kit (Roche, Penzberg, Germany) according to manufacturer's instruction.
Caspase activation
The 106 LoVo cells were treated for 24 hrs with tested compounds, harvested and homogenized in a lysis buffer [1% Triton X-100, 320 nM sucrose, 5 mM EDTA, 10 mM Tris-HCl and 2 mM DTT buffer (pH 7.6)]. Protein aliquots (100 mg) were stained at 37°C for 60 min with fluorescent caspase substrates. Substrate hydrolysis was measured after 60 min by monitoring the release of AMC using a spectrofluorometer (excitation at 370 nm, emission at 460 nm).
Cytochrome c release
LoVo cells (30 × 106) were treated for 12 hrs with tested compounds at IC50 concentration. Afterwards, cells were processed and cytochrome c was assessed as previously described [28].
Mitochondrial membrane potential depletion
Mitochondrial membrane potential (MMP) depletion was determined by measuring the fluorescence of cells stained with tetramethylrhodamine methyl ester (TMRM) as previously described [9].
Microscopy analysis
Phase contrast microscopy
LoVo cells (5 × 104 cells/well) were plated on eight-well slides. Following 12 hrs CP treatment, cells were washed with PBS, observed under a phase-contrast microscope (Olympus BX41).
Transmission electron microscopy analyses
The 106 LoVo cells were treated for 24 hrs with IC50 of CP and subjected to transmission electron microscopy (TEM) analyses as previously described [29].
Immunofluorescence
LoVo cells, seeded into eight-well slides at 5 × 104 cells/well, were treated with IC50 of tested complexes, fixed in 4% ice-cold paraformaldehyde, permeabilized in 0.2% Triton X-100 in PBS and air-dried. Slides were then stained with primary antibody (mouse monoclonal anti-ubiquitin, rabbit polyclonal anti-calnexin, anti-pPERK and anti-pIRE1) and incubated with fluorescein-isothiocyanate-conjugated secondary antibody and then visualized by means of a fluorescence microscopy (Olympus BX41).
Western blot analyses
The 106 LoVo cells were treated, harvested and lysed in RIPA buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% SDS). Afterwards, cells were centrifuged at 13,000 × g for 15 min. at 4°C. b-Actin was used as a loading control. An equal amount of proteins for each sample was electrophoresed on a 12% SDS-PAGE and blotted to a nitrocellulose membrane. The membrane was incubated for 1 hr in PBS-Tween20 (0.05%) containing 5% non-fat milk and then at 37°C for 1 hr with primary antibodies (mouse monoclonal anti-ubiquitin and anti-β-actin, rabbit polyclonal anti-pPERK and anti-pIRE1). The membranes were stained with the corresponding peroxidase-conjugated secondary antibodies for 1 hr at room temperature and detected by ECL according to the manufacturer's protocol (GE).
Proteasome activity
Purified cell extracts
Seventy per cent confluent LoVo cells were harvested, homogenized in lysis buffer and ultracentrifuged for two hrs at 300,000 × g. Protein aliquots (100 mg) were incubated for 60 min. at 37°C with tested compounds. Afterwards, fluorogenic peptides were added and substrate hydrolysis was measured after 30 min. by monitoring spectrofluorometrically the release of AMC (excitation at 370 nm, emission at 460 nm).
Intact cells
The 106 LoVo cells were treated, harvested and homogenized in a lysis buffer (50 mM Tris-HCl, pH 7.5, 250 mM sucrose, 5 mM MgCl2, 1 mM DTT, 0.5 mM EDTA). Protein aliquots (100 μg) were stained at 37°C for 30 min. with fluorescent proteasome substrates and their hydolysis was measured as described above.
Statistical analysis
All the values are the means ± S.D. of not less than three measurements. Multiple comparisons were made by anova followed by Tukey–Kramer multiple comparison test (**P < 0.01; *P < 0.05), using GraphPad Software.
Results
Cytotoxicity assays
The CP ability to induce colon cancer cell death was evaluated in a panel of colon carcinoma cell lines corresponding to different stages of disease progression. LoVo, DLD1, SW480, HCT-15 and CaCo-2 cells were treated for 72 hrs with CP, OxPt or CisPt and cell viability was evaluated by MTT test (Table 1). These colon cancer cells had differential responses to CisPt, being LoVo and DLD1 quite sensitive, whereas HCT-15, CaCo-2 and SW480 intrinsically resistant (for the latter, IC50 were about up to three- to fourfold higher than those of the sensitive cells). OxPt elicited IC50 from 2- to 30-fold lower than CisPt and induced a different pattern of response across the cell lines, being mainly active against LoVo and CaCo-2 and showing a 10 times lower efficacy against SW480 and DLD1 cells. CP showed the greatest antitumour efficacy over all cell lines, with IC50 values mainly in the submicromolar range. CaCo-2 cells were about 40-fold more sensitive to treatment with CP compared to CisPt. IC50 values calculated for CP, besides being similar in LoVo and CaCo-2 cells, were considerably lower than OxPt in the other cell lines. In particular, apoptosis-resistant SW480 and DLD1 cells were 3- and 15-fold more sensitive to CP than OxPt, respectively.
Table 1.
Responsiveness of colon cancer cell lines
| Compound | CaCo-2 | LoVo | IC50 (μM) ± S.D. HCT-15 | DLD1 | SW480 |
|---|---|---|---|---|---|
| CP | 0.48 ± 0.41 | 0.23 ± 0.11 | 0.54 ± 0.20 | 0.37 ± 0.11 | 1.33 ± 0.16 |
| CisPt | 18.31 ± 2.21 | 9.02 ± 1.34 | 16.65 ± 2.63 | 8.01 ± 2.22 | 25.25 ± 3.33 |
| OxPt | 1.02 ± 0.25 | 0.33 ± 0.17 | 2.37 ± 0.58 | 5.81 ± 1.93 | 4.20 ± 0.75 |
Cells treated for 72 hrs with increasing concentrations of tested compounds. Cytotoxicity assessed by MTT test. IC50 values calculated by four parameter logistic model (P < 0.05).
The causes of the clinical failure of chemotherapy are numerous but resistance represents a key determinant for the variable efficacy of platinum-based anticancer therapy [1, 2]. Although OxPt induces DNA crosslinks as CisPt, it shows activity in cell lines resistant to CisPt, suggesting that the two complexes may have different mechanism of action and/or resistance [30]. To evaluate CP ability in overcoming OxPt resistance, we developed a resistant cell line by growing LoVo cells in increasing concentrations of OxPt. The degree of resistance was evaluated by means of resistant factor (RF), which is defined as the ratio between IC50 (obtained with MTT assay after 48 hrs drug exposure) calculated for the resistant cells and those arising from the sensitive ones. The LoVo–OxPt cells, obtained following 9 months of selection, were fivefold resistant to OxPt (Table 2). Cytotoxicity assays testing CP against LoVo–OxPt allowed the calculation of a RF value fivefold lower than that obtained with OxPt, thus suggesting that CP circumvented OxPt resistance. Very often, metal complexes have limited clinical use as a result of their systemic toxicity caused by very low selectivity towards cancer cells. The ability to discriminate between normal and malignant cells is of paramount importance for developing clinically applicable chemotherapics. Cytotoxic activity of CP was evaluated on three non-tumour cell lines: MRC-5, HFF-1 and CCD-18Co human fibroblasts (Table 3). Notably, CP displayed lower cytotoxicity over the non-tumour cells as compared with the reference drugs, being CP mean IC50 about 1.5-fold higher than those calculated with CisPt and OxPt. The selectivity index value (SI = the quotient of the average IC50 towards normal cells divided by the average IC50 for the malignant cells) of CP was 56, respectively 35- and 10-fold higher than those obtained with CisPt and OxPt, thus confirming a noticeable preferential cytotoxicity of CP versus neoplastic cells. To evaluate the long-term effects of CP, clonogenic assays were performed in LoVo colon cancer and CCD-18Co non-tumour colon cells (Fig. 2). Analysis of the stained colonies revealed that CP efficiently abrogated the clonogenic ability of LoVo cells in a dose-dependent manner. Conversely, treatment of CCD-18Co cells with CP did not significantly affect their capacity to form viable colonies and the decrease of survival fraction of CCD-18Co cells was considerably lower than that obtained in LoVo tumour cells.
Table 2.
Oxaliplatin cross-resistance profile
| Compound | LoVo | IC50 (μM) ± S.D. LoVo–OxPt | RF |
|---|---|---|---|
| CP | 1.37 ± 0.35 | 1.46 ± 0.27 | 1.0 |
| OxPt | 2.05 ± 0.43 | 10.89 ± 1.34 | 5.3 |
Cells treated for 48 hrs with increasing concentrations of tested compounds. Cytotoxicity assessed by MTT test. IC50 values calculated by four parameter logistic model (P < 0.05). RF = IC50 resistant/IC50 sensitive.
Table 3.
Cytotoxicity in non-tumour cells
| Cell lines | CP | IC50 (μM) ± S.D. CisPt | OxPt |
|---|---|---|---|
| MRC-5 | 32.67 ± 1.34 | 19.66 ± 1.31 | 23.93 ± 1.35 |
| CCD-18Co | 38.98 ± 3.33 | 31.32 ± 2.12 | 31.31 ± 2.23 |
| HFF-1 | 30.97 ± 2.11 | 23.41 ± 1.97 | 27.05 ± 1.29 |
Cells treated for 72 hrs with increasing concentrations of tested com-pounds. Cytotoxicity assessed by MTT test. IC50 values calculated by four parameter logistic model (P < 0.05).
Fig 2.
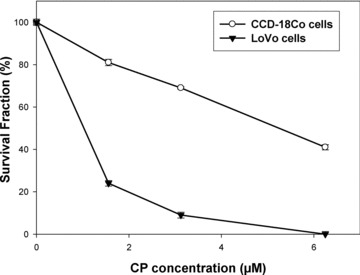
Clonal growth. Fraction of surviving cells determined by clonogenic assay in LoVo and CCD-18Co cells after CP exposure.
Cell death induced by CP in colon cancer cells is independent from DNA-fragmentation, caspase activation, cytochrome c release and MMP depletion
We had previously demonstrated that CP induced a caspase-3-independent paraptotic cell death in ovarian cancer cells [9]. The assessment in LoVo cells of the genomic DNA fragmentation (Fig. 3A) and of the apoptosome complex formation (Fig. 3B), confirmed the absence of chromatin condensation and DNA fragmentation in the cell death pathway triggered by CP. Indeed, CP cell treatment did not influence neither the electrophoretic migration of the DNA fragments in single-cell gel electrophoresis assay nor mono- and oligo-nucleosome formation. Interestingly, the extent of nucleosomes was even markedly lower than that quantified in control cells. Conversely, OxPt induced a significant increase in comet appearance and a roughly 30-fold increase in histone-DNA formation in 12-hr-treated LoVo cells compared to controls. The activity of the two initiator caspases (-8 and -9), and the downstream effectors (-3/-7 and -6) was analysed to elucidate if caspase(s) mediated CP-induced cell death. As shown in Figure 3C, OxPt provoked a substantial activation of all caspases. No increase in free AMC has been detected by using substrates for all caspases in cells exposed to CP for 12 or 24 hrs. Interestingly, a sensible decrease testing caspase-3/-7 and -9 activities could be underlined in CP-treated cells, thus hypothesizing that the copper complex caused a reduction of pro-caspase-3/-7 and -9 cleavage by upstream proteases or elicited a direct inhibition of these caspases. The results presented in the inserts of Figure 3, showing a reduced DNA fragmentation (Fig. 3B, b) and caspase induction (Fig. 3C, c) after treatment with OxPt, clearly attested for LoVo–OxPt cells an increase of resistance to apoptosis.
Fig 3.
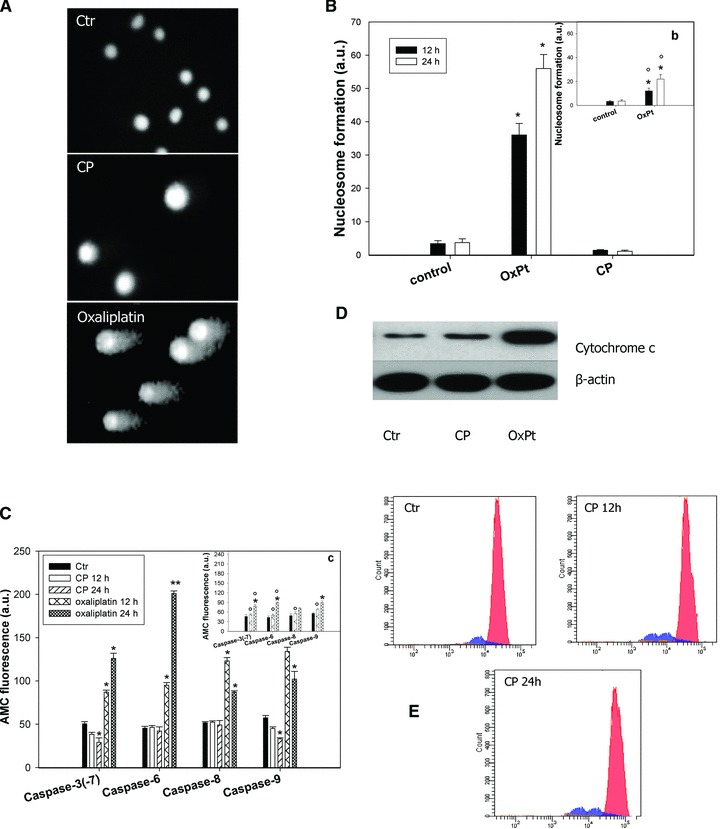
Non-apoptotic cell death induction. (A) Comet assay. LoVo cells treated for 12 hrs with IC50 of CP or O×Pt. (B) Nuclear DNA fragmentation. LoVo cells were treated for 12 or 24 hrs with IC50 of CP or O×Pt. Quantitative estimation of DNA fragmentation was obtained with an ELISA test. *P < 0.05 compared to control. Insert (b) LoVo–O×Pt cells treated for 12 or 24 hrs with IC50 of O×Pt. Data are the means of five independent experiments. Error bars indicate S.D. *P < 0.05 compared to control and °P < 0.05 compared to O×Pt treated LoVo cells. (C) Caspase activity. LoVo cells incubated for 12 and 24 hrs with CP or O×Pt and processed for caspase-3/-7, -6, -8, -9 activity. Insert (c): LoVo–O×Pt cells treated for 12 or 24 hrs with IC50 of O×Pt. Data are the means of at least three independent experiments. Error bars indicate S.D. *P < 0.05 compared to control and °P < 0.05 compared to O×Pt treated LoVo cells. (D) Cytochrome c release. LoVo cells treated with IC50 of tested compounds for 12 hrs and cytochrome c was estimated by Western blotting. (E) Flow cytometric profiles of LoVo cells untreated and treated with IC50 of CP for 12 and 24 hrs and stained with TMRM.
Although the mitochondrial pathway of apoptosis requires the release of cytochrome c from the mitochondria [31, 32], the mechanisms underlying mitochondria involvement in paraptosis have not been fully elucidated. Information concerning mitochondrial contribution to paraptosis have been reported by some authors which demonstrated that paraptosis may be characterized by a process of vacuolisation beginning with a physical enlargement of mitochondria and of ER [33]. Monitoring cytochrome c release in LoVo cells after 6, 12 and 24 hrs of treatment with IC50 of CP, no release of this haeme-protein has been detected for all exposure times. Figure 3D shows results obtained after 12 hrs treatment. On the contrary, as previously well documented by literature [34], OxPt determined a substantial cytohrome c release after 12 hrs exposure (Fig. 3D). MMP depletion generally precedes apoptotic cell death [31]. However, recent studies underlined a key role of mitochondrial permeability transition pore opening in necrosis and mitotic catastrophe [35, 36]. By treating LoVo cells with IC50 of CP for 12 and 24 hrs, TMRM fluorescence intensity did not decrease but, in contrast, continuously augmented by increasing exposure times (Fig. 3E).
CP induces massive cytoplasmatic vacuoles originated from ER
LoVo colon cancer cells treated for 24 hrs with IC50 of CP and analysed by phase contrast microscopy, revealed evident morphological changes, primarily associated with intracellular vacuolization (Fig. 4A, a′). Interestingly, neither Bafilomycin A, a macrolide antibiotic which is a potent inhibitor of vacuolar-type H1-ATPase, nor 3-methyladenine and monensin, both autophagy inhibitors, repressed cell vacuolisation (data not shown). These findings confirmed the non-autophagic nature of these vacuoles. To better characterize cellular morphological changes induced by CP, LoVo cells exposed to IC50 of CP for 12 and 24 hrs were subjected to TEM analysis (Fig. 4B). No signs of classic apoptotic changes, such as cell shrinkage, chromatin condensation and apoptotic body formation were evidenced for both exposure times (Fig. 4B, b′, b′, b″). In addition, mitochondria appeared conserved in shape and internal structures and no swelling features (increase in size and decrease in turbidity) could be detected. LoVo CP-treated cells showed striking morphological changes, such as ER swelling, previously characterized as hallmarks of paraptotic cell death [9, 16, 37]. ER cisternae at first began to increase in abundance and dilated, subsequently an increase of cisternae inner volume and a radical change in ER shape occurred. Interestingly, following 24-hr treatment, most of the ribosomes were no longer apposed to the ER membrane. Calnexin immunostaining of 12-hr-treated LoVo cells revealed that most of the intracellular vacuoles induced by CP were positive for calnexin, an ER-resident protein (Fig. 4C). Overall, these results clearly confirmed CP-induced vacuoles originated from the ER cisternae as a consequence of ER swelling.
Fig 4.
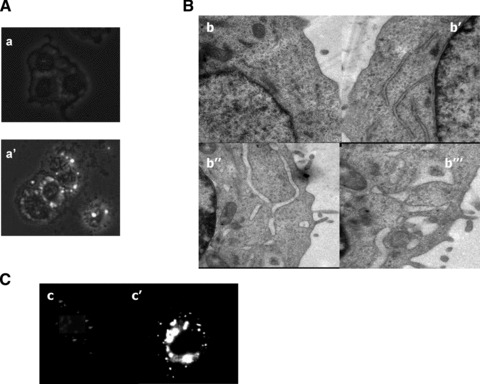
Morphological changes in CP-treated LoVo cells. (A) Phase-contrast micropho-tographs (100×) of control (a) or treated for 24 hrs with IC50 of CP (a′) LoVo cells. (B) TEM analysis of control (b), or CP-treated LoVo cells for 12 hrs (b′) or 24 hrs (b″ and b‴). (C) Calnexin staining of control LoVo cells (c) or exposed to CP for 12 hrs (c′).
CP induces the impairment of the UPS
ER swelling has been increasingly documented as a morphological alteration symptomatic of an ER-proteasome cooperative activity disturbance [18, 19]. Very recently, Dou et al. reported that certain copper complexes based on mixtures of dithiocarbamate and Cu(II) salts act as proteasome inhibitors in cancer cells [38]. To examine whether CP is capable of inhibiting the proteasome activity, we first tested the functioning of each individual active site of the 26S proteasome under cell-free conditions. CP, tested at increasing concentrations on a purified 26S proteasome extract from LoVo cells, inhibited all the proteolitic activities with IC50 values (μM) of 23, 34 and 21 concerning chymotrypsin-like (CT-L), trypsin-like (T-L) and caspase-like (C-L) activity, respectively (Fig. 5A). To further evaluate CP anti-proteasome activity, LoVo cells were treated with increasing concentrations of CP for 24 hrs and then submitted to proteasome activity measurement and estimation of poly-ubiquitinated proteins. The proteasomal CT-L, T-L and C-L activities were decreased by 50% with 31, 42 and 37 μM of CP, respectively, thus confirming that CP is able to inhibit all three proteasome activities also in intact cells (Fig. 5B). Consistent with the inhibition of the proteasomal activities, Western blot analysis demonstrated time-dependent accumulation of poly-ubiquitinated proteins in lysates prepared from LoVo cells treated for different exposure times (6, 12 and 24 hrs) with IC50 CP (Fig. 5C). Poly-ubiquitinated aggregosomes were confirmed in 24 hrs CP-treated LoVo cells by means of an immunofluorescence staining (Fig. 5D).
Fig 5.
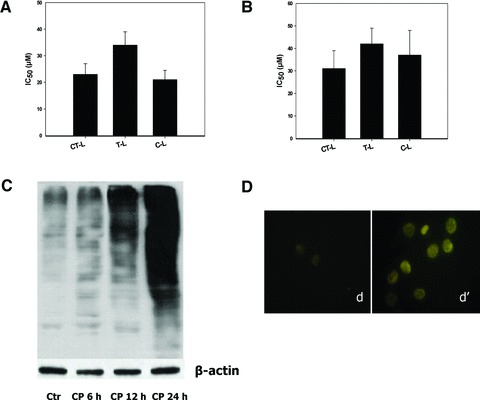
Effect of CP on proteasome inhibition. Inhibition of human 26S proteasome purified from LoVo cells (A) and in intact LoVo cells (B) estimated by means of specific fluorogenic substrates. IC50 values calculated by four-parameter logistic model (P < 0.05). In (B), LoVo cells following 24-hr treatment with increasing concentrations of CP. Detection of poly-ubiquitinated proteins in LoVo cells (C and D). In (C), cells treated for 6, 12 and 24 hrs with IC50 of CP and ubiquitinated proteins estimated by Western blotting. In (D), cells treated for 24 hrs with IC50 of CP and submitted to immunofluorescence analysis.
Induction of ER stress response by CP
Intracellular accumulation of misfolded proteins resulting from proteasome inhibition is known to activate a signalling cascade called UPR [39]. Misfolded proteins within the ER activate by autophosphorilation ER-resident transmembrane proteins such as PERK (PKR-like ER kinase), IRE1 (inositol requiring enzyme) and ATF6 (activating transcription factor 6), which mediate cytoplasmic and nuclear signalling pathways acting as ER stress sensors. We considered the effects of CP on the ER stress response by monitoring ER chaperones PERK and IRE1 in LoVo cells by means of immunofluorescence staining and Western blotting (Fig. 6A and B). Immunofluorescence analysis revealed a high enhance in phosphorilation of both ER sensors in LoVo cells treated for 12 hrs with CP, compared to control cells. Furthermore, as evident from Western blotting, treatment with increasing exposure-times of CP at IC50, provoked a time-dependent increase of both p-PERK and p-IRE1. In particular, following 12 hrs treatment, p-PERK and p-IRE1 increased four and eight times, respectively.
Fig 6.
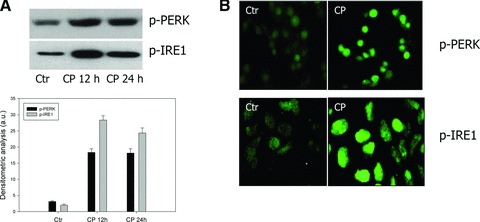
Effect of CP on UPR. (A) Western blot analysis of p-PERK and p-IRE1 in LoVo cells treated with IC50 of CP for 12 and 24 hrs. *P < 0.05 compared to control. (B) Immunofluorescence staining of LoVo cells treated with IC50 of CP for 24 hrs for p-PERK and p-IRE1 detection.
Discussion
In this study we demonstrated that CP induced paraptotic cell death in human colon cancer cells. In contrast with caspase-dependent apoptosis induced by OxPt, our results confirmed that the cell death process triggered by CP did not encompass DNA fragmentation or activation of caspases, but on the contrary, an inhibition of caspase-3(7) activity was detected. Mitochondria, organelles that serve as a gatekeeper to trap a variety of pro-apoptotic proteins whose release into the cytosol induces apoptosis, seemed to be marginally involved in CP-induced cell death pathway because no release in cytochrome c and MMP depletion have been recorded in treated cells. Moreover, TEM analysis revealed that mitochondria conserved their morphology (both internal structural features and external shape) indicating that mitochondria swelling could not be deemed as a CP-induced paraptosis hallmark. Nevertheless, the mitochondrial membrane hyperpolarization, previously underlined in human ovarian adenocarcinoma cells [9], has been elicited by CP also in colon cancer cells. Although implications of mitochondrial function and of redox equilibria involved in CP-induced paraptotic cell death are currently under investigation, increase of MMP may be properly indicative of substantial changes in mitochondrial metabolic activity. Morphological analysis of CP-treated cells revealed a ribosomal dissociation from ER membrane that is triggered during UPR to stop new protein synthesis and to relieve stress on the ER [40]. Moreover, a time-dependent formation of cytoplasmatic vacuoles derived from ER swelling. The latter was paralleled by a strong increase in the levels of phosphorilated ER sensors, PERK and IRE1, confirming the concomitant induction of the UPR.
ER dilatation and vacuolization, processes that define paraptotic-like cell death induced by CP, could be attributed to its ability to directly inhibit the 26S proteasome activity.
Hypothesizing hence a mechanistic description of CP-induced cell death, when a critical amount of ubiquitinated unfolded proteins has been achieved following proteasome inhibition, ER stress and swelling consequently occur and cells undergo UPR. The UPR cytoprotective pathway temporarily suspends protein production to restore ER homeostasis. If the offending ER substance is eliminated, the UPR pathway shuts down and normal protein translation and folding resume. Conversely, if the UPR pathway remains functionally active, it leads to the inhibition of bulk protein synthesis, cell cycle arrest and cells undergo cell death [39].
From our results, it clearly emerged that the CP treatment hindered pro-apoptotic signal transducer thus allowing the supposition that paraptosis takes place following UPR initiation as an alternative cell death, bypassing apoptosis machinery. Because one of the major factor contributing to drug resistance is the failure of colorectal tumours to undergo apoptosis, there is a continuous and urgent need to develop novel therapeutic approaches that can target apoptosis-resistant cells. Consequently, it is not surprising that toxicants that can induce alternative cell death pathways including autophagic cell death [41] and paraptosis [42] have begun to receive much attention. In this work we have highlighted the marked ability of CP to inhibit cell viability of colon cancer cell, mostly characterized by the predominance of antiapoptotic factors or defects in apoptosis effectors. In particular, among all cell lines, CP was highly effective even against SW480 and DLD1 colon cancer cells, in which mutation of p53 gene or high expression of mitochondrial COX-2, respectively, conferred resistance to apoptosis [43, 44]. CP ability to trigger an apoptosis-independent stress response represents an interesting property that may expand its spectrum of action towards aggressive cancers characterized by apoptosis resistance. In this view, the high cytotoxic activity showed by CP against LoVo–OxPt cells appears of great importance. Once more, targeting ER stress pathway is confirmed as an attractive strategy for anticancer therapeutics [45]. Not of less importance, CP demonstrated a high selectivity against tumour cells compared with non-tumour cells. CP selectivity index was even three times higher than that calculated for OxPt. This special tropism for neoplastic cells may be related to CP ability to inhibit all three catalytic activities of 26S proteasome. To date, the anti- proteasome activity of copper complexes has only been studied by monitoring CT-L activity. For the first time, we have described a phosphine copper(I) complex as an inhibitor of all three catalytic activities of human 26S proteasome. The relative importance of the different proteolytic sites in mammalian proteasome in protein degradation has not been systematically studied. Because investigations in yeast suggested a primary role of the CT-L site, up to now the various synthetic proteasome inhibitors had been optimized according to their capacity to specifically block the CT-L sites. However, recent studies demonstrated that the reduction in proteolysis also requires inhibition of other activities (C-L and T-L) [46]. In conclusion, our results provide molecular evidence to validate the potential of CP as a new therapeutic tool for selectively killing colon cancer cells through paraptosis.
Acknowledgments
This work was financially supported by MIUR (PRIN 20078EWK9B). We are grateful to CIRCMSB (Consorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 2.Zhang CX, Lippard SJ. New metal complexes as potential therapeutics. Curr Opin Chem Biol. 2003;7:481–9. doi: 10.1016/s1367-5931(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 3.Simon TM, Kunishima DH, Vibert GJ, et al. Screening trial with the coordinated gold compound auranofin using mouse lymphocyte leukemia P388. Cancer Res. 1981;41:94–7. [PubMed] [Google Scholar]
- 4.Berners-Price SJ, Johnson RK, Mirabelli CK, et al. Copper(I) complexes with bidentate tertiary phosphine ligands: solution chemistry and antitumor activity. Inorg Chem. 1987;26:3383–7. [Google Scholar]
- 5.Berners-Price SJ, Mirabelli CK, Johnson RK, et al. In vivo antitumor activity and in vitro cytotoxic properties of bis [1,2-bis(diphenylphosphino)ethane]gold(I) chloride. Cancer Res. 1986;46:5486–9. [PubMed] [Google Scholar]
- 6.Hoke GD, Macia RA, Meunier PC. In vivo and in vitro cardiotoxicity of a gold-containing antineoplastic drug candidate in the rabbit. Toxicol Appl Pharmacol. 1989;100:293–306. doi: 10.1016/0041-008x(89)90315-3. [DOI] [PubMed] [Google Scholar]
- 7.Adwankar MK, Wycliff C, Samuelson AG. In vitro cytotoxic effect of new diphenylphosphinoethane-copper(I) complexes on human ovarian carcinoma cell lines. Indian J Exp Biol. 1997;34:810–4. [PubMed] [Google Scholar]
- 8.Sanghamitra NJ, Phatak P, Das S, et al. Mechanism of cytotoxicity of copper(I) complexes of 1,2-bis(diphenylphosphino)ethane. J Med Chem. 2005;48:977–85. doi: 10.1021/jm049430g. [DOI] [PubMed] [Google Scholar]
- 9.Marzano C, Gandin V, Pellei M, et al. In vitro antitumor activity of the water soluble copper(I) complexes bearing the tris(hydroxymethyl)phosphine ligand. J Med Chem. 2008;51:798–808. doi: 10.1021/jm701146c. [DOI] [PubMed] [Google Scholar]
- 10.Marzano C, Pellei M, Colavito D, et al. Synthesis, characterization and in vitro antitumor properties of tris(hydroxymethyl)phosphine copper(I) complexes containing the new bis(1,2,4-triazol-1-yl)acetate Ligand. J Med Chem. 2006;49:7317–24. doi: 10.1021/jm0601248. [DOI] [PubMed] [Google Scholar]
- 11.Tisato F, Marzano C, Porchia M, et al. Copper in diseases and treatments, and copper-based anticancer strategies. Med Res Rev. 2010;30:708–49. doi: 10.1002/med.20174. [DOI] [PubMed] [Google Scholar]
- 12.Marzano C, Pellei M, Tisato F, et al. Copper complexes as anticancer agents. Anticancer Agents Med Chem. 2009;9:185–211. doi: 10.2174/187152009787313837. [DOI] [PubMed] [Google Scholar]
- 13.Tardito S, Marchiò L. Copper compounds in anticancer strategies. Curr Med. Chem. 2009;16:1325–48. doi: 10.2174/092986709787846532. [DOI] [PubMed] [Google Scholar]
- 14.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 15.Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–81. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tardito S, Isella C, Medico E, et al. The thioxotriazole copper(II) complex A0 induces endoplasmic reticulum stress and paraptotic death in human cancer cells. J Biol Chem. 2009;284:24306–19. doi: 10.1074/jbc.M109.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obeng EA, Carlson LM, Gutman DM, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ustundag Y, Bronk SF, Gores GJ. Proteasome inhibition-induces endoplasmic reticulum dysfunction and cell death of human cholangiocarcinoma cells. World J Gastroenterol. 2007;13:851–7. doi: 10.3748/wjg.v13.i6.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hindo SS, Frezza M, Tomco D, et al. Metals in anticancer therapy: copper(II) complexes as inhibitors of the 20S proteasome. Eur J Med Chem. 2009;44:4353–61. doi: 10.1016/j.ejmech.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padhye S, Yang H, Jamadar A, et al. New difluoro Knoevenagel condensates of curcumin, their Schiff bases and copper complexes as proteasome inhibitors and apoptosis inducers in cancer cells. Pharm Res. 2009;26:1874–80. doi: 10.1007/s11095-009-9900-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding WX, Ni HM, Yin XM. Absence of Bax switched MG132-induced apoptosis to non-apoptotic cell death that could be suppressed by transcriptional or translational inhibition. Apoptosis. 2007;12:2233–44. doi: 10.1007/s10495-007-0142-0. [DOI] [PubMed] [Google Scholar]
- 22.Jemal A, Center MM, Desantis C, et al. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010 doi: 10.1158/1055-9965.EPI-10-0437. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 23.Huerta S, Goulet EJ. Livingston EH. Colon cancer and apoptosis. Am J Surg. 2006;191:517–26. doi: 10.1016/j.amjsurg.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Gourdier I, Del Rio M, Crabbé L, et al. Drug specific resistance to oxaliplatin is associated with apoptosis defect in a cellular model of colon carcinoma. FEBS Lett. 2001;529:232–6. doi: 10.1016/s0014-5793(02)03347-1. [DOI] [PubMed] [Google Scholar]
- 25.Raymond E, Faivre S, Chaney S, et al. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–35. [PubMed] [Google Scholar]
- 26.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marzano C, Mazzega Sbovata S, Gandin V, et al. A new class of antitumor trans-amine-amidine-Pt(II) cationic complexes: influence of chemical structure and solvent on in vitro and in vivo tumour cell proliferation. J Med Chem. 2010 doi: 10.1021/jm1006534. doi: 10.1021/jm1006534. [DOI] [PubMed] [Google Scholar]
- 28.Marzano C, Gandin V, Folda A, et al. Inhibition of thioredoxin reductase by auranofin induces apoptosis in CisPt-resistant human ovarian cancer cells. Free Radic Biol Med. 2007;42:872–81. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 29.Caicci F, Zaniolo G, Burighel P, et al. Differentiation of papillae and rostral sensory neurons in the larva of the ascidian Botryllus schlosseri (Tunicata) J Comp Neurol. 2010;518:547–66. doi: 10.1002/cne.22222. [DOI] [PubMed] [Google Scholar]
- 30.Gatti L, Perego P. Cellular resistance to oxaliplatin and drug accumulation defects. In: Bonetti A, editor. Cancer drug discovery and development platinum and other heavy metal compounds in cancer chemotherapy. New York: Humana Press; 2009. pp. 115–24. [Google Scholar]
- 31.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–300. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 32.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–4. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Li X, Wang L, et al. An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J Cell Sci. 2004;117:1525–32. doi: 10.1242/jcs.00994. [DOI] [PubMed] [Google Scholar]
- 34.Arango D, Wilson AJ, Shim Q. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br J Cancer. 2004;91:1931–46. doi: 10.1038/sj.bjc.6602215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zamzami N, Larochette N, Kroemer G. Mitochondrial permeability transition in apoptosis and necrosis. Cell Death Diff. 2005;12:1478–80. doi: 10.1038/sj.cdd.4401682. [DOI] [PubMed] [Google Scholar]
- 36.Jo WS, Jeong MH, Jin YH, et al. Loss of mitochondrial membrane potential and caspase activation enhance apoptosis in irradiated K562 cells treated with herbimycin A. Int J Radiat Biol. 2005;81:531–43. doi: 10.1080/09553000500303773. [DOI] [PubMed] [Google Scholar]
- 37.Pourahmad J, Ross S, O’Brien PJ. Lysosomal involvement in hepatocyte cytotoxicity induced by Cu(21) but not Cd(21) Free Radic Biol Med. 2001;30:89–97. doi: 10.1016/s0891-5849(00)00450-0. [DOI] [PubMed] [Google Scholar]
- 38.Cvek B, Milacic V, Taraba J, et al. Ni(II), Cu(II), and Zn(II) diethyldithiocarbamate complexes show various activities against the proteasome in breast cancer cells. J Med Chem. 2008;51:6256–8. doi: 10.1021/jm8007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 40.Blobel G, Dobberstein B. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J Cell Biol. 1975;67:835–51. doi: 10.1083/jcb.67.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res. 2010;70:3431–4. doi: 10.1158/0008-5472.CAN-09-4027. [DOI] [PubMed] [Google Scholar]
- 42.Bröker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11:3155–62. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 43.Yagi S, Oda-Sato E, Uehara I, et al. 5-Aza-2’-deoxycytidine restores proapoptotic function of p53 in cancer cells resistant to p53-induced apoptosis. Cancer Invest. 2008;26:680–8. doi: 10.1080/07357900701840212. [DOI] [PubMed] [Google Scholar]
- 44.Liou JY, Aleksic N, Chen SF, et al. Mitochondrial localization of cyclooxygenase-2 and calcium-independent phospholipase A2 in human cancer cells: implication in apoptosis resistance. Exp Cell Res. 2005;306:75–84. doi: 10.1016/j.yexcr.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Linder S, Shoshan MC. Lysosomes and endoplasmic reticulum: targets for improved, selective anticancer therapy. Drug Resist Updat. 2005;8:199–204. doi: 10.1016/j.drup.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 46.Kisselev A, Callard A, Goldberg AL. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem. 2006;281:8582–90. doi: 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]