

Abstract
Common cardiovascular diseases, such as atherosclerosis and congestive heart failure, are exceptionally complex, involving a multitude of environmental and genetic factors that often show nonlinear interactions as well as being highly dependent on sex, age, and even the maternal environment. Although focused, reductionistic approaches have led to progress in elucidating the pathophysiology of cardiovascular diseases, such approaches are poorly powered to address complex interactions. Over the past decade, technological advances have made it possible to interrogate biological systems on a global level, raising hopes that, in combination with computational approaches, it may be possible to more fully address the complexities of cardiovascular diseases. In this Review, we provide an overview of such systems-based approaches to cardiovascular disease and discuss their translational implications.
Introduction
The past several decades of cardiovascular research have been dominated by reductionistic approaches. These strategies have led to the identification of many molecules and regulatory mechanisms involved in normal and pathological cardiovascular states, and the roles of hundreds of these molecules have been studied in vivo using targeted pharmacological or genetic manipulation in various animal models. Nonetheless, many important questions about cardiovascular diseases, particularly those relating to its biological complexity, remain unanswered. For instance, the fundamental causes of common complex forms of cardiovascular disease are still unknown. An illustration of our ignorance comes from genome-wide association studies (GWAS) in patients with coronary artery disease (CAD) that have revealed about 30 genetic loci or genes that are likely to be the most-important genetic disease-susceptibility factors.1 Some of these genes had been identified previously, but the majority were not connected to any known risk factor or signaling pathway that contributes to the development of atherosclerosis. A similar challenge exists for heart failure, for which GWAS revealed only a few genetic loci or genes, of which only a small proportion had a clear biological role in the disease.
Over the past one or two decades, scientists have witnessed a revival in interest in systems-biology approaches to the study of multicomponent, biological processes.2–8 This revival is, in part, a result of the Human Genome Project and related technological developments, such as gene-expression arrays, that have enabled researchers to interrogate biological systems at a global level. In addition, new computational and mathematical approaches, such as network modeling, are being developed to extract biological information from data obtained by high-throughput analyses and other data. In addition, the realization is growing that reductionistic approaches alone will not allow us to fully address phenomena such as the beating of the heart or the development of an athero-sclerotic plaque. For those working in the field of cardiovascular medicine, the current interest in systems-based approaches was preceded by Denis Noble’s realization several decades ago that the narrowly focused analysis of individual transporters could never explain the rhythms of a beating heart.9–11
The urgent need for systems-based approaches becomes apparent as we wrestle with the clinical burden of CAD and congestive heart failure (CHF). The lifetime risk of CAD is about 50% in Western countries and, although effective preventative treatments for CAD (such as cholesterol-lowering drugs) are widely used, the incidence of the disease has decreased only slightly over the past two decades.12 Notably, the current obesity epidemic is predicted to increase the incidence of one of the major risk factors for CAD—type 2 diabetes mellitus—and, therefore, the incidence of atherosclerosis will also increase. CHF affects one in five individuals in the USA during their lifetime, and the incidence of CHF is rising because of the rapidly aging population.13 The high prevalence of CAD and CHF impose enormous human, social, and economic costs in both developed and developing countries. In this Review, we explain ‘systems-based’ approaches to cardiovascular disease and discuss their translational implications.
Systems-based studies
Basic principles
The fundamental principle underlying systems biology is that the whole is greater than the sum of the parts—that is, that a complex system has intrinsic novel properties that cannot be derived directly from the additive effects of its individual parts.4 Take, for example, the action potential of a cardiomyocyte, which requires the coordinated action of more than 20 different ion transporters and channels. Studying these individual components might provide information about their role in a specific aspect of the action potential, but to fully appreciate action-potential generation, scientists require an understanding of how these components function together over time and need to integrate them into a quantitative mathematical model.14–17 A striking example of the importance of studying multiple components simultaneously is the discovery of induced pluripotent stem cells.18 In their groundbreaking study, Takahashi and Yamanaka hypothesized that multiple factors would be required for reprogramming an adult somatic cell into a pluripotent stem cell, and they successfully identified the required factors by examining various combinations of candidate transcription factors. Their approach would not have worked if they had tested individual candidates separately.
A typical systems-based study involves the following five steps.4,19 The first objective is to define the ‘system’ to be examined. Such a system could be an organelle, organ, or organism. The second step is to identify the components of the study system, which could include mRNA transcripts, noncoding RNAs, small interfering RNAs, proteins, small molecule metabolites, membrane potentials, or other physiological or pathological parameters that are relevant to the study. Thirdly, investigators need to determine how these components interact with each other, either by conducting experiments or by searching published literature. For example, protein–protein interactions can be defined by co-immunoprecipitation (Figure 1) and functional relationships between genes can be investigated using co-expression studies (see below). The fourth step is to model the dynamics of the network mathematically to understand how the interactions between network components change over time or in response to various perturbations, and to attribute emerging properties to the targeted system. Fifth, the models should be validated using specific experimental perturbations, which can also be used iteratively to refine the models.4
Figure 1.
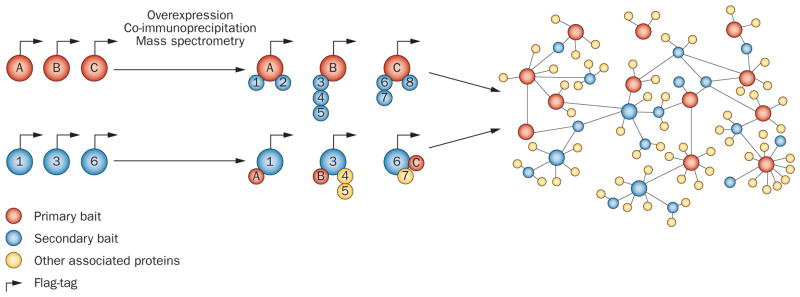
Construction of a protein-interaction network. The following approach was used by Behrends et al. to study autophagy,109 a cellular process that has been linked to various cardiovascular diseases.110 Proteins previously known to be involved in authophagy (red circles with letters) were retrovirally expressed as Flag–HA52 fusion proteins in cultured cells. Proteins and their binding partners (numbered blue circles) were then precipitated using anti-HA antibodies and identified by mass spectrometry. An additional set of interacting proteins that were identified in the first experiments acted as secondary ‘baits and was processed identically. This ‘reciprocal’ analysis allowed validation of the original findings and identified connections between the baits of the first set of experiments as well as with other proteins (numbered yellow circles). The network was then further refined using mutagenesis and RNA interference. Abbreviation: HA, hemaglutinin.
One of the hallmarks of systems biology has been the utilization of mathematics and computation to develop models and analyze large datasets.20–23 In general, multi-parametric data are collected after an experimental perturbation or during multiple states in which the system can exist, and are subjected to various kinds of computational filtering (such as removal of outliers and normalization). Statistical analyses are often required to identify significant differences between states. The resulting lists or matrices can then be subjected to various kinds of mathematical analyses to identify biologically meaningful patterns. For example, a particularly important application of systems biology has been the modeling of oscillating systems, such as the cell cycle and biological clocks.24,25 Perturbations to a system and interactions between its elements can be modeled using various mathematical equations, or they can be represented as simplified networks.25 With the flood of genomics data from various high-throughput technologies, much emphasis has been placed on the development of statistical methods that can be used to identify functional relationships. For instance, one can ask whether lists of biological parameters (such as changes in gene expression in pathologic states) are enriched for known pathways.19
Role of high-throughput techniques
One of the most-important factors fueling the current interest in systems-based studies was the development of a variety of powerful high-throughput technologies that can generate large datasets (Table 1). The first of these technologies was the gene-expression array, which was developed in the 1990s and allows global quantification of mRNA transcripts.26–33 Gene-expression arrays were originally expensive, but the cost of performing an array experiment has now fallen to nearly that of studying a handful of genes by common methods such as quantitative PCR. Similarly, advances in high-throughput mass spectrometry have made it possible to examine the levels of hundreds or thousands of proteins or metabolites in a single experiment.34–37 A striking example of the utility of ‘metabolomics’ is the discovery in 2011 of a novel molecule that promotes atherosclerosis.38
Table 1.
High-throughput technologies in biology
| Assessed parameter | Technique |
|---|---|
| DNA sequence | NGS |
| DNA methylation | Microarrays, bisulfite treatment followed by NGS |
| Chromatin states | ChIP followed by sequencing (ChIP-Seq) |
| Exome sequence | NGS after capture of specific DNA regions using hybridization |
| Levels of mRNA and mRNA splicing | Gene-expression arrays or high-throughput RNA sequencing |
| Levels of microRNAs | Microarrays, quantitative PCR, or NGS |
| Protein levels | LC–MS |
| Protein–protein interactions | Co-immunoprecipitation followed by LC–MS |
| Metabolite levels | LC–MS, NMR imaging |
| Gut flora | PCR of ribosomal RNA regions followed by NGS |
| Cellular processes | Imaging with reporter systems |
| Clinical phenotypes (in vivo studies) | Imaging, multiplex immunoassays |
Abbreviations: ChIP, Chromatin immunoprecipitation; LC–MS, liquid chromatography combined with mass spectrometry; NGS, next-generation sequencing; NMR, nuclear magnetic resonance.
GWAS are another form of ‘high-throughput biology’ that is relevant to systems-based approaches. In GWAS, the statistical association between common genetic variants in the population and complex phenotypical traits is examined. Alone, GWAS do not have sufficient power to demonstrate interactions among genes or between genes and the environment. However, GWAS can be combined with other techniques, such as expression quantitative trait locus (eQTL) mapping, to provide information about the functions of the loci and genes associated with disease (Figure 2). The power of eQTL mapping was demonstrated in a study by Small and colleagues, who showed how an eQTL acts as a ‘master regulator’ that controls a variety of loci associated with metabolic syndrome.39
Figure 2.

Principles of eQTL analysis. Loci that control transcript levels are referred to as eQTL. Typically, eQTL analysis of humans or mice reveals thousands of eQTL, which are classified as either ‘local’ or ‘distant’, depending on the distance of the locus from the gene that an eQTL regulates. a. A local eQTL most likely acts in cis, meaning it affects the expression of a gene on the same chromosome (for example, an eQTL in a promoter only influences expression of the contiguous gene). b. Distal loci act in trans, affecting both copies of the genes they regulate (for example, an eQTL may encode a transcription factor that controls the expression of the regulated gene on both chromosomes). c. This panel illustrates possible causal relationships between a SNP, a transcript (RNA) and a physiological or pathologic trait (phenotype). Using the SNP as a ‘causal anchor’, causal relationships between the three can be modeled.71 Abbreviations: eQTL, expression quantitative trait locus; SNP, single nucleotide polymorphism.
Particularly important for the progress of systems biology have been the advances in DNA sequencing that, since the time of the Human Genome Project, have resulted in an over 100,000-fold decrease in cost of sequencing per base. The reduction in cost has even enabled researchers to use next-generation sequencing, which is based on massively parallel sequence analysis, to search for rare genetic variants that contribute to complex traits. Indeed, sequencing the entire exome (that is, all protein-coding exons) after isolating coding regions with hybridization techniques is now fairly straightforward.40 The sequencing revolution has also made examination of DNA methylation (the ‘methylome’) or chromatin states (using chromatin immunoprecipitation followed by sequencing, also known as ChIP-Seq) possible on a global scale.40
Biological networks
An important concept that has emerged over the past decade is that biological elements, such as macromolecules and metabolites, do not function in isolation, but instead form ‘networks’ with characteristic topologies.2,7,20,41–45 Network science simplifies complex systems by summarizing the elements (known as ‘nodes’) and their interactions (termed ‘edges’), and by identifying functional groups (called ‘modules’). The concept of biological networks first arose when metabolic pathways were represented as graphs, with metabolites as nodes and their interactions (enzymatic conversions) as edges.45
Biological networks can include interactions between molecules (DNA, transcripts, proteins, metabolites), cell organelles, cells, tissues, and even between individuals and the diseases they have.19,42 A challenge of systems biology is to model networks at each scale—from gene to organism to disease—and then connect orthogonal datasets.
Networks can be constructed on the basis of published biological literature, from results of unbiased experimental studies, or by a combination of the two approaches. In experimental studies, researchers can aim to identify physical interactions between components (as in protein–protein complexes; Figure 1) or to examine correlations between components (for example, between mRNA levels after a series of experimental perturbations; Figure 3).
Figure 3.

Construction of a co-expression network. a. Identify a system of interest (for example, a cell or an animal model) and perturb it in multiple ways (for example, using siRNA or via a series of genetic knockouts). b. Quantitate mRNA transcripts using gene-expression arrays or high-throughput RNA sequencing. c. Identify transcripts that have similar patterns of expression and d. construct a correlation matrix for all transcripts. e. Transform data to a topographical overlap matrix using appropriate algorithms for hierarchical clustering of transcripts (‘modules’). f. Represent data as a graph with genes as circles (‘nodes’) and correlations in expression levels as lines between nodes (‘edges’). Highly connected genes, or ‘hubs’, are indicated by blue circles. In this example, three ‘modules’ of highly connected genes are shown.
Many biological networks seem to have a topology described as ‘scale-free’, in which most nodes have few links, but a small subset of nodes has many links (these subsets are also known as ‘hubs’). Scale-free networks are found throughout biology, and some investigators believe that their architecture confers an evolutionary advantage in that it exhibits biological robustness because multiple alternative pathways exist to get from one node to another.46,47 This redundancy also makes networks adaptable to changing environments, for example, by minimizing the number of steps required for interconversion of various food substances.
Another important consideration in network studies is the degree to which networks are preserved across various phenotypic conditions (such as between sexes, tissues, and species). This problem has been addressed in several studies,48–50 and algorithms to examine network preservation have been developed.51 For instance, whereas networks are largely conserved between sexes, they diverge across species, even in species as closely related as humans and chimpanzees.51 Network divergence can provide useful insights into the similarity or differences between model systems that might not be obvious from examination of their component parts.
Studying cardiovascular diseases
As a result of the diversity in developmental origin, structure, and function of the heart and blood vessels, mapping the complex interactions within the cardiovascular system can be challenging. This task is further complicated by the hierarchy within each component, considering the potential interactions between genes,38 proteins, macro-molecular complexes, organelles, cells, and organs in the cardiovascular system.17,52
Pathogenesis of cardiovascular diseases
Atherosclerosis, a chronic inflammatory disease of the large arteries, is the primary cause of CAD.12,53,54 Most forms of CAD result from metabolic disturbances that include elevated blood pressure, increased levels of low-density lipoproteins, decreased high-density lipoprotein levels, and insulin resistance (Figure 4). The accumulation of oxidized LDL in the subendothelial space initiates an inflammatory response that involves the recruitment of monocytes and, subsequently, lymphocytes to the vessel wall. The development of advanced atherosclerotic lesions is a chronic process that generally takes decades. The most important clinical consequence of atherosclerosis is myocardial infarction, a thrombotic occlusion of a coronary blood vessel that is often precipitated by the rupture or denudation of a lesion and the exposure of prothrombotic tissue factor. If an individual survives a myocardial infarction, the damage can result in cardiac abnormalities, such as arrhythmia or pathological remodeling, with eventual CHF.12,53,54
Figure 4.

Complex interactions in CAD. This schematic diagram depicts our current knowledge of the factors that determine CAD and CHF and their complex interactions. The blue boxes contain risk factors or initiating factors for the disease, orange boxes stand for disease-related traits, and the red-brown boxes represent diseases. The yellow arrow on the left indicates that all these interactions depend on genetic background and patient sex, and that changes are chronic. Abbreviations: CAD, coronary artery disease; CHF, chronic heart failure; EC, endothelial cell; ECM extracellular matrix; ROS, reactive oxygen species; TG, triglycerides; TMA, trimethylamine; TMAO, TMA N-oxide.
The past decade has seen an explosion of research into the pathogenesis of CHF.13,55,56 CHF can result from endogenous defects within the cardiomyocytes themselves or from external factors that place excessive demands upon the heart (Figure 4). Examples of intrinsic factors are diseases such as genetically mediated dilated cardiomyopathy or hypertrophic cardiomyopathy. External factors that can lead to heart failure include myocardial infarction, uncontrolled hypertension, metabolic disorders, or hormonal disorders such as hyperthyroidism. Collectively, these factors increase biomechanical stress and lead to a host of changes at the molecular and cellular level (Figure 4). β-adrenergic blockade and inhibition of the renin–angiotensin–aldosterone system are now the standard regimens in the management of patients with CHF. Despite much progress, CHF remains a leading cause of morbidity and mortality, and no therapy is available that can effectively reverse disease progression.
The many environmental factors that affect the development of CAD and CHF play on the background of hundreds of genetic factors that, although individually exerting modest effects, together dramatically influence disease susceptibility.12 Additional complexities arise when considering age-related effects and differences between sexes or ethnic groups (Figure 4). If the diseases resulted simply from the sum of the individual contributing factors, a reductionistic approach might eventually lead to a detailed understanding of susceptibility and therapeutic responses. However, given the prevalence of interactions among genes or between genes and the environment observed in simple model organisms or cultured cells, more-complex interactions are a certainty.57 Systems biology offers an approach to meet such challenges by attempting to decipher the global principles that govern these complex biological systems.
Cardiovascular networks
A study by Ramsey and co-workers illustrates the approach of experimentally perturbing a biological system and subsequent mathematical modeling of the interactions in the system.58 The investigators explored the macrophage transcriptional network mediated by stimulation of Toll-like receptors (TLRs), which have been implicated in the development of atherosclerosis.53 TLRs recognize a variety of pathogen-associated molecules and possibly certain endogenous molecules. Upon ligand binding, TLRs signal through adaptor molecules, such as Toll–interleukin-1 receptor domain-containing adapter molecule 1 (TICAM-1; also known as TRIF) and Myd88, and then parallel cross-talking signaling pathways. These signaling pathways initiate a cellular program that leads to the differential expression of over 1,000 genes, including hundreds of transcription factors. Although these differentially expressed genes were known, the network of interactions between transcription factors and mRNA levels has proved difficult to address for several reasons. Firstly, the network is combinatorial, involving multiple large datasets. Secondly, many transcription factors are regulated by post-translational modifications, such as phosphorylation. Thirdly, unlike in yeast, creating targeted genetic perturbations in mammals is not feasible on the large scale that is required for some systems-biology studies and siRNA-mediated knockdown of proteins tends to stimulate TLR signaling pathways. Instead, the investigators combined two types of data to infer the network. First, they performed computational scanning of promoter sequences of clusters of co-expressed genes for known transcription-factor binding sites. They then used a signal processing technique called ‘expression dynamics analysis’ and modeled time-course expression data to best fit mRNA levels of a transcription factor with the expression of its potential target genes.
The team used a series of single-gene perturbations to obtain the primary expression data. They treated primary bone-marrow-derived macrophages from five strains of mice (the wild type and four strains with genetic mutations) with six different TLR agonists and performed whole-genome expression-array-analyses at multiple time points after stimulation (0–48 h). In total, 95 different datasets from various combinations of strains, stimuli, and time points were obtained. The set of differentially expressed genes was clustered, promoter sequences of each gene were scanned for transcription-factor binding sites, and the temporal expression patterns of transcription factors and target genes were compared to identify potential causal influences. When integrated, the results provided a broad picture of the dynamic transcriptional program of the TLR network. Many previously known interactions were identified, and a small subset was confirmed experimentally. Altogether, the connections between 36 transcription factors and 27 clusters of target genes were recognized. The integration of time course data with promoter scanning clearly provided a more-detailed picture of the interactions than either technique would have provided alone. This general approach has now been applied to numerous mammalian systems, including cell signaling, oncogenesis, development, and common complex diseases.24,25,59–62
The concept of networks can also be applied to very complex traits, such as the physiological relationships between normal biological features and those observed in disease. In a proof-of-concept paper, Nadeau and colleagues illustrated how genetically randomized populations can be used to construct interaction networks for cardiovascular traits.63 The researchers first showed that significant variation existed in traits such as blood pressure, heart rate, ventricular shortening, ventricular mass, and end-diastolic dimension among inbred strains of mice. To understand the functional relationships between these traits, the team examined a series of recombinant inbred strains. The traits in these strains differed because of subtle, naturally occurring genetic variations. If traits shared genetic factors or showed homeostatic interactions, correlations would be observed among the set of recombinant inbred strains studied, allowing for the modeling of a network in which the nodes are ‘traits’ and the edges are ‘correlations’. The network was consistent with previous physiological studies10 and was further elaborated by examining the effects of certain pathologic perturbations using genetically engineered mice or mice treated with pharmacologic agents.63 This study emphasizes the utility of multiple genetic perturbations, rather than single-gene perturbations, for the analysis of complex systems. Single-gene perturbations, such as in transgenic mice, allow causality to be established, but have limited power to resolve the pleiotrophic and homeostatic interactions resulting from the perturbation.
Systems genetics of cardiovascular traits
Any natural population contains many thousands of variations that perturb gene expression or gene sequence. Some of these variations contribute to disease susceptibility. The concept of ‘systems genetics’ is simple: monitor the effects of natural variations at molecular levels (for example, mRNA, protein, or metabolite levels) and then integrate these data with clinical phenotypes (Figure 5).19,64,65 For example, one might examine gene expression in monocytes from 100 individuals with atherosclerosis and 100 healthy controls, and then ask whether statistically significant differences exist between the two groups. With a dense single-nucleotide polymorphism (SNP) map of the DNA from both groups, one could also map eQTLs and investigate whether these loci match disease susceptibility loci identified in GWAS (Figure 2). Furthermore, one could use the gene-expression data to model co-expression networks and determine which modules are correlated with atherosclerosis. Systems genetics, therefore, allows the integration of molecular and clinical data in a powerful way. Transcript levels have been examined as a function of genetics in a number of clinical and animal studies and have yielded promising results.8,31,65–75
Figure 5.
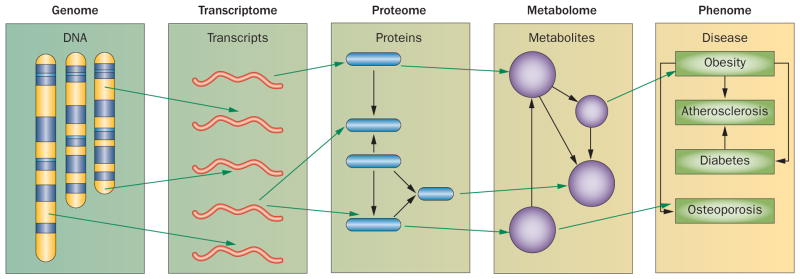
Systems genetics for analysis of complex disease. Although traditional genetic approaches (including genome-wide association studies) attempt to directly relate genetic variation to clinical traits, system genetics monitors various molecular phenotypes, such as levels of mRNA (transcriptome), proteins (proteome), and metabolites (metabolome) as a function of genetics. The molecular data can then be used to identify genetic loci that control their levels (such as expression quantitative trait loci), and such loci can be intersected with loci for clinical traits (phenome). Alternatively, the molecular data can help to model biological networks.
Causality is an important concept in network modeling. For example, co-expression networks are based simply on correlation and, as such, are considered ‘undirected’. However, by making certain assumptions (as described below) or by introducing specific perturbations of a network component, investigators can model causal interactions and derive a ‘directed’ network. For instance, in the above networks8,31,65–75, siRNA knockdown or gene targeting was used to establish causality with respect to certain nodes in the network. Given such causal anchors, and knowing the nodes in a system as well as their variances, deriving a set of equations that describe the causal relationships of the various elements is theoretically possible using structural equation modeling or Bayesian analysis.76–78
In systems genetics, DNA can serve as a causal anchor in that information flows from DNA to mRNA transcripts, proteins, metabolites, and clinical traits, and this concept has been used to broadly model directed biological networks (Figure 3).66,79 An example is the study by Gargalovic and colleagues,80 who used gene co-expression analysis to model an inflammatory network for responses of human endothelial cells to oxidized phospholipids, which are thought to promote atherosclerosis by stimulating endothelial cells to produce inflammatory cytokines such as interleukin 8 (IL-8). Gargalovic et al. exploited common genetic variations in the human population.80 In preliminary studies, the researchers investigated common variations in inflammatory responses of endothelial cells isolated from various individuals using IL-8 immuno-assays. The results indicated that striking differences in responses existed, with some individuals producing essentially no IL-8 in response to oxidized phospholipids, whereas other individuals showed a more than 10-fold induction of IL-8. The investigators then used microarrays to measure global gene expression in endothelial cells isolated from 12 different individuals and observed that over 1,000 genes were differentially regulated by the treatment with oxidized phospholipids. These variations in the sampled population were then used to construct a gene co-expression network comprised of 15 modules of highly connected genes. The identified modules were significantly enriched in genes for previously known pathways (although the role of these genes in this context was not previously recognized), including modules corresponding to two different arms of the unfolded protein response. In addition to confirming key pathways involved in the inflammatory response, the network also predicted regulatory mechanisms and identified previously unknown gene functions.70,80
Romanoski and colleagues extended the study by examining a total of 166 primary endothelial cell lines that they genotyped using high-density SNP arrays.81 This larger dataset enabled the team to integrate genetic associations, including novel trans-acting regulatory pathways, into their network model. Their predictions on the basis of this model were validated by siRNA knockdown and overexpression of the relevant genes, such as predicted regulators of other genes.81
In a systems-genetics study of CHF, Cook and collaborators examined the regulation of left ventricular mass (LVM) in a panel of recombinant inbred rat strains derived from the Brown Norway rat and the spontaneously hypertensive rat (SHR).82,83 The hearts were examined for LVM, and global gene-expression profiles were determined. The genetic loci contributing to LVM were mapped using linkage analysis (that is, by asking which regions of the genome were significantly associated with the traits of interest). The researchers identified a region on rat chromosome 17p14 that was associated with LVM. The many genes contained in this region were screened for those that showed strong cis-eQTL characteristics. The gene encoding osteoglycin stood out as a strong candidate, and subsequent knockout studies in mice confirmed its role in LVM. The investigators then went on to examine mRNA levels in human heart biopsies from patients with aortic stenosis. Of all candidates tested, osteoglycin expression had the highest correlation with LVM measured by echocardiography.82,83 These studies82,83 emphasize the utility of combining animal models with clinical investigations for understanding pathways that contribute to complex traits.
Systems biology and GWAS
Large GWAS have been performed for CAD, CHF, and various related traits such as hyperlipidemia and hypertension.1,84,85 A large number of genetic loci have been identified but, collectively, these loci explain only a small fraction of cases, and the underlying causal gene often cannot be unambiguously identified owing to correlation structure (or linkage disequilibrium) among SNPs at a locus (that is, because of population history, SNPs at a particular locus are often highly correlated). Examining eQTL (Figure 2) at a genetic locus associated with a clinical trait can help prioritize gene candidates and suggest causal mechanisms of disease. For example, a novel genetic locus strongly associated with the regulation of LDL-cholesterol levels was identified on human chromosome 1 in several GWAS.86 The locus contained several candidate genes in linkage disequilibrium with the peak SNP. To narrow the search for the responsible gene, Musunuru and colleagues examined the genes for potential eQTL controlled by the SNP.86 Combining eQTL mapping with various molecular analyses, the researchers identified SORT1 as the gene underlying the phenotype of this locus and validated their findings by overexpressing SORT1 in mice using an adeno-associated viral vector. Their results indicated that SORT1 overexpression decreased VLDL and apolipoprotein B secretion.86
Another example of the use of eQTL mapping to help provide mechanistic information is a study of the Kruppel-like factor 14 (KLF14) transcription factor locus, which was originally found in GWAS to be implicated in certain traits associated with metabolic syndrome.39 Reasoning that KLF14 variation might control target genes, Small et al. identified 10 additional genes with transcript levels that correlated with those of KLF14 in adipose tissue of patients with metabolic syndrome and showed that these transcripts partly mediated the effects of KLF14. Several of these new loci proved to contain SNPs that were also associated with traits of metabolic syndrome.
Beyond eQTL analyses, network modeling can be combined with GWAS to provide information about novel GWAS ‘hits’. A case in point is the large GWAS for CAD conducted by Schunkert and colleagues, who identified a total of 26 loci.1 Most of these genes had no connection to any signaling pathway or risk factor previously associated with the disease, but several of the genes were found in networks modeled for endothelial cells under oxidative stress, immediately providing candidate pathways and interacting genes.57,80 For instance, the poorly understood NT5C2 gene encodes a nucleoside monophosphate phosphohydrolase which, among other substrates, can dephosphorylate noncyclic AMP. How NT5C2 may contribute to atherosclerosis is totally unknown, but the finding that it occurs in an inflammatory module of a human endothelial cell network (Figure 6) suggest possible mechanisms or interactions with other genes through which NT5C2 might affect atherosclerosis progression.
Figure 6.

Network analysis provides context to hits in GWAS. a. A GWAS study for atherosclerosis (involving more than 100,000 individuals) identified genetic loci containing many novel genes contributing to atherosclerosis.1 b. One of these genes, NT5C2, formed a ’hub’ in the ‘green’ module of a human endothelial-cell inflammatory co-expression network. c. The detailed view of the ‘green’ hub reveals interaction partners of NT5C2. Abbreviation: GWAS, genome-wide association studies. Permission for panel a obtained from Nature Publishing Group © Schunkert, H. et al. Nat. Genet. 43, 333–338 (2011).
Clinical implications
Network medicine
The concept of ‘network medicine’ is built upon the hypothesis that perturbations of biological networks, owing to injury, infection or other pathological triggers, are the underlying causes of complex human diseases.5,42 Some investigators believe that systems-based approaches might not only complement reductionistic approaches but that they will be essential to effectively translate the vast amounts of currently available biological information into treatment modalities for common diseases.
How will these novel insights translate into improved health? The translation will likely take several forms. For instance, systems-based approaches might predict new disease-causing genes, identify individuals who are at increased risk of disease or complications, optimize existing therapies in target populations, or lead to the design of completely novel therapies that could not have been developed without a systems-based approach. Whereas disease-associated genes can often be identified with ‘traditional’ genetic approaches, system-based approaches have the potential to identify novel disease genes based on disease-network classification. Disease networks identify the proteins that interact directly with known disease-associated genes and with the genes and proteins (or cellular components), the expression, function, or localization of which is regulated along with the disease-associated gene. Such a network can then be used to predict new disease-causing genes since proteins within a disease network that interact with (or are regulated by) disease-associated genes have an increased likelihood of being disease-causing genes themselves.
Using known gene mutations as edges to link disease nodes that share the same mutations, Barabási and collaborators have built the first human disease network, which has many interesting features and led to some unexpected findings.87 One of these features is that a large number of human diseases cluster in a single component in the disease network, suggesting a common underlying mechanism of a diverse collection of human diseases that involves abnormalities in a specific, critical subset of genes. Unexpectedly, the network also suggests that most disease-causing genes are nonessential and do not preferentially encode hub proteins.
Alternatively, genes associated with a particular disease can be analyzed in a disease-specific gene network. In both general disease-network analyses and disease-specific gene-network analyses, investigators can evaluate the potential interdependence of cardiovascular diseases and CHF with other diseases that might share common pathogenic mechanisms, which could lead to the discovery of previously unknown causal genes associated with cardiovascular disease and CHF.52
Diagnostics
Although molecular diagnosis is less common in cardiology than in oncology, several examples of the successful development of assays that aid in clinical decision-making exist for this field. For instance, approximately 40% of patients who undergo heart transplantation experience at least one episode of acute rejection in the first year after transplantation. In most cases, these rejection episodes are reversible when immunosuppression is intensified. However, immunosuppressive drugs have substantial adverse effects, and rejection can only be diagnosed by endomyocardial biopsy—an invasive procedure that can be painful, expensive, and damaging to the transplanted heart. To circumvent this problem, investigators used microarrays to analyze gene-expression patterns in peripheral leukocytes of patients who had undergone a heart transplantation and revealed specific patterns that identified individuals who were experiencing a rejection.88 The network analysis identified 11 critical genes, the expression of which can be measured in the commercially available AlloMap® assay (XDX, Brisbane, CA, USA) using quantitative reverse transcriptase PCR. AlloMap® testing can reduce the number of invasive biopsies required to monitor selected patients and has a major impact on the way immunosuppression of transplant recipients is monitored.89 Similar efforts are ongoing in patients with CAD or CHF to determine whether gene-expression monitoring can identify disease state and optimize treatment.90
Genetic testing
Classically, genetic information is used to identify individuals who are genetically predisposed to disease so that interventions can be devised to reduce the incidence and severity of disease. Given that consumers can now purchase a direct-to-consumer (DTC) genome-wide disease-risk-profiling kit at their local pharmacy, has the era of personalized genetic risk prediction arrived? These kits quantify genetic risk of a host of medical conditions, but what will the public do with the information, and will it trigger an epidemic of needless screening, treatment, or both? Disease penetrance can vary highly within at-risk populations, presumably secondary to variation in modifier genes or exposure to environmental factors that precipitate the disease (for example, smoking or elevated LDL-cholesterol levels, among others), and their response to interventions aimed at reducing disease risk. Even more concerning is the lack of regulatory oversight for these self-administered tests. Substantial concerns exist within the scientific community about the validity, clinical utility, and marketing claims of many commercially-available DTC test kits. One could envision a utopia where individuals use this genetic disease-risk information to improve their health-related behavior, but is this expectation realistic? Bloss et al. surveyed over 2,000 individuals who voluntarily purchased a DTC genetic risk kit and found that genetic testing did not result in any statistically significant short-term changes in psychological health, dietary fat intake, or exercise behavior.91 Overall, the testing did not increase anxiety levels, but the severity of test-related distress correlated with increasing estimated average lifetime risk of any condition, suggesting the response was proportional to the perceived risk. Worryingly, individuals with higher composite measures of all-disease risk indicated their intention to use more medical testing in the future, validating concerns that this form of testing could open the flood gates to inappropriate disease screening and the costs associated with these tests. Given concerns regarding over-testing and over-utilization of medical resources by most Americans, easily accessible and unregulated tests could burden an already overwhelmed medical system. Although DTC genome-wide disease-risk profiling might seem a revolutionary advance in our approach to medicine, the impacts of such testing on psychology, health, and society are uncertain. However, one issue experts agree on is that if this genetic information is to be applied in a cost-effective way, models that predict high-risk individuals more accurately will be required, which stresses the importance of developing better disease-specific networks.
Pharmacogenomics
Even if the use of the vast sums of genetic information that is currently available is problematic, surely such data could be used to optimize existing therapies in target populations. Integrating genetic information with therapeutic efficacy is the foundation of personalized or stratified medicine, of which pharmacogenomics is the classic example.92,93 Pharmacogenomically-guided dosing has been proposed as an ideal way to improve drug efficacy and reduce adverse effects. Numerous sequence variations in specific genes are associated with altered sensitivity or metabolism of common medication. The best example of this is pharmacogenetically-guided improvements in warfarin dosing, since monitoring anticoagulation therapy is time consuming and expensive, and warfarin overdoses are associated with a large number of adverse reactions.94,95 However, dosing based on polymorphisms in the gene encoding the vitamin K epoxide reductase complex 1 (VKORC1), which partially determines sensitivity of patients to warfarin, neither reduced the incidence of adverse events nor reduced out-of-range blood-clotting times (measured using the international normalized ratio). These results might be a moot point, as new direct thrombin inhibitors are likely to replace the routine use of warfarin in anticoagulation therapy.96
Efforts to develop pharmacogenetic tools in CHF have also encountered problems. One of the fundamental life-prolonging therapies for patients with CHF is the administration of β-blockers, but predicting which patients will respond to this class of medications is impossible with currently available tools. Furthermore, why some β-blockers work whereas others do not is unclear. Conflicting data have been reported on the utility of using polymorphisms within genes that are involved in the metabolism or action of β-blockers to predict therapeutic outcome. However, most investigators have not been able to demonstrate differences in survival among patients with CHF who were stratified on the basis of genetic polymorphisms associated with the pharmacokinetics or pharmacodynamics of β-blocker therapy.97–99
Similarly, given the prevalence of hyperlipidemia in the general population and the common use of statins, clinicians hoped that pharmacogenomics would be a useful tool to distinguish patients in whom statin therapy is beneficial from those who have serious adverse effects.100 Although genetic polymorphisms have been identified that are associated with reductions in cardiovascular events and lipoprotein levels, the fairly small magnitude of these reductions have not influenced clinical decision making as it relates to statin therapy.101,102
Although pharmacogenomically-guided dosing of cardiovascular medications has the potential to improve patient health while decreasing healthcare costs compared with standard dosing regimens, this potential has yet to be realized. The reasons for the limited success of this approach are speculative, but perhaps attempts to adjust drug doses on the basis of a single polymorphism are subject to the same limitations as single-gene approaches are to other aspects of biology. High-throughput approaches that integrate information from GWAS can be used to identify multiple genetic variations that predict drug responses better. For example, Shu et al. integrated diverse data to demonstrate that genetic variation in the organic cation transporter 1 (OCT1) affects the action of metformin, a drug widely used to treat patients with diabetes.103 Metformin enters hepatocytes and enterocytes predominantly via OCT1 and a number of human variants in OCT1 that are associated with reduced transporter activity have been characterized.103 The clinical significance of the interaction between OCT1 polymorphisms and metformin activity has yet to be fully characterized, but this general approach could be used to determine responders to a specific pharmacological therapy and, ultimately, allow personalized tailoring of therapy on the basis of a patient’s genotype.104 Nonetheless, these data provide a cautionary note that the application of pharmacogenomic information in clinical practice is unlikely to be as straightforward as investigators and physicians alike had hoped, and that systems-based approaches may be required here as well to ensure successful translation to the clinics.
Drug discovery
In 1996, 53 new molecular entities were approved by the FDA. Ten years later, this number had dropped to 18 and has remained at similarly low levels ever since.105 The explanation for this drop in approval of new therapeutics is likely to be multifactorial, but might largely be because of the limited number of novel drug targets that are being identified as scientific fields mature and a shift in emphasis to biological therapies. Drug discovery has traditionally been ‘target-focused’. However, complex diseases can sometimes only be understood when viewed from the perspective of the multiple genetic and environmental factors, and—importantly—when taking into account their interactions. Techniques that integrate genetic, expression, and clinical data to elucidate networks underlying disease thus provide a powerful approach to developing novel therapeutic interventions, since they identify critical genes and interactions between genes that might serve as drug targets or reveal adverse effects.8 Analysis of these networks can lead to the discovery of novel drugs or novel uses of existing drugs based on targeting multiple genes within a network.
An example of how a systems-based approach that integrates data from multiple sources can lead to the design of a new drug can be found in the field of cardiac arrhythmia.106 Ventricular arrhythmia, and ventricular fibrillation in particular, is a leading cause of sudden cardiac death. Fibrillation is generated by the deterioration of organized ventricular electrical activity into fragmented electrical waves that lead to ineffective contraction. By integrating experimental evidence and computer modeling of electrical activity, Garfinkel et al. concluded that fibrillation is created and sustained by the property of restitution of the cardiac action-potential duration.107 The restitution properties of the cardiac action-potential duration and conduction velocity contribute to the breakup of re-entrant wavefronts during cardiac fibrillation, independent of pre-existing electrophysiological heterogeneities in the tissue. This inference enabled the team to identify the ‘ideal’ drug to correct the abnormalities in restitution that drive cardiac fibrillation. The investigators subsequently showed that bretylium, a rarely-used antiarrhythmic drug that flattens restitution curves in cardiac myocytes, prevents wavebreak and, therefore, fibrillation.107 The development of therapies that favorably alter these cardiac electrical restitution properties is a promising new approach to preventing fibrillation.
Several other systems-based approaches to drug discovery are moving away from the traditional target-centric approach and instead focus on disease-related pathways and networks. This general approach has the advantage of not requiring specific knowledge of the actual target, or mechanism of action, of the drug candidate. One such approach uses quantitative network structure–activity relationships to identify molecules that shift disease pathways in a direction that is considered to be beneficial.108 This approach involves the development of a disease-based network that can be used to model the effects of module perturbation and couple it with high-throughput profiling of the effects that specific molecules have on individual pathways and on the network as a whole. These datasets are then integrated to identify candidate compounds that have the desired impact on the network and an acceptable safety profile. This approach remains theoretical at this point, but given the progress over the past 5 years in defining disease networks, such an approach is likely to have a major role in drug design in the future.
Conclusions
Common, complex cardiovascular diseases involve nonlinear genetic and environmental interactions that are difficult to dissect using reductionistic approaches. Technological advances have made global interrogation of these biologic systems possible, as well as application of the concepts of systems biology (particularly mathematical modeling) to address their complexity. Systems-based approaches have already revealed novel insights into cardiovascular disease and help to develop new diagnostic and therapeutic modalities. ‘Systems-genetics’ approaches that integrate natural genetic variation with high-throughput technologies and mathematical modeling have been particularly informative. These novel approaches will clearly dominate efforts to improve the health care of patients with cardiovascular disease in the future.
Key Points.
The cardiovascular system is a complex network of organs and cell types each with specialized, but highly coordinated, functions; therefore, cardiovascular disease is complex in etiology and manifestation
A systems-based approach aims to reveal the architecture and the emerging properties of a complex network by uncovering the relationships among the constituents and establishing global governing principles
Current advances in bioinformatics, genomics, proteomics, and metabolomics offer an excellent opportunity to use systems-based analysis to dissect complex networks involved in cardiovascular physiology and diseases
In the study of cardiovascular diseases, systems biology compliments genetic analyses, such as genome-wide association studies, by establishing the underlying mechanisms and the functional significance of the candidate genes
‘Systems genetics’ is a new approach based on systems-based analysis of genetic variants and phenotypic spectra at various levels, spanning from gene expression to organ physiology
By establishing the molecular components and gene networks for the cardiovascular system, systems biology can help to develop effective and personalized diagnostic tools and therapies for cardiovascular diseases
Review criteria.
The articles on which this Review is based were identified primarily through searches for specific investigators in the PubMed database, or were known to the authors from their work in the field. No date or language limits have been applied.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
All authors contributed equally to all aspects of the article, including researching data, discussion of content, and writing, reviewing and editing the manuscript before submission
Contributor Information
W. Robb MacLellan, Cardiovascular Research Laboratories, University of California Los Angeles, 675 Charles E. Young Drive South, MRL 3645, University of California Los Angeles, Los Angeles, CA 90095-1760, USA.
Yibin Wang, Department of Anesthesiology, BH-569 CHS, University of California Los Angeles, Los Angeles, CA 90095-7115, USA.
Aldons J. Lusis, David Geffen School of Medicine, University of California Los Angeles, 695 Charles E. Young Drive South, 6524A Gonda Building, Los Angeles, CA 90095-1679, USA
References
- 1.Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barabasi AL. Linked: The new science of networks. Penguin Books; London: 2002. [Google Scholar]
- 3.Bousquet J, et al. Systems medicine and integrated care to combat chronic noncommunicable diseases. Genome Med. 2011;3:43. doi: 10.1186/gm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 5.Nadeau JH, Subramaniam S. Systems biology and medicine: a new take on an old paradigm. Wiley Interdiscip Rev Syst Biol Med. 2009;1:1–3. doi: 10.1002/wsbm.34. [DOI] [PubMed] [Google Scholar]
- 6.Nadeau JH, Subramaniam S. Systems biology—old wine in a new bottle or is the bottle changing the wine? Wiley Interdiscip Rev Syst Biol Med. 2010;2:1–2. doi: 10.1002/wsbm.91. [DOI] [PubMed] [Google Scholar]
- 7.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- 8.Schadt EE, Lum PY. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Reverse engineering gene networks to identify key drivers of complex disease phenotypes. J Lipid Res. 2006;47:2601–2613. doi: 10.1194/jlr.R600026-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Noble D. The surprising heart: a review of recent progress in cardiac electrophysiology. J Physiol. 1984;353:1–50. doi: 10.1113/jphysiol.1984.sp015320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble D. Modeling the heart—from genes to cells to the whole organ. Science. 2002;295:1678–1682. doi: 10.1126/science.1069881. [DOI] [PubMed] [Google Scholar]
- 11.Noble D. The music of life: biology beyond genes. Oxford University Press; Oxford: 2006. [Google Scholar]
- 12.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 14.Bondarenko VE, Szigeti GP, Bett GC, Kim SJ, Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1378–1403. doi: 10.1152/ajpheart.00185.2003. [DOI] [PubMed] [Google Scholar]
- 15.Greenstein JL, Winslow RL. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys J. 2002;83:2918–2945. doi: 10.1016/S0006-3495(02)75301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss JN, et al. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98:1244–1253. doi: 10.1161/01.RES.0000224540.97431.f0. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 19.Lusis AJ, Weiss JN. Cardiovascular networks: systems-based approaches to cardiovascular disease. Circulation. 2010;121:157–170. doi: 10.1161/CIRCULATIONAHA.108.847699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman MEJ. Networks: an introduction. Oxford University Press; Oxford: 2010. [Google Scholar]
- 21.Shipley B. Cause and correlation: a user’s guide to path analysis, structural equations and causal inference. Cambridge University Press; Cambridge: 2002. [Google Scholar]
- 22.Subramaniam S, Nadeau JH. Systems medicine—viewed through the real and computing lenses. Wiley Interdiscip Rev Syst Biol Med. 2010;2:383–384. doi: 10.1002/wsbm.103. [DOI] [PubMed] [Google Scholar]
- 23.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrell JE, Jr, Tsai TY, Yang Q. Modeling the cell cycle: why do certain circuits oscillate? Cell. 2011;144:874–885. doi: 10.1016/j.cell.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Yosef N, Regev A. Impulse control: temporal dynamics in gene transcription. Cell. 2011;144:886–896. doi: 10.1016/j.cell.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giallourakis C, Henson C, Reich M, Xie X, Mootha VK. Disease gene discovery through integrative genomics. Annu Rev Genomics Hum Genet. 2005;6:381–406. doi: 10.1146/annurev.genom.6.080604.162234. [DOI] [PubMed] [Google Scholar]
- 28.Stoll M, et al. A genomic-systems biology map for cardiovascular function. Science. 2001;294:1723–1726. doi: 10.1126/science.1062117. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keller MP, Attie AD. Physiological insights gained from gene expression analysis in obesity and diabetes. Annu Rev Nutr. 2010;30:341–364. doi: 10.1146/annurev.nutr.012809.104747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su WL, Kleinhanz RR, Schadt EE. Characterizing the role of miRNAs within gene regulatory networks using integrative genomics techniques. Mol Syst Biol. 2011;7:490. doi: 10.1038/msb.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong H, Yang X, Kaplan LM, Molony C, Schadt EE. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am J Hum Genet. 2010;86:581–591. doi: 10.1016/j.ajhg.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng MC, et al. Noninvasive discrimination of rejection in cardiac allograft recipients using gene expression profiling. Am J Transplant. 2006;6:150–160. doi: 10.1111/j.1600-6143.2005.01175.x. [DOI] [PubMed] [Google Scholar]
- 34.Peri S, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerszten RE, Asnani A, Carr SA. Status and prospects for discovery and verification of new biomarkers of cardiovascular disease by proteomics. Circ Res. 2011;109:463–474. doi: 10.1161/CIRCRESAHA.110.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis GD, Gerszten RE. Toward metabolomic signatures of cardiovascular disease. Circ Cardiovasc Genet. 2010;3:119–121. doi: 10.1161/CIRCGENETICS.110.954941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang TJ, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Small KS, et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lander ES. Genome-sequencing anniversary. The accelerator. Science. 2011;331:1024. doi: 10.1126/science.1204037. [DOI] [PubMed] [Google Scholar]
- 41.Neto EC, Keller MP, Attie AD, Yandell BS. Causal graphical models in systems genetics: a unified framework for joint inference of causal network and genetic architecture for correlated phenotypes. Ann Appl Stat. 2010;4:320–339. doi: 10.1214/09-aoas288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell. 2011;144:986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barabasi AL, Bonabeau E. Scale-free networks. Sci Am. 2003;288:60–69. doi: 10.1038/scientificamerican0503-60. [DOI] [PubMed] [Google Scholar]
- 44.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 45.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 46.Albert R, Jeong H, Barabasi AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–382. doi: 10.1038/35019019. [DOI] [PubMed] [Google Scholar]
- 47.Barabasi AL. Scale-free networks: a decade and beyond. Science. 2009;325:412–413. doi: 10.1126/science.1173299. [DOI] [PubMed] [Google Scholar]
- 48.Cai C, et al. Is human blood a good surrogate for brain tissue in transcriptional studies? BMC Genomics. 2010;11:589. doi: 10.1186/1471-2164-11-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci USA. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Nas A, et al. Elucidating the role of gonadal hormones in sexually dimorphic gene coexpression networks. Endocrinology. 2009;150:1235–1249. doi: 10.1210/en.2008-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loscalzo J, Kohane I, Barabasi AL. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 54.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 55.Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–2018. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 56.Creemers EE, Wilde AA, Pinto YM. Heart failure: advances through genomics. Nat Rev Genet. 2011;12:357–362. doi: 10.1038/nrg2983. [DOI] [PubMed] [Google Scholar]
- 57.Romanoski CE, et al. Systems genetics analysis of gene-by-environment interactions in human cells. Am J Hum Genet. 2010;86:399–410. doi: 10.1016/j.ajhg.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramsey SA, et al. Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics. PLoS Comput Biol. 2008;4:e1000021. doi: 10.1371/journal.pcbi.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ideker T, Dutkowski J, Hood L. Boosting signal-to-noise in complex biology: prior knowledge is power. Cell. 2011;144:860–863. doi: 10.1016/j.cell.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lander AD. Pattern, growth, and control. Cell. 2011;144:955–969. doi: 10.1016/j.cell.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Becker L, et al. A macrophage sterol-responsive network linked to atherogenesis. Cell Metab. 2010;11:125–135. doi: 10.1016/j.cmet.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brehme M, Vidal M. A global protein-lipid interactome map. Mol Syst Biol. 2010;6:443. doi: 10.1038/msb.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nadeau JH, et al. Pleiotropy, homeostasis, and functional networks based on assays of cardiovascular traits in genetically randomized populations. Genome Res. 2003;13:2082–2091. doi: 10.1101/gr.1186603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nadeau JH, Dudley AM. Genetics. Systems genetics. Science. 2011;331:1015–1016. doi: 10.1126/science.1203869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghazalpour A, et al. Genomic analysis of metabolic pathway gene expression in mice. Genome Biol. 2005;6:R59. doi: 10.1186/gb-2005-6-7-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghazalpour A, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2:e130. doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keller MP, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18:706–716. doi: 10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meng H, et al. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc Natl Acad Sci USA. 2007;104:4530–4535. doi: 10.1073/pnas.0607620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS, Lusis AJ. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J Immunol. 2009;182:466–476. doi: 10.4049/jimmunol.182.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schadt EE, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37:710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang SS, et al. Identification of pathways for atherosclerosis in mice: integration of quantitative trait locus analysis and global gene expression data. Circ Res. 2007;101:e11–e30. doi: 10.1161/CIRCRESAHA.107.152975. [DOI] [PubMed] [Google Scholar]
- 73.Yang X, et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet. 2009;41:415–423. doi: 10.1038/ng.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hagg S, et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: the Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet. 2009;5:e1000754. doi: 10.1371/journal.pgen.1000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dobrin R, et al. Multi-tissue coexpression networks reveal unexpected subnetworks associated with disease. Genome Biol. 2009;10:R55. doi: 10.1186/gb-2009-10-5-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beaumont MA, Rannala B. The Bayesian revolution in genetics. Nat Rev Genet. 2004;5:251–261. doi: 10.1038/nrg1318. [DOI] [PubMed] [Google Scholar]
- 77.Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR. A primer on learning in Bayesian networks for computational biology. PLoS Comput Biol. 2007;3:e129. doi: 10.1371/journal.pcbi.0030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Emilsson V, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 79.Ferrara CT, et al. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet. 2008;4:e1000034. doi: 10.1371/journal.pgen.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gargalovic PS, et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA. 2006;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Romanoski CE, et al. Network for activation of human endothelial cells by oxidized phospholipids: a critical role of heme oxygenase 1. Circ Res. 2011;109:e27–e41. doi: 10.1161/CIRCRESAHA.111.241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petretto E, et al. Integrated genomic approaches implicate osteoglycin (Ogn) in the regulation of left ventricular mass. Nat Genet. 2008;40:546–552. doi: 10.1038/ng.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sarwar R, Cook SA. Genomic analysis of left ventricular remodeling. Circulation. 2009;120:437–444. doi: 10.1161/CIRCULATIONAHA.108.797225. [DOI] [PubMed] [Google Scholar]
- 84.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 85.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 86.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goh KI, et al. The human disease network. Proc Natl Acad Sci USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horwitz PA, et al. Detection of cardiac allograft rejection and response to immunosuppressive therapy with peripheral blood gene expression. Circulation. 2004;110:3815–3821. doi: 10.1161/01.CIR.0000150539.72783.BF. [DOI] [PubMed] [Google Scholar]
- 89.Pham MX, et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med. 2010;362:1890–1900. doi: 10.1056/NEJMoa0912965. [DOI] [PubMed] [Google Scholar]
- 90.Eagle KA, et al. Identifying patients at high risk of a cardiovascular event in the near future: current status and future directions: report of a national heart, lung, and blood institute working group. Circulation. 2010;121:1447–1454. doi: 10.1161/CIRCULATIONAHA.109.904029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bloss CS, Schork NJ, Topol EJ. Effect of direct-to-consumer genomewide profiling to assess disease risk. N Engl J Med. 2011;364:524–534. doi: 10.1056/NEJMoa1011893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ginsburg GS, Donahue MP, Newby LK. Prospects for personalized cardiovascular medicine: the impact of genomics. J Am Coll Cardiol. 2005;46:1615–1627. doi: 10.1016/j.jacc.2005.06.075. [DOI] [PubMed] [Google Scholar]
- 93.Nadeau JH, Topol EJ. The genetics of health. Nat Genet. 2006;38:1095–1098. doi: 10.1038/ng1006-1095. [DOI] [PubMed] [Google Scholar]
- 94.Anderson JL, et al. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation. 2007;116:2563–2570. doi: 10.1161/CIRCULATIONAHA.107.737312. [DOI] [PubMed] [Google Scholar]
- 95.Krynetskiy E, McDonnell P. Building individualized medicine: prevention of adverse reactions to warfarin therapy. J Pharmacol Exp Ther. 2007;322:427–434. doi: 10.1124/jpet.106.117952. [DOI] [PubMed] [Google Scholar]
- 96.Connolly SJ, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139–1151. doi: 10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 97.Baudhuin LM, et al. Relation of ADRB1, CYP2D6, and UGT1A1 polymorphisms with dose of, and response to, carvedilol or metoprolol therapy in patients with chronic heart failure. Am J Cardiol. 2010;106:402–408. doi: 10.1016/j.amjcard.2010.03.041. [DOI] [PubMed] [Google Scholar]
- 98.Sehnert AJ, et al. Lack of association between adrenergic receptor genotypes and survival in heart failure patients treated with carvedilol or metoprolol. J Am Coll Cardiol. 2008;52:644–651. doi: 10.1016/j.jacc.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 99.Sehrt D, Meineke I, Tzvetkov M, Gultepe S, Brockmöller J. Carvedilol pharmacokinetics and pharmacodynamics in relation to CYP2D6 and ADRB pharmacogenetics. Pharmacogenomics. 2011;12:783–795. doi: 10.2217/pgs.11.20. [DOI] [PubMed] [Google Scholar]
- 100.SEARCH Collaborative Group et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 101.Chasman DI, et al. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA. 2004;291:2821–2827. doi: 10.1001/jama.291.23.2821. [DOI] [PubMed] [Google Scholar]
- 102.Thompson JF, et al. An association study of 43 SNPs in 16 candidate genes with atorvastatin response. Pharmacogenomics J. 2005;5:352–358. doi: 10.1038/sj.tpj.6500328. [DOI] [PubMed] [Google Scholar]
- 103.Shu Y, et al. Evolutionary conservation predicts function of variants of the human organic cation transporter, OCT1. Proc Natl Acad Sci USA. 2003;100:5902–5907. doi: 10.1073/pnas.0730858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reitman ML, Schadt EE. Pharmacogenetics of metformin response: a step in the path toward personalized medicine. J Clin Invest. 2007;117:1226–1229. doi: 10.1172/JCI32133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hughes B. 2009 FDA drug approvals. Nat Rev Drug Discov. 2010;9:89–92. doi: 10.1038/nrd3101. [DOI] [PubMed] [Google Scholar]
- 106.Weiss JN, Garfinkel A, Chen PS. Novel approaches to identifying antiarrhythmic drugs. Trends Cardiovasc Med. 2003;13:326–330. doi: 10.1016/j.tcm.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 107.Garfinkel A, et al. Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA. 2000;97:6061–6066. doi: 10.1073/pnas.090492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baggs JE, Hughes ME, Hogenesch JB. The network as the target. Wiley Interdiscip Rev Syst Biol Med. 2010;2:127–133. doi: 10.1002/wsbm.57. [DOI] [PubMed] [Google Scholar]
- 109.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang ZV, Rothermel BA, Hill JA. Autophagy in hypertensive heart disease. J Biol Chem. 2010;285:8509–8514. doi: 10.1074/jbc.R109.025023. [DOI] [PMC free article] [PubMed] [Google Scholar]