

Abstract
The aim of the present review is to discuss the participation of mast cells in the pathogenesis of erosion and rupture of atherosclerotic plaques, the major causes behind acute coronary syndromes and myocardial infarction. We present ex vivo observations describing mast cells and their activation in human atherosclerotic plaques and discuss in vitro and in vivo data showing that mast cells are potential regulators of inflammation, immunity and adverse remodeling, including matrix remodeling and cell death. Furthermore, we focus on studies that have been performed with human tissues and human mast cells, but when appropriate, we also discuss observations made in animal models. Finally, we present potential pharmacological means to modulate mast cell responses in the arterial vessel walls.
Keywords: apoptosis, atherosclerosis, endothelium, erosion, mast cell, pericellular matrix, plaque rupture, proteases, smooth muscle cell
Mast cells – an introduction
The origin of mast cells
Mature mast cells are progeny of multipotent haematopoietic stem cells that commit to the mast cell lineage already in the bone marrow, being positive for CD34 and c-kit, but negative for FcɛRI[1–3]. The mast cell-committed progenitors leave the bone marrow, circulate in the blood as CD34+ and c-kit+ pre-cursor cells [4], and adhere to activated endothelial cells viaα4β1 integrins, vascular cell adhesion molecule-1 (VCAM-1) and E-selectin [5]. In the presence of specific chemotactic signals, such as stem cell factor (SCF) and eotaxin [6], the mast cell precursors migrate into tissues using as sensors of chemotaxis different chemokine receptors, such as CXCR2, CCR3, CXCR4 and CCR5 [7]. Recently, the expression of transcription factor T-bet by dendritic cells has been shown to regulate the homing of mast cell pre-cursors [8]. In the tissues, the proliferation and differentiation of mast cell precursors depend on the presence of local growth factors and cytokines, notably SCF, IL-3, IL-4, IL-6, IL-9 and NGF [9–12], which are secreted by various tissue cells. The crucial requirement of a functional SCF and c-kit signaling system for mast cell growth and development is underscored by the fact that lack of c-kit signaling in mice results in mast cell deficiency [13–15], whereas an elevated expression of c-kit in patients induces mastocytosis [16, 17]. Similarly, injection of SCF into the skin of human beings results in local accumulation of mast cells [18]. Depending on the site of tissue infiltration, the precursor cells proliferate and differentiate either into connective tissue-type or mucosal-type mast cells, two well-described subsets of mast cells that differ in the quality and quantity of their stored mediators and also in their physiological functions [19].
Mast cell subtypes
The connective tissue-type and mucosal mast cells can be morphologically distinguished using histochemical, electron microscopic, biochemical or immunological criteria that determine their tissue distribution, specific structural features and mediator content [19]. Using a more simplistic approach, human mast cells have been divided into subtypes depending on their variable content of two neutral serine proteases, tryptase and chymase. Thus, mast cells containing only tryptase (MCT) represent the mucosal mast cells and are typically present in the lungs and in the intestinal mucosa. Mast cells containing both tryptase and chymase (MCTC) represent the connective tissue-type mast cells and are typically found in the skin, synovium and perivascular tissue. However, the two tissue subtypes can interchange, their ultimate phenotype (MCT or MCTC) being determined by their microenvironment. Indeed, by varying the culture conditions in vitro human mast cells of different phenotypes may be obtained [20]. The presence of IL-4 [21] or IL-6 [10] has been shown to induce the differentiation of CD34+ precursor cells into chymase-containing human MCTC.
Mast cell localization and physiological function
Mast cells are generally found at the boundaries between the outside world and the internal milieu of the body, such as in the skin and in the mucosa of the pulmonary, gastrointestinal and genitourinary system. Moreover, they are prevalent in the conjunctivas and the mucosa of the nose. At these sites, they act as surveillance antennae of the local microenvironment and direct immune responses by regulating innate and adaptive immune mechanisms [22, 23]. In addition, mast cells are present in most vascularized tissues, where they reside in the vicinity of blood vessels and lymphatic vessels [24]. Although mast cells are best known for their ability to release histamine and to induce IgE-mediated type I hypersensitivity reactions [25, 26], they also initiate and regulate inflammatory responses, defend the host against bacterial and parasitic pathogens, regulate vascular functions, participate in wound healing and neovascularization and recruit and activate other types of inflammatory cells, and stromal cells, as well [27–30].
Based on the above findings, the mast cells may be considered 'sentinels' or 'friends' that play an important role in the normal homeostasis of the body. Indeed, several studies in mice have indicated that mast cell deficiency may be harmful and even lethal if the mast cell-deficient host is pre-disposed to exogenous insults, such as bacterial infections, acute septic peritonitis, ET-1 or snake and honeybee venom [31–34]. However, chronic local activation of mast cells in diseases, such as atherosclerosis [35], rheumatoid arthritis [36] and congestive heart failure [37], may result in a 'foe' response that is unregulated, and if not properly terminated, may turn out to be harmful and even lethal to the host.
Mast cell – a potent effector cell
Mast cells contain a wide variety of preformed mediators that are secreted acutely upon mast cell activation with ensuing degranulation and that participate in the mast cell-mediated 'friend or foe' responses. The preformed mediators can roughly be divided into five classes of effector molecules, notably histamine, proteoglycans, proteases, growth factors and cytokines, all of which may have an impact on the vulnerability of an atherosclerotic plaque. In addition to the preformed mediators, the activated mast cells also produce newly formed lipid mediators of which prostaglandins and leukotrienes are the major ones, as well as cytokines and chemokines. Below, we shortly discuss the mast cell-derived mediators and their possible relation to the pathogenesis of plaque erosion and rupture.
Histamine
Histamine is a biogenic amine that exerts its functions via four different histamine receptors (H1, H2, H3 and H4 receptors) differently expressed in various cells and tissues. The physiological and pathophysiological effects of histamine include blood vessel dilatation, increased vasopermeability and translocation of P-selectin to the endothelial cell surface with subsequent induction of leukocyte rolling in vivo[38]. Thus, the histamine-mediated effects lead to local edema (swelling) and attraction of inflammatory cells, which may then increase the susceptibility of plaque erosion and rupture. Furthermore, histamine has been shown to induce the expression of Toll-like receptors 2 and 4 in endothelial cells, which may be of relevance in local innate immune responses in the vulnerable plaque [39]. In addition, histamine induces the expression of tissue factor by smooth muscle and endothelial cells [40], which may promote thrombosis in atherosclerotic arteries. Since atherosclerotic coronary arteries contain more histamine than healthy coronary arteries and also are hyperreactive to it, histamine has a potential of inducing vasospasm in human athero-sclerotic coronaries [41].
Mast cell-derived proteoglycans
Human mast cell granules contain heparin and chondroitin sulphate proteoglycans that form the macro-complexes in which proteases and growth factors are embedded. Upon degranulation, the exocytosed mast cell proteoglycans exert antithrombotic effects [42] and a variety of functions on neighboring cells, in particular the smooth muscle cells [43–45]. Heparin may also bind lipoproteins, and thus play a role in foam cell formation, i.e. in the accumulation of low-density lipoprotein (LDL) cholesterol in macrophages typically seen in the early stages of atherogenesis [46]. Heparin is also important for the activity of mast cell serine proteases, as it stabilizes the active tetrameric form of tryptase [47] and protects chymase and cathepsin G from their natural inhibitors present in the interstitial fluid [48–50]. Thus, in contrast to many proteases released by other inflammatory cells, the mast cell-derived proteolytic enzymes are secreted bound to proteoglycans that partially protect them from inactivation in the presence of natural protease inhibitors. Mast cell heparin also binds several growth factors, attenuates their inactivation and allows their prolonged presence in the extracellular space.
Mast cell-derived proteases
Tryptase is a mast cell-specific neutral serine protease, which is found in two isoforms, α- and β-tryptase, the former being released from mast cells constitutively and the latter upon degranulation [51]. The tetrameric form of β-tryptase is proteolytically active when bound to heparin, but an active monomeric form has also been described [52]. When released by the mast cells, tryptase is capable of activating neighboring cells by cleaving and activating protease-activated receptor (PAR)-2 [53], and thrombin receptors [54]. In addition, tryptase has been shown to degrade neuropeptides, such as vasoactive intestinal peptide (VIP) and calcitonin gene related peptide (CGRP) [55], and the pericellular matrix components fibronectin and vitronectin [56, 57], and to activate pro-matrix metalloproteinases (MMP)-1, -2 and -3 [58, 59]. Interestingly, there are no known natural inhibitors of the tetrameric form of ß-tryptase [60]; however, the disruption of tryptase–proteoglycan interactions by lactoferrin can inactivate tryptase by inducing its monomerization [61]. Also, the reassembly and activity of monomers can be inhibited by a variety of endogenous inhibitors, such as antithrombin III and α-2-macroglobulin [52]. Furthermore, tryptases isolated from different tissues are very heterogeneous and show variation in size, charge and proteolytic activity [62, 63].
Chymase is a neutral serine protease secreted exclusively by MCTC and it appears that chymase plays a major role in the proteolytic activation of MMP-1, -2 and -9 [59, 64, 65] as well as in the physiologic degradation of fibronectin and thrombin in tissues [66]. Chymase may also act as an angiotensin-converting enzyme [67–69], release latent TGF-ß1 from the extracellular matrix [70], inhibit smooth muscle growth [71] and induce apoptosis of arterial smooth muscle cells [72] and endothelial cells [73]. In addition, mast cell proteases and endotoxin synergistically activate human coronary artery endothelial cells to generate interleukin-6 and interleukin-8, which may enhance the inflammatory response in atherosclerotic lesions [74].
Cathepsin G is an elastolytic, angiotensin II-forming serine protease that is present in the granules of human MCTC[75]. Our recent observations show that mast cells are a major local source of cathepsin G in the coronary arteries and that cathepsin G can degrade both fibronectin and VE-cadherin [76], which are necessary for adhesion of endothelial cells to their basement membrane and to each other.
Renin, an angiotensin I-forming enzyme has recently been shown to be present in cardiac mast cells of rats and guinea pigs, as well as in the human mastocytoma cell line, HMC-1 [77, 78]. If present also in mast cells of the arterial wall, renin may participate in the local production of angiotensin II, and participate in the adverse vascular effects mediated by the reninangiotensin system (RAS). Angiotensin II has been shown to induce atherosclerosis through various processes, such as by inducing endothelial dys-function, cellular proliferation and inflammation [79].
Mast cells are also known to produce MMPs, notably MMP-1 [80] and MMP-9 [81, 82], that may actively participate in plaque remodeling and increase the susceptibility to plaque erosion and rupture. Although mast cells have been shown to contain tissue inhibitor metalloproteinase 4 (TIMP-4) [83], the mast cell-derived proteases may promote the activity of MMPs by degrading the TIMPs [84]. The important role of various MMPs in plaque destabilization has been demonstrated in mouse models of atherosclerosis [85].
Growth factors and preformed cytokines
Human mast cells have also been shown to contain several different growth factors, such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF) and transforming growth factor (TGF)-β, that upon secretion may recruit effector cells and enhance angiogenesis [86], and thus, participate in the pathogenesis of plaque erosion and rupture. Tumour necrosis factor (TNF)-α is a preformed cytokine that is stored in the granule compartment and released with the granules during mast cell activation and degranulation, and also secreted after mast cell activation as a newly formed cytokine [87]. TNF-α is known to induce either directly or indirectly endothelial dysfunction and expression of the endothelial cell adhesion molecules P-selectin, E-selectin and VCAM [88], which are responsible for leukocyte infiltration. TNF-α is also known to increase endothelial permeability at very low concentrations [89, 90], an effect that is mediated via TNF receptor 1 [91]. Interestingly, the TNF-α-mediated endothelial effects may be potentiated by interferon (IFN)-γ[92], a cytokine also found in atherosclerotic lesions.
Newly formed cytokines and chemokines
Upon activation, mast cells produce a plethora of different cytokines and chemokines that may actively participate in the pathogenesis of the atherosclerotic plaques. These include at least the following: TNF-α, IFN-α, IFN-β, IL-1α IL-1β, IL-6, IL-18, granulocyte macrophage-colony stimulating factor (GM-CSF), and leukaemia inhibitory factor (LIF), all of which are involved in the induction of inflammation [86]. Furthermore, mast cells also secrete cytokines that are capable of inducing either T helper 1-type (Th1) polarization (IL-12 and IFN-γ) or T helper 2-type (Th2) polarization (IL-3, IL-4, IL-5, IL-9, IL-13, IL-15 and IL-16) [93]. The activated mast cells also secrete IL-10 and TGF-β, which are known to attenuate the inflammatory response in the atherosclerotic lesions [94]. In addition to releasing cytokines, the activated mast cells are able to secrete both –chemokines (CXCL1, CXCL2, CXCL8, CXCL9, CXCL10, CXCL11) and β-chemokines (CCL2, CCL3, CCL4, CCL5, CCL11, CCL20) that recruit additional effector cells and regulate immune responses [29, 86, 95].
Newly formed lipid mediators
Activated mast cells also mobilize arachidonic acid through the activation of cytosolic phospholipase A2, with ensuing rapid generation and secretion of both prostaglandin D2 and leukotriene C4[96]. The mast cell-derived eicosanoids interact with their respective cellular receptors and are known to serve diverse functions in vasoconstriction, cell trafficking, antigen presentation, immune cell activation, matrix deposition and fibrosis [96]. Interestingly, atherosclerotic arteries are hyperresponsive to the constricting effects of leukotrienes [97], whereas prostanoids, by being important mediators of inflammation and endothelial dysfunction are also likely to contribute to the hyperresponsiveness of the endothelium in atherosclerosis lesions [98].
Mast cells in the vessel wall
In the arterial vessel wall, mast cells localize preferentially to the subendothelial space of the intimal layer, i.e. to the close vicinity of the endothelium (ECs), which likely reflects the fact that ECs are a major source of SCF [99, 100], a vital growth factor for mast cells in the vessel wall. In the normal human aortic intima, approximately 60% of all mast cells (15 mast cells/mm2) are of the MCT subtype, and approximately 40% are of the MCTC subtype [101]. It is evident that the microenvironment in the arterial wall is effectively influencing the mast cell phenotype, since both subtypes can be found in the same atherosclerotic plaques [101]. In healthy human arteries, there are no mast cells present in the medial layer, i.e. the layer that constitutes of concentric layers of contractile smooth muscle cells, and is responsible for the regulation of the arterial tone. However, the outer adventitial layer of a normal coronary artery contains high amounts of mast cells (19 mast cells/mm2). Even larger numbers of mast cells were found to be present in the adventitia of ruptured plaques (98 mast cells/mm2) than in the adventitia of non-ruptured plaques (41 mast cells/mm2) [102]. The majority of the adventitial mast cells contained both tryptase and chymase, and were the only cells in the coronary adventitia that contained histamine. Interestingly, the amounts of adventitial macrophages and T-lymphocytes were also found to be increased in the segments with plaque rupture, indicating that the inflammatory process is not only restricted to the intimal layer of the lesion. Long-term cocaine abusers have an increased level of atherosclerosis and increased numbers of adventitial mast cells in their coronary arteries, suggesting that adventitial mast cells may potentiate atherosclerosis, vasospasm, thrombosis and even premature sudden death [103]. Interestingly, while the normal coronary intima has only low numbers of mast cells, the intima of healthy aortas contains significant numbers of mast cells [101]. In sharp contrast to human beings, in the normal mouse no mast cells are present in the aortic intima or media. However, since the distance between the intima and adventitia is small in mice, the adventitial mast cells may well be capable of influencing the atherogenic processes that occur within the intima.
Mast cells and atherosclerosis
A short history
Originally, mast cells were suggested to participate in the pathogenesis of atherosclerosis by Constantinides in 1953 [104, 105]. Three years later McGovern suggested that, by residing in the immediate vicinity of endothelial surfaces, human mast cells may regulate arterial thrombus formation by releasing the antithrombotic mediator, heparin [106]. Shortly thereafter, the increased numbers of adventitial mast cells were found to correlate with the progression of ather-osclerosis, their numbers being particularly high in areas of arterial thrombosis [107]. Importantly, the highest numbers of intimal mast cells have been found at sites of rupture and/or erosion in infarct-related coronary arteries [35]. Similar to the coronary arteries, the numbers of mast cells in the carotid arteries increased as the atherosclerotic lesions became more advanced [108, 109]. However, in contrast to coronary and carotid lesions, the number of intimal mast cells in the aorta seemed to decline as the atherosclerotic disease progressed [101]. Interestingly, a general role for mast cells in atherosclerosis has been suggested in a study with Brown Norway rats, in which the authors showed that, after induction of atherosclerosis by immunization with ovalbumin, the intimal thickness correlated positively with the level of chymase [110]. In addition, preliminary data in our laboratory suggest that mast cell-deficient KitW-sh/W-sh mice on a LDLR−/− background develop less atherosclerosis when fed an atherogenic western-type diet (Fig. 1). Thus, increasing in vitro and in vivo evidence suggests that mast cells contribute to the early pathogenesis of atherosclerosis by promoting intimal lipid accumulation and foam cell formation by multiple mechanisms [46] (Fig. 2).
1.

Diet-induced atherosclerosis is attenuated in mast cell-deficient mice. To test the role of mast cells in the pathogenesis of atherosclerosis in vivo, mast cell-deficient mice (KitW-sh/W-sh) were crossed with LDLR-deficient mice (LDLR−/−) to obtain a LDLR−/−/KitW-sh/W-sh double knockout mouse model. As shown in Figure 1, the extent of aortic atherosclerosis that is induced in an LDLR−/−/Kit+/+ mouse (A) by a Western-type diet for 9 weeks is slightly attenuated in an LDLR−/−/KitW-sh/W-sh mouse (B).
2.
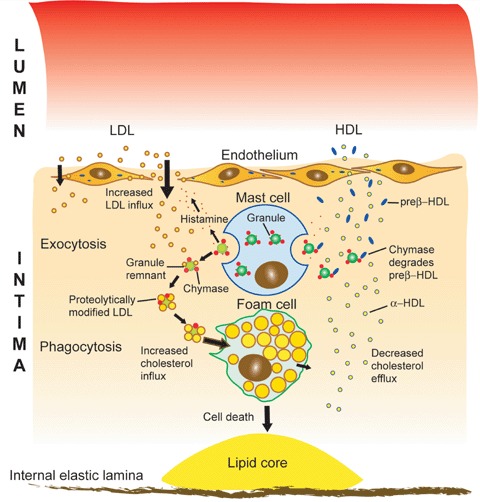
A role for mast cells in the intra- and extracellular accumulation of low density lipoprotein (LDL) cholesterol in atherogenesis. Upon activation, the mast cell secretes its preformed granules by exocytosis into the subendothelial space of the arterial intima. Histamine, a mast cell-derived soluble mediator increases the transendothelial transport of plasma LDL into the subendothelial space, where it is bound by the heparin component of the insoluble granule remnants and degraded by chymase, a granule remnant neutral serine psrotease. The proteolytically modified LDL particles become unstable and fuse on the remnant surface, after which the macrocomplex is phagocytozed and degraded by the macrophage with the subsequent formation of a foam cell, a hallmark of an early atherosclerotic lesion. High density lipoprotein (HDL), being responsible for the efflux of LDL-derived cholesterol from the macrophage foam cell, is also proteolyzed by mast cell chymase and the high-affinity component of the HDL-dependent cholesterol efflux is so impaired. As a result, the balance between cholesterol influx and efflux is disturbed and a cholesterol-filled foam cell is formed. The foam cell may eventually die and so contribute to the formation of an extracellular lipid core, which is a hallmark of an advanced atherosclerotic lesion.
Mast cells have also been shown to associate with neovessels formed within the atherosclerotic plaques [111–113]. Although neovessels are a common feature of advanced human atherosclerotic plaques, it is unclear whether angiogenesis, by causing intraplaque hemorrhage and growth of lipid core, is responsible for plaque instability [114]. Interestingly, data from clinical cancer trials have shown that prevention of angiogenesis with antibodies against VEGF, bevacizumab (Avastin®), leads to an increased risk of thromboembolic complications, including cerebrovascular events, myocardial infarction and deep vein thrombosis [115]. Furthermore, current evidence from clinical trials of both proangiogenic and antiangiogenic therapies suggests that inhibition of angiogenesis is not a viable therapeutic strategy for cardiovascular diseases, and that VEGF-induced angiogenesis in human arteries may rather play a protective role [116].
Activated mast cells in human atherosclerotic plaques
When compared with normal intima, the number of activated mast cells in atherosclerotic plaques is especially high in the shoulder regions prone to plaque rupture [117, 118]. Upon activation, mast cells release their preformed mediators in an active exocytotic process called degranulation, and most importantly, patients who died of acute myocardial infarction had an increased number of degranulated mast cells at the actual site of plaque erosion or rupture [35]. In general, mast cells can be activated by a wide variety of physiological and non-physiological mediators [86]. In atherosclerotic plaques, the accumulating modified lipids, other inflammatory components or the neighboring cells may be responsible for mast cell activation. Interestingly, the level of mast cell degranulation was elevated in intimal areas that contained an increased number of macrophages and T-lymphocytes, suggesting that factors responsible for mast cell degranulation in vivo may be derived from neighboring inflammatory cells. Since the complement anaphylatoxins, C5a and C3a are capable of inducing mast cell activation [119], and since the complement cascade is a functional component of the inflammatory process in advanced human coronary plaques [120], it is possible that they trigger mast cell-mediated responses in the coronary plaques. Recently, oxidized LDL that are present in high amounts in atherosclerotic lesions, have been shown to induce mast cell activation and secretion of IL-8 [121]. The mechanisms involved in this activation are still unclear, but may involve Toll-like receptors and some type of molecular mimicry. Smoking is also a risk factor for atherosclerosis, and nicotine present in smoke extract has been shown to activate human mast cells [122], and moreover chronic exposure to cigarette smoke induces the expression of mast cell proteases [123].
The activation of adventitial mast cells may occur by similar mechanisms. In addition, sensory nerves that are able to secrete substance P and CGRPs, are potential activators of the adventitial mast cells. Indeed, the number of mast cells were higher in the adventitia of advanced atherosclerotic coronary lesions (104 mast cells/mm2) compared to normal intima (31 mast cells/mm2), and the number of mast cell–nerve contacts (30 nerve contacts/mm2) were significantly greater than in normal intima (four nerve contacts/mm2) [124]. In atherosclerotic lesions, neurogenic activation of adventitial mast cells may involve the release of vasoactive compounds, such as histamine and leukotrienes, which can induce coronary vasoconstriction [124].
Mast cell activation without degranulation
Previously, activation of mast cells was thought to exclusively involve the process of degranulation.
However, an evolving concept suggests that activation of mast cells in tissues during disease progression may occur without signs of degranulation [125–127]. Recently, both IL-1 and TNF-α have been shown to induce mast cell-mediated secretion of cytokines in the absence of degranulation, i.e. without secretion of histamine and other granule components. Since the physiological and pathophysiological compounds capable of activating mast cells are so diverse [86], it is also likely that different mechanisms of mast cell activation exist. These would then range from selective secretion of individual cytokines or chemokines to anaphylaxis with exhaustive degranulation and acute secretion of a multitude of cytokines and chemokines.
Mast cell – an inflammatory cell
Due to their strategic localization in the close vicinity of endothelial cells, mast cells are involved in the regulation of innate and acquired immune responses, such as the infiltration of T-cells, macrophages and neutrophils [23, 86]. In human atherosclerotic plaques, T-cells co-localize with mast cells, suggesting a close interaction between these cell types. The direct contact between the two cell types may not only influence the functions of the individual cells, but may exert a more general immunoregulatory function that affects their microenvironment [128–130]. For instance, inflammatory areas of ruptured human atherosclerotic plaques have been shown to contain CD30 [131], and CD30–CD30L interaction is known to induce degranulation-independent secretion of chemokines by mast cells [127]. In addition, mast cells may activate T-cells by releasing TNF-α[132] and by expressing the costimulatory OX40L, which induces a direct cell–cell interaction between mast cells and the OX40 receptor on T-cells [133].
How could the Th1/Th2 polarization in atherosclerotic lesions affect the local development and activation of mast cells? Since human mast cells obey SCF-dependent, cytokine-driven (IL-3, IL-5, IL-6, IL-9 and granulocyte/macrophage colony-stimulating factor) mitogenic responses, and since they have a unique profile of chemokine receptors (e.g. CCR3), an increased infiltration and differentiation of mast cells reflects a Th2-type polarization [7]. In contrast, a Th1-type polarization, involving high expression of IFN-γ is inhibitory for human mast cell growth and differentiation [134]. Since atherosclerosis is generally considered to be a Th1-driven disease, which at the early stages of fatty streak formation may be counteracted by Th2-mechanisms [135], the role of mast cells may change with the progression of the disease. Indeed, in advanced stages of atherosclerosis, the Th2-driven processes might also be proatherosclerotic [136, 137]. Indeed, at least in the mouse severe hypercholesterolaemia is associated with advanced atherosclerotic plaques and with a switch from Th1 to Th2-driven processes. Since Th1 and Th2 cells counteract each other, the appearance of Th2 cytokines may have important consequences for the inflammatory/immune process in atherosclerosis [138], and especially in terms of mast cell proliferation and differentiation.
Mast cells also induce infiltration of monocytes and neutrophils [139] by secreting various chemokines, such as MCP-1 and IL-8. The participation of monocyte/ macrophages in plaque remodeling that pre-dispose to rupture is well established [140]. In addition, the mast cell-dependent recruitment of neutrophils may be of special importance in the infarct-related coronary plaques, which, in contrast to the more innocent stable plaques, often contain neutrophils [141]. Indeed, mast cells participate in the induction of antigen- and Th17 cell-dependent neutrophil-rich inflammatory responses [142], and IL-17, the main mediator of Th17-driven inflammation, is induced in unstable angina and acute myocardial infarction [143].
Coronary mast cells in acute human myocardial infarction
By pre-disposing atherosclerotic plaques to erosion and rupture, mast cells are thought to contribute actively to acute coronary syndromes, including myocardial infarction. The mechanisms by which mast cells may induce atherothrombotic events include adverse remodeling of the atherosclerotic plaque [144], induction of smooth muscle cell death [72, 145], induction of death [73] or detachment of vulnerable endothelial cells [76], and triggering coronary artery spasm [41, 102, 146] (Fig. 3).
3.
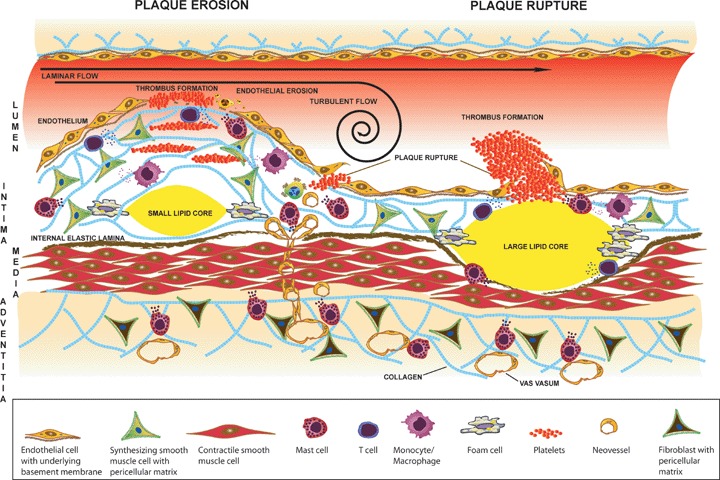
Activated mast cells may contribute to plaque erosion and rupture. The number of activated intimal and adventitial mast cells is increased in atherosclerotic plaques. Two types of plaques are shown: (1) a stenotic plaque with a thick fibrous cap and a small necrotic lipid core (left), and (2) a non-stenotic plaque with a thin fibrous cap and a large lipid core (right). The stenotic plaque is prone to erosion and the non-stenotic plaque is prone to rupture. Activated mast cells secrete cytokines and chemokines that regulate innate and acquired immune responses. Mast cells also secrete neutral serine proteases and MMPs that may degrade the components of the pericellular and extracellular matrices, and also the internal elastic lamina. Proteolytic degradation of collagen, elastin and proteoglycan-containing matrices weakens the fibrous cap and renders the plaque susceptible to erosion and rupture. Mast cells, as a source of proangiogenic growth factors and proteases may also induce the formation of neovessels that traverse the medial layer into the intima and weaken the plaque, particularly when they rupture and cause intraplaque haemorrhage. Furthermore, histamine-induced coronary spasm or high shear stress may further precipitate formation of plaque erosions and ruptures. Exposure of the subendothelial thrombogenic tissue at the site of plaque erosion and rupture triggers the formation of an arterial thrombus.
The number of plaque erosions and the proportion of mast cells associated with erosions increases markedly as the disease becomes more severe [76].
Mast cells are also known to cause endothelial dys-function and activation of endothelial adhesion molecules [30], effects that also may pre-dispose to plaque erosion and rupture. Mast cell-derived histamine increases the permeability of EC by binding to histamine receptors (H1 receptors) on the endothelial cell surface so increasing phosphorylation of adherent junction molecules and loosening of vascular endothelial (VE)-cadherin-mediated endothelial cell–cell adhesions [147–149]. PAR-2 cleavage and activation by mast cell-derived tryptase, chymase and cathepsin G may also induce loosening of VE-cadherin-mediated cell–cell adhesions [150–152].
Activation of adventitial mast cells by nervous stimuli may regulate vessel tone and also various functions of vasa vasorum[124], and so participate in events leading to plaque erosion and rupture. Indeed, histamine released from activated adventitial mast cells may reach the media, where it could act on smooth muscle cells, and by provoking a local spasm, histamine could contribute to the onset of acute cardiac events [146, 153]. Interestingly, the proportion of activated and degranulated adventitial mast cells was found to be highest in the segments with ruptured plaques [102]. However, it is presently not known whether activation and degranulation of the adventitial mast cells precede the rupture or occur as a consequence of plaque rupture.
Animal models of plaque erosion and rupture
Studies with experimental animals have shown that most species spontaneously develop fatty streaks and some even more advanced atherosclerotic lesions with time, but that plaque rupture with subsequent thrombosis is exceedingly rare. In most animal models, dietary addition of fat and cholesterol have induced an accelerated development of advanced lesions, but again, without an increased frequency of plaque rupture [154]. Although some studies have shown that artificial induction of neointima by ligation of the common carotid artery [155], by non-constrictive cuffing of the femoral artery [156], or by a combination of both [157, 158] may lead to endothelial damage and thrombus formation, they are not optimal models to study mechanisms involved in the rupture of vulnerable atherosclerotic plaques in man. So far, only one group has showed that fatfed apoE knockout mice develop advanced atherosclerotic plaques in their brachiocephalic arteries with frequent rupture and formation of luminal thrombi [159]. According to these data, the ruptured plaques show many of the characteristics of vulnerable plaques in human beings [159], and the ruptures can be prevented by statin treatment [160]. Thus, by crossing the above apoE-deficient model with a mast cell-deficient mouse model (KitW-sh/W-sh), one would potentially create an animal model that would clarify the in vivo role of mast cells in plaque erosion and rupture. However, one should keep in mind that rodent mast cells are different from human mast cells in terms of both mediator contents and functions [161, 162], and that observations made in such animal models may not truly reflect the human disease. Indeed, one of the major obstacles in the use of animal models for studying the role of mast cells in atherosclerosis is the lack of intimal mast cells in many commonly used laboratory animals.
Mice do have mast cells in the aortic adventitial layer, and since human adventitial mast cells are activated in the areas of atherosclerotic plaques [102], the adventitial mouse mast cells may be able to affect pathological processes in the arterial intima. Interestingly, another experimental approach to induce accelerated atherosclerosis in animal models has been the use of arterial cuffs [163, 164], that may activate the adventitial mast cells. In addition, recent data show that activation of adventitial mast cells promote atherogenesis and induce plaque destabilization in apoE-deficient mice [165]. A targeted activation of adventitial mast cells in advanced carotid athero-sclerotic plaques sharply increases the incidence of intraplaque haemorrhage, macrophage apoptosis, vascular leakage and CXCR2/VLA-4-mediated recruitment of leukocytes to the plaque [165].
Clinical approaches to stabilize mast cells in the atherosclerotic plaque
Pathological vasoconstriction or spasm of inflamed atherosclerotic coronary segments plays an important role in acute coronary syndromes and has also been suggested to pre-dispose to plaque rupture and erosion [41, 166, 167]. Since mast cell-derived histamine and leukotrienes have been suggested to trigger coronary spasm [124, 146] and since both anti-histamines and leukotriene receptor antagonists are available for human use [168], clinical studies regarding their use in the treatment of atherosclerosis may be feasible in the future [169]. Interestingly, an H1-antihistamine (desloratadine) has been shown to inhibit mast cell activation in vitro[170] and simultaneous inhibition of H1 and H2 receptors may be even more efficient in mast cell stabilization than inhibition of either receptor alone [171]. Taken together, antihistamines and leukotriene receptor antagonists might provide means to inhibit progression of atherosclerosis by reducing plaque inflammation. These drugs may potentially also prevent coronary spasms.
A major mechanism pre-disposing to plaque rupture is the increased degradation of extracellular and pericellular matrix components in the fibrous cap. The activated mast cells secrete proteases that either directly or indirectly can induce degradation of collagen, elastin and proteoglycans [172]. Degradation of the extracellular and pericellular matrices induces apoptosis and weakens the fibrous cap rendering it more susceptible to rupture and pre-cipitation of an acute coronary syndrome. Although pharmacological inhibition of MMPs and cathepsins has been proposed in the treatment of atherosclerosis [173, 174], many of these proteases are crucial for normal vessel homeostasis [85, 175]. In addition, since the mast cells present in the plaque are filled with the neutral proteases, tryptase, chymase and cathepsin G, which they upon activation avidly secrete into the extracellular space, a therapeutic need to inhibit their activity in vivo may exist. However, although tryptase inhibitors have been used in the treatment of asthma and ulcerative colitis [176], they have not yet been tested in atherosclerosis. Heparin antagonists, such as protamine and Polybrene® also inhibit tryptase activity by causing dissociation of active tetrameric tryptase into inactive monomers [61]. Several orally active inhibitors of chy-mase are currently also available [177], and animal data support their benefits in a variety of cardiovascular diseases [178, 179]. However, human clinical data regarding the effects of chymase inhibitors in the pathogenesis of atherosclerosis are still lacking. Taken together, the key enzymes responsible for the pathological matrix degradation should be identified before specific antiprotease therapy can be envisioned, and even then the redundancy of proteases may render this approach unsuccessful.
For the time being, anti-inflammatory therapies aiming at lowering the numbers of mast cells or stabilizing mast cells may be the best way of lessening their proteolytic burden in the vulnerable cap. Although sodium cromolyn has successfully been used to stabilize human mast cells locally in the eye, it is presently not suitable for systemic use in human beings. Interestingly, intravenously and intraperitoneally given cromolyn treatment during dinitrophenyl-albumin (DNP) challenge of collar-induced carotid artery lesions in apoE-deficient mice normalized the extent of mast cell degranulation in the adventitia, while preventing intraplaque hemorrhage [165]. An orally absorbable phosphodiesterase (PDE5) inhibitor zaprinast has also been shown to stabilize mast cells and to possess a moderate bronchodilator effect [180]. PDE5 inhibitors were tested also for treatment of angina pectoris, but proved not to have any beneficial effect over nitrates [181]. Since SCF is necessary for mast cell development, proliferation and survival, and participates in homing and adhesion of mast cells, it may also be a target of therapy [182]. Indeed, drugs targeting SCF and/or Kit, including anti-SCF antibodies, antisense oligonucleotides and Kit inhibitors have been studied for their anti-allergic properties [182], and thus, may be useful in stabilizing mast cells present in atherosclerotic plaques.
Furthermore, statins have been shown, at least in vitro, to inhibit the secretion of MMPs by macrophages [183], but no data on the effects of statins on the release of mast cell proteases appear to be available. However, previous studies in a rat mast cell line (RBL-2H3) have shown that statin-treatment (lovastatin) resulted in impaired tyrosine phosphorylation and in inhibition of the IgE-mediated degranulation [184]. In addition, recent results have shown that cerivastatin and atorvastatin, and partially also lovastatin act as inhibitors of growth and function of human mast cells [185]. However, simvastatin and pravastatin did not affect mediator release or growth of human mast cells, revealing that the inhibitory effects were not a class effect, but peculiarly specific for particular statins [185].
Conclusions
Accumulating evidence clearly suggests a role for mast cells in the pathogenesis of plaque erosion and rupture [144] (Fig. 3). Their strategic tissue location, vast content of powerful mediators ranging from vasoactive substances and proinflammatory cytokines to proteolytic enzymes, ability to rapidly become activated upon contact with specific triggers, and ability to regulate innate and acquired immune responses, are attributes which set the scene for their view to a kill. However, we eagerly await more animal models that would produce proof of concept and give us the confidence to develop means to treat against mast cell infiltration and activation in the pathogenesis of plaque erosion and rupture. Although we presently think that chronic activation of mast cells in the atherosclerotic lesions pre-disposes to plaque rupture, it may well be that the initial activation of the intimal and adventitial mast cells are necessary for the onset of immunological defense mechanisms in the arterial wall [135, 186]. Accordingly, mast cells have been shown important for the healing responses of injured arterial walls, in that they participate in the initiation of thrombus formation [187], as well as in the resolution of thrombus formation and in the process of neovascularization [29, 188]. Thus, mast cells, by being powerful regulators of immune responses and inflammation, may represent a double-edged sword in the pathogenesis of plaque erosion and rupture [135].
References
- 1.Kirshenbaum AS, Kessler SW, Goff JP, Metcalfe DD. Demonstration of the origin of human mast cells from CD34+ bone marrow progenitor cells. J Immunol. 1991;146:1410–5. [PubMed] [Google Scholar]
- 2.Rottem M, Okada T, Goff JP, Metcalfe DD. Mast cells cultured from the peripheral blood of normal donors and patients with mastocytosis originate from a CD34+/Fc epsilon RI- cell population. Blood. 1994;84:2489–96. [PubMed] [Google Scholar]
- 3.Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13) Blood. 1999;94:2333–42. [PubMed] [Google Scholar]
- 4.Kitamura Y, Ito A. Mast cell-committed progenitors. Proc Natl Acad Sci USA. 2005;102:11129–30. doi: 10.1073/pnas.0505073102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyce JA, Mellor EA, Perkins B, Lim YC, Luscinskas FW. Human mast cell progenitors use alpha4-integrin, VCAM-1, and PSGL-1 E-selectin for adhesive interactions with human vascular endothelium under flow conditions. Blood. 2002;99:2890–6. doi: 10.1182/blood.v99.8.2890. [DOI] [PubMed] [Google Scholar]
- 6.Haley KJ, Lilly CM, Yang JH, Feng Y, Kennedy SP, Turi TG, Thompson JF, Sukhova GH, Libby P, Lee RT. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation. 2000;102:2185–9. doi: 10.1161/01.cir.102.18.2185. [DOI] [PubMed] [Google Scholar]
- 7.Ochi H, Hirani WM, Yuan Q, Friend DS, Austen KF, Boyce JA. T helper cell type 2 cytokinemediated comitogenic responses and CCR3 expression during differentiation of human mast cells in vitro. J Exp Med. 1999;190:267–80. doi: 10.1084/jem.190.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alcaide P, Jones TG, Lord GM, Glimcher LH, Hallgren J, Arinobu Y, Akashi K, Paterson AM, Gurish MA, Luscinskas FW. Dendritic cell expression of the transcription factor T-bet regulates mast cell progenitor homing to mucosal tissue. J Exp Med. 2007;204:431–9. doi: 10.1084/jem.20060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito H, Ebisawa M, Tachimoto H, Shichijo M, Fukagawa K, Matsumoto K, Iikura Y, Awaji T, Tsujimoto G, Yanagida M, Uzumaki H, Takahashi G, Tsuji K, Nakahata T. Selective growth of human mast cells induced by Steel factor, IL-6, and prostaglandin E2 from cord blood mononuclear cells. J Immunol. 1996;157:343–50. [PubMed] [Google Scholar]
- 10.Kinoshita T, Sawai N, Hidaka E, Yamashita T, Koike K. Interleukin-6 directly modulates stem cell factor-dependent development of human mast cells derived from CD34(+) cord blood cells. Blood. 1999;94:496–508. [PubMed] [Google Scholar]
- 11.Matsuzawa S, Sakashita K, Kinoshita T, Ito S, Yamashita T, Koike K. IL-9 enhances the growth of human mast cell progenitors under stimulation with stem cell factor. J Immunol. 2003;170:3461–7. doi: 10.4049/jimmunol.170.7.3461. [DOI] [PubMed] [Google Scholar]
- 12.Kanbe N, Kurosawa M, Miyachi Y, Kanbe M, Saitoh H, Matsuda H. Nerve growth factor prevents apoptosis of cord blood-derived human cultured mast cells synergistically with stem cell factor. Clin Exp Allergy. 2000;30:1113–20. doi: 10.1046/j.1365-2222.2000.00866.x. [DOI] [PubMed] [Google Scholar]
- 13.Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447–52. [PubMed] [Google Scholar]
- 14.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–9. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 15.Copeland NG, Gilbert DJ, Cho BC, Donovan PJ, Jenkins NA, Cosman D, Anderson D, Lyman SD, Williams DE. Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell. 1990;63:175–83. doi: 10.1016/0092-8674(90)90298-s. [DOI] [PubMed] [Google Scholar]
- 16.Worobec AS, Semere T, Nagata H, Metcalfe DD. Clinical correlates of the presence of the Asp816Val c-kit mutation in the peripheral blood mononuclear cells of patients with mastocytosis. Cancer. 1998;83:2120–9. doi: 10.1002/(sici)1097-0142(19981115)83:10<2120::aid-cncr10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 17.Castells MC, Friend DS, Bunnell CA, Hu X, Kraus M, Osteen RT, Austen KF. The presence of membrane- bound stem cell factor on highly immature nonmetachromatic mast cells in the peripheral blood of a patient with aggressive systemic mastocytosis. J Allergy Clin Immunol. 1996;98:831–40. doi: 10.1016/s0091-6749(96)70133-1. [DOI] [PubMed] [Google Scholar]
- 18.Costa JJ, Demetri GD, Harrist TJ, Dvorak AM, Hayes DF, Merica EA, Menchaca DM, Gringeri AJ, Schwartz LB, Galli SJ. Recombinant human stem cell factor (kit ligand) promotes human mast cell and melanocyte hyperplasia and functional activation in vivo. J Exp Med. 1996;183:2681–6. doi: 10.1084/jem.183.6.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oskeritzian CA, Zhao W, Min HK, Xia HZ, Pozez A, Kiev J, Schwartz LB. Surface CD88 functionally distinguishes the MCTC from the MCT type of human lung mast cell. J Allergy Clin Immunol. 2005;115:1162–8. doi: 10.1016/j.jaci.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toru H, Eguchi M, Matsumoto R, Yanagida M, Yata J, Nakahata T. Interleukin-4 promotes the development of tryptase and chymase double-positive human mast cells accompanied by cell maturation. Blood. 1998;91:187–95. [PubMed] [Google Scholar]
- 21.Xia HZ, Du Z, Craig S, Klisch G, Noben-Trauth N, Kochan JP, Huff TH, Irani AM, Schwartz LB. Effect of recombinant human IL-4 on tryptase, chymase, and Fc epsilon receptor type I expression in recombinant human stem cell factor-dependent fetal liver-derived human mast cells. J Immunol. 1997;159:2911–21. [PubMed] [Google Scholar]
- 22.Galli SJ, Maurer M, Lantz CS. Mast cells as sentinels of innate immunity. Curr Opin Immunol. 1999;11:53–9. doi: 10.1016/s0952-7915(99)80010-7. [DOI] [PubMed] [Google Scholar]
- 23.Frossi B, De Carli M, Pucillo C. The mast cell: an antenna of the microenvironment that directs the immune response. J Leukoc Biol. 2004;75:579–85. doi: 10.1189/jlb.0603275. [DOI] [PubMed] [Google Scholar]
- 24.Sacchi G, Weber E, Agliano M, Lorenzoni P, Rossi A, Caruso AM, Vernillo R, Gerli R, Lorenzi M. Lymphatic vessels in colorectal cancer and their relation with inflammatory infiltrate. Dis Colon Rectum. 2003;46:40–7. doi: 10.1007/s10350-004-6494-4. [DOI] [PubMed] [Google Scholar]
- 25.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–79. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz LB. Effector cells of anaphylaxis: mast cells and basophils. Novartis Found Symp. 2004;257:65,74. [PubMed] [Google Scholar]
- 27.De Pater-Huijsen FL, De Riemer MJ, Reijneke RM, Pompen M, Lutter R, Jansen HM, Out TA. Human mast cells modulate proliferation and cytokine production by CD8+ T lymphocytes. Int Arch Allergy Immunol. 1997;113:287–8. doi: 10.1159/000237575. [DOI] [PubMed] [Google Scholar]
- 28.Egozi EI, Ferreira AM, Burns AL, Gamelli RL, Dipietro LA. Mast cells modulate the inflammatory but not the proliferative response in healing wounds. Wound Repair Regen. 2003;11:46–54. doi: 10.1046/j.1524-475x.2003.11108.x. [DOI] [PubMed] [Google Scholar]
- 29.Norrby K. Mast cells and angiogenesis. APMIS. 2002;110:355–71. doi: 10.1034/j.1600-0463.2002.100501.x. [DOI] [PubMed] [Google Scholar]
- 30.Dileepan KN, Stechschulte DJ. Endothelial cell activation by mast cell mediators. Methods Mol Biol. 2006;315:275–94. doi: 10.1385/1-59259-967-2:275. [DOI] [PubMed] [Google Scholar]
- 31.Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 32.Echtenacher B, Mannel DN, Hultner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–7. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 33.Maurer M, Wedemeyer J, Metz M, Piliponsky AM, Weller K, Chatterjea D, Clouthier DE, Yanagisawa MM, Tsai M, Galli SJ. Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature. 2004;432:512–6. doi: 10.1038/nature03085. [DOI] [PubMed] [Google Scholar]
- 34.Metz M, Piliponsky AM, Chen CC, Lammel V, Abrink M, Pejler G, Tsai M, Galli SJ. Mast cells can enhance resistance to snake and honeybee venoms. Science. 2006;313:526–30. doi: 10.1126/science.1128877. [DOI] [PubMed] [Google Scholar]
- 35.Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92:1084–8. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- 36.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–92. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 37.Hara M, Ono K, Hwang MW, Iwasaki A, Okada M, Nakatani K, Sasayama S, Matsumori A. Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med. 2002;195:375–81. doi: 10.1084/jem.20002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kubes P, Granger DN. Leukocyte-endothelial cell interactions evoked by mast cells. Cardiovasc Res. 1996;32:699–708. [PubMed] [Google Scholar]
- 39.Talreja J, Kabir MH, B Filla M, Stechschulte DJ, Dileepan KN. Histamine induces Toll-like receptor 2 and 4 expression in endothelial cells and enhances sensitivity to Gram-positive and Gram-negative bacterial cell wall components. Immunology. 2004;113:224–33. doi: 10.1111/j.1365-2567.2004.01946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steffel J, Akhmedov A, Greutert H, Luscher TF, Tanner FC. Histamine induces tissue factor expression: implications for acute coronary syndromes. Circulation. 2005;112:341–9. doi: 10.1161/CIRCULATIONAHA.105.553735. [DOI] [PubMed] [Google Scholar]
- 41.Kalsner S, Richards R. Coronary arteries of cardiac patients are hyperreactive and contain stores of amines: a mechanism for coronary spasm. Science. 1984;223:1435–7. doi: 10.1126/science.6701530. [DOI] [PubMed] [Google Scholar]
- 42.Lassila R, Lindstedt K, Kovanen PT. Native macro-molecular heparin proteoglycans exocytosed from stimulated rat serosal mast cells strongly inhibit platelet-collagen interactions. Arterioscler Thromb Vasc Biol. 1997;17:3578–87. doi: 10.1161/01.atv.17.12.3578. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Kovanen PT. Heparin proteoglycans released from rat serosal mast cells inhibit proliferation of rat aortic smooth muscle cells in culture. Circ Res. 1999;84:74–83. doi: 10.1161/01.res.84.1.74. [DOI] [PubMed] [Google Scholar]
- 44.Hedin U, Daum G, Clowes AW. Heparin inhibits thrombin-induced mitogen-activated protein kinase signaling in arterial smooth muscle cells. J Vasc Surg. 1998;27:512–20. doi: 10.1016/s0741-5214(98)70326-x. [DOI] [PubMed] [Google Scholar]
- 45.Clowes AW. Regulation of smooth muscle cell function by heparin. J Vasc Surg. 1992;15:911–3. doi: 10.1016/0741-5214(92)90746-u. [DOI] [PubMed] [Google Scholar]
- 46.Kovanen PT. Mast cells in human fatty streaks and atheromas: implications for intimal lipid accumulation. Curr Opin Lipidol. 1996;7:281–6. doi: 10.1097/00041433-199610000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Lindstedt KA, Kokkonen JO, Kovanen PT. Regulation of the activity of secreted human lung mast cell tryptase by mast cell proteoglycans. Biochim Biophys Acta. 1998;1425:617–27. doi: 10.1016/s0304-4165(98)00115-9. [DOI] [PubMed] [Google Scholar]
- 48.Selwood T, Smolensky H, McCaslin DR, Schechter NM. The interaction of human tryptase-beta with small molecule inhibitors provides new insights into the unusual functional instability and quaternary structure of the protease. Biochemistry. 2005;44:3580–90. doi: 10.1021/bi047765u. [DOI] [PubMed] [Google Scholar]
- 49.Lindstedt L, Lee M, Kovanen PT. Chymase bound to heparin is resistant to its natural inhibitors and capable of proteolyzing high density lipoproteins in aortic intimal fluid. Atherosclerosis. 2001;155:87–97. doi: 10.1016/s0021-9150(00)00544-x. [DOI] [PubMed] [Google Scholar]
- 50.Ermolieff J, Boudier C, Laine A, Meyer B, Bieth JG. Heparin protects cathepsin G against inhibition by protein proteinase inhibitors. J Biol Chem. 1994;269:29502–8. [PubMed] [Google Scholar]
- 51.Buckley MG, Walters C, Wong WM, Cawley MI, Ren S, Schwartz LB, Walls AF. Mast cell activation in arthritis: detection of alpha- and beta-tryptase, histamine and eosinophil cationic protein in synovial fluid. Clin Sci. 1997;93:363–70. doi: 10.1042/cs0930363. [DOI] [PubMed] [Google Scholar]
- 52.Fukuoka Y, Schwartz LB. Human beta-tryptase: detection and characterization of the active monomer and prevention of tetramer reconstitution by protease inhibitors. Biochemistry. 2004;43:10757–64. doi: 10.1021/bi049486c. [DOI] [PubMed] [Google Scholar]
- 53.Compton SJ, Renaux B, Wijesuriya SJ, Hollenberg MD. Glycosylation and the activation of proteinase-activated receptor 2 (PAR(2)) by human mast cell tryptase. Br J Pharmacol. 2001;134:705–18. doi: 10.1038/sj.bjp.0704303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA, Schechter N, Woolkalis M, Brass LF. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–9. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- 55.Tam EK, Caughey GH. Degradation of airway neuropeptides by human lung tryptase. Am J Respir Cell Mol Biol. 1990;3:27–32. doi: 10.1165/ajrcmb/3.1.27. [DOI] [PubMed] [Google Scholar]
- 56.Vartio T, Seppa H, Vaheri A. Susceptibility of soluble and matrix fibronectins to degradation by tissue proteinases, mast cell chymase and cathepsin G. J Biol Chem. 1981;256:471–7. [PubMed] [Google Scholar]
- 57.Lohi J, Harvima I, Keski-Oja J. Pericellular substrates of human mast cell tryptase: 72,000 dalton gelatinase and fibronectin. J Cell Biochem. 1992;50:337–49. doi: 10.1002/jcb.240500402. [DOI] [PubMed] [Google Scholar]
- 58.Gruber BL, Marchese MJ, Suzuki K, Schwartz LB, Okada Y, Nagase H, Ramamurthy NS. Synovial pro-collagenase activation by human mast cell tryptase dependence upon matrix metalloproteinase 3 activation. J Clin Invest. 1989;84:1657–62. doi: 10.1172/JCI114344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson JL, Jackson CL, Angelini GD, George SJ. Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 1998;18:1707–15. doi: 10.1161/01.atv.18.11.1707. [DOI] [PubMed] [Google Scholar]
- 60.Pereira PJ, Bergner A, Macedo-Ribeiro S, Huber R, Matschiner G, Fritz H, Sommerhoff CP, Bode W. Human beta-tryptase is a ring-like tetramer with active sites facing a central pore. Nature. 1998;392:306–11. doi: 10.1038/32703. [DOI] [PubMed] [Google Scholar]
- 61.Hallgren J, Estrada S, Karlson U, Alving K, Pejler G. Heparin antagonists are potent inhibitors of mast cell tryptase. Biochemistry. 2001;40:7342–9. doi: 10.1021/bi001988c. [DOI] [PubMed] [Google Scholar]
- 62.Peng Q, McEuen AR, Benyon RC, Walls AF. The heterogeneity of mast cell tryptase from human lung and skin. Eur J Biochem. 2003;270:270–83. doi: 10.1046/j.1432-1033.2003.03385.x. [DOI] [PubMed] [Google Scholar]
- 63.Hallgren J, Pejler G. Biology of mast cell tryptase. An inflammatory mediator. FEBS J. 2006;273:1871–95. doi: 10.1111/j.1742-4658.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 64.Saarinen J, Kalkkinen N, Welgus HG, Kovanen PT. Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84 bond by mast cell chymase. J Biol Chem. 1994;269:18134–40. [PubMed] [Google Scholar]
- 65.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metallo-protease- 9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280:9291–6. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 66.Tchougounova E, Pejler G, Abrink M. The chy-mase, mouse mast cell protease 4, constitutes the major chymotrypsin-like activity in peritoneum and ear tissue. A role for mouse mast cell protease 4 in thrombin regulation and fibronectin turnover. J Exp Med. 2003;198:423–31. doi: 10.1084/jem.20030671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urata H, Nishimura H, Ganten D. Chymase-dependent angiotensin II forming systems in humans. Am J Hypertens. 1996;9:277–84. doi: 10.1016/0895-7061(95)00349-5. [DOI] [PubMed] [Google Scholar]
- 68.Miyazaki M, Takai S. Local angiotensin II-generating system in vascular tissues: the roles of chymase. Hypertens Res. 2001;24:189–93. doi: 10.1291/hypres.24.189. [DOI] [PubMed] [Google Scholar]
- 69.Kovanen PT, Lindstedt MK, Shiota N, Kokkonen JO. Chymase-dependent angiotensin II formation in human vascular tissue. Circulation. 2000;102:E32. doi: 10.1161/01.cir.102.4.e32. [DOI] [PubMed] [Google Scholar]
- 70.Lindstedt KA, Wang Y, Shiota N, Saarinen J, Hyytiainen M, Kokkonen JO, Keski-Oja J, Kovanen PT. Activation of paracrine TGF-beta1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. 2001;15:1377–88. doi: 10.1096/fj.00-0273com. [DOI] [PubMed] [Google Scholar]
- 71.Wang Y, Shiota N, Leskinen MJ, Lindstedt KA, Kovanen PT. Mast cell chymase inhibits smooth muscle cell growth and collagen expression in vitro: transforming growth factor-beta1-dependent and -independent effects. Arterioscler Thromb Vasc Biol. 2001;21:1928–33. doi: 10.1161/hq1201.100227. [DOI] [PubMed] [Google Scholar]
- 72.Leskinen MJ, Lindstedt KA, Wang Y, Kovanen PT. Mast cell chymase induces smooth muscle cell apoptosis by a mechanism involving fibronectin degradation and disruption of focal adhesions. Arterioscler Thromb Vasc Biol. 2003;23:238–43. doi: 10.1161/01.atv.0000051405.68811.4d. [DOI] [PubMed] [Google Scholar]
- 73.Latti S, Leskinen M, Shiota N, Wang Y, Kovanen PT, Lindstedt KA. Mast cell-mediated apoptosis of endothelial cells in vitro: A paracrine mechanism involving TNF-alpha-mediated down-regulation of bcl-2 expression. J Cell Physiol. 2003;195:130–8. doi: 10.1002/jcp.10235. [DOI] [PubMed] [Google Scholar]
- 74.Jehle AB, Li Y, Stechschulte AC, Stechschulte DJ, Dileepan KN. Endotoxin and mast cell granule proteases synergistically activate human coronary artery endothelial cells to generate interleukin-6 and interleukin-8. J Interferon Cytokine Res. 2000;20:361–8. doi: 10.1089/107999000312298. [DOI] [PubMed] [Google Scholar]
- 75.Schechter NM, Irani AM, Sprows JL, Abernethy J, Wintroub B, Schwartz LB. Identification of a cathepsin G-like proteinase in the MCTC type of human mast cell. J Immunol. 1990;145:2652–61. [PubMed] [Google Scholar]
- 76.Mayranpaa MI, Heikkila HM, Lindstedt KA, Walls AF, Kovanen PT. Desquamation of human coronary artery endothelium by human mast cell proteases: implications for plaque erosion. Coron Artery Dis. 2006;17:611–21. doi: 10.1097/01.mca.0000224420.67304.4d. [DOI] [PubMed] [Google Scholar]
- 77.Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D, Levi R. Mast cells: a unique source of renin. Proc Natl Acad Sci USA. 2004;101:13607–12. doi: 10.1073/pnas.0403208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mackins CJ, Kano S, Seyedi N, Schafer U, Reid AC, Machida T, Silver RB, Levi R. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest. 2006;116:1063–70. doi: 10.1172/JCI25713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brasier AR, Recinos A, 3rd, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257–66. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- 80.Di Girolamo N, Wakefield D. In vitro and in vivo expression of interstitial collagenase/MMP-1 by human mast cells. Dev Immunol. 2000;7:131–42. doi: 10.1155/2000/82708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baram D, Vaday GG, Salamon P, Drucker I, Hershkoviz R, Mekori YA. Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-alpha. J Immunol. 2001;167:4008–16. doi: 10.4049/jimmunol.167.7.4008. [DOI] [PubMed] [Google Scholar]
- 82.Di Girolamo N, Indoh I, Jackson N, Wakefield D, McNeil HP, Yan W, Geczy C, Arm JP, Tedla N. Human mast cell-derived gelatinase B (matrix metal-loproteinase- 9) is regulated by inflammatory cytokines: role in cell migration. J Immunol. 2006;177:2638–50. doi: 10.4049/jimmunol.177.4.2638. [DOI] [PubMed] [Google Scholar]
- 83.Koskivirta I, Rahkonen O, Mayranpaa M, Pakkanen S, Husheem M, Sainio A, Hakovirta H, Laine J, Jokinen E, Vuorio E, Kovanen P, Jarvelainen H. Tissue inhibitor of metalloproteinases 4 (TIMP4) is involved in inflammatory processes of human cardiovascular pathology. Histochem Cell Biol. 2006 doi: 10.1007/s00418-006-0163-8. [DOI] [PubMed] [Google Scholar]
- 84.Frank BT, Rossall JC, Caughey GH, Fang KC. Mast cell tissue inhibitor of metalloproteinase-1 is cleaved and inactivated extracellularly by alpha-chy-mase. J Immunol. 2001;166:2783–92. doi: 10.4049/jimmunol.166.4.2783. [DOI] [PubMed] [Google Scholar]
- 85.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci USA. 2005;102:15575–80. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol. 2004;4:787–99. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 87.Gordon JR, Galli SJ. Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature. 1990;346:274–6. doi: 10.1038/346274a0. [DOI] [PubMed] [Google Scholar]
- 88.Norman MU, Lister KJ, Yang YH, Issekutz A, Hickey MJ. TNF regulates leukocyte-endothelial cell interactions and microvascular dysfunction during immune complex-mediated inflammation. Br J Pharmacol. 2005;144:265–74. doi: 10.1038/sj.bjp.0706081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lejeune FJ, Lienard D, Matter M, Ruegg C. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun. 2006;6:6. [PubMed] [Google Scholar]
- 90.Lejeune FJ. Clinical use of TNF revisited: improving penetration of anti-cancer agents by increasing vascular permeability. J Clin Invest. 2002;110:433–5. doi: 10.1172/JCI16493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Curnis F, Sacchi A, Corti A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J Clin Invest. 2002;110:475–82. doi: 10.1172/JCI15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11:397–408. doi: 10.1634/theoncologist.11-4-397. [DOI] [PubMed] [Google Scholar]
- 93.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–81. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 94.Robertson AK, Hansson GK. T cells in atherogenesis: for better or for worse? Arterioscler Thromb Vasc Biol. 2006;26:2421–32. doi: 10.1161/01.ATV.0000245830.29764.84. [DOI] [PubMed] [Google Scholar]
- 95.Zudaire E, Martinez A, Garayoa M, Pio R, Kaur G, Woolhiser MR, Metcalfe DD, Hook WA, Siraganian RP, Guise TA, Chirgwin JM, Cuttitta F. Adrenomedullin is a cross-talk molecule that regulates tumor and mast cell function during human carcinogenesis. Am J Pathol. 2006;168:280–91. doi: 10.2353/ajpath.2006.050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boyce JA. Eicosanoid mediators of mast cells: receptors, regulation of synthesis, and pathobiologic implications. Chem Immunol Allergy. 2005;87:59–79. doi: 10.1159/000087571. [DOI] [PubMed] [Google Scholar]
- 97.Allen SP, Yacoub MH. Role of leukotrienes in coronary artery surgery. Curr Opin Cardiol. 1995;10:605–13. doi: 10.1097/00001573-199511000-00009. [DOI] [PubMed] [Google Scholar]
- 98.Reiss AB, Edelman SD. Recent insights into the role of prostanoids in atherosclerotic vascular disease. Curr Vasc Pharmacol. 2006;4:395–408. doi: 10.2174/157016106778521652. [DOI] [PubMed] [Google Scholar]
- 99.Miyamoto T, Sasaguri Y, Sasaguri T, Azakami S, Yasukawa H, Kato S, Arima N, Sugama K, Morimatsu M. Expression of stem cell factor in human aortic endothelial and smooth muscle cells. Atherosclerosis. 1997;129:207–13. doi: 10.1016/s0021-9150(96)06043-1. [DOI] [PubMed] [Google Scholar]
- 100.Mierke CT, Ballmaier M, Werner U, Manns MP, Welte K, Bischoff SC. Human endothelial cells regulate survival and proliferation of human mast cells. J Exp Med. 2000;192:801–11. doi: 10.1084/jem.192.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kaartinen M, Penttila A, Kovanen PT. Mast cells of two types differing in neutral protease composition in the human aortic intima. Demonstration of tryptase-and tryptase/chymase-containing mast cells in normal intimas, fatty streaks, and the shoulder region of atheromas. Arterioscler Thromb. 1994;14:966–72. doi: 10.1161/01.atv.14.6.966. [DOI] [PubMed] [Google Scholar]
- 102.Laine P, Kaartinen M, Penttila A, Panula P, Paavonen T, Kovanen PT. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation. 1999;99:361–9. doi: 10.1161/01.cir.99.3.361. [DOI] [PubMed] [Google Scholar]
- 103.Kolodgie FD, Virmani R, Cornhill JF, Herderick EE, Smialek J. Increase in atherosclerosis and adventitial mast cells in cocaine abusers: an alternative mechanism of cocaine-associated coronary vasospasm and thrombosis. J Am Coll Cardiol. 1991;17:1553–60. doi: 10.1016/0735-1097(91)90646-q. [DOI] [PubMed] [Google Scholar]
- 104.Constantinides P. Mast cells and susceptibility to experimental atherosclerosis. Science. 1953;117:505–6. doi: 10.1126/science.117.3045.505. [DOI] [PubMed] [Google Scholar]
- 105.Cairns A, Constantinides P. Mast cells in human atherosclerosis. Science. 1954;120:31–2. doi: 10.1126/science.120.3105.31. [DOI] [PubMed] [Google Scholar]
- 106.McGovern VJ. Mast cells and their relationship to endothelial surfaces. J Pathol Bacteriol. 1956;71:1–6. [PubMed] [Google Scholar]
- 107.Pomerance A. Periarterial mast cells in coronary atheroma and thrombosis. J Pathol Bacteriol. 1958;76:55–70. doi: 10.1002/path.1700760106. [DOI] [PubMed] [Google Scholar]
- 108.Lehtonen-Smeds EM, Mayranpaa M, Lindsberg PJ, Soinne L, Saimanen E, Jarvinen AA, Salonen O, Carpen O, Lassila R, Sarna S, Kaste M, Kovanen PT. Carotid plaque mast cells associate with atherogenic serum lipids, high grade carotid stenosis and symptomatic carotid artery disease. Results from the helsinki carotid endarterectomy study. Cerebrovasc Dis. 2005;19:291–301. doi: 10.1159/000084497. [DOI] [PubMed] [Google Scholar]
- 109.Jeziorska M, McCollum C, Woolley DE. Mast cell distribution, activation, and phenotype in atherosclerotic lesions of human carotid arteries. J Pathol. 1997;182:115–22. doi: 10.1002/(SICI)1096-9896(199705)182:1<115::AID-PATH806>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 110.Nishizono S, Kusaba M, Adan Y, Imaizumi K. Induction of atherosclerosis in Brown Norway rats by immunization with ovalbumin. Biosci Biotechnol Biochem. 1999;63:379–83. doi: 10.1271/bbb.63.379. [DOI] [PubMed] [Google Scholar]
- 111.Kaartinen M, Penttila A, Kovanen PT. Mast cells accompany microvessels in human coronary atheromas: implications for intimal neovascularization and haemorrhage. Atherosclerosis. 1996;123:123–31. doi: 10.1016/0021-9150(95)05794-3. [DOI] [PubMed] [Google Scholar]
- 112.Jeziorska M, Woolley DE. Local neovascularization and cellular composition within vulnerable regions of atherosclerotic plaques of human carotid arteries. J Pathol. 1999;188:189–96. doi: 10.1002/(SICI)1096-9896(199906)188:2<189::AID-PATH336>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 113.Lappalainen H, Laine P, Pentikainen MO, Sajantila A, Kovanen PT. Mast cells in neovascularized human coronary plaques store and secrete basic fibroblast growth factor, a potent angiogenic mediator. Arterioscler Thromb Vasc Biol. 2004;24:1880–5. doi: 10.1161/01.ATV.0000140820.51174.8d. [DOI] [PubMed] [Google Scholar]
- 114.Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, Narula J, Finn AV, Virmani R. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–25. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 115.Ratner M. Genentech discloses safety concerns over Avastin. Nat Biotechnol. 2004;22:1198. doi: 10.1038/nbt1004-1198. [DOI] [PubMed] [Google Scholar]
- 116.Khurana R, Simons M, Martin JF, Zachary IC. Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation. 2005;112:1813–24. doi: 10.1161/CIRCULATIONAHA.105.535294. [DOI] [PubMed] [Google Scholar]
- 117.Kaartinen M, Penttila A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. 1994;90:1669–78. doi: 10.1161/01.cir.90.4.1669. [DOI] [PubMed] [Google Scholar]
- 118.Kaartinen M, Penttila A, Kovanen PT. Mast cells in rupture-prone areas of human coronary atheromas produce and store TNF-alpha. Circulation. 1996;94:2787–92. doi: 10.1161/01.cir.94.11.2787. [DOI] [PubMed] [Google Scholar]
- 119.El-Lati SG, Dahinden CA, Church MK. Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J Invest Dermatol. 1994;102:803–6. doi: 10.1111/1523-1747.ep12378589. [DOI] [PubMed] [Google Scholar]
- 120.Laine P, Pentikainen MO, Wurzner R, Penttila A, Paavonen T, Meri S, Kovanen PT. Evidence for complement activation in ruptured coronary plaques in acute myocardial infarction. Am J Cardiol. 2002;90:404–8. doi: 10.1016/s0002-9149(02)02498-0. [DOI] [PubMed] [Google Scholar]
- 121.Kelley J, Hemontolor G, Younis W, Li C, Krishnaswamy G, Chi DS. Mast cell activation by lipoproteins. Methods Mol Biol. 2006;315:341–8. doi: 10.1385/1-59259-967-2:341. [DOI] [PubMed] [Google Scholar]
- 122.Helske S, Syvaranta S, Kupari M, Lappalainen J, Laine M, Lommi J, Turto H, Mayranpaa M, Werkkala K, Kovanen PT, Lindstedt KA. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006;27:1495–504. doi: 10.1093/eurheartj/ehi706. [DOI] [PubMed] [Google Scholar]
- 123.Small-Howard A, Turner H. Exposure to tobacco-derived materials induces overproduction of secreted proteinases in mast cells. Toxicol Appl Pharmacol. 2005;204:152–63. doi: 10.1016/j.taap.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 124.Laine P, Naukkarinen A, Heikkila L, Penttila A, Kovanen PT. Adventitial mast cells connect with sensory nerve fibers in atherosclerotic coronary arteries. Circulation. 2000;101:1665–9. doi: 10.1161/01.cir.101.14.1665. [DOI] [PubMed] [Google Scholar]
- 125.Dvorak AM, Kissell S. Granule changes of human skin mast cells characteristic of piecemeal degranulation and associated with recovery during wound healing in situ. J Leukoc Biol. 1991;49:197–210. doi: 10.1002/jlb.49.2.197. [DOI] [PubMed] [Google Scholar]
- 126.Kandere-Grzybowska K, Letourneau R, Kempuraj D, Donelan J, Poplawski S, Boucher W, Athanassiou A, Theoharides TC. IL-1 induces vesicular secretion of IL-6 without degranulation from human mast cells. J Immunol. 2003;171:4830–6. doi: 10.4049/jimmunol.171.9.4830. [DOI] [PubMed] [Google Scholar]
- 127.Fischer M, Harvima IT, Carvalho RF, Moller C, Naukkarinen A, Enblad G, Nilsson G. Mast cell CD30 ligand is upregulated in cutaneous inflammation and mediates degranulation-independent chemokine secretion. J Clin Invest. 2006;116:2748–56. doi: 10.1172/JCI24274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mekori Y, Baram D. Heterotypic adhesion-induced mast cell activation: biologic relevance in the inflammatory context. Mol Immunol. 2002;38:1363. doi: 10.1016/s0161-5890(02)00089-5. [DOI] [PubMed] [Google Scholar]
- 129.Biedermann T, Kneilling M, Mailhammer R, Maier K, Sander CA, Kollias G, Kunkel SL, Hultner L, Rocken M. Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J Exp Med. 2000;192:1441–52. doi: 10.1084/jem.192.10.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J, Strom TB, Zheng XX, Noelle RJ. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- 131.Boyle JJ. Association of coronary plaque rupture and atherosclerotic inflammation. J Pathol. 1997;181:93–9. doi: 10.1002/(SICI)1096-9896(199701)181:1<93::AID-PATH696>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 132.Nakae S, Suto H, Kakurai M, Sedgwick JD, Tsai M, Galli SJ. Mast cells enhance T cell activation: Importance of mast cell-derived TNF. Proc Natl Acad Sci USA. 2005;102:6467–72. doi: 10.1073/pnas.0501912102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nakae S, Suto H, Iikura M, Kakurai M, Sedgwick JD, Tsai M, Galli SJ. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J Immunol. 2006;176:2238–48. doi: 10.4049/jimmunol.176.4.2238. [DOI] [PubMed] [Google Scholar]
- 134.Kirshenbaum AS, Worobec AS, Davis TA, Goff JP, Semere T, Metcalfe DD. Inhibition of human mast cell growth and differentiation by interferon gamma-1b. Exp Hematol. 1998;26:245–51. [PubMed] [Google Scholar]
- 135.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 136.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor−/−mice. Arterioscler Thromb Vasc Biol. 2002;22:456–61. doi: 10.1161/hq0302.104905. [DOI] [PubMed] [Google Scholar]
- 138.Zhou X, Paulsson G, Stemme S, Hansson GK. Hypercholesterolaemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101:1717–25. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen R, Fairley JA, Zhao ML, Giudice GJ, Zillikens D, Diaz LA, Liu Z. Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol. 2002;169:3987–92. doi: 10.4049/jimmunol.169.7.3987. [DOI] [PubMed] [Google Scholar]
- 140.Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT. Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol. 1996;7:330–5. doi: 10.1097/00041433-199610000-00012. [DOI] [PubMed] [Google Scholar]
- 141.Naruko T, Ueda M, Haze K, Van Der Wal AC, Van Der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–900. doi: 10.1161/01.cir.0000042674.89762.20. [DOI] [PubMed] [Google Scholar]
- 142.Nakae S, Suto H, Berry GJ, Galli SJ. Mast cell-derived TNF can promote Th17 cell-dependent neutrophil recruitment in ovalbumin-challenged OTII mice. Blood. 2006;109:3640–8. doi: 10.1182/blood-2006-09-046128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hashmi S, Zeng QT. Role of interleukin-17 and inter-leukin- 17-induced cytokines interleukin-6 and inter-leukin- 8 in unstable coronary artery disease. Coron Artery Dis. 2006;17:699–706. doi: 10.1097/01.mca.0000236288.94553.b4. [DOI] [PubMed] [Google Scholar]
- 144.Lindstedt KA, Kovanen PT. Mast cells in vulnerable coronary plaques: potential mechanisms linking mast cell activation to plaque erosion and rupture. Curr Opin Lipidol. 2004;15:567–73. doi: 10.1097/00041433-200410000-00011. [DOI] [PubMed] [Google Scholar]
- 145.Leskinen M, Wang Y, Leszczynski D, Lindstedt KA, Kovanen PT. Mast cell chymase induces apoptosis of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:516–22. doi: 10.1161/01.atv.21.4.516. [DOI] [PubMed] [Google Scholar]
- 146.Forman MB, Oates JA, Robertson D, Robertson RM, Roberts LJ, 2nd, Virmani R. Increased adventitial mast cells in a patient with coronary spasm. N Engl J Med. 1985;313:1138–41. doi: 10.1056/NEJM198510313131807. [DOI] [PubMed] [Google Scholar]
- 147.Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–97. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 148.Shasby DM, Ries DR, Shasby SS, Winter MC. Histamine stimulates phosphorylation of adherens junction proteins and alters their link to vimentin. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1330–8. doi: 10.1152/ajplung.00329.2001. [DOI] [PubMed] [Google Scholar]
- 149.Winter MC, Shasby SS, Ries DR, Shasby DM. Histamine selectively interrupts VE-cadherin adhesion independently of capacitive calcium entry. Am J Physiol Lung Cell Mol Physiol. 2004;287:L816–23. doi: 10.1152/ajplung.00056.2004. [DOI] [PubMed] [Google Scholar]
- 150.Itoh Y, Sendo T, Oishi R. Physiology and pathophysiology of proteinase-activated receptors (PARs): role of tryptase/PAR-2 in vascular endothelial barrier function. J Pharmacol Sci. 2005;97:14–9. doi: 10.1254/jphs.fmj04005x3. [DOI] [PubMed] [Google Scholar]
- 151.Schechter NM, Brass LF, Lavker RM, Jensen PJ. Reaction of mast cell proteases tryptase and chy-mase with protease activated receptors (PARs) on keratinocytes and fibroblasts. J Cell Physiol. 1998;176:365–73. doi: 10.1002/(SICI)1097-4652(199808)176:2<365::AID-JCP15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 152.Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor- 2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol. 2003;28:339–46. doi: 10.1165/rcmb.4908. [DOI] [PubMed] [Google Scholar]
- 153.Clejan S, Japa S, Clemetson C, Hasabnis SS, David O, Talano JV. Blood histamine is associated with coronary artery disease, cardiac events and severity of inflammation and atherosclerosis. J Cell Mol Med. 2002;6:583–92. doi: 10.1111/j.1582-4934.2002.tb00456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rosenfeld ME, Carson KG, Johnson JL, Williams H, Jackson CL, Schwartz SM. Animal models of spontaneous plaque rupture: the holy grail of experimental atherosclerosis research. Curr Atheroscler Rep. 2002;4:238–42. doi: 10.1007/s11883-002-0025-3. [DOI] [PubMed] [Google Scholar]
- 155.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–44. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 156.Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, Huang PL. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J Clin Invest. 1998;101:1225–32. doi: 10.1172/JCI1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sasaki T, Kuzuya M, Cheng XW, Nakamura K, Tamaya-Mori N, Maeda K, Kanda S, Koike T, Sato K, Iguchi A. A novel model of occlusive thrombus formation in mice. Lab Invest. 2004;84:1526–32. doi: 10.1038/labinvest.3700171. [DOI] [PubMed] [Google Scholar]
- 158.Sasaki T, Kuzuya M, Nakamura K, Cheng XW, Shibata T, Sato K, Iguchi A. A simple method of plaque rupture induction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:1304–9. doi: 10.1161/01.ATV.0000219687.71607.f7. [DOI] [PubMed] [Google Scholar]
- 159.Williams H, Johnson JL, Carson KG, Jackson CL. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2002;22:788–92. doi: 10.1161/01.atv.0000014587.66321.b4. [DOI] [PubMed] [Google Scholar]
- 160.Johnson J, Carson K, Williams H, Karanam S, Newby A, Angelini G, George S, Jackson C. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation. 2005;111:1422–30. doi: 10.1161/01.CIR.0000158435.98035.8D. [DOI] [PubMed] [Google Scholar]
- 161.Tkaczyk C, Okayama Y, Metcalfe DD, Gilfillan AM. Fcgamma receptors on mast cells: activatory and inhibitory regulation of mediator release. Int Arch Allergy Immunol. 2004;133:305–15. doi: 10.1159/000077213. [DOI] [PubMed] [Google Scholar]
- 162.Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol. 2007;7:93–104. doi: 10.1038/nri2018. [DOI] [PubMed] [Google Scholar]
- 163.Kockx MM, De Meyer GR, Andries LJ, Bult H, Jacob WA, Herman AG. The endothelium during cuffinduced neointima formation in the rabbit carotid artery. Arterioscler Thromb. 1993;13:1874–84. doi: 10.1161/01.atv.13.12.1874. [DOI] [PubMed] [Google Scholar]
- 164.Castellino FJ, Ganopolsky JG, Noria F, Sandoval-Cooper MJ, Ploplis VA. Focal arterial inflammation is augmented in mice with a deficiency of the protein C gene. Thromb Haemost. 2006;96:794–801. [PubMed] [Google Scholar]
- 165.Bot I, De Jager SCA, Zernecke A, Lindstedt KA, Weber C, Van Berkel TJC, Biessen EAL. Perivascular Mast Cells Promote Atherogenesis and Induce Plaque Destabilization in ApoE Deficient Mice. Circulation. 2007;115:2516–25. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 166.Hackett D, Davies G, Chierchia S, Maseri A. Intermittent coronary occlusion in acute myocardial infarction. Value of combined thrombolytic and vasodilator therapy. N Engl J Med. 1987;317:1055–9. doi: 10.1056/NEJM198710223171704. [DOI] [PubMed] [Google Scholar]
- 167.Kalsner S. Coronary artery spasm. Multiple causes and multiple roles in heart disease. Biochem Pharmacol. 1995;49:859–71. doi: 10.1016/0006-2952(94)00447-t. [DOI] [PubMed] [Google Scholar]
- 168.Simons FE, Simons KJ. Clinical pharmacology of H1- antihistamines. Clin Allergy Immunol. 2002;17:141–78. [PubMed] [Google Scholar]
- 169.Sharma JN, Mohammed LA. The role of leukotrienes in the pathophysiology of inflammatory disorders: is there a case for revisiting leukotrienes as therapeutic targets? Inflammopharmacology. 2006;14:10–6. doi: 10.1007/s10787-006-1496-6. [DOI] [PubMed] [Google Scholar]
- 170.Wang YH, Tache Y, Harris AG, Kreutner W, Daly AF, Wei JY. Desloratadine prevents compound 48/80- induced mast cell degranulation: visualization using a vital fluorescent dye technique. Allergy. 2005;60:117–24. doi: 10.1111/j.1398-9995.2004.00641.x. [DOI] [PubMed] [Google Scholar]
- 171.Kurosawa M, Amano H, Kanbe N, Akimoto S, Takeuchi Y, Yamashita T, Hashimoto T, Kurimoto F, Miyachi Y. Heterogeneity of mast cells in mastocytosis and inhibitory effect of ketotifen and ranitidine on indolent systemic mastocytosis. J Allergy Clin Immunol. 1997;100:S25–32. doi: 10.1016/s0091-6749(97)70001-0. [DOI] [PubMed] [Google Scholar]
- 172.Lindstedt KA, Leskinen MJ, Kovanen PT. Proteolysis of the pericellular matrix: a novel element determining cell survival and death in the pathogenesis of plaque erosion and rupture. Arterioscler Thromb Vasc Biol. 2004;24:1350–8. doi: 10.1161/01.ATV.0000135322.78008.55. [DOI] [PubMed] [Google Scholar]
- 173.George SJ. Therapeutic potential of matrix metallo-proteinase inhibitors in atherosclerosis. Expert Opin Investig Drugs. 2000;9:993–1007. doi: 10.1517/13543784.9.5.993. [DOI] [PubMed] [Google Scholar]
- 174.Liu J, Sukhova GK, Sun JS, Xu WH, Libby P, Shi GP. Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1359–66. doi: 10.1161/01.ATV.0000134530.27208.41. [DOI] [PubMed] [Google Scholar]
- 175.Johnson JL, Fritsche-Danielson R, Behrendt M, Westin-Eriksson A, Wennbo H, Herslof M, Elebring M, George SJ, McPheat WL, Jackson CL. Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc Res. 2006;71:586–95. doi: 10.1016/j.cardiores.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 176.Tremaine WJ, Brzezinski A, Katz JA, Wolf DC, Fleming TJ, Mordenti J, Strenkoski-Nix LC, Kurth MC AXYS Ulcerative Colitis Study Group. Treatment of mildly to moderately active ulcerative colitis with a tryptase inhibitor (APC 2059): an open-label pilot study. Aliment Pharmacol Ther. 2002;16:407–13. doi: 10.1046/j.1365-2036.2002.01194.x. [DOI] [PubMed] [Google Scholar]
- 177.Doggrell SA, Wanstall JC. Vascular chymase: pathophysiological role and therapeutic potential of inhibition. Cardiovasc Res. 2004;61:653–62. doi: 10.1016/j.cardiores.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 178.Miyazaki M, Takai S, Jin D, Muramatsu M. Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol Ther. 2006;112:668–76. doi: 10.1016/j.pharmthera.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 179.Uehara Y, Urata H, Ideishi M, Arakawa K, Saku K. Chymase inhibition suppresses high-cholesterol dietinduced lipid accumulation in the hamster aorta. Cardiovasc Res. 2002;55:870–6. doi: 10.1016/s0008-6363(02)00458-3. [DOI] [PubMed] [Google Scholar]
- 180.Rudd RM, Gellert AR, Studdy PR, Geddes DM. Inhibition of exercise-induced asthma by an orally absorbed mast cell stabilizer (M & B 22,948) Br J Dis Chest. 1983;77:78–86. [PubMed] [Google Scholar]
- 181.Boswell-Smith V, Spina D, Page CP. Phosphodiesterase inhibitors. Br J Pharmacol. 2006;147:S252–7. doi: 10.1038/sj.bjp.0706495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jensen BM, Metcalfe DD, Gilfillan AM. Targeting kit activation: a potential therapeutic approach in the treatment of allergic inflammation. Inflamm Allergy Drug Targets. 2007;6:57–62. doi: 10.2174/187152807780077255. [DOI] [PubMed] [Google Scholar]
- 183.Bellosta S, Via D, Canavesi M, Pfister P, Fumagalli R, Paoletti R, Bernini F. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol. 1998;18:1671–8. doi: 10.1161/01.atv.18.11.1671. [DOI] [PubMed] [Google Scholar]
- 184.Shakarjian MP, Eiseman E, Penhallow RC, Bolen JB. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibition in a rat mast cell line. Impairment of tyro-sine kinase-dependent signal transduction and the subsequent degranulation response. J Biol Chem. 1993;268:15252–9. [PubMed] [Google Scholar]
- 185.Krauth MT, Majlesi Y, Sonneck K, Samorapoompichit P, Ghannadan M, Hauswirth AW, Baghestanian M, Schernthaner GH, Worda C, Muller MR, Sperr WR, Valent P. Effects of various statins on cytokine-dependent growth and IgE-dependent release of histamine in human mast cells. Allergy. 2006;61:281–8. doi: 10.1111/j.1398-9995.2006.00997.x. [DOI] [PubMed] [Google Scholar]
- 186.Malaviya R, Abraham SN. Mast cell modulation of immune responses to bacteria. Immunol Rev. 2001;179:16–24. doi: 10.1034/j.1600-065x.2001.790102.x. [DOI] [PubMed] [Google Scholar]
- 187.Wojta J, Huber K, Valent P. New aspects in thrombotic research: complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol Haemost Thromb. 2003;33:438–41. doi: 10.1159/000083842. [DOI] [PubMed] [Google Scholar]
- 188.Valent P, Baghestanian M, Bankl HC, Sillaber C, Sperr WR, Wojta J, Binder BR, Lechner K. New aspects in thrombosis research: possible role of mast cells as profibrinolytic and antithrombotic cells. Thromb Haemost. 2002;87:786–90. [PubMed] [Google Scholar]