

Abstract
Although J2N-k strain of cardiomyopathic hamsters is an excellent model of dilated cardiomyopathy, the presence and mechanisms of apoptosis in the hearts of these genetically modified animals have not been investigated. This study examined the hypothesis that cardiac dysfunction and apoptosis in the cardiomyopathic hamsters were associated with tumour necrosis factor-alpha (TNF-α)-mediated signalling pathway involving the activation of some pro-apoptotic proteins and/or deactivation of some antiapoptotic proteins. Echocardiographic assessment of 31-week-old hamsters indicated an increase in the internal dimension of the left ventricle as well as decreases in the ejection fraction, fractional shortening and cardiac output without any evidence of cardiac hypertrophy. Increased level of TNF-α and apoptosis in cardiomyopathic hearts were accompanied by increased protein content for protein kinase C (PKC) -α and -ɛ isozymes as well as caspases 3 and 9. Phosphorylated protein content for p38 MAPK and NFκB was increased whereas that for Erk1/2, BAD and Bcl-2 was decreased in cardiomyopathic hearts. These results support the view that TNF-α and PKC isozymes may promote apoptosis due to the activation of p38 MAPK and deactivation of Erk1/2 pathways, and these changes may contribute toward the development of cardiac dysfunction in dilated cardiomyopathy.
Keywords: cardiomyopathic hearts, TNF-α and NFκB, PKC and MAPK, BAD and Bcl-2, caspases 3 and 9
Introduction
It is now becoming evident that programmed cell death (apoptosis) is one of the important determinants of cardiac contractile dysfunction under different pathological conditions [1–4]. Several investigators [5–9] have shown that different factors such as intracellular Ca2+-overload, oxidative stress and cytokines including tumour necrosis factor-alpha (TNF-α) play a critical role in the development of cardiomyocyte apoptosis. In particular, the up-regulation or down-regulation of some proteins such as protein kinase C (PKC) isozymes, caspases 3 and 9, p38 mitogen activated protein kinase (p38 MAPK), nuclear factor (NFκB), extracellular protein kinases (Erk1/2), BAD, and Bcl-2 are considered to be intimately involved in the genesis of cell death or cell survival [10–18]. Furthermore, the activation of some proteins such as p38 MAPK and NFκB as well as deactivation of Erk1/2, BAD and Bcl-2 due to phosphorylation/dephosphorylation process determine the extent of activation of mitochondrial caspases for the occurrence of apoptosis [14–23]. Thus, in spite of the complexities of signal transduction mechanisms involved in the genesis of cardiomyocyte apoptosis, the process of cellular death appears to represent an important feature for remodelling (changes in the size and shape) of the myocardium and cardiac dysfunction [24–26].
The occurrence of apoptosis in myocardium has been identified in failing human hearts as well as in experimental models subsequent to myocardial infarction [27–29]. Heart failure due to pressure overload [1, 30] or volume overload [31] was also observed to show the presence of cardiomyocyte apoptosis. Both ischaemia-reperfusion injury and induction of Ca2+-paradox have been reported to result in the loss of contractile function as well as development of apoptosis in the isolated perfused hearts [32, 33]. Likewise, contractile dysfunction due to drug-induced cardiomyopathy has been associated with the presence of apoptosis in the heart [34, 35]. Furthermore, cardiomyocyte apoptosis has been observed in heart failure in dilated cardiomyopathy due to oxidative stress in dogs [36]. Although the J2N-k strain of hamsters has been shown to form an excellent model of dilated cardiomyopathy [37–40], no information regarding the occurrence of apoptosis or the mechanisms of its development in these genetically modified animals is available in the literature. This study was therefore undertaken to investigate if dilated cardiomyopathy in the J2N-k strain of hamsters was associated with contractile dysfunction and apoptosis. Furthermore, alterations in protein content for TNF-α, PKC isozymes and caspases were determined to gain some insight regarding the mechanisms of apoptosis in the cardiomyopathic hamsters. In addition, the phosphorylated and unphosphorylated content of p38 MAPK, Erk1/2, NFκB, BAD and Bcl-2 were measured to test if the phosphorylation status of these effectors is associated with apoptosis in the cardiomyopathic hamster hearts.
Materials and methods
All experimental protocols used in this study were approved by the Animal Care Committee of the University of Manitoba following the guidelines established by the Canadian Council on Animal Care.
Cardiomyopathic hamsters
The animal model used in this study was the cardiomyopathic J2N-k male hamsters along with age-matched healthy controls, the J2N-n male hamsters. These animals were obtained from the Jikei University Aoto Hospital in Japan and housed in humidity- and temperature-controlled rooms. The hamsters until the age of 31 week were allowed free access to water and standard chow, assessed for cardiac function by echocardiography and then sacrificed.
Echocardiographic assessment
This method is the same as described previously [41]. Briefly, cardiac ultrasound study was carried out using the SONOS 5500 ultrasonograph (Agilent Technologies, Andover, MA, USA). The hamsters were anaesthetized with 2.5% isoflurane gas in 2 l/min oxygen and were allowed to breathe spontaneously. The chest was shaved with electric clippers and echocardiographic readings were recorded with the hamsters lying on their left side. A 12-MHz annular array ultrasound transducer was gently positioned on the coupling gel over the, allowing appropriate contact without an excessive pressure on the chest. Transthoracic short axis measurements were performed in the left lateral decubitus position, while the transducer was gently rotated to achieve the best position. In the parasternal short axis orientation, the probe recorded left ventricular diastolic and systolic measurements. The M-mode echocardiograms at the papillary muscle level measured the following parameters: interventricular septum thickness diastole/systole (IVSd, IVSs), left ventricular internal dimension diastole/systole (LVIDd, LVIDs), left ventricular posterior wall thickness diastole/systole (LVPWd, LVPWs), ejection fraction (EF), per cent fractional shortening (FS), cardiac output (CO) and heart rate (HR). The hearts of the control and cardiomyopathic animals were then quickly excised, washed in cold 0.9% saline, weighed and placed directly into liquid nitrogen. The ventricles were then stored at −80°C until further use.
Measurement of TNF-α
Ventricular tissue was homogenized in 10 volumes of phosphate buffered saline (PBS), which contained 1% Triton X-100 (Sigma–Aldrich, Oakville, ON, Canada) along with a protease inhibitor cocktail (Roche Applied Science, Laval, PQ, Canada). The homogenate was centrifuged at 2500 g for 20 min. at 4°C. The supernatant was collected and the TNF-α level was measured using a sandwich ELISA kit for rat TNF-α with a 12.5 pg/ml detection limit (R&D Systems, Minneapolis, MN, USA). The assay was performed according to the manufacturer’s instructions. Absorbance of standards and samples was determined spectrophotometrically (SPECTRAmax PLUS384, Molecular Devices, Sunnyvale, CA, USA) at 450 nm. Results were calculated from the standard curve and were reported as pg/g protein.
Preparation of tissue extract for Western blot analysis
Ventricular tissue (50 mg) was homogenized at setting 8 twice for 30 sec. each (Polytron PT 3000; Brinkmann Instruments, Mississauga, ON, Canada) in 1 ml of buffer A, which contained 50 mM Tris-HCl, 0.25 M sucrose, 10 mM EGTA, 4 mM EDTA, pH 7.5, and protease inhibitor cocktail (Roche Applied Science). The suspension was sonicated twice for 15 sec. each and then centrifuged at 100,000 g for 60 min. in an ultracentrifuge (Model L70; Beckman Instruments, Fullerton, CA, USA), and the supernatant was then collected as the cytosolic fraction. The pellet was resuspended in 1 ml of buffer B (buffer A plus 1% Triton X-100), incubated on ice for 60 min. and then centrifuged at 100,000 g for 60 min. It is pointed out that both buffer A and buffer B had no phosphatase inhibitors. This supernatant, which contained the dissolved membrane protein, was labelled as the particulate fraction. Another piece (50 mg) of ventricular tissue was suspended in buffer B after homogenization (twice for 30 sec. each) and sonication (twice for 15 sec. each). The homogenate was incubated on ice for 60 min. and then centrifuged at 100,000 g for 60 min. This supernatant was labelled as the homogenate fraction. The protein content was determined using a modified Bradford method and bovine serum albumin was used as the standard. This method for studying subcellular distribution of an enzyme or protein is the same as described earlier [42].
Western blot method
Total protein (50 μg) in the Clontech Extraction buffer was added to an equal volume of sodium dodecyl sulphate (SDS) buffer and boiled for 10 min. before being separated on 7–15% SDS polyacrylamide gels in running buffer (25 mM Tris, 192 mM glycine, 0.1% (w/v) SDS, pH 8.3)at 200 V. The prestained protein marker (10 μl) (Invitrogen Life Technologies, Carlsbad, CA, USA) was used as molecular weight standards. The gel was transferred onto a nitrocellulose membrane (Bio-Rad Laboratories, Mississauga, ON, Canada) at 100 V for 1 hr in transfer buffer (25 mM Tris base, 192 mM glycine, 20% (v/v) methanol, pH 8.3). After blocking the membranes for 1 hr in Tris-buffered saline (TBS-T) (50 mM Tris, pH 7.5, 150 mM NaCl) containing 0.1% (v/v) Tween-20 and 5% (w/v) non-fat dry milk, blots were incubated overnight at 4°C with the primary antibody. All antibodies were purchased from Cell Signaling Technology, Mississauga, ON, Canada and were used at manufacturer’s recommended dilutions. Membranes were washed three times in TBS-T prior to incubation for 1 hr with horseradish peroxide (HRP)-conjugated secondary antibody diluted 1:2000 in TBS-T and 5% (w/v) non-fat dry milk. Western blots were developed with the ECL Detection System (Amersham-Pharmacia Biotech, Baie d’Urfe, PQ, Canada) and exposed to Kodak X-OMAT film.
Detection of cardiomyocyte apoptosis
Cardiomyocyte apoptosis was measured using a Cell Death Detection ELISA Plus kit (Roche Applied Science). Briefly, 20 μl of homogenate sample and 80 μl of immunoreagent (supplied by manufacturer) were loaded into each well. The plate was then covered with foil and incubated under gentle agitation for 2 hrs at 15–25°C. The samples were aspirated and the wells were rinsed three times with 250–300 μl of incubation buffer (supplied by manufacturer); the buffer was removed each time by suction. One hundred microlitres of ABTS solution (supplied by manufacturer) was added to each well and incubated for 15 min. at room temperature under gentle agitation. The degree of apoptosis was colorimetrically assessed at 405 nm against ABTS solution as the blank (reference wavelength approximately 490 nm).
Statistical analysis
The data were expressed as means ± SE. Differences between the control and experimental groups were analysed using an unpaired Student’s t-test. The data from more than two groups were evaluated by one-way ANOVA followed by the Newman–Keuls test. A P level of <0.05 was considered as the threshold for statistical significance.
Results
Cardiac performance of cardiomyopathic hamsters
The data for general characteristics and echocardiographic assessment of control (J2N-n strain) and cardiomyopathic (J2N-k strain) are shown in Fig. 1 and Table 1. Figure 1 shows the M-Mode echocardiographic images of control (Fig. 1A) and cardiomyopathic (Fig. 1B) hamster hearts. Significant depressions in EF (Fig. 1C), FS (Fig. 1D), CO (Fig. 1E) and HR (Fig. 1F) in the cardiomyopathic hamsters suggest impaired cardiac performance. Although cardiac performance parameters such as EF, FS and CO are interrelated, it can be seen from Fig. 1 that the depressions in EF and FS are less than that in CO in the cardiomyopathic hamsters. Because CO is a product of stroke volume and HR, a small change in HR can be seen to make a great change in CO and thus may account for the differences in the degree of depression in various parameters. The dimensions of the heart chamber and ventricular walls (Table 1) show no change in IVSd, LVPWd and LVPWs in cardiomyopathic hearts. No changes in the body weight or heart weight/body weight ratio were apparent in cardiomyopathic hamsters (Table 1). Neither the wet/dry lung weight ratio was increased (Table 1) nor the presence of ascites was evident in cardiomyopathic animals.
Fig 1.
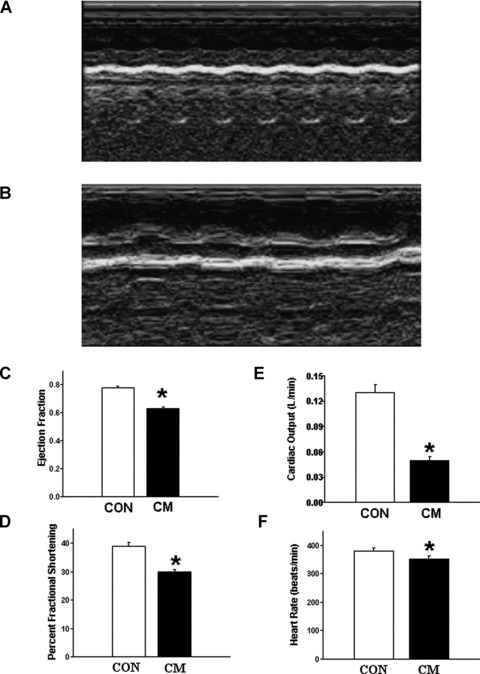
Echocardiographic assessment of control (CON) and cardiomyopathic (CM) hamsters. (A) M-Mode echocardiographic image of cardiomyopathic J2N-k hamster heart. (B) M-Mode echocardiographic image of control J2N-n hamster heart. (C) Ejection fraction. (D) Per cent fractional shortening. (E) Cardiac output. (F) Heart rate. *P < 0.05 versus control; (n= 20).
Table 1.
General characteristics as well as ventricular systolic and diastolic dimensions of 31-week-old control and J2N-k strain of cardiomyopathic hamsters
| Parameter | Control | Cardiomyopathic |
|---|---|---|
| Body weight (g) | 120 ± 1.67 | 134 ± 3.13 |
| Heart/body weight ratio (×10−3) | 4.26 ± 0.33 | 4.60 ± 0.42 |
| Wet/dry lung weight ratio | 4.22 ± 0.08 | 4.31 ± 0.11 |
| Interventricular septum diastole (cm) | 0.19 ± 0.02 | 0.16 ± 0.02 |
| Interventricular septum systole (cm) | 0.25 ± 0.02 | 0.25 ± 0.01 |
| Left ventricular internal dimension diastole (cm) | 0.42 ± 0.02 | 0.62 ± 0.02* |
| Left ventricular internal dimension systole (cm) | 0.24 ± 0.01 | 0.43 ± 0.01* |
| Left ventricular posterior wall diastole (cm) | 0.22 ± 0.02 | 0.24 ± 0.01 |
| Left ventricular posterior wall systole (cm) | 0.24 ± 0.02 | 0.30 ± 0.002 |
Values are means ± SE; n= 20. *P < 0.05 versus control.
Protein content of various effectors in cardiomyopathic hearts
Western blot analysis revealed an increased protein content of PKC-α (Fig. 2A) and PKC-ɛ (Fig. 2C) isoforms in the cardiomyopathic hearts compared to control hearts, whereas the protein content of PKC-β (Fig. 2B) and PKC-γ (Fig. 2D) isoforms in cardiomyopathic hearts was not significantly different from the control group. In cardiomyopathic hamsters, the phosphorylated p38 MAPK content in the heart was increased significantly compared to the control group but there was no difference in the unphosphorylated p38 MAPK content (Fig. 3A and B). As shown in Fig. 3C and D, the levels of phosphorylated Erk1/2 content decreased significantly by 47% and 51% in cardiomyopathic hearts compared to the control animals, respectively, whereas there was no difference in protein content of unphosphorylated Erk1/2. Figure 4A shows a significant decrease of phosphorylated BAD content by 158% in cardiomyopathic hamsters in comparison to the control animals without any difference in the level of unphosphorylated BAD content (Fig. 4A). The phosphorylated Bcl-2 protein content was also decreased significantly in cardiomyopathic hearts compared to control groups but no difference was evident in the unphosphorylated Bcl-2 protein level (Fig. 4B).
Fig 2.
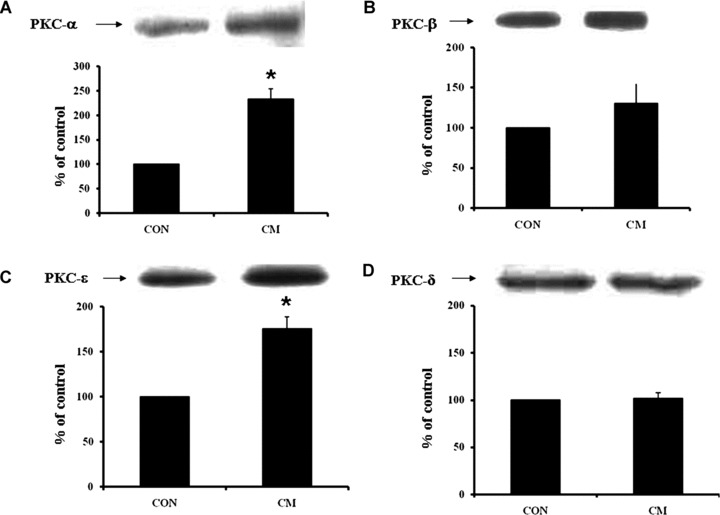
PKC protein levels of isoform α (A), isoform β (B), isoform ɛ (C) and isoform γ (D) in control (CON) and cardiomyopathic (CM) hamsters. *P < 0.05 versus control; (n= 6).
Fig 3.
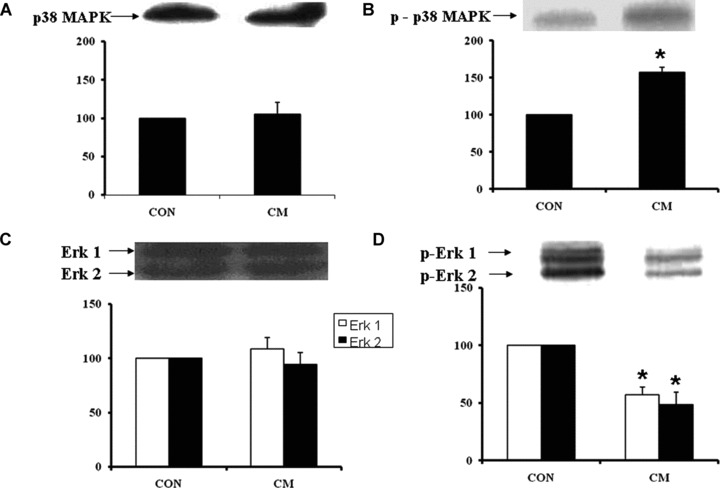
Western blot analysis of p38 MAPK (A), phosphorylated p38 MAPK (B), Erk1/2 (C) and phosphorylated Erk 1/2 (D) in control (CON) and cardiomyopathic (CM) hamster hearts. *P < 0.05 versus control; (n= 6).
Fig 4.
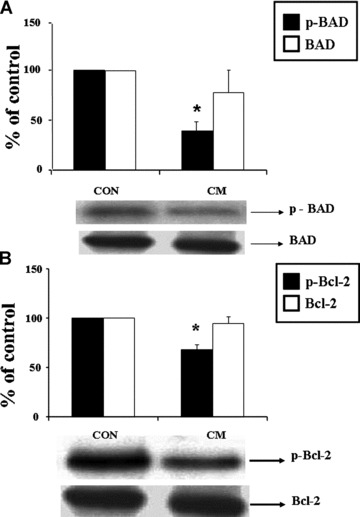
Western blot analysis of phosphorylated BAD and BAD (A), phosphorylated BCl-2 and BCl-2 (B), and the levels of phosphorylated and unphosphorylated BAD as well as phosphorylated and unphosphorylated BCl-2 in control (CON) and cardiomyopathic (CM) hamster hearts. *P < 0.05 versus control; (n= 6).
Protein content for NFκB and caspases 3 and 9, cardiomyocyte apoptosis and TNF-α production
As shown in Fig. 5A, the phosphorylated NFκB content was about 3.5-fold increased without any change in the protein content of unphosphorylated NFκB content (Fig. 5B) in cardiomyopathic hearts compared to control group. The 3-fold increase in the ratio of phosphorylated NFκB and total NFκB content (Fig. 5C) indicate a marked activation of NFκB in the cardiomyopathic heart. Western blot analysis also revealed significant increases in protein content of caspase 9 (Fig. 6A) as well as caspase 3 (Fig. 6B) in cardiomyopathic hearts compared to control group. For caspase 9, the increase was almost 2-fold and for caspase 3, the increase was 3-fold. Furthermore, a marked increase in TNF-α level was detected in the myocardium of the cardiomyopathic hamsters compared to the control animal groups (Fig. 7A). The incidence of apoptosis, as seen by the Cell Death Detection ELISA assay, was well pronounced in the cardiomyopathic hearts (Fig. 7B).
Fig 5.
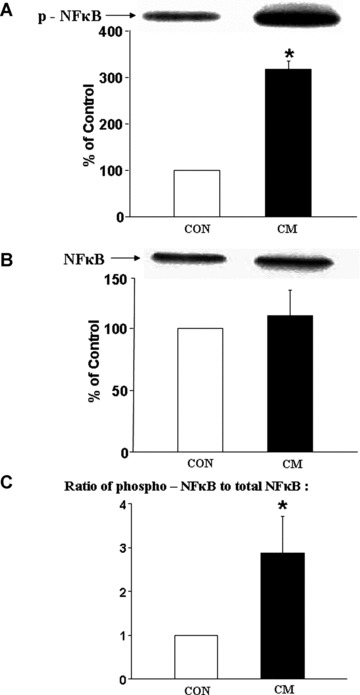
Western blot analysis of phosphorylated NFκB (A), NFκB (B) and the ratio of phosphorylated NFκB to NFκB (C) in control (CON) and cardiomyopathic (CM) hamster hearts. *P < 0.05 versus control; (n= 6).
Fig 6.
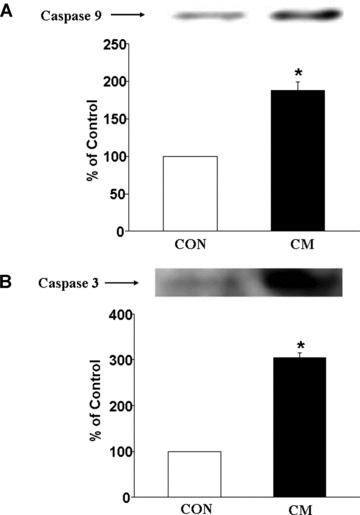
Western blot analysis of caspase 9 (A) and caspase 3 (B) in control (CON) and cardiomyopathic (CM) hamster hearts. *P < 0.05 versus control; (n= 6).
Fig 7.
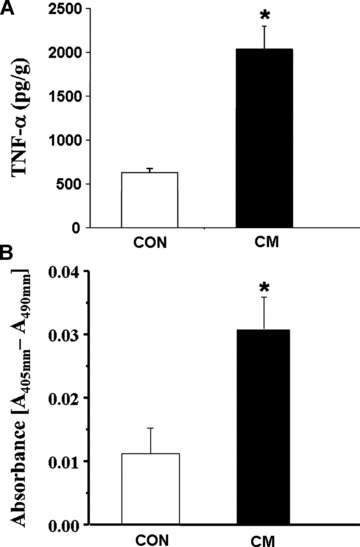
Protein levels of TNF-α (A) and cardiomyocyte apoptosis as detected by the ELISA kit (B) in control (CON) and cardiomyopathic (CM) hamster hearts. *P < 0.05 versus control; (n= 6).
Discussion
In this study, we have shown that the 31-week-old JN2-K cardiomyopathic hamsters had impaired cardiac function because the EF, FS and HR were depressed significantly and the CO was decreased markedly. These alterations in cardiac performance were not associated with cardiac hypertrophy because the heart/body weight ratio, left ventricular wall thickness (LVPWd and LVPWs) and intraventricular septum thickness (IVSd and IVSs) in cardiomyopathic hamsters were not different from the control group. Furthermore, these cardiomyopathic animals did not exhibit any clinical signs of congestive heart failure because neither the wet/dry lung weight ratio (an index of lung congestion) was increased nor was the presence of ascites evident. On the other hand, a marked increase in ventricular dimensions (LVIDd and LVIDs) indicating ventricular dilatation was observed in the cardiomyopathic hamsters. These observations suggest that cardiac dysfunction in the 31-week-old JN2-k strain of hamsters may be due to cardiac dilatation and are in agreement with our earlier report for the 36-week-old JN2-k hamsters [41]. It is pointed out that the lifespan of the JN2-k cardiomyopathic hamsters has been described to be 248 ± 18 days [40]; both cardiac dilatation and cardiac dysfunction in these animals begin at about 20 weeks [39]. Furthermore, the JN2-k strain of hamsters exhibits dilated cardiomyopathy, whereas the BIO 14.6 strain or UM-X7.1 strain of hamsters shows hypertrophic cardiomyopathy [12, 43–46]. The observations reported here support the view that the JN2-k genetically modified hamsters are an excellent model of dilated cardiomyopathy [37–40]. It should be mentioned that the JN2-k cardiomyopathic hamsters have been shown to experience a slow progression of interstitial fibrosis that parallels the focal necrosis in the heart throughout their life [47].
Cardiac dysfunction in the J2N-k hamster model of dilated cardiomypathy was associated with a marked increase in the level of TNF-α in the myocardium. TNF-α has also been reported to be increased in patients with dilated cardiomyopathy [48–51]. Because the cardiac contractile failure due to ischaemia-reperfusion injury [9, 32, 52, 53] as well as due to the induction of Ca2+-paradox [8, 33] is associated with an increased level of TNF-α, it is likely that this cytokine may be involved in the development of cardiac dysfunction in the hamster-dilated cardiomyopathy. This view is supported by the findings that heart failure in both patients and different experimental animals has been observed to be associated with elevated levels of TNF-α in the myocardium [5, 10, 24, 25, 27, 31]. Although high concentrations of TNF-α have been reported to depress cardiac contractile force development [5, 10], the indirect effects of TNF-α for inducing cardiac dysfunction in dilated cardiomyopathy cannot be ruled out. Furthermore, cardiac dysfunction in dilated cardiomyopathy in the J2N-k cardiomyopathic hamsters has been reported to be associated with abnormalities in the sarcoplasmic reticulum and extracellular matrix [41, 44, 47], whereas that in the Bio 53.58 strain of cardiomyopathic hamsters is associated with remodelling of myofibrils and collagen [43, 54]. Cardiac dysfunction in dog-dilated cardiomyopathy induced by rapid pacing has been attributed to the development of oxidative stress [36]. Thus, there appears to be several types of defects, which may account for the occurrence of cardiac dysfunction in the J2N-k hamster-dilated cardiomyopathy.
Although evaluation of the J2N-k cardiomyopathic hamster hearts by ELISA Cell Death Detection assay has revealed the presence of apoptosis, this study does not provide any information regarding the cause–effect relationship between cardiomyocyte loss due to apoptosis and cardiac dysfunction in this experimental model. Because extensive cardiomyocyte necrosis has been reported to be present in these cardiomyopathic hamster hearts (37–40, 47), no effort was made to determine the percentage of apoptotic cells in the myocardium. Nonetheless, a marked increase in the protein content of caspases 3 and 9, which are known to play a critical role in the apoptotic cell death [35], also provides evidence for the occurrence of apoptosis in the J2N-k cardiomyopathic hamster hearts. Because caspases 3 and 9 are activated by proteolytic cleavage, information on protein content of procaspases 3 and 9 would serve as a useful reference point. However, in the absence of such data, some caution should be used for interpreting results on caspases 3 and 9. Nonetheless, the activation of caspases 3 and 9 as well as cardiomyocyte apoptosis have been reported in dog-dilated cardiomyopathy due to rapid pacing [36]. In fact, the presence of cardiomyocyte apoptosis has also been observed in different types of failing hearts in patients as well as several experimental models of heart failure [1, 27–31].
Because TNF-α level as well as protein content of PKC-α, PKC-ɛ, phosphorylated p38 MAPK and phosphorylated NFκB were increased in the cardiomyopathic hamster hearts, it is likely that apoptosis in dilated cardiomyopathy is a result of the up-regulation of TNF-α– PKC – p38 MAPK – NFκB – caspases proapoptotic pathway. Such a view is consistent with the cell death signal transduction pathway, which has been implicated for apoptosis in different other types of heart failure [6, 10–17, 21, 22, 28–31, 34, 35, 42]. Although we do not have any information regarding a specific isoform of p38 MAPK which is phosphorylated, it is possible that PKC-α may be involved in this proapoptotic pathway. On the other hand, decreased levels of phosphorylated Erk1/2, phosphorylated BAD, phosphorylated Bcl-2 in the cardiomyopathic hearts suggest the down-regulation of TNF-α/PKC-mediated Erk1/2 – Bcl-2/BAD signal transduction pathway, which will activate caspases and result in apoptosis. The down-regulation of such an antiapoptotic pathway has also been observed in different types of failing hearts [11, 15, 18–23, 28–31, 34]. These observations suggest that both the up-regulation of apoptotic signal pathway and the down-regulation of apoptotic signal pathway are involved in the development of cardiomyocyte apoptosis in the JN2k hamster dilated cardiomyopathy. Although extensive studies are needed to establish the exact PKC isozyme, which may carry out the phosphorylation of Erk1/2, Bcl-2 and BAD proteins, it is possible that PKC-ɛ may be involved in this antiapoptotic pathway. Nonetheless, depressed level of phosphorylated Erk1/2, BAD and Bcl-2 proteins in the failing heart may be due to augmented dephosphorylation, which may account for down-regulation of the antiapoptotic pathway. Thus, in view of the marked increased in the level of TNF-α and significant alterations in the phosphorylated proapoptotic and antiapoptotic proteins observed in this study, it appears that TNF-α-mediated signal transduction mechanisms play an important role in both cardiac apoptosis and cardiac dysfunction during the development of dilated cardiomyopathy. Although we did not determine the circulating levels of TNF-α in hamsters with dilated cardiomyopathy, it is pointed out that the circulating level of TNF-α has been shown to be elevated in heart failure and anti-TNF-α antibody has been reported to attenuate heart failure in a transgenic model [55].
Acknowledgments
The research work reported in this article was supported by a grant from the Canadian Institute of Health Research as well as CHFNET/IHRT program grant from the Institute of Circulatory and Respiratory Health. The infrastructure for this project was provided by the St. Boniface General Hospital Research Foundation. AB was a graduate student supported by the University of Manitoba Fellowship. DR-L was a Visiting Scientist from Cardiovascular Research Division, V.I. Lenin University Hospital, Holguin, Cuba.
References
- 1.Bing DH. Hypothesis: apoptosis may be mechanism for the transition to heart failure with chronic pressure overload. J Mol Cell Cardiol. 1994;26:943–8. doi: 10.1006/jmcc.1994.1115. [DOI] [PubMed] [Google Scholar]
- 2.Narrula J, Haider N, Virmani R, et al. Apoptosis in myocytes in end-stage heart failure. N Eng J Med. 1996;335:1182–9. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 3.Olivetti G, Abbi R, Quaini F, et al. Apoptosis in the failing human heart. N Eng J Med. 1997;336:1131–41. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 4.Regula KM, Kirshenbaum LA. Apoptosis of ventricular myocytes: a means to an end. J Mol Cell Cardiol. 2005;38:3–13. doi: 10.1016/j.yjmcc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Torre-Amione G, Kapadia S, Lee J, et al. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995;92:1487–93. doi: 10.1161/01.cir.92.6.1487. [DOI] [PubMed] [Google Scholar]
- 6.Brown KA, Page MT, Nguyen C, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–65. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhalla NS, Elmoselhi AB, Hata N, et al. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res. 2000;47:446–56. doi: 10.1016/s0008-6363(00)00078-x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Xu Y-J, Saini HK, et al. TNF-alpha as a potential mediator of cardiac dysfunction due to intracellular Ca2+-overload. Biochem Biophys Res Commun. 2005;327:57–63. doi: 10.1016/j.bbrc.2004.11.131. [DOI] [PubMed] [Google Scholar]
- 9.Saini HK, Xu Y-J, Zhang M, et al. Role of tumour necrosis factor-alpha and other cytokines in ischemia-reperfusion-induced injury in the heart. Exp Clin Cardiol. 2005;10:213–22. [PMC free article] [PubMed] [Google Scholar]
- 10.Sack M. Tumor necrosis factor-alpha in cardiovascular biology and the potential role for anti-tumor necrosis factor-alpha therapy in heart disease. Pharmacol Ther. 2002;94:123–35. doi: 10.1016/s0163-7258(02)00176-6. [DOI] [PubMed] [Google Scholar]
- 11.Das DK. Protein kinase C isozymes signaling in the heart. J Mol Cell Cardiol. 2003;35:887–9. doi: 10.1016/s0022-2828(03)00172-x. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Liu X, Arneja AS, et al. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can J Cardiol. 1999;15:683–90. [PubMed] [Google Scholar]
- 13.Plo I, Lautier D, Levade T, et al. Phosphatidylcholine-specific phospholipase C and phospholipase D are respectively implicated in mitogen-activated protein kinase and nuclear factor kappa B activation in tumor-necrosis-factor-alpha-treated immature acute-myeloid-leukaemia cells. Biochem J. 2000;351:459–67. [PMC free article] [PubMed] [Google Scholar]
- 14.Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307–14. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 15.Badrichani AZ, Stroka DM, Bilbao G, et al. Bcl-2 and Bcl-XL serve an anti-inflammatory function in endothelial cells through inhibition of NF-kappaB. J Clin Invest. 1999;103:543–53. doi: 10.1172/JCI2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol. 1999;31:1429–34. doi: 10.1006/jmcc.1999.0979. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Huang S, Sah VP, et al. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–8. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 18.Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–81. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 19.Yang E, Zha J, Jockel J, et al. Bad, a heterodimeric partner for Bcl-XL and Bcl2, displaces Bax and promotes cell death. Cell. 1995;80:285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 20.Scheid MP, Duronio V. Dissociation of cytokine-induced phosphorylation of BAD and activation of PKB/akt: involvement of MEK upstream of bad phosphorylation. Proc Natl Acad Sci USA. 1998;95:7439–44. doi: 10.1073/pnas.95.13.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 22.Nicolson WD, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;257:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 23.Ruvolo PP, Deng X, May WS. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia. 2001;15:515–22. doi: 10.1038/sj.leu.2402090. [DOI] [PubMed] [Google Scholar]
- 24.Irwin MW, Mak S, Mann DL, et al. Tissue expression and immunolocalization of tumor necrosis factor-α in postinfarction dysfunctional myocardium. Circulation. 1999;99:1492–8. doi: 10.1161/01.cir.99.11.1492. [DOI] [PubMed] [Google Scholar]
- 25.Nian M, Lee P, Khaper N, et al. Inflammatory cytokine and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 26.Dhalla NS, Saini-Chohan HK, Rodriguez-Levya D, et al. Subcellular remodeling may induce cardiac dysfunction in congestive heart failure. Cardiovasc Res. 2009;81:429–38. doi: 10.1093/cvr/cvn281. [DOI] [PubMed] [Google Scholar]
- 27.Torre-Amione G, Kapadia S, Lee J, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation. 1996;93:704–11. doi: 10.1161/01.cir.93.4.704. [DOI] [PubMed] [Google Scholar]
- 28.Baldi A, Abbate A, Bussani R, et al. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol. 2002;34:165–74. doi: 10.1006/jmcc.2001.1498. [DOI] [PubMed] [Google Scholar]
- 29.Chang W, Kajstura J, Nitahara JA, et al. Programmed cell death affects the viable myocardium after infarction in rats. Exp Cell Res. 1996;226:316–27. doi: 10.1006/excr.1996.0232. [DOI] [PubMed] [Google Scholar]
- 30.Condorelli G, Morisco C, Stassi G, et al. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and Bcl-2 during left ventricular adaptation to chronic pressure overload in the rat. Circulation. 1999;99:3071–8. doi: 10.1161/01.cir.99.23.3071. [DOI] [PubMed] [Google Scholar]
- 31.Dent MR, Das S, Dhalla NS. Alterations in both death and survival signals for apoptosis in heart failure due to volume overload. J Mol Cell Cardiol. 2007;43:726–32. doi: 10.1016/j.yjmcc.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Zhang M, Xu Y-J, Saini HK, et al. Pentoxifylline attenuates cardiac dysfunction and reduces TNF-α level in ischemic-reperfused heart. Am J Physiol Heart Circ Physiol. 2005;289:H832–9. doi: 10.1152/ajpheart.00178.2005. [DOI] [PubMed] [Google Scholar]
- 33.Xu Y-J, Saini HK, Zhang M, et al. MAPK activation and apoptotic alterations in hearts subjected to calcium paradox are attenuated by taurine. Cardiovasc Res. 2006;72:173–74. doi: 10.1016/j.cardiores.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 34.Zhu W, Zou Y, Aikawa R, et al. MAPK superfamily plays an important role in daunomycin-induced apoptosis of cardiac myocytes. Circulation. 1999;100:2100–7. doi: 10.1161/01.cir.100.20.2100. [DOI] [PubMed] [Google Scholar]
- 35.Lou H, Danelisen I, Singal PK. Involvement of mitogen-activated protein kinases in adriamycin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol. 2005;288:H1925–30. doi: 10.1152/ajpheart.01054.2004. [DOI] [PubMed] [Google Scholar]
- 36.Cesselli D, Jakoniuk I, Barlucchi L, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–86. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 37.Takeda N. Cardiomyopathy: molecular and immunological aspects. Int J Mol Med. 2003;11:13–6. [PubMed] [Google Scholar]
- 38.Sakamoto A, Ono K, Abe M, et al. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: an animal model of disrupted dystrophin-associated glycoprotein complex. Proc Natl Acad Sci USA. 1997;94:13873–8. doi: 10.1073/pnas.94.25.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitsuhashi S, Saito N, Watano K, et al. Defect of delta-sarcoglycan gene is responsible for development of dilated cardiomyopathy of a novel hamster strain, J2N-k: calcineurin/PP2B activity in the heart of J2N-k hamster. J Biochem. 2003;134:269–76. doi: 10.1093/jb/mvg140. [DOI] [PubMed] [Google Scholar]
- 40.Takagi C, Urasawa K, Yoshida I, et al. Enhanced GRK5 expression in the hearts of cardiomyopathic hamsters, J2N-k. Biochem. Biophys. Res. Commun. 1999;262:206–10. doi: 10.1006/bbrc.1999.1179. [DOI] [PubMed] [Google Scholar]
- 41.Babick A, Cantor E, Babick J, et al. Cardiac contractile dysfunction in J2N-k cardiomyopathic hamsters is associated with impaired SR function and regulation. Am J Physiol Cell Phyiol. 2004;287:C1202–8. doi: 10.1152/ajpcell.00155.2004. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Liu X, Sentex E, et al. Increased expression of protein kinase C isoforms in heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;284:H2277–87. doi: 10.1152/ajpheart.00142.2002. [DOI] [PubMed] [Google Scholar]
- 43.Makino N, Masutomo K, Nishimura M. Cardiac collagen expression in the development of two types of cardiomyopathic hamsters (Bio 14.6 and Bio 53.58) In: Nagano M, Takeda N, Dhalla NS, et al., editors. The cardiomyopathic heart. New York: Raven Press, Ltd; 1994. pp. 57–64. [Google Scholar]
- 44.Araki T, Shimizu M, Sugihara N. Effect of angiotensin-converting enzyme inhibitor on myocardial collagen metabolism in cardiomyopathic hamsters. In: Nagano M, Takeda N, Dhalla NS, et al., editors. The cardiomyopathic heart. New York: Raven Press, Ltd; 1994. pp. 137–43. [Google Scholar]
- 45.Panagia V, Lee SL, Singh A, et al. Impairment of mitochondrial and sarcoplasmic reticular functions during the development of heart failure in cardiomyopathic (UMX7.1) hamsters. Can J Cardiol. 1986;2:236–47. [PubMed] [Google Scholar]
- 46.Sethi R, Bector N, Takeda N, et al. Alterations in G−proteins in congestive heart failure in cardiomyopathic (UM-X7.1) hamsters. Mol Cell Biochem. 1994;140:163–70. doi: 10.1007/BF00926754. [DOI] [PubMed] [Google Scholar]
- 47.Kato M, Takeda N, Yang J, et al. The effects of angiotensin converting enzyme inhibitors and the role of the renin-angiotensin-aldosterone system in J-2-N cardiomyopathic hamsters. Jpn Circ J. 1992;56:46–51. doi: 10.1253/jcj.56.46. [DOI] [PubMed] [Google Scholar]
- 48.Satoh M, Nakamura M, Akatsu T, et al. C-reactive protein co-expresses with tumor necrosis factor-alpha in the myocardium in human dilated cardiomyopathy. Eur J Heart Fail. 2005;7:748–54. doi: 10.1016/j.ejheart.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 49.Ohtsuka T, Hamada M, Hiasa G, et al. Effect of beta-blockers on circulating levels of inflammatory and anti-inflammatory cytokines in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2001;37:412–7. doi: 10.1016/s0735-1097(00)01121-9. [DOI] [PubMed] [Google Scholar]
- 50.Satoh M, Nakamura M, Tamura G, et al. Inducible nitric oxide synthase and tumor necrosis factor-alpha in myocardium in human dilated cardiomyopathy. J Am Coll Cardiol. 1997;29:716–24. doi: 10.1016/s0735-1097(96)00567-0. [DOI] [PubMed] [Google Scholar]
- 51.Habib FM, Springall DR, Davies GJ, et al. Tumour necrosis factor and inducible nitric oxide synthase in dilated cardiomyopathy. Lancet. 1996;347:1151–5. doi: 10.1016/s0140-6736(96)90610-8. [DOI] [PubMed] [Google Scholar]
- 52.Shames BD, Barton HH, Reznikov LL, et al. Ischemia alone is sufficient to induce TNF-alpha mRNA and peptide in the myocardium. Shock. 2002;17:114–9. doi: 10.1097/00024382-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 53.Gurevitch J, Frolkis I, Yuhas Y, et al. Tumor necrosis factor-alpha is released from the isolated heart undergoing ischemia and reperfusion. J Am Coll Cardiol. 1996;28:247–52. doi: 10.1016/0735-1097(96)00105-2. [DOI] [PubMed] [Google Scholar]
- 54.Malhotra A. Contractile and regulatory proteins in hamster cardiomyopathy. In: Nagano M, Takeda N, Dhalla NS, editors. The cardiomyopathic heart. New York: Raven Press, Ltd; 1994. pp. 31–9. [Google Scholar]
- 55.Kadokami T, Frye C, Lemster B, et al. Anti-tumor necrosis factor-α antibody limits heart failure in a transgenic model. Circulation. 2001;104:1094–7. doi: 10.1161/hc3501.096063. [DOI] [PubMed] [Google Scholar]