

Abstract
Tyrosine kinase fusion genes represent an important class of oncogenes associated with leukaemia and solid tumours. They are produced by translocations and other chromosomal rearrangements of a subset of tyrosine kinase genes, including ABL, PDGFRA, PDGFRB, FGFR1, SYK, RET, JAK2 and ALK. Based on recent findings, this review discusses the common mechanisms of activation of these fusion genes. Enforced oligomerization and inactivation of inhibitory domains are the two key processes that switch on the kinase domain. Activated tyrosine kinase fusions then signal via an array of transduction cascades, which are largely shared. In addition, the fusion partner provides a scaffold for the recruitment of proteins that contribute to signalling, protein stability, cellular localization and oligomerization. The expression level of the fusion protein is another critical parameter. Its transcription is controlled by the partner gene promoter, while translation may be regulated by miRNA. Several mechanisms also prevent the degradation of the oncoprotein by proteasomes and lysosomes, leading to its accumulation in cells. The selective inhibition of the tyrosine kinase activity by adenosine-5′-triphosphate competitors, such as imatinib, is a major therapeutic success. Imatinib induces remission in leukaemia patients that are positive for BCR-ABL or PDGFR fusions. Recently, crizotinib produced promising results in a subtype of lung cancers with ALK fusion. However, resistance was reported in both cases, partially due to mutations. To tackle this problem, additional levels of therapeutic interventions are suggested by the complex mechanisms of fusion tyrosine kinase activation. New approaches include allosteric inhibition and interfering with oligomerization or chaperones.
Keywords: receptor tyrosine kinase, BCR-ABL, kinase inhibitors, chromosomal translocations
Introduction
The study of the t(9;22) translocation associated with chronic myelogenous leukaemia (CML) led to the discovery of the first protein tyrosine kinase (TK) fusion gene, BCR-ABL, more than 25 years ago (reviewed in Ref. [1]). Later on, several other TK fusion genes were identified in haematological malignancies. They involved both cytosolic TK, such as JAK2 or SYK, and receptor TK, including PDGFRA, PDGFRB, FGFR1 and ALK [2, 3]. Tyrosine kinase fusion genes were found in solid tumours at a lower rate, which may be underestimated due to the lack of systematic cytogenetic analysis. Nevertheless, papillary thyroid carcinoma frequently harbours activated RET fusion genes and the EML4-ALK rearrangement is found in about 5% of non–small cell lung carcinomas [4, 5].
Among the 90 TK genes that are present in the human genome, at least 14 were found rearranged with various partner genes in cancer (Fig. 1, Table S1 and databases, Refs. [6, 7]). Some fusions are tightly associated with a particular neoplasm, while others were only reported in one patient. A few more TK genes have the potential of forming activated fusion oncogenes, even though they have not been found in cancer patients yet (Table S1 and Refs. [8, 9]). Remarkably, no fusion has been reported so far for some TK genes that frequently harbour other types of cancer mutations, such as the epidermal growth factor receptor family.
Fig 1.
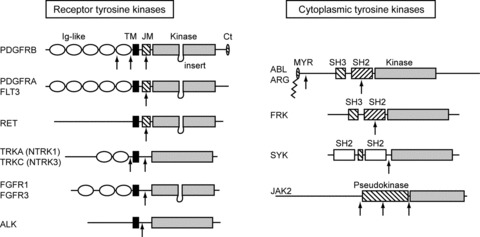
Structure of the tyrosine kinases involved in TK fusions. Domains that stabilize the inactive conformation are hatched. TM: transmembrane domain (black box); JM: juxtamembrane domain; MYR: myristoylation motif. Breakpoints are indicated by arrows.
Tyrosine kinase domains share a conserved bilobal structure. The N-terminal lobe binds adenosine-5’-triphosphate (ATP), while the active site is at the hinge between the two lobes. In the inactive conformation, the activation loop of the C-terminal lobe prevents substrate binding. Upon phosphorylation, this loop undergoes an important conformational change that allows substrate binding and phosphate transfer from ATP. The phosphorylation of the activation loop of ABL is normally mediated by another kinase. In the case of receptor TKs and JAKs, trans-autophosphorylation is triggered by dimerization and conformational changes induced by ligand binding to the extracellular part of the receptor. In addition, inhibitory domains that keep the TK domain silent in the absence of stimulus have been identified in most TK proteins. The oncogenic activation of TK fusions invariably involves enforced dimerization and/or inactivation of inhibitory domains, as discussed later. The partner gene fused to the TK gene plays an important role by controlling the oligomerization and the expression level of the fusion oncoprotein. Additional roles of the partners will also be discussed in this review.
Tyrosine kinase inhibitors
Achieving specific inhibition of TK enzymes with ATP competitors was initially though to be unlikely. The success of imatinib, designed as a selective inhibitor of ABL, thus came as a surprise. Leukaemic cells survival turned out to be highly dependent on BCR-ABL signalling, a process sometimes referred to as oncogene addiction [10]. Imatinib monotherapy is now the first line treatment of CML and induces long-term remission in the majority of patients, although resistance does occur in part due to BCR-ABL mutations [1]. Imatinib is also effective in BCR-ABL-positive acute lymphoblastic leukaemia but the rate of relapse is much higher than in CML [11]. Novel inhibitors such as nilotinib and dasatinib are active against imatinib-resistant BCR-ABL mutants, except the so-called ‘gatekeeper’ residue mutation T315I. Like imatinib, nilotinib binds only to the inactive ABL conformation, which is destabilized by T315I. Dasatinib binds to the active conformation but the T315I substitution introduces a steric clash in the ATP pocket. Molecules such as DCC-2036, which binds to the ATP pocket and to residues that control the switch between the inactive and the active conformation may overcome this resistance [12]. Other types of inhibitors will be discussed later.
Imatinib is an even more potent inhibitor of platelet-derived growth factor (PDGF) receptors. Accordingly, myeloproliferative neoplasms carrying a PDGF receptor fusion are extremely sensitive to low-dose imatinib [13]. These major successes have prompted the development of inhibitors of other TK, such as FGFR1, JAK2 and ALK (Table 1). Some of these molecules are now tested in clinical trials (for a review, see Refs. [14, 15]). Remarkably, the ALK inhibitor crizotinib produced promising results in a subset of non–small cell lung cancer patients that are positive for the EML4-ALK rearrangement [5]. Again, resistant ALK mutations were identified in treated patients [15], calling for alternative strategies that could be used as a complement of ATP competition.
Table 1.
TK fusion inhibitors
| Target process | Molecule | TK fusion | Current status | Resistance | Reference |
|---|---|---|---|---|---|
| ATP competition | Imatinib, nilotinib, dasatinib | X-ABL, X-PDGFRA and X-PDGFRB | Approved | Mutations | [11, 13] |
| DCC-2036 | BCR-ABL | Mouse model | † | [12] | |
| Crisotinib (PF02341066) | EML4-ALK | Clinical trial | Mutations | [5] | |
| CH5424802 | EML4-ALK | Mouse model | † | [109] | |
| Dovitinib (TKI258) | X-FGFR1 | In vitro* | [110, 111] | ||
| Tasocitinib (CP690550) and Ruxolitinib (INCB018424) | X-JAK2 | In vitro* | Mutations | [69] | |
| Oligomerization | Helix-2 | BCR-ABL | In vitro | † | [40] |
| Conformation | GNF-2, GNF-5 (allosteric inhibitors) | BCR-ABL | Mouse model | Mutations‡ | [62] |
| Expression and chaperones | Tanespimycin (17-AAG) | BCR-ABL | In vitro | [25, 112, 113] | |
| Alvespimycin (17-DMAG) | BCR-ABL | In vitro* | [104] | ||
| EC141 | BCR-ABL | In vitro | [114] | ||
| Novobiocin | BCR-ABL | In vitro | [115] | ||
| Ascorbate + menadione | BCR-ABL | In vitro | [105] | ||
| siRNA | BCR-ABL | In vitro | † | [25, 26] |
Clinical trials are ongoing for other indications.
Active against mutants that are resistant to conventional ATP competitors.
The combination of GNF-2 with helix-2 or nilotinib is active against resistant mutations.
The success of kinase inhibitors is not restricted to TK fusions. Some molecules are also active against receptor TKs activated by point mutations, such as c-KIT in gastrointestinal stromal tumours, and mutated serine/threonine kinases. For instance, the B-RAF inhibitor PLX4032 (vemurafenib) was shown to improve survival of metastatic melanoma patients with a B-RAF V600E mutation [16]. This molecule binds to the ATP pocket as well as to a distinct allosteric site, leading to conformational changes within the kinase domain [17].
Fusion gene structure and expression
Tyrosine kinase fusion genes were first discovered at chromosomal translocation breakpoints revealed by cytogenetic analysis, such as t(9;22) for BCR-ABL. It is now clear that other types of chromosomal rearrangements can generate fusions. One of the best examples is FIP1L1-PDGFRA, which results from a cryptic deletion on chromosome 4 [18]. DNA damage, in particular double strand breaks, and repair via the non-homologous end-joining pathway are likely to play a role in chromosomal rearrangements but the detailed mechanism is unknown. Several reports have suggested that fusions may preferentially occur at chromosome fragile sites, which are prone to DNA breakage [19, 20]. These large regions scattered in the human genome include PDGFRA and RET, as well as several partner genes, such as FIP1L1[20]. No particular sequence has been identified at breakpoints, which seem to occur preferentially in larger introns, suggesting random breakage within the fragile sites [21].
In most TK proteins, the TK domain is located in the C-terminus while inhibitory domains are rather N-terminal (Fig. 1). Accordingly, the partner gene always replaces the N-terminal part in the hybrid oncoprotein, retaining the C-terminal TK domain. The entire extracellular ligand-binding domain of RTK is thus lost in most fusion products. An important consequence of this organization is that the expression level of the fusion product is driven by the gene promoter of the partner gene (Fig. 2). This means that the wild-type TK gene may not be normally expressed in the cell type that is transformed by the fusion product. For instance, platelet-derived growth factor receptors (PDGFRA and PDGFRB) are poorly expressed in normal haematopoietic cells [2]. The fusion of these genes in myeloid malignancies not only results in constitutive TK activity but also in aberrant overexpression, which can be monitored as a clue of gene fusion [22].
Fig 2.
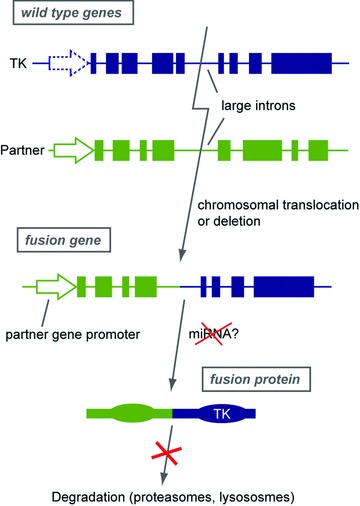
Structure and expression of TK hybrid genes and proteins. In wild-type and fusion genes, arrows depict the gene promoter and filled boxes represent exons. The fusion breakpoint is usually located in a large intron, with a few exceptions such as PDGFRA. The partner is in green and the tyrosine kinase in blue. See text for details.
The importance of the expression pattern of TK fusions is illustrated by experiments performed with inducible BCR-ABL transgenic mice. Indeed, CML arises in these mice only if the oncogene is expressed specifically in haematopoietic stem cells, while a BCR-ABL transgene under the control of an inappropriate promoter leads to other types of haematopoietic neoplasms [1, 23].
In addition, BCR-ABL expression was shown to be controlled by small regulatory RNA molecules. MiR-203 expression is lost in several haematopoietic tumours including CML due to its localization in a fragile chromosomal region and to DNA hypermethylation. Because miR-203 reduces ABL and BCR-ABL protein levels and inhibits ABL-dependent tumour cell proliferation, it may act as a tumour suppressor and control the disease development [24]. Experimental treatments based on artificial siRNA are being developed to decreased BCR-ABL expression in human leukaemic cells [25, 26].
Several other mechanisms were shown to enhance the protein expression level of fusion TKs, which can interact with chaperones and escape the normal degradation pathways, as described in a dedicated section later.
When the fusion is caused by a reciprocal translocation, a reciprocal product may encode a hybrid protein, which is devoid of TK activity and is expressed only if the TK promoter is active in the target cell. Accordingly, such reciprocal transcripts have not been detected in the case of PDGF receptor translocations. Nevertheless, the t(9;22) reciprocal translocation product ABL-BCR can be detected and may contribute to the development of acute lymphoblastic leukaemia, according to a recent study [27].
Loss of wild-type alleles
In addition to the oncogenic effect of the activated TK fusion, the loss of one normal allele of the partner gene was suggested to contribute to the disease in a number of cases, although its importance is still a matter of debate. For instance, SSBP2, KANK1 and PRKAR1A (fused to JAK2, PDGFRB and RET, respectively) are potential tumour suppressor genes [4, 28]. In several instances, the expression of the second allele is also abrogated as a consequence of an additional genetic alteration or epigenetic modification, leading to the complete loss of expression of the normal partner protein in cancer cells. This was suggested for KANK1 [28] and ETV6 (initially named TEL). ETV6 is a transcriptional repressor that is essential for haematopoiesis. It is a frequent partner gene of TK, including ABL, PDGFRB, JAK2, FLT3 and FGFR1 (Table S1). In addition, ETV6 is often deleted or inactivated in cells harbouring ETV6 translocations in acute myeloid leukaemia [29]. A point mutation that abolishes DNA binding of ETV6 was reported in the non-rearranged allele of T lineage acute lymphoblastic leukaemia cells that express the ETV6–ABL2 fusion [30]. In another report, Vu et al. showed that the expression of the endogenous ETV6 protein was completely lost in a patient who harboured an ETV6–FLT3 hybrid [31]. The absence of wild-type ETV6 protein may be a secondary genetic event implicated in leukaemogenesis.
In addition, the endogenous normal partner protein can act as an inhibitor of the fusion TK oligomerization, as mentioned later. In this respect, the loss of the wild-type allele could thus provide an additional selective advantage even if it is not a tumour suppressor gene.
Oligomerization triggers TK activation
Many TK hybrids are fused to partner proteins that harbour potential multimerization domains. By bringing hybrid proteins close to each over, these dimerization motifs can induce the constitutive activation of the TK domain, mimicking receptor TK activation. The best-studied example of oligomerization domain in TK fusions is the pointed (PNT) domain of ETV6. Different reports showed that this domain, also named helix-loop-helix or SAM, is required for cell transformation driven by the fusion of ETV6 with ABL, PDGFRB, JAK2 and TRKC [3, 32–34]. Such a pointed domain is not present in other TK fusion partners.
The most frequent oligomerization domains in TK fusion are coiled coils, which are found in more than 60% of TK fusion products, compared to 9% in the human proteome, as defined in the Ensembl database (Table S1). The importance of coiled coils has been studied in a limited number of cases. For instance, deletion of the EML1-coiled coil domain abrogates the EML1-ABL transforming activity [35]. The coiled coil of BCR is also essential for BCR-ABL-induced oligomerization and cell transformation [36]. It can be replaced by another dimerization domain, such as the leucine zipper of the yeast transcription factor GCN4 [37]. It was shown that the BCR-ABL coiled coil disrupts the autoinhibited conformation through oligomerization and intermolecular autophosphorylation [38]. However, He et al. reported that a BCR-ABL mutant devoid of coiled coil domain still exhibits elevated phosphotyrosine activity and stimulated cell growth in vitro[39]. Nevertheless, this mutant failed to induce a myeloproliferative disease in mice. These observations led to the development of a new type of inhibitors. Indeed, a 40 amino-acid peptide derived from the helix 2 of the BCR CC domain was shown to inhibit BCR-ABL oligomerization and decrease cell transformation [40]. Interestingly, this peptide is active against the BCR-ABL T315I mutant, which is resistant to all ATP competitor drugs. Whether this strategy can be translated into a useful therapy is not yet clear.
In a number of cases, unique oligomerization domains have been identified in fusion proteins. In a study of the HIP–PDGFRB fusion, homodimerization was not driven by its coiled coil/leucine-zipper domain but by a sequence that shares homology with talin [41]. In line with this observation, we recently demonstrated that KANK1 coiled coils are dispensable for KANK1-PDGFRB oligomerization [42].
Different oligomerization levels have been reported for TK fusion complexes. ZNF198-FGFR1 is a dimer [43], KANK1-PDGFRB is a trimer [42], while BCR-ABL forms a tetramer [44] and ETV6 fusions may adopt a helicoidal polymeric structure [33]. Tognon et al. suggested that only polymeric—but not dimeric—ETV6-NTRK3 can transform cells [33].
Beside direct oligomerization of the fusion protein, inclusion in a larger protein complex is thought to produce the same effect (Fig. 3). This is illustrated by the NUP214–ABL fusion in T cell acute lymphoblastic leukaemia. The NUP214 protein localizes to the cytoplasmic side of the nuclear pore complex and participates to the nuclear export of molecules. The two central NUP214 coiled coil motifs do not mediate the protein oligomerization. Instead, they bind to NUP88, thereby targeting the NUP214–ABL fusion to the nuclear pore complex, a process that is required for cell transformation [45].
Fig 3.
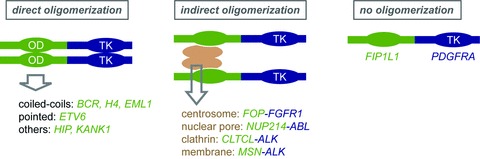
Role of oligomerizatioon in TK fusion activation. OD: oligomerization domain; TK: tyrosine kinase domain. The indicated partner genes harbour an oligomerization domain that was confirmed experimentally.
Several partner genes encode centrosomal proteins, namely FOP, CEP110, NIN, PDE4DIP, PCM1 and TRIP11. FOP–FGFR1 interacts with another centrosomal protein CAP350 through FOP, thereby targeting the hybrid to the centrosome, which seems to be essential for haematopoietic cell transformation [46, 47].
The ITK–SYK hybrid transforming properties requires the ITK PH domain, which binds to phosphatidylinositol-triphophate [48]. Constitutive association of ITK-SYK with lipid rafts in T cells is enough to trigger its phosphorylation and mimic signalling by the T cell receptor [49]. Again, concentration in a particular structure seems to be the key to activation. Similar mechanisms were suggested to govern the activation of the ALK fusion products with moesin (MSN) and the clathrin subunit CLTCL [50, 51].
Receptor studies have shown that dimerization of the intracellular domain is not enough to switch on signalling. The two TK domains must be precisely oriented, presumably to favour trans-autophosphorylation [52, 53]. It is likely that a similar orientation constraint applies to fusion TK proteins and is determined by sequences located between the oligomerization and TK domains. In ETV6–PDGFRB, we have shown that the transmembrane domain of PDGFRB, which is retained most PDGFRB fusion products, is required to adopt a conformation that is optimal for signalling [54]. This is a unique example of a hydrophobic helix that acts as a transmembrane domain in the wild-type protein and plays a completely different role in the fusion product.
In conclusion, TK fusion oligomerization is induced directly through oligomerization motifs that are present in the fusion partner protein, or indirectly through the recruitment of additional proteins that integrate the hybrid oncogene in a larger multimeric complex.
Inhibitory domain deletion
As mentioned earlier, most TK proteins comprise inhibitory domains that dampen the kinase activity in the absence of stimuli by stabilizing the inactive conformation (Fig. 1 and Table S1). Such inhibitory domains are frequently deleted in fusion proteins.
In receptor TK, the juxtamembrane domain, namely the domain located between the transmembrane helix and the kinase fold, often plays an inhibitory role. Its structure was best characterized in FLT3, in which it was shown to contact several key amino acids of the kinase domain [55]. A similar mechanism has been described in PDGFRA, PDGFRB and RET. Cancer point mutations in this domain are enough to constitutively activate these receptors [2, 4]. In FIP1L1–PDGFRA, this appears as the principal mechanism of activation. In this fusion, the breakpoint in PDGFRA is located within the juxtamembrane region, which was suggested to adopt a WW-like domain structure. The truncation of this domain is sufficient to constitutively activate the PDGFRA kinase [56]. A similar mechanism activates the PRKG2–PDGFRB fusion [57]. However, most breakpoints within PDGFRB fall before the transmembrane domain keeping the juxtamembrane domain intact [54]. Stover et al. showed that enforced dimerization overcomes the inhibition by an intact juxtamembrane domain. Nevertheless, the combination of dimerization and juxtamembrane domain deletion may produce a more potent PDGFR oncogene [56]. In ETV6–FLT3, the first inhibitory loop of the juxtamembrane domain is disrupted, as in PDGFRA fusions [31]. Interestingly, the remaining part of the juxtamembrane domain is required for signalling and cell transformation by ETV6–FLT3 [58].
In PDGF receptors, the C-terminal tail was also shown to play a negative role [59, 60]. We observed that the deletion of this domain in ETV6–PDGFRB enhances its transformation potential (unpublished data).
The N-terminal part of ABL and ABL2 (also called ARG) contain a myristoylation site, one SH2 and one SH3 domain. In the inactive conformation, these domains are assembled in an autoinhibited structure, in which they function as a clamp that switches off the kinase activity [1]. A partial deletion of these domains is enough to activate ABL. In particular, the myristoyl group binds to a hydrophobic pocket within the kinase domain [61]. The N-terminal myristoylation site is lost in all ABL and ARG fusions, contributing to constitutive activation. This mechanism does not seem to be conserved in other cytosolic TK. Interestingly, a ligand that mimics a myristoyl group and binds to the hydrophobic pocket acts as an allosteric inhibitor of BCR-ABL by restoring its inactive conformation [62]. Such molecules constitute a novel class of TK inhibitors (Table 1). Interestingly, a synergy between ATP competitors, oligomerization inhibitors and allosteric inhibitors has been observed and may be useful to overcome resistance [40, 62].
In addition, the SH3 and SH2 domains are deleted in a minority of ABL fusion products, including SFPQ-ABL and RCSD1-ABL, but not in BCR-ABL [63]. Remarkably, the SH2 domain of BCR-ABL is required to induce CML but not a lymphoid disease in mice [64, 65]. Recently RIN1, a RAS effector protein, was found to associate with the ABL SH2 and SH3 domains. These multiple interactions maintain the kinase domain in its active form and enhance the hybrid activity [66]. In line with this observation, RIN1 was found overexpressed in some leukaemias [67]. The SH3 and SH2 domains also participate in the inactive conformation of other cytosolic TK. Deletion of inhibitory domains was also found in FRK and SYK fusions (Table S1).
JAK kinases share a pseudokinase domain, also called JH2, which presents a significant homology with a TK domain but lacks a few key amino acids and is devoid of activity [68]. This JH2 domain interacts with and negatively regulates the JAK TK domain. In most cases, the fusion of ETV6 with JAK2 results in the truncation of the JH2 pseudokinase domain [3]. Although this truncation is not enough to activate JAK2 [69], it remains to be tested whether it increases the activity of the oligomerized hybrid.
In conclusion, the deletion of inhibitory domains is frequent among TK fusion products and contributes to the constitutive TK activity. This is however not an absolute requirement as oligomerization by itself can destabilize the autoinhibited TK conformation. In a number of cases, the combination of oligomerization and deletion of inhibitory motifs was shown to synergistically enhance the kinase activity.
Signalling pathways
Cell transformation resulting from TK hybrids expression is the consequence of the activation of signalling pathways that control cell proliferation and apoptosis inhibition. Most TK fusions, like their wild-type counterparts, use a common set of signalling pathways: phosphatidylinositol-3-kinase (PI3K) and its downstream effector PKB, the MAP kinase pathways and the transcription factors signal transducer and activator of transcription (STAT) and NF-κB. Activating mutations in RAS, RAF or PI3K, which are commonly found in cancer, and TK fusions are mutually exclusive, indicating that they constitutively activate signalling pathways in a similar manner and that there is no further advantage for tumour cells in combining such mutations. The importance of these cascades in cancer has been extensively reviewed and it is impossible to mention here all the reports that studied signalling by TK fusions (reviewed in Refs. [1, 70–72]).
Tyrosine kinase fusions activate aberrant signalling pathways compared to the normal TK form, which may contribute to oncogene addiction. For instance, Voss et al. provided evidence that ABL oncogenic hybrids (BCR-ABL and ETV6-ABL) harbour catalytic specificities that activate distinct signalling pathways compare to wild-type ABL [73]. Similarly, NUP214-ABL and BCR-ABL do not phosphorylate exactly the same set of peptide substrates [74]. Differences were also reported between fusion and wild-type PDGF receptors, in particular regarding signal transducer and activator of transcription (STAT) activation [75].
STAT transcription factors are activated by most TK fusions [1, 2, 76–78]. It was demonstrated that STAT5 is particularly important for leukaemogenesis induced by BCR-ABL and ETV6-PDGFRB in mice [79, 80], and for FIP1L1-PDGFRA in human primary haematopoietic cells [81]. ZNF198-FGFR1 also activates STAT5 leading to cell cycle progression and apoptosis inhibition [82].
MAP kinases and PI3K are other mediators shared by TK fusions. Most studies addressed the role of these pathways using pharmalogical inhibitors whose specificity has been largely debated. Nevertheless, additional experiments have confirmed their importance. In BCR-ABL, tyrosine 177 within the BCR part is phosphorylated and binds to GRB2, which in turn recruits GAB2 (GRB2-associated binding protein 2) and Son of Sevenless homolog (SOS), a guanine-nucleotide exchanger of RAS. The phosphorylation-dependent formation of this complex leads to the activation of RAS and PI3K [83, 84]. The Y177F mutation in BCR-ABL abolishes GRB2 binding without affecting the kinase activity of ABL [83, 84]. In a mouse bone marrow transplantation model of CML, the Y177F mutant showed a reduced ability to induce a myeloproliferative disorder. Accordingly, the targeted deletion of GAB2 or PI3Kγ, the major haematopoietic PI3K isoform, reduces the leukaemic potential of BCR-ABL-expressing cells [85, 86].
Even though targeting the TK fusion itself seems more efficient than targeting downstream signalling, combining both approaches may be a way to overcome resistance. For instance, MAP kinase pathway inhibitors sensitize imatinib-resistant CML cells to the BCR-ABL inhibitor dasatinib [87]. In this respect, a number of inhibitors of the MAP kinase and PI3K pathways have now entered the clinic and may be tested in cancers associated with rearrangement of TK genes.
Recruitment of additional molecules by the fusion partner
Early studies suggested that the role of the partner is limited to oligomerization because it can be replaced by an artificial dimerization domain, at least when proliferation of a model cell line, such as Ba/F3, is the readout. However, accumulating evidence highlights additional roles for the fusion partner by recruiting proteins involved in signalling or protein stabilization, for instance (Fig. 4). This is illustrated by the FIP1L1 part of FIP1L1–PDGFRA, which does not promote oligomerization but is required for optimal proliferation of human CD34+ haematopoietic progenitors [81]. By contrast, this part is dispensable to sustain the proliferation of the mouse Ba/F3 cell line [56].
Fig 4.
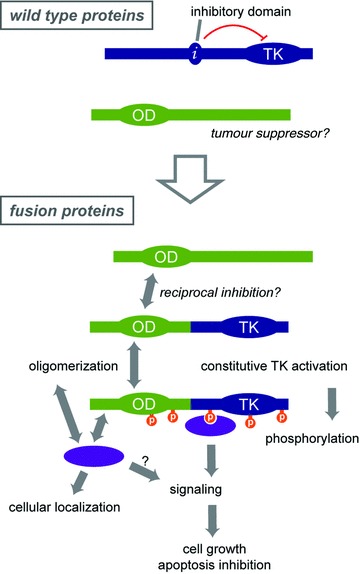
Overview of the mechanisms of cell transformation by TK fusions. See text for details.
As mentioned earlier, tyrosine residues located in the partner part may be phosphorylated and act as docking site for SH2-containing signalling mediators. The partner parts of BCR-ABL (including Y177), ETV6-PDGFRB and FIP1L1-PDGFRA have been shown to be phosphorylated, although the role of this event in PDGFR fusion signalling remains unclear [88, 89].
Fusion partners are also able to recruit the endogenous form of the partner protein through their oligomerization domain. The fusion thereby acts as a dominant-negative form of the wild-type endogenous partner, which may contribute to disease development. By using the yeast two-hybrid approach, HHR6, an ubiquitin-conjugating DNA repair enzyme, was found to associate with ZNF198 and ZNF198–FGFR1 [90]. RAD18, another DNA damage repair protein, was also found in the complex. The fact that cells expressing ZNF198–FRGR1 show an increased sensitivity to UVB irradiations indicated that the hybrid acts as a dominant-negative form of ZNF198 by affecting DNA repair. Kunapuli et al. suggested that heterodimerization of ZNF198–FGFR1 hybrid with the endogenous form may impair ZNF198 sumoylation required for its function in DNA damage repair [91].
Another example of dominant negative effect of the fusion is ETV6–FRK, which was shown to inhibit ETV6-mediated transcriptional repression [92]. This was also demonstrated for the fusion of the non-muscular tropomyosine TPM3 with ALK. TPM3–ALK expressing cells displayed a highest migratory and invasive capacities compared to cells expressing other ALK fusions [93]. It was demonstrated that TPM3–ALK has the ability to interact with endogenous tropomyosine, possibly impairing tropomyosine cellular function and actin cytoskeleton organization [94].
Although the hybrid may act as a dominant negative on the endogenous protein, the endogenous partner can also prevent the fusion dimerization and thereby limits its activity, as shown for ETV6–NTRK3 and ZNF198–FGFR1 [33, 43].
Additional proteins, such as chaperones, may also be recruited to the fusion partner independently of phosphorylation and regulate various processes such as hybrid protein stability and signalling. ZNF198 and ZNF198–FGFR1 proteins are found in complex with HSPA1A, a protein that belongs to the heat shock protein HSP70 genes family. HSPA1A expression was increased in presence of wild type or hybrid ZNF198. HSPA1A stabilizes ZNF198 and ZNF198–FGFR1 and contributes to the activation of STAT3 and to cell transformation [95]. Chaperones are also interacting with BCR-ABL and NPM-ALK as discussed later, but it is not clear whether they interact with the partner part in these cases.
Stabilization and degradation
As already stated earlier, the level of TK expression is critical for cell transformation [96]. Decreasing degradation of the oncoprotein is one way to achieve a higher expression. Receptor TK are quickly degraded upon activation, a process that limits the duration of growth stimulation. Receptor TK degradation occurs mainly in lysosomes after endocytosis, although proteasomes may also play a role. Protein degradation is initiated by ubiquitination by ubiquitin ligase complexes. In particular, the E3 subunit c-CBL recruits many TK, such as BCR-ABL, to initiate ubiquitination [97]. The importance of CBL is illustrated by the discovery of mutations that inactivate its ligase activity in cancer cells. In the TRP–MET fusion, the CBL-binding site of MET is lost, which decreases TRP–MET degradation and contributes its oncogenic activity [98]. Similarly, we demonstrated that chimeric receptors (ETV6–PDGFRB, FIP1L1–PDGFRA and ZNF198–FGFR1) escape ubiquitination and degradation, again leading to the accumulation of the oncoprotein [75]. The cytosolic localization of most TK fusions may by itself prevent the entry into the degradation route followed by activated membrane receptors.
Chaperone proteins, such as HSP90, enable the correct folding and prevent proteolytic degradation of various oncoproteins. Recently, Tsukahara et al. presented a model in which newly synthesized BCR-ABL proteins are stabilized by HSC70 and then passed on to HSP90 for maturation. Interference with this pathway triggers newly synthesized BCR-ABL degradation in a process regulated by Bag1 and the E3 ubiquitin ligase CHIP, while mature phosphorylated BCR-ABL proteins are targeted to degradation by c-CBL [99]. HSP90 also modulates maturation and activity of NPM–ALK fusion oncoprotein [100]. When association between HSP90 and NPM-ALK is impaired the fusion protein is rapidly degraded through HSP70-assisted ubiquitin-dependent proteasomal degradation [101]. We previously mentioned that ZNF198–FGFR1 also bind to chaperones [95].
Because of its large client repertoire, including TKs, anti-cancer agents were developed against HSP90, including geldanamycin derivatives, and are now being tested in clinical trials [102]. These compounds effectively kill BCR-ABL expressing cells, in synergy with other inhibitors (Table 1 and Refs. [25, 103, 104]). Alternatively, ascorbate and menadione produce a tumour-specific oxidative stress associated with HSP90 cleavage and BCR-ABL degradation [105].
SOCS proteins are potent inhibitors of JAK-STAT signalling, at least in part by targeting JAKs for degradation. ETV6–JAK2 remains sensitive to the effect of SOCS1, and even up-regulates its expression [106]. The forced expression of SOCS1-induced apoptosis of Ba/F3 cells expressing ETV6-JAK2 via the ubiquitin-dependant hybrid proteolysis [107]. By contrast, other TK fusions, such as ZNF198–FGFR1, ETV6–ABL and ETV6–PDGFRB, are insensitive to SOCS1 [43, 106], most likely because these fusions do not require JAKs for signalling, do not bind directly to SOCS1 and induce the phosphorylation of SOCS on tyrosine residues [108].
Conclusion
While new cancer subtypes with TK fusions are constantly discovered, understanding how these oncogenes work is critical to improve treatments. The independent analysis of the mechanisms of cell transformation by BCR-ABL, ETV6-PDGFRB, NPM-ALK, ZNF198–FGFR1 and a few other recurrent hybrid oncogenes has led to a model that may apply to most if not all TK fusions (Fig. 4). The understanding of the mechanisms that govern TK fusion activation is now being translated into innovative therapeutic approaches to improve the treatment of TK fusion-associated cancers.
Acknowledgments
This work was supported by grants from the Salus Sanguinis foundation and Action de Recherches Concertées (Communauté Française de Belgique). We thank Dr Hélène Schoemans and Laura Noel for critical reading of the manuscript and the members of the Experimental Medicine Unit for constant support. We apologize to the authors whose excellent work could not be cited due to space limitations.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Tyrosine kinase gene fusions in cancer
References
- 1.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–83. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 2.Toffalini F, Demoulin JB. New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood. 2010;116:2429–37. doi: 10.1182/blood-2010-04-279752. [DOI] [PubMed] [Google Scholar]
- 3.Lacronique V, Boureux A, Valle VD, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–12. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 4.Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155:645–53. doi: 10.1530/eje.1.02289. [DOI] [PubMed] [Google Scholar]
- 5.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitelman F, Johansson B, Mertens F. Mitelman database of chromosome aberrations and gene fusions in cancer http://cgapncinihgov/Chromosomes/Mitelman 2011.
- 7.Novo FJ, de Mendibil IO, Vizmanos JL. TICdb: a collection of gene-mapped translocation breakpoints in cancer. BMC Genomics. 2007;8:33. doi: 10.1186/1471-2164-8-33. doi: 10.1186/1471-2164-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lacronique V, Boureux A, Monni R, et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 2000;95:2076–83. [PubMed] [Google Scholar]
- 9.Lierman E, Van Miegroet H, Beullens E, et al. Identification of protein tyrosine kinases with oncogenic potential using a retroviral insertion mutagenesis screen. Haematologica. 2009;94:1440–4. doi: 10.3324/haematol.2009.007328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. ; discussion 80. [DOI] [PubMed] [Google Scholar]
- 11.Gruber F, Mustjoki S, Porkka K. Impact of tyrosine kinase inhibitors on patient outcomes in Philadelphia chromosome-positive acute lymphoblastic leukaemia. Br J Haematol. 2009;145:581–97. doi: 10.1111/j.1365-2141.2009.07666.x. [DOI] [PubMed] [Google Scholar]
- 12.Chan WW, Wise SC, Kaufman MD, et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell. 2011;19:556–68. doi: 10.1016/j.ccr.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.David M, Cross NC, Burgstaller S, et al. Durable responses to imatinib in patients with PDGFRB fusion gene-positive and BCR-ABL-negative chronic myeloproliferative disorders. Blood. 2007;109:61–4. doi: 10.1182/blood-2006-05-024828. [DOI] [PubMed] [Google Scholar]
- 14.Pardanani A, Tefferi A. Targeting myeloproliferative neoplasms with JAK inhibitors. Curr Opin Hematol. 2011;18:105–10. doi: 10.1097/MOH.0b013e3283439964. [DOI] [PubMed] [Google Scholar]
- 15.Gerber DE, Minna JD. ALK inhibition for non-small cell lung cancer: from discovery to therapy in record time. Cancer Cell. 2010;18:548–51. doi: 10.1016/j.ccr.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–14. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 19.Gandhi M, Dillon LW, Pramanik S, et al. DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene. 29:2272–80. doi: 10.1038/onc.2009.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burrow AA, Williams LE, Pierce LC, et al. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10:59. doi: 10.1186/1471-2164-10-59. doi: 10.1186/1471-2164-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novo FJ, Vizmanos JL. Chromosome translocations in cancer: computational evidence for the random generation of double-strand breaks. Trends Genet. 2006;22:193–6. doi: 10.1016/j.tig.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Erben P, Gosenca D, Muller MC, et al. Screening for diverse PDGFRA or PDGFRB fusion genes is facilitated by generic quantitative reverse transcriptase polymerase chain reaction analysis. Haematologica. 2010;95:738–44. doi: 10.3324/haematol.2009.016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koschmieder S, Gottgens B, Zhang P, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood. 2005;105:324–34. doi: 10.1182/blood-2003-12-4369. [DOI] [PubMed] [Google Scholar]
- 24.Bueno MJ, Perez de Castro I, Gomez de Cedron M, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Futami M, Hatano T, Soda Y, et al. RNAi-mediated silencing of p190Bcr-Abl inactivates Stat5 and cooperates with imatinib mesylate and 17-allylamino-17-demetoxygeldanamycin in selective killing of p190Bcr-Abl-expressing leukemia cells. Leukemia. 2008;22:1131–8. doi: 10.1038/leu.2008.60. [DOI] [PubMed] [Google Scholar]
- 26.Koldehoff M, Kordelas L, Beelen DW, et al. Small interfering RNA against BCR-ABL transcripts sensitize mutated T315I cells to nilotinib. Haematologica. 2010;95:388–97. doi: 10.3324/haematol.2009.016063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng X, Oancea C, Henschler R, et al. Reciprocal t(9;22) ABL/BCR fusion proteins: leukemogenic potential and effects on B cell commitment. PLoS One. 2009;4:e7661. doi: 10.1371/journal.pone.0007661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medves S, Duhoux FP, Ferrant A, et al. KANK1, a candidate tumour suppressor gene, is fused to PDGFRB in an imatinib-responsive myeloid neoplasm with severe thrombocythemia. Leukemia. 2010;24:1052–5. doi: 10.1038/leu.2010.13. [DOI] [PubMed] [Google Scholar]
- 29.Barjesteh van Waalwijk van Doorn-Khosrovani S, Spensberger D, de Knegt Y, et al. Somatic heterozygous mutations in ETV6 (TEL) and frequent absence of ETV6 protein in acute myeloid leukemia. Oncogene. 2005;24:4129–37. doi: 10.1038/sj.onc.1208588. [DOI] [PubMed] [Google Scholar]
- 30.Griesinger F, Janke A, Podleschny M, et al. Identification of an ETV6-ABL2 fusion transcript in combination with an ETV6 point mutation in a T-cell acute lymphoblastic leukaemia cell line. Br J Haematol. 2002;119:454–8. doi: 10.1046/j.1365-2141.2002.03850.x. [DOI] [PubMed] [Google Scholar]
- 31.Vu HA, Xinh PT, Masuda M, et al. FLT3 is fused to ETV6 in a myeloproliferative disorder with hypereosinophilia and a t(12;13)(p13;q12) translocation. Leukemia. 2006;20:1414–21. doi: 10.1038/sj.leu.2404266. [DOI] [PubMed] [Google Scholar]
- 32.Golub TR, Goga A, Barker GF, et al. Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol Cell Biol. 1996;16:4107–16. doi: 10.1128/mcb.16.8.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tognon CE, Mackereth CD, Somasiri AM, et al. Mutations in the SAM domain of the ETV6-NTRK3 chimeric tyrosine kinase block polymerization and transformation activity. Mol Cell Biol. 2004;24:4636–50. doi: 10.1128/MCB.24.11.4636-4650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jousset C, Carron C, Boureux A, et al. A domain of TEL conserved in a subset of ETS proteins defines a specific oligomerization interface essential to the mitogenic properties of the TEL-PDGFR beta oncoprotein. EMBO J. 1997;16:69–82. doi: 10.1093/emboj/16.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Keersmaecker K, Graux C, Odero MD, et al. Fusion of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32) Blood. 2005;105:4849–52. doi: 10.1182/blood-2004-12-4897. [DOI] [PubMed] [Google Scholar]
- 36.McWhirter JR, Galasso DL, Wang JY. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol. 1993;13:7587–95. doi: 10.1128/mcb.13.12.7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McWhirter JR, Wang JY. Effect of Bcr sequences on the cellular function of the Bcr-Abl oncoprotein. Oncogene. 1997;15:1625–34. doi: 10.1038/sj.onc.1201342. [DOI] [PubMed] [Google Scholar]
- 38.Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell. 2003;12:27–37. doi: 10.1016/S1097-2765(03)00274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He Y, Wertheim JA, Xu L, et al. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcr/abl. Blood. 2002;99:2957–68. doi: 10.1182/blood.v99.8.2957. [DOI] [PubMed] [Google Scholar]
- 40.Mian AA, Oancea C, Zhao Z, et al. Oligomerization inhibition, combined with allosteric inhibition, abrogates the transformation potential of T315I-positive BCR/ABL. Leukemia. 2009;23:2242–7. doi: 10.1038/leu.2009.194. [DOI] [PubMed] [Google Scholar]
- 41.Ross TS, Gilliland DG. Transforming properties of the Huntingtin interacting protein 1/platelet-derived growth factor beta receptor fusion protein. J Biol Chem. 1999;274:22328–36. doi: 10.1074/jbc.274.32.22328. [DOI] [PubMed] [Google Scholar]
- 42.Medves S, Noel LA, Montano-Almendras CP, et al. Multiple oligomerization domains of KANK1-PDGFRB are required for JAK2-independent hematopoietic cell proliferation and signaling via STAT5 and ERK. Haematologica. 2011;96 doi: 10.3324/haematol.2011.040147. in press: doi: 10.3324/haematol.2011.040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baumann H, Kunapuli P, Tracy E, et al. The oncogenic fusion protein-tyrosine kinase ZNF198/fibroblast growth factor receptor-1 has signaling function comparable with interleukin-6 cytokine receptors. J Biol Chem. 2003;278:16198–208. doi: 10.1074/jbc.M300018200. [DOI] [PubMed] [Google Scholar]
- 44.Zhao X, Ghaffari S, Lodish H, et al. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol. 2002;9:117–20. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
- 45.De Keersmaecker K, Rocnik JL, Bernad R, et al. Kinase activation and transformation by NUP214-ABL1 is dependent on the context of the nuclear pore. Mol Cell. 2008;31:134–42. doi: 10.1016/j.molcel.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 46.Lelievre H, Chevrier V, Tassin AM, et al. Myeloproliferative disorder FOP-FGFR1 fusion kinase recruits phosphoinositide-3 kinase and phospholipase Cgamma at the centrosome. Mol Cancer. 2008;7:30. doi: 10.1186/1476-4598-7-30. doi: 10.1186/1476-4598-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mikolajka A, Yan X, Popowicz GM, et al. Structure of the N-terminal domain of the FOP (FGFR1OP) protein and implications for its dimerization and centrosomal localization. J Mol Biol. 2006;359:863–75. doi: 10.1016/j.jmb.2006.03.070. [DOI] [PubMed] [Google Scholar]
- 48.Rigby S, Huang Y, Streubel B, et al. The lymphoma-associated fusion tyrosine kinase ITK-SYK requires pleckstrin homology domain-mediated membrane localization for activation and cellular transformation. J Biol Chem. 2009;284:26871–81. doi: 10.1074/jbc.M109.034272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pechloff K, Holch J, Ferch U, et al. The fusion kinase ITK-SYK mimics a T cell receptor signal and drives oncogenesis in conditional mouse models of peripheral T cell lymphoma. J Exp Med. 2010;207:1031–44. doi: 10.1084/jem.20092042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tort F, Pinyol M, Pulford K, et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab Invest. 2001;81:419–26. doi: 10.1038/labinvest.3780249. [DOI] [PubMed] [Google Scholar]
- 51.Touriol C, Greenland C, Lamant L, et al. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like) Blood. 2000;95:3204–7. [PubMed] [Google Scholar]
- 52.Constantinescu SN, Huang LJ, Nam H, et al. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol Cell. 2001;7:377–85. doi: 10.1016/s1097-2765(01)00185-x. [DOI] [PubMed] [Google Scholar]
- 53.Bell CA, Tynan JA, Hart KC, et al. Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol Biol Cell. 2000;11:3589–99. doi: 10.1091/mbc.11.10.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toffalini F, Hellberg C, Demoulin JB. Critical role of the platelet-derived growth factor receptor (PDGFR) beta transmembrane domain in the TEL-PDGFRbeta cytosolic oncoprotein. J Biol Chem. 2010;285:12268–78. doi: 10.1074/jbc.M109.076638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffith J, Black J, Faerman C, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–78. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 56.Stover EH, Chen J, Folens C, et al. Activation of FIP1L1-PDGFRalpha requires disruption of the juxtamembrane domain of PDGFRalpha and is FIP1L1-independent. Proc Natl Acad Sci USA. 2006;103:8078–83. doi: 10.1073/pnas.0601192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lahortiga I, Akin C, Cools J, et al. Activity of imatinib in systemic mastocytosis with chronic basophilic leukemia and a PRKG2-PDGFRB fusion. Haematologica. 2008;93:49–56. doi: 10.3324/haematol.11836. [DOI] [PubMed] [Google Scholar]
- 58.Vu HA, Xinh PT, Kano Y, et al. The juxtamembrane domain in ETV6/FLT3 is critical for PIM-1 up-regulation and cell proliferation. Biochem Biophys Res Commun. 2009;383:308–13. doi: 10.1016/j.bbrc.2009.03.157. [DOI] [PubMed] [Google Scholar]
- 59.Chiara F, Bishayee S, Heldin CH, et al. Autoinhibition of the platelet-derived growth factor beta-receptor tyrosine kinase by its C-terminal tail. J Biol Chem. 2004;279:19732–8. doi: 10.1074/jbc.M314070200. [DOI] [PubMed] [Google Scholar]
- 60.Demoulin JB, Seo JK, Ekman S, et al. Ligand-induced recruitment of Na+/H+-exchanger regulatory factor to the PDGF (platelet-derived growth factor) receptor regulates actin cytoskeleton reorganization by PDGF. Biochem J. 2003;376:505–10. doi: 10.1042/BJ20030385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hantschel O, Nagar B, Guettler S, et al. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–57. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Adrian FJ, Jahnke W, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–6. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duhoux FP, Auger N, De Wilde S, et al. The t(1;9)(p34;q34) fusing ABL1 with SFPQ, a pre-mRNA processing gene, is recurrent in acute lymphoblastic leukemias. Leuk Res. 2011;35:e114–7. doi: 10.1016/j.leukres.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 64.Roumiantsev S, de Aos IE, Varticovski L, et al. The src homology 2 domain of Bcr/Abl is required for efficient induction of chronic myeloid leukemia-like disease in mice but not for lymphoid leukemogenesis or activation of phosphatidylinositol 3-kinase. Blood. 2001;97:4–13. doi: 10.1182/blood.v97.1.4. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, Wong R, Hao SX, et al. The SH2 domain of bcr-Abl is not required to induce a murine myeloproliferative disease; however, SH2 signaling influences disease latency and phenotype. Blood. 2001;97:277–87. doi: 10.1182/blood.v97.1.277. [DOI] [PubMed] [Google Scholar]
- 66.Thai M, Ting PY, McLaughlin J, et al. ABL fusion oncogene transformation and inhibitor sensitivity are mediated by the cellular regulator RIN1. Leukemia. 2011;25:290–300. doi: 10.1038/leu.2010.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morikawa J, Li H, Kim S, et al. Identification of signature genes by microarray for acute myeloid leukemia without maturation and acute promyelocytic leukemia with t(15;17)(q22;q12)(PML/RARalpha) Int J Oncol. 2003;23:617–25. [PubMed] [Google Scholar]
- 68.Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19:385–93. doi: 10.1016/j.semcdb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 69.Van Roosbroeck K, Cox L, Tousseyn T, et al. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood. 2011;117:4056–64. doi: 10.1182/blood-2010-06-291310. [DOI] [PubMed] [Google Scholar]
- 70.Hazlehurst LA, Bewry NN, Nair RR, et al. Signaling networks associated with BCR-ABL-dependent transformation. Cancer Control. 2009;16:100–7. doi: 10.1177/107327480901600202. [DOI] [PubMed] [Google Scholar]
- 71.Turner SD, Alexander DR. Fusion tyrosine kinase mediated signalling pathways in the transformation of haematopoietic cells. Leukemia. 2006;20:572–82. doi: 10.1038/sj.leu.2404125. [DOI] [PubMed] [Google Scholar]
- 72.Wong S, Witte ON. The BCR-ABL story: bench to bedside and back. Annu Rev Immunol. 2004;22:247–306. doi: 10.1146/annurev.immunol.22.012703.104753. [DOI] [PubMed] [Google Scholar]
- 73.Voss J, Posern G, Hannemann JR, et al. The leukaemic oncoproteins Bcr-Abl and Tel-Abl (ETV6/Abl) have altered substrate preferences and activate similar intracellular signalling pathways. Oncogene. 2000;19:1684–90. doi: 10.1038/sj.onc.1203467. [DOI] [PubMed] [Google Scholar]
- 74.De Keersmaecker K, Versele M, Cools J, et al. Intrinsic differences between the catalytic properties of the oncogenic NUP214-ABL1 and BCR-ABL1 fusion protein kinases. Leukemia. 2008;22:2208–16. doi: 10.1038/leu.2008.242. [DOI] [PubMed] [Google Scholar]
- 75.Toffalini F, Kallin A, Vandenberghe P, et al. The fusion proteins TEL-PDGFRbeta and FIP1L1-PDGFRalpha escape ubiquitination and degradation. Haematologica. 2009;94:1085–93. doi: 10.3324/haematol.2008.001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Essaghir A, Toffalini F, Knoops L, et al. Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucleic Acids Res. 2010;38:e120. doi: 10.1093/nar/gkq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Palmer RH, Vernersson E, Grabbe C, et al. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420:345–61. doi: 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilbanks AM, Mahajan S, Frank DA, et al. TEL/PDGFbetaR fusion protein activates STAT1 and STAT5: a common mechanism for transformation by tyrosine kinase fusion proteins. Exp Hematol. 2000;28:584–93. doi: 10.1016/s0301-472x(00)00138-7. [DOI] [PubMed] [Google Scholar]
- 79.Cain JA, Xiang Z, O’Neal J, et al. Myeloproliferative disease induced by TEL-PDGFRB displays dynamic range sensitivity to Stat5 gene dosage. Blood. 2007;109:3906–14. doi: 10.1182/blood-2006-07-036335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoelbl A, Schuster C, Kovacic B, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2:98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buitenhuis M, Verhagen LP, Cools J, et al. Molecular mechanisms underlying FIP1L1-PDGFRA-mediated myeloproliferation. Cancer research. 2007;67:3759–66. doi: 10.1158/0008-5472.CAN-06-4183. [DOI] [PubMed] [Google Scholar]
- 82.Heath C, Cross NC. Critical role of STAT5 activation in transformation mediated by ZNF198-FGFR1. J Biol Chem. 2004;279:6666–73. doi: 10.1074/jbc.M308743200. [DOI] [PubMed] [Google Scholar]
- 83.Pendergast AM, Quilliam LA, Cripe LD, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–85. [PubMed] [Google Scholar]
- 84.Puil L, Liu J, Gish G, et al. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994;13:764–73. doi: 10.1002/j.1460-2075.1994.tb06319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zebedin E, Simma O, Schuster C, et al. Leukemic challenge unmasks a requirement for PI3Kdelta in NK cell-mediated tumour surveillance. Blood. 2008;112:4655–64. doi: 10.1182/blood-2008-02-139105. [DOI] [PubMed] [Google Scholar]
- 86.Sattler M, Mohi MG, Pride YB, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1:479–92. doi: 10.1016/s1535-6108(02)00074-0. [DOI] [PubMed] [Google Scholar]
- 87.Nguyen TK, Rahmani M, Harada H, et al. MEK1/2 inhibitors sensitize Bcr/Abl+ human leukemia cells to the dual Abl/Src inhibitor BMS-354/825. Blood. 2007;109:4006–15. doi: 10.1182/blood-2006-09-045039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sjoblom T, Boureux A, Ronnstrand L, et al. Characterization of the chronic myelomonocytic leukemia associated TEL-PDGF beta R fusion protein. Oncogene. 1999;18:7055–62. doi: 10.1038/sj.onc.1203190. [DOI] [PubMed] [Google Scholar]
- 89.Goss VL, Lee KA, Moritz A, et al. A common phosphotyrosine signature for the Bcr-Abl kinase. Blood. 2006;107:4888–97. doi: 10.1182/blood-2005-08-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kunapuli P, Somerville R, Still IH, et al. ZNF198 protein, involved in rearrangement in myeloproliferative disease, forms complexes with the DNA repair-associated HHR6A/6B and RAD18 proteins. Oncogene. 2003;22:3417–23. doi: 10.1038/sj.onc.1206408. [DOI] [PubMed] [Google Scholar]
- 91.Kunapuli P, Kasyapa CS, Chin SF, et al. ZNF198, a zinc finger protein rearranged in myeloproliferative disease, localizes to the PML nuclear bodies and interacts with SUMO-1 and PML. Exp Cell Res. 2006;312:3739–51. doi: 10.1016/j.yexcr.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 92.Hosoya N, Qiao Y, Hangaishi A, et al. Identification of a SRC-like tyrosine kinase gene, FRK, fused with ETV6 in a patient with acute myelogenous leukemia carrying a t(6;12)(q21;p13) translocation. Genes Chromosomes Cancer. 2005;42:269–79. doi: 10.1002/gcc.20147. [DOI] [PubMed] [Google Scholar]
- 93.Armstrong F, Duplantier MM, Trempat P, et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene. 2004;23:6071–82. doi: 10.1038/sj.onc.1207813. [DOI] [PubMed] [Google Scholar]
- 94.Armstrong F, Lamant L, Hieblot C, et al. TPM3-ALK expression induces changes in cytoskeleton organisation and confers higher metastatic capacities than other ALK fusion proteins. Eur J Cancer. 2007;43:640–6. doi: 10.1016/j.ejca.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 95.Kasyapa CS, Kunapuli P, Cowell JK. HSPA1A is an important regulator of the stability and function of ZNF198 and its oncogenic derivative, ZNF198-FGFR1. J Cell Biochem. 2007;102:1308–17. doi: 10.1002/jcb.21362. [DOI] [PubMed] [Google Scholar]
- 96.Barnes DJ, Schultheis B, Adedeji S, et al. Dose-dependent effects of Bcr-Abl in cell line models of different stages of chronic myeloid leukemia. Oncogene. 2005;24:6432–40. doi: 10.1038/sj.onc.1208796. [DOI] [PubMed] [Google Scholar]
- 97.Mao JH, Sun XY, Liu JX, et al. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc Natl Acad Sci USA. 2010;107:21683–8. doi: 10.1073/pnas.1016311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mak HH, Peschard P, Lin T, et al. Oncogenic activation of the Met receptor tyrosine kinase fusion protein, Tpr-Met, involves exclusion from the endocytic degradative pathway. Oncogene. 2007;26:7213–21. doi: 10.1038/sj.onc.1210522. [DOI] [PubMed] [Google Scholar]
- 99.Tsukahara F, Maru Y. Bag1 directly routes immature BCR-ABL for proteasomal degradation. Blood. 2010;116:3582–92. doi: 10.1182/blood-2009-10-249623. [DOI] [PubMed] [Google Scholar]
- 100.Bonvini P, Gastaldi T, Falini B, et al. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res. 2002;62:1559–66. [PubMed] [Google Scholar]
- 101.Bonvini P, Dalla Rosa H, Vignes N, et al. Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res. 2004;64:3256–64. doi: 10.1158/0008-5472.can-03-3531. [DOI] [PubMed] [Google Scholar]
- 102.Porter JR, Fritz CC, Depew KM. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr Opin Chem Biol. 2010;14:412–20. doi: 10.1016/j.cbpa.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 103.Trepel J, Mollapour M, Giaccone G, et al. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–49. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nguyen TK, Rahmani M, Gao N, et al. Synergistic interactions between DMAG and mitogen-activated protein kinase kinase 1/2 inhibitors in Bcr/abl+ leukemia cells sensitive and resistant to imatinib mesylate. Clin Cancer Res. 2006;12:2239–47. doi: 10.1158/1078-0432.CCR-05-2282. [DOI] [PubMed] [Google Scholar]
- 105.Beck R, Verrax J, Gonze T, et al. Hsp90 cleavage by an oxidative stress leads to its client proteins degradation and cancer cell death. Biochem Pharmacol. 2009;77:375–83. doi: 10.1016/j.bcp.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 106.Frantsve J, Schwaller J, Sternberg DW, et al. Socs-1 inhibits TEL-JAK2-mediated transformation of hematopoietic cells through inhibition of JAK2 kinase activity and induction of proteasome-mediated degradation. Mol Cell Biol. 2001;21:3547–57. doi: 10.1128/MCB.21.10.3547-3557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kamizono S, Hanada T, Yasukawa H, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. 2001;276:12530–8. doi: 10.1074/jbc.M010074200. [DOI] [PubMed] [Google Scholar]
- 108.Cacalano NA, Sanden D, Johnston JA. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 2001;3:460–5. doi: 10.1038/35074525. [DOI] [PubMed] [Google Scholar]
- 109.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 19:679–90. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 110.Wasag B, Lierman E, Meeus P, et al. The kinase inhibitor TKI258 is active against the novel CUX1-FGFR1 fusion detected in a patient with T-lymphoblastic leukemia/lymphoma and t(7;8)(q22;p11) Haematologica. 2011;96:922–6. doi: 10.3324/haematol.2010.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chase A, Grand FH, Cross NC. Activity of TKI258 against primary cells and cell lines with FGFR1 fusion genes associated with the 8p11 myeloproliferative syndrome. Blood. 2007;110:3729–34. doi: 10.1182/blood-2007-02-074286. [DOI] [PubMed] [Google Scholar]
- 112.George P, Bali P, Annavarapu S, et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood. 2005;105:1768–76. doi: 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- 113.Radujkovic A, Schad M, Topaly J, et al. Synergistic activity of imatinib and 17-AAG in imatinib-resistant CML cells overexpressing BCR-ABL—inhibition of P-glycoprotein function by 17-AAG. Leukemia. 2005;19:1198–206. doi: 10.1038/sj.leu.2403764. [DOI] [PubMed] [Google Scholar]
- 114.Tong WG, Estrov Z, Wang Y, et al. Invest New Drugs. The synthetic heat shock protein 90 (Hsp90) inhibitor EC141 induces degradation of Bcr-Abl p190 protein and apoptosis of Ph-positive acute lymphoblastic leukemia cells. in press: doi: 10.1007/s10637-010-9465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu LX, Xu JH, Zhang KZ, et al. Disruption of the Bcr-Abl/Hsp90 protein complex: a possible mechanism to inhibit Bcr-Abl-positive human leukemic blasts by novobiocin. Leukemia. 2008;22:1402–9. doi: 10.1038/leu.2008.89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tyrosine kinase gene fusions in cancer