

Abstract
Therapeutically validated oncoproteins in myeloproliferative neoplasms (MPN) include BCR-ABL1 and rearranged PDGFR proteins. The latter are products of intra- (e.g. FIP1L1-PDGFRA) or inter-chromosomal (e.g.ETV6-PDGFRB) gene fusions. BCR-ABL1 is associated with chronic myelogenous leukaemia (CML) and mutant PDGFR with an MPN phenotype characterized by eosinophilia and in addition, in case of FIP1L1-PDGFRA, bone marrow mastocytosis. These genotype-phenotype associations have been effectively exploited in the development of highly accurate diagnostic assays and molecular targeted therapy. It is hoped that the same will happen in other MPN with specific genetic alterations: polycythemia vera (JAK2V617F and other JAK2 mutations), essential thrombocythemia (JAK2V617F and MPL515 mutations), primary myelofibrosis (JAK2V617F and MPL515 mutations), systemic mastocytosis (KITD816V and other KIT mutations) and stem cell leukaemia/lymphoma (ZNF198-FGFR1 and other FGFR1 fusion genes). The current review discusses the above-listed mutant molecules in the context of their value as drug targets.
Keywords: V617F, polycythemia, thrombocythemia, myelofibrosis, eosinophilia, mastocytosis
Introduction
Myeloid malignancies are stem cell-derived clonal disorders and include three broad clinicopathologic categories: acute myeloid leukaemia (AML), myelodysplastic syndrome (MDS) and myeloproliferative neoplasms (MPN). Such classification is however operational and not precise; for example, some patients present with histologic features that are reminiscent of both MPN and MDS and are assigned the diagnosis of ‘MDS/MPN’ overlap [1]. The history of MPN dates back to 1951 when William Dameshek coined the term ‘myeloproliferative disorders (MPD)’ as a clinico-pathologic category that included chronic myelogenous leukaemia (CML), polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) [2, 3]. These ‘classic’ MPD are now included in an expanded category of ‘MPN’, according to the World Health Organization (WHO) classification system, which also includes systemic mastocytosis (SM) and chronic neutrophilic (CNL) and eosinophilic (CEL) leukaemias (Table 1) [4]. Current classification of chronic myeloid neoplasms is semi-molecular although primarily based on myeloid cell morphology and presence or absence of effective haematopoiesis; Fig. 1 illustrates the histological hallmarks that distinguish MDS, MPN and MDS/MPN.
1.
Classification of chronic myeloid neoplasms modified from the 2008 World Health Organization classification scheme [313]
| 1. Myelodysplastic syndromes (MDS) |
| 1.1. Refractory cytopenia (RC) with uni-lineage dysplasia (RCUD) |
| 1.1.1. Refractory anaemia (RA) |
| 1.1.2. RA with ring sideroblasts (RARS) |
| 1.1.3. Refractory neutropenia |
| 1.1.4. Refractory thrombocytopenia |
| 1.2. RC with multi-lineage dysplasia (RCMD) |
| 1.3. RA with excess blasts (RAEB) |
| 1.4. MDS with isolated del(5q) |
| 1.5. MDS, unclassifiable |
| 1.6. Childhood MDS |
| 2. Myeloproliferative neoplasms (MPN) |
| 2.1. Classic |
| 2.1.1. Chronic myelogenous leukaemia, BCR-ABL1 positive (CML) |
| 2.1.2. Polycythemia vera (PV) |
| 2.1.3. Essential thrombocythemia (ET) |
| 2.1.4. Primary myelofibrosis (PMF) |
| 2.2. Non-classic |
| 2.2.1. Chronic neutrophilic leukaemia |
| 2.2.2. Chronic eosinophilic leukaemia, not otherwise specified (CEL-NOS) |
| 2.2.3. Mastocytosis |
| 2.2.4. MPN, unclassifiable |
| 3. MDS/MPN |
| 3.1. Chronic myelomonocytic leukaemia (CMML) |
| 3.2. Juvenile myelomonocytic leukaemia (JMML) |
| 3.3. Atypical chronic myeloid leukaemia, BCR-ABL1-negative |
| (aCML) |
| 3.4. MDS/MPN, unclassifiable |
| 3.4.1. Provisional entity: RARS and thrombocytosis (RARS-T) |
| 4. Myeloid and/or lymphoid neoplasms with eosinophilia and abnor-malities of, |
| 4.1. PDGFRA |
| 4.2. PDGFRB |
| 4.3. FGFR1 (8p11 myeloproliferative syndrome; a. k. a. stem cell leukaemia/lymphoma) |
1.
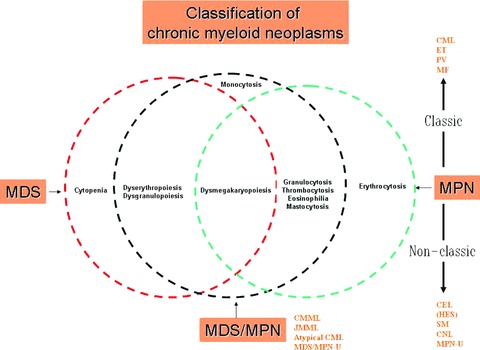
Histological hallmarks that distinguish myelodysplastic syndrome (MDS) from myelo-proliferative neoplasm (MPN) and MDS/MPN. CML, chronic myelogenous leukaemia; PV, polycythemia vera; ET, essential thrombocythemia; MF, primary myelofibrosis; CEL, chronic eosinophilic leukaemia; HES, hypereosinophilic syndrome; SM, systemic mastocytosis; CNL, chronic neutrophilic leukaemia; MPN-U, MPN, unclassifiable; CMML, chronic myelomonocytic leukaemia; JMML, juvenile myelomono-cytic leukaemia; MDS/MPN-U, MDS/MPN, unclassifiable.
Pathogenetic breakthroughs in MPN began in 1960 when the Philadelphia (Ph) chromosome was described in CML [5, 6]. This historic discovery later (1980-1990) led to the identification of BCR-ABL1 as the disease-causing mutation in CML [7–9]. In 1993 and 1994, stem cell factor receptor (KIT) and platelet-derived growth factor receptor-β (PDGFRB) mutations were associated with SM (KITD816V and KITV560G) [10] and an MPN phenotype characterized by eosinophilia and monocytosis (ETV6-PDGFRB) [11]. In 1998, a fibroblast growth factor 1 (FGFR1) mutation was described in stem cell leukaemia/lymphoma (SCLL; ZNF198-FGFR1). In 2003, FIP1L1-PDGFRA, a karyotypically occult platelet-derived growth factor receptor-a (PDGFRA) mutation, was described in association with an MPN phenotype characterized by eosinophilia and mastocytosis [12]. The molecular pathogenesis of BCR-ABL1-negative classic MPN remained elusive until early 2005 [13–16] when several groups reported a Janus kinase 2 (JAK2)gain-of-function (GOF) mutation (JAK2V617F) in PV, ET and PMF. In 2006, a GOF thrombopoietin receptor (MPL) mutation (MPLW515L) was reported in JAK2V617F-negative PMF [17]. In 2007, other JAK2 mutations (exon 12 mutations) in JAK2V617F-negative patients with PV were described [18]. Additional MPL and JAK2 exon 12 mutations have since been added to the list [19–21].
The above-listed revelations in putative disease-causing or disease-promoting genetic changes have ignited much interest in the development of molecular targeted therapy in MPN. Proof-of-principle in this regard has already been accomplished with the use of imatinib mesylate (IM) in CML [22] and PDGFR-rearranged MPN [12, 23]. On the other hand, therapeutic targeting of KITD816V or mutant FGFR1has not been as successful whereas phase I/II clinical trials evaluating anti-JAK2 drugs are currently ongoing. In this review, I will provide a clinically relevant overview of mutant molecules of interest in adult MPN and discuss the current state of affairs in regards to targeted therapy.
BCR-ABL1
The stage for the discovery of BCR-ABL1 in CML was set in 1960 when Peter Nowell and David Hungerford described the Ph chromosome [5]. In 1967, Philip Fialkow and colleagues applied polymorphisms in the X-linked glucose-6-phosphate dehydrogenase (G-6-PD) locus to establish CML as a stem cell-derived clonal disorder [24]. In 1972, Janet Rowley clarified the constitution of the Ph chromosome as a reciprocal translocation between chromosomes 9 and 22; t(9; 22)(q34; q11) [25].
In 1982, the human homologue (ABL1; 225 kb total gene size) of v-abl was mapped to chromosome 9 [26] and shown to be involved in the Ph translocation [27]. In 1984, the chromosome 22 breakpoint was mapped to a 5.8 kb area and named the ‘breakpoint cluster region (bcr)’, which is part of the BCR gene (135 kb total gene size) [28, 29]. In 1990, retroviral infection of haematopoietic stem cells with BCR-ABL was shown to induce CML-like disease in mice [7–9].
ABL1
ABL1 is a cytoplasmic protein tyrosine kinase (PTK) that plays a role in non-erythroid myelopoiesis [30], cytoskeletal rearrangement and inhibition of cell migration [31]. Wild-type ABL1 exists in two isoforms that can localize to both the cytoplasm and nucleus, influencing cell proliferation/survival and apoptosis [32–34]. ABL1 contains both an SH2 and an SH3 (autoregulatory) domain in addition to the catalytic kinase domain and undergoes a treatment-relevant conformational change when activated by phosphorylation of the activation loop tyrosine residues [35].
BCR-ABL1
The chromosome 9 breakpoints in CML involve a large, ∼200 kb region within the ABL1 alternative first exons (1a and 1b), but invariably result in fusion genes that incorporate ABL1 exon 2 [36]. In contrast, the breakpoints on chromosome 22 are clustered within three much smaller regions of the BCR gene [37]; the major breakpoint cluster region (M-bcr; a 5.8 kb region spanning exons 12–16 and resulting in a p210 fusion protein) [28], the minor breakpoint cluster region (m-bcr; upstream of M-bcr and involving the first intron and resulting in a p190 fusion protein) [38, 39] and μ-bcr involving intron 19 that is downstream of M-bcr and resulting in a p230 fusion protein [40]. By far the most frequent chromosome 22 breakpoint in CML is M-bcr and the other two, in the context of CML, are extremely rare. There are usually two junction variants of M-bcr; b2a2 and b3a2, without any documented clinical relevance [41].
BCR-ABL1 gets transcribed as a chimeric mRNA (8.5-kb) as opposed to the normal ABL1 mRNA (a 6- or 7-kb) [42] and subsequently translated to an activated BCR-ABL1 gene product (most commonly 210-kD) instead of the normal ABL1 gene product (145-kD) [43]. BCR-ABL1 localizes to the cytoskeleton and displays an up-regulated tyrosine kinase activity [44] that leads to the recruitment of downstream effectors of cell proliferation and cell survival and consequently leukaemogenesis, as has been demonstrated in cell lines, primary cells and mouse transplant or transgenic models [7, 8, 45–47]. BCR-ABL1 signal transduction involves several adapter molecules (e.g. GRB2, GAB2, CRKL, etc.) and signalling pathways (e.g. Ras, PI3K, JAK-STAT, etc.) that are all thought to contribute to the pathogenesis of CML [35, 48, 49].
Anti-BCR-ABL1 targeted therapy in CML
In 1996, Brian Druker and his colleagues described the in vitro, anti-BCR-ABL1 activity of imatinib mesylate (IM) [22], a 2-phenylaminopyrimidine class kinase inhibitor that targets ABL1 [50], PDGFR [51], ARG [52] and KIT [53]; the mechanism of action involves competitive inhibition of the ATP binding site of the kinase [54]. Consequently, orally administered IM has been shown to be effective in the treatment of neoplastic diseases with mutations involving the aforementioned kinases: CML [55–61], Ph-positive acute lymphoblastic leukaemia (ALL) [56, 62], gastrointestinal stromal tumours (known to harbour KIT or PDGFR mutations) [63, 64] and PDGFR-rearranged MPN [12, 23, 65–69].
In newly diagnosed patients with chronic phase CML (CP-CML), IM is now recommended as the initial treatment of choice [70]. In the International Randomized Study of Interferon and STI571 (IRIS), interferon alpha/cytarabine combination was compared with IM in 1106 newly diagnosed CP-CML patients. The results of this trial were recently updated in 2006 [71]. IM was found to be superior to combination chemotherapy in terms of both response rates and progression-free survival; the 553 patients initially assigned to IM therapy had been followed for at least 5 years and the best observed rates for complete haematological and cytogenetic remissions were 97% and 82%, respectively. Five-year survival was 89%.
Imatinib resistance and second-generation anti-BCR-ABL1 kinase inhibitors
IM is not as effective in advanced phase CML as it is in CP-CML [56]. Furthermore, approximately 20% of CP-CML patients either fail treatment, for one reason or another, or transform into advanced phase disease. IM-resistance has operationally been classified into primary (not achieving complete cytogenetic remission) and secondary (loss of response). In each instance, several underlying mechanisms have been implicated and include altered BCR-ABL1/IM cellular concentration ratio due to increased BCR-ABL1 expression or abnormal cellular drug transport, activation of alternative signal pathways, inherent IM-resistance by BCR-ABL1 leukaemia stem cells and emergence of IM-resistant BCR-ABL1 mutations. The latter is by far the most frequent and best understood mechanism of IM resistance.
The crystal structure of IM bound to the ABL1 indicates that the drug stabilizes the oncoprotein in an enzymatically inactive conformation [54]. BCR-ABL1 mutations induce IM resistance by hindering drug-oncoprotein binding at critical contact points or prevent the formation of inactive structural conformation that is required for IM activity [72]. At present, more than 50 BCR-ABL1 mutations have been described and most affect the ABL1 kinase domain; in general four categories of mutations are identified and seven mutations account for two-thirds of the cases (G250, Y253, E255, T315, M351, F359, H396): phosphate-binding loop (e.g. E255, Y253, Q252, G250; affecting ATP binding site); IM binding site (e.g. T315); catalytic domain (e.g. F359); and activation loop (e.g. H396; forcing an active conformation and thus making IM unable to bind) [73]. Not all these mutations are IM-insensitive and some (e.g. T315, E255, Y253) are more resistant than others (e.g. M244, G250, Q252, F317, E355, F359, V379, H396) [74].
The development of second generation kinase inhibitors and better understanding of mutant ABL1 conformation dynamics have had some success in addressing the aforementioned challenges of IM resistance [75–80]. New drugs include more potent ABL1 inhibitors such as nilotinib, dual ABL1/Src inhibitors such as dasatinib and bosutinib, dual ABL1/Lyn inhibitors such as INNO-406, ABL1-substrate binding inhibitors (i. e. non-ATP competitive) such as ONO12380. Most but not all (e.g. T315) ABL1 mutations are sensitive to one or more of the above listed new drugs whose value as salvage therapy for IM-refractory disease is being validated [75–80]. Finally, there is more and more discussion about IM-resistant BCR-ABL1-positive leukaemia stem cells and their dependence on both altered BCR-ABL1 expression and other signalling pathways [81]. Whether or not going after such quiescent leukaemia stem cells is therapeutically essential, in patients achieving complete molecular remissions with currently available drugs, remains to be seen.
Other ABL1 mutations
In the context of myeloid malignancies, ETV6 is the only currently known non-BCR fusion partner for ABL1 although other ABL1 fusion proteins have been described in acute lymphoblastic leukaemia (ALL) [82]. ETV6-ABL1, which corresponds to t(9; 12)(q34; p13), has been associated with ALL [83–86], AML (with transient response to IM) [87–89] and atypical CML (with minor response to imatinib) [85, 90–92]. Like BCR-ABL1, ETV6-ABL1 encodes for an activated and transforming ABL1 [87, 93]. The PNT oligomerization motif of ETV6 is thought to activate the ABL1 kinase by a similar mechanism to the BCR coiled coil oligomerization domain in the context of the BCR-ABL1.
JAK2 and MPL mutations
Like ABL1, JAK2 is a cytoplasmic PTK and mutant JAK2, like mutant ABL1, has now been associated with classic MPN. JAK2 is a member of the Janus family of kinases that were incidentally discovered around 1989 and set aside as ‘just another kinase’[94]. Their name was later modified to Janus kinases (JAKs) [95] after the Roman god with two faces because they contain two symmetrical kinase-like domains; the C-terminal JAK homology 1 (JH1) domain possesses tyrosine kinase function while the immediately adjacent JH2 domain is enzymatically inert but is believed to regulate the activity of JH1. The possible role of JAK2 mutations in haematological malignancy was first suspected after the observation that a dominant mutation in HOP, a JAK homologue in Drosophila, induced leukaemia-like defects [96].
Janus kinases
There are four JAKs (JAK1, JAK2, JAK3 and tyrosine kinase 2) and they all contain seven JH domains organized into four regions; kinase (JH1), pseudo-kinase (JH2), FERM (the N-terminal JH7, JH6, JH5, and part of JH4) and SH2-like (JH3 and part of JH4) [97]. In human beings, JAK1is located on chromosome 1p31.3, JAK2on 9p24, JAK3on 19p13.1 and TYK2on 19p13.2. At present, human cancer-relevant JAK mutations have been described for JAK1 (T-ALL, AML, breast cancer, lung cancer) [98–101], JAK3 (AMKL cell lines and primary cells, breast cancer, gastric cancer) [98, 102] and JAK2 (MPN primarily and other myeloid malignancies infrequently, trisomy 21 associated ALL) [13–16, 103–105]. However, other than JAK2mutations in MPN and JAK1 mutations in T-ALL, the other associations are rare.
JAK2 fusion mutations
GOF JAK2 fusion mutations have been associated with T or pre-B ALL, T-cell lymphoma, AML and MPN-U; these include ETV6-JAK2, t(9; 12)(p24; p13), PCM1-JAK2, t(8; 9)(p22; p24), BCR-JAK2, t(9; 22)(p24; q11.2) and SSBP2-JAK2, t(5; 9)(q14.1; p24.1) (Table 2) [106–114]. PCM1 displays multiple coiled-coil motifs and it is believed that these are preserved and function as a dimerization domain in PCM1-JAK2. In ETV-6-JAK2, the chimeric protein includes the Helix-Loop-Helix oligomerization domain of ETV6 fused to the kinase domain of JAK2 and the fusion oncogene induces both T and B cell lymphoid neoplasms in transgenic mice [106, 107, 115, 116]. In a more recent study, expression of ETV-6-JAK2in human cord blood cells resulted in Epo-independent erythroid differentiation in vitro and induction of myelofibrosis in an in vivoxenotransplantation model [117]. In BCR-JAK2, the fusion protein includes the coiled-coil dimerization domain of BCR and the JAK2 kinase domain [112]. The patient in this particular instance had MPN-U and did not respond to imatinib. Another patient with BCR-JAK2had AML [114]. The BCR-JAK2fusion in this instance had a different BCR breakpoint, although it was similar to that of BCR-ABL1. The most recently described JAK2 fusion partner, SSBP2, is located at 5q14.1 and the associated phenotype was pre-B ALL [118]. The NH2 terminus of the SSBP2 protein contains a LisH motif that is believed to function as the dimerization domain.
2.
Mutations of putative pathogenetic relevance in myeloproliferative neoplasms
| JAK2 | MPL | PDGFRA | PDGFRB | FGFR1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (phenotype) | (phenotype) | (phenotype) | (phenotype) | (phenotype) | |||||||
| JAK2V617F (PV, ET, PMF) | MPLW515L/K (PMF, ET) | FIP1L1-PDGFRA | ETV6-PDGFRB | ZNF 198-FGFR1 | |||||||
| del(14q12) (CEL with mas-tocytosis) | t(5; 12)(q33; p13) (CMML with eosinophilia) | t(8; 13)(p11; q12) (SCLL) | |||||||||
| JAK2Exon 12 mutations | BCR-PDGFRA | RABAPTIN-5-PDGFRB | F0P-FGFR1 | ||||||||
| F537-K539delinsL | t(4; 22)(q12; q11) (CEL, MPN-U) | t(5; 17)(q33; p13) (CMML) | t(6; 8)(q27; p11) (SCLL) | ||||||||
| H538QK539L K539L | HCMOGT-1-PDGFRB | ||||||||||
| N542-E543del (PV) | t(5; 17)(q33; p11.2) (JMML with eosinophilia) | ||||||||||
| ETV6-JAK2 | KIF5B-PDGFRA | CEV14-PDGFRB | FGFR10P2-PDGFRA | ||||||||
| t(9; 12)(p24; p13) (AML, MPN-U) | t(4; 10)(q12; p11) (CEL) | t(5; 14)(q33; q32) (AML with eosinophilia) NIN-PDGFRBt(5; 14)(q33; q24) (MPN with eosinophilia) KIAA1509-PDGFRB t(5; 14)(q31; q32) (CMML with eosinophilia) | Ins(12; 8)(p11; p11p22) (SCLL) | ||||||||
| BCR-JAK2 | CDK5RAP2-PDGFRA | TP53BP1-PDGFRB | TIF1-FGFR1 | ||||||||
| t(9; 22)(p24; q11.2) (MPN-U, AML) | Ins(9; 4)(q33; q12q25) (CEL) | t(5; 15)(q33; q22) (MPN-U with eosinophilia) | t(7; 8)(q34; p11) (SCLL) | ||||||||
| PCM1-JAK2 | PDE4DIP-PDGFRB | MY018A-FGFR1 | |||||||||
| t(8; 9)(p22; p24) (AML, MPN-U) | t(1; 5)(q23; q33) (MPN-U with eosinophilia) | t(8; 17)(p11; q23) (SCLL) | |||||||||
| SSBP2-JAK2 | HIP1-PDGFRB | HERV-K-FGFR1 | |||||||||
| t(5; 9)(q14.1; p24.1) (pre-B ALL) | t(5; 7)(q33; q11.2) (CMML with eosinophilia) | t(8; 19)(p12; q13.3) (SCLL) | |||||||||
| H4-PDGFRB | BCR-FGFR1 | ||||||||||
| t(5; 10)(q33; q22) (MPN-U) | t(8; 22)(p11; q11) (CML-like MPN) | ||||||||||
| CEP110-FGFR1 | |||||||||||
| t(8; 9)(p12; q33) (SCLL) | |||||||||||
| CPSF6-FGFR1 | |||||||||||
| t(8; 12)(p11; q15) (SCLL) | |||||||||||
PV, polycythemia vera; ET, essential thrombocythemia; PMF, primary myelofibrosis; AML, acute myeloid leukaemia; MPN-U, unclassified MPN; CEL-SM, chronic eosinophilic leukaemia associated with systemic mastocytosis; CMML, chronic myelomonocytic leukaemia; JMML, juvenile myelomonocytic leukaemia; SCLL, stem cell leukaemia-lymphoma syndrome.
JAK2V617F
JAK2V617F is an exon 14 G to T somatic mutation. The nucleotide change at position 1849 results in the substitution of valine to phenylalanine at codon 617. JAK2V617F was first described in 2005 in patients with PV, ET and PMF [13–16]. Subsequently, the mutation has also described in other myeloid neoplasms [103, 119]. As of the time of this writing, JAK2V617F has not been reported in lymphoid disorders [120–123], solid tumour [124–126] or secondary myeloproliferation [127, 128]. In general, mutational frequency is estimated at over 95% in PV, 50% in ET or PMF, 20% in certain other MPNs including refractory anaemia with ringed sideroblasts and thrombocytosis (RARS-T) and less than 5% in AML or MDS [104, 129–131].
JAK2V617F induces a PV-like phenotype in murine transplant models: erythrocytosis but not thrombocytosis, low serum Epo level, splenomegaly, extramedullary haematopoiesis, granulocytosis, megakaryocytic hyperplasia and ultimately bone marrow fibrosis and anaemia [14, 132, 133]. However, in a recent study of JAK2V617F transgenic mice, manipulation of mutant gene expression resulted in either an ET (lower expression compared to wild-type allele) or PV phenotype with (equal expression) or without (higher expression) thrombocytosis [134]. Such mutant allele burden-dependent phenotypic variation in transgenic mice was also demonstrated by another study [135].
Current information regarding mutant allele burden in human beings with MPN partially recapitulates what was seen in the above-mentioned experiments with transgenic mice. In other words, mutant allele burden in patients with ET is significantly lower than that seen in patients with either PV or PMF [136–140]. At least in PV, a higher allele burden is the result of JAK2V617F homozygosity, which is accomplished by mitotic recombination [13, 16, 141]. Some have suggested that such homologous recombination and genomic instability in general is facilitated by JAK2V617F [142]. However, the interaction between JAK2V617F homozygosity and mutant allele burden, in terms of phenotypic determination, is currently not clear [143, 144].
In human beings, JAK2V617F occurs at a primitive stem cell level and is chronologically an early event [145–147]. Some but not all [148] studies have suggested JAK2V617F clonal involvement of NK [149], T [150] and B [150] lymphocytes. Regardless, there is evidence to suggest that JAK2V617F may not be the initial clonogenic event in either PV or other MPNs and that its presence might not be mandatory for endogenous colony formation [151–153]. The demonstration of JAK2V617F-negative leukaemia clones arising in JAK2V617F-positive MPN patients lends further support in this regard [154, 155]. In the latter instance, both mutation positive and negative cells were shown to share the same cytogenetic abnormality and thus probably arose from a common ancestral clone [154]. On the other hand, JAK2V617F or other JAK2 mutations might be a necessary component of the PV phenotype, because they are detected in all patients with the disease [20]. Furthermore, recent studies suggest that germline genetic variation [156] and/or the occurrence of other concomitant mutations [19] might explain why the same mutation is present in apparently different disease phenotypes.
Other reported JAK2 exon 14 mutations include D620E (PV, MPN, unclassifiable), E627E (MPN, unclassifiable), K607N (AML), L611S (B-ALL), JAK2DeltaIREED (DS-associated B-ALL), C616Y (PV), V617F from c. 1848_1849delinsCT (post-ET MF) and V617F/C618R from c. 1849-1852GTCT > TTTC (PV) [125, 157–163]. In addition, an activating JAK2T875N was described in an AMKL cell line [164]. There is no doubt that the list of such mutations will grow with time and, interestingly, some of the new mutations identified in PV co-existed with JAK2V617F.
JAK2 exon 12 mutations
In 2007, a set of JAK2 exon 12 mutations were described in JAK2V617F-negative patients with PV in whom erythrocytosis was the predominant feature [165]. Because of the latter feature, some of the cases were assigned the diagnosis of ‘idiopathic’ erythrocytosis although their serum Epo level was almost always below the reference range and EECs were demonstrated in every instance when tested. The majority of the cases (10 of 11) in the original report [165] were found to harbour one of four exon 12 JAK2 mutant alleles: N542-E543del (4 cases), F537-K539delinsL (3 cases), K539L (2 cases), H538QK539L (1 case). All four exon 12 mutant alleles induced cytokine-independent/hypersensitive proliferation in erythropoietin receptor-expressing cell lines and constitutive activation of JAK-STAT signalling [165]. In addition, JAK2K539L induced a PV phenotype in a mouse transplant model. Also, in this first report, the four newly described JAK2 exon 12 mutations, which include both in-frame deletions and tandem point mutations, appeared to be specific to either PV or ‘idiopathic’ erythrocytosis. Unlike the case with PV-associated JAK2V617F, exon 12 JAK2 mutations were heterozygous but associated with stronger abnormal JAK2 activation.
Many other studies have now confirmed the observations from the above-mentioned study [20, 166–170] and in the process, most [20, 166, 167] but not all [168, 170] of the studies suggested that exon 12 mutations occurred in virtually all JAK2V617F-negative PV cases (i. e. approximately 3% of all PV cases). Furthermore, several other exon 12 mutation variants were added to the list including R541-E543delinsK, I540–E543delinsMK, V536–I546dup11, F537-I546dup10 + 547L and E543–D544del [167–169]. Among all currently described exon 12 mutations, the most frequent was N542–E543del comprising 17 of 52 cases reported at the time of the latest report [168]. In general, many JAK2 exon 12 mutation-positive patients present with apparently ‘isolated’ erythrocytosis but because of their association with subnormal serum Epo level and the presence of EECs, they fulfil the 2008 WHO diagnostic criteria for PV [171]. Finally, information presented at the 2007 American Society of Hematology (ASH) meeting suggested that JAK2exon 12 mutations, as is the case with JAK2V617F, might clonally involve both B and NK lymphocytes [172] and also occur in ET and ‘idiopathic’ abdominal vein thrombosis [173, 174]. Obviously, more studies are needed to fully appreciate the phenotypic spectrum of these mutations.
JAK2 mutations associated with trisomy 21 associated acute lymphoblastic leukaemia
A recent report in Lancet described somatically acquired GOF JAK2 mutations (JAK2R683G/S/K; exon 16 and mostly heterozygous) in 16 of 88 (18%) patients with Down's syndrome-associated B-ALL but only 1 of 216 patients with sporadic ALL [105]. The latter patient had an isochromosome 21q. The mutations caused constitutive JAK-STAT activation and cytokine-independ-ent growth of BaF3 cells that was inhibited by JAK inhibitor I. Overall prognosis did not appear to be affected.
MPL mutations
MPL (myeloproliferative leukaemia virus oncogene homologue) belongs to the haematopoietin receptor superfamily and enables its ligand, thrombopoietin (Tpo), to facilitate both global haematopoiesis (early expansion of differentiating clones) and megakaryocyte growth and differentiation [175–177]. The gene for MPL maps to chromosome 1p34 and contains 12 exons [178]. Germline MPL mutations have been associated with either familial thrombocytosis (GOF transmembrane domain mutation; S505N) [179] or congenital amegakaryocytic thrombocytopenia (LOF mutations) that often progresses to aplastic anaemia [180–182]. In addition, an MPL single nucleotide polymorphism (G1238T) that results in a K39N substitution is found in approximately 7% of African Americans [183]. Such patients display higher platelet counts and lower MPL expression, when compared to patients without the specific MPL polymorphism [183].
In 2006, a somatic GOF MPLW515L mutation (a G to T transition at nucleotide 1544 resulting in a tryptophan to leucine substitution at codon 515 of the transmembrane region) was described in JAK2V617F-negative PMF [17]. Subsequently, an additional MPL mutation involving the same 515 codon (MPLW515K) was incidentally discovered during screening for MPLW515L and the prevalence of both mutations was determined at approximately 5% in PMF and 1% in ET [184]. More recent studies have identified other somatic MPL mutations including MPLW515S and MPLS505N (mentioned above in association with familial thrombocytosis) and higher prevalence rates: approximately 4% in ET (9% in JAK2V617F-negative cases) and up to 11% in PMF [185–188]. Most recently, using SNP-array karyotyping, biallelic MPLW515L mutation and uniparental disomy 1p was demonstrated in some patients with RARS-T, thus providing a mechanism of homozygosity similar to that of JAK2V617F [189].
As is the case with JAK2V617F, MPL515 mutations are early, stem cell-derived events involving both myeloid and lymphoid progenitors [190–192] and MPLW515L has been shown to transform cell lines in terms of both cytokine-independent growth and Tpo hypersensitivity, activate JAK-STAT/ERK/Akt and induce PMF-like disease in mice that is characterized by a rapid fatal course, marked thrombocytosis, leukocytosis, hepatosplenomegaly and bone marrow fibrosis [17]. Interestingly, some patients display multiple MPL mutations and others a minor JAK2V617F cloner together with an MPL mutation [187, 193]. Again, these observations support the secondary nature of such mutations and underscore the complexity of pathogenetic mechanisms in MPNs. Finally, preliminary information suggests that MPL mutations favour megakaryocytic/myeloid as opposed to the erythroid skewed proliferation/differentiation seen with JAK2 mutations [151, 191].
Clinical and prognostic correlates of JAK2 and MPL mutations
Virtually all patients with PV carry a JAK2 mutation (97% with JAK2V617F and the rest with JAK2 exon 12 mutations) [20]. In PV, JAK2V617F ‘homozygous’ as opposed to ‘heterozygous’ state has been associated with a higher haemoglobin level, higher leukocyte count, lower platelet count and presence of pruritus [139, 194]. A somewhat similar set of correlations were made for higher mutant allele burden in PV, measured by quantitative assays [137, 140, 195]. The small number of patients with JAK2 exon 12 mutations are more likely to present with isolated erythrocytosis but there is substantial phenotypic overlap with JAK2V617F-positive PV [18, 168].
In ET, the presence of JAK2V617F has been associated with advanced age, higher haemoglobin level, increased leukocyte count and decreased platelet count [136, 196–198]. In mutation-positive patients with ET, JAK2V617F allele burden has been directly correlated with leukocyte count, platelet count and the presence of palpable splenomegaly [136, 139, 140]. Similarly in PMF, the presence of JAK2V617F has been associated with an older age at diagnosis and higher leukocyte count [199]. In addition, JAK2V617F ‘homozygous’ PMF patients displayed an even higher incidence of leukocytosis, marked splenomegaly and pruritus [200]. Both PMF and ET patients with MPL mutations were found to be older and more anaemic than their MPL mutation-negative counterparts [186, 188].
JAK2 or MPL mutations in MPNs might not have prognostic relevance. In ET, overall or leukaemia-free survival was not affected by either the presence of JAK2V617F or its allele burden [136, 196]; the impact on the risk of thrombosis or fibrotic transformation is less clear [136, 139, 196, 201]. Equally unclear is the prognostic relevance of JAK2V617F allele burden in PV where a higher mutant allele burden is implicated by some but not by others as an adverse prognostic factor for fibrotic transformation, thrombosis and need for chemotherapy [137, 139, 194, 195]. In PMF, JAK2V617F presence was associated with inferior survival in one but not in another study [199, 202]. Similarly divergent results were reported in terms of leukaemic transformation rate and need for chemotherapy or splenectomy [200, 203]. The most recent study on the subject matter revealed shortened overall and leukaemia-free survival in PMF patients with lower as opposed to higher quartile JAK2V617F allele burden [203].
Anti-JAK2 small molecule therapy
A number of anti-JAK2 drugs have undergone preclinical testing and some have already been introduced into clinical trials [204]. Amongst them, some are JAK2 selective ATP-mimetic small molecules: e.g. TG101209, TG101348, INCB018424, XL019, CEP701 and SB1518 [205–208]. TG101209, an orally available small molecule JAK2-selective kinase inhibitor, is one of the first compounds to undergo extensive preclinical testing [209]. The drug's anti-JAK2 kinase activity was estimated at an IC50 of 6 nM, compared to 169 nM for JAK3 [209]. In HEL cells (homozygous for JAK2V617F) and other JAK2V617F-transduced cell lines, TG101209 induced apoptosis and inhibited phosphorylation of JAK2V617F, STAT5 and STAT3 (IC50 of approximately 600 nM) [209]. At similar or lesser drug concentrations, the drug also inhibited colony growth of primary cells from JAK2V617F-positive PV patients and MPLW515L-positve PMF patients [209]. Furthermore, the growth inhibitory effect of TG101209 was relatively selective to mutated-colonies. TG101348, a drug that is very similar to TG101209, was also shown to display similar in vitroas well as in vivo anti-PV and anti-MPN activity in the context of exon 12, exon 14 JAK2 and MPL mutations [205, 210–212]. In a murine model of polycythemia vera, oral TG101348 therapy resulted in significant reduction of haematocrit, leukocyte count, extramedullary haematopoiesis, myelofibrosis and JAK2V617F-expressing clones [211]. TG101348, but not TG101209, is currently undergoing phase I/II clinical trial in PMF and post-PV/ET M F.
Several other potent JAK2 inhibitors have also been reported to have in vitro or in vivo (i. e. mouse models) activity against JAK2V617F-driven cell proliferation and signal transduction. Among them, INCB018424, XL019, CEP-701 and SB1518 are currently undergoing clinical trials in patients with advanced stages of PMF or post-PV/ET MF (INCB018424, XL019, CEP701), PV (XL019, CEP701), JAK2V617F-positive ET (CEP701) and acute and chronic haematologic malignancies (SB1518) [206–208, 213, 214]. All of these drugs display potent JAK2 inhibitory activity but their in vitroJAK2 selectivity, in the context of the JAK family of kinases, is different. For example, JAK3 is spared by TG101348, XL019 and INCB018424 but not by CEP701. Similarly, JAK1 is spared by TG101348 and XL019 but not by INCB018424.
Preliminary results regarding INCB018424 in PMF and post-PV/ET MF are encouraging in terms of the drug's toxicity profile and its activity against splenomegaly and constitutional symptoms [213]. More follow-up is required to determine the drug's effect on anaemia, leukoerythroblastosis, myelofibrosis, cytogenetic abnormalities or JAK2V617F allele burden. Results regarding other JAK2 inhibitors are too early to comment on. Nevertheless, the expected anti-inflammatory cytokine effect of this class of drugs is expected to be a confounding variable in terms of both toxicity and efficacy assessment.
KIT mutations
In 1993, Furitsu and colleagues described a mutant KIT allele with two-point mutations as being responsible for the ligand-independent constitutive activation of KIT in a human mast cell leukaemia cell line (HMC-1); one mutation was located at the JM region at codon 560 and nucleotides 1699–1701 (V560G; GTT→GGT) and the other at the kinase domain at codon 816 and nucleotides 2467–2469 (D816V; GAC→GTC) [10]. One year later (1994), Tsujimura et al. reported a kinase domain point mutation at nucleotide 2468 (GC to TA) resulting in an amino acid substitution at codon 814 (KITD814Y), in a murine mastocytoma cell line, P-815 [215]. In both instances, either the mutations themselves [215] or their corresponding murine constructs (V559G and D814V) [10] were shown to produce constitutive activation of KIT signalling in cell lines.
Nagata and colleagues were the first to describe one of the aforementioned mutations (KITD816V) in blood mononuclear cells from patients with SM and an associated non-mast cell myeloid neoplasm but not in those with indolent or aggressive SM, solitary mastocytoma or chronic myelomonocytic leukaemia [216]. However, subsequent studies have shown the presence of KITD816V or other KIT mutations (KITD820G, KITV560G, KITD816Y, KITE839K, KITD816F, KITF522C, etc.) in other categories of mast cell disease (MCD) including indolent and aggressive SM [217–222]. It is currently touted that all patients with MCD carry a KIT mutation but its laboratory detection might be hampered by the use of non-informative cell source or inadequate assay sensitivity [223, 224]. JM KIT mutations have also been described in canine mastocytomas [225].
KIT
KIT is located at chromosome 4q12 and encodes KIT, a class III receptor tyrosine kinase [226, 227]. The type III class also includes PDGFR, CSF-1 and Flt3 and is characterized by an extracellular component of five immunoglobulin-like domains, a trans-membrane segment, a juxtamembrane (JM) domain and a cytoplasmic kinase domain with a 70-100 amino acid kinase insert near its centre [228]. Normally, KIT is activated when bound to its ligand, the stem cell factor (SCF), which is encoded by SCF on chromosome 12q22 [229–231]. KIT (CD117) is notably expressed by mast cells, haematopoietic stem cells, germ cells, melanocytes and Cajal cells of the gastrointestinal tract and is therefore functionally relevant for mast cell development, haematopoiesis, gametogenesis and melanogenesis [232]. KIT signalling and the corresponding cellular response depends on the specific cell type involved and the putative downstream effectors include PI3K-Akt, Src family of kinases, Ras-Erk, phospholipase C/y, MAPK and JAK-STAT [233].
Mutant KIT
Activating KIT mutations have been described in a spectrum of haematologic (e.g. mastocytosis, acute leukaemia) and non-haematologic (e.g. gastrointestinal stromal cell tumour, germ cell tumour) malignancies [232]. Among these, SM is considered an MPN by virtue of its clonal derivation from the haematopoietic stem cell [234–236]. Observations from several laboratory studies support the oncogenic role of KIT mutations. For example, in murine experiments, D814Y has been shown to induce ligand-independent mast cell growth in vitro, tumourigenicity in vivo, and mast cell differentiation [237]. Similarly, retroviral infection of haematopoietic progenitors with mouse KITD814Y and KITV559G mutants induces autonomous myeloid and mast cell colony formation as well acute leukaemia in murine transplant models [238]. Furthermore, transgenic mice expressing KITD816V restricted to their mast cells display an SM phenotype that closely resembles the clinically heterogeneous disease in man [239]. This is in contrast to another report [240] that suggested a differentiating but not transforming potential for the mutation, which nevertheless probably participates in enhancing mast cell chemotaxis [241] and clustering [236]. Mutant Kit signalling might involve both PI3K [242] and Src [243] participation although utilization of pathway molecules might be different between the wild-type and mutant protein [244].
Systemic mastocytosis
The 2008 World Health Organization (WHO) classification system for myeloid malignancies considers SM as a myeloproliferative neoplasm (MPN) [4]. WHO-defined SM should be distinguished from PDGFR-mutated (FIP1L1-PDGFRA, PRKG2-PDGFRB) myeloid malignancy associated with bone marrow mastocytosis and either eosinophilia or basophilia [69, 245]. The latter but not the former are effectively treated by IM. SM is sometimes associated with a clonally related second myeloid neoplasm, which is not surprising considering its origin as a stem cell disease with multilineage clonal involvement.
Targeted therapy in systemic mastocytosis
Although several drugs have shown in vitro anti-KIT activity [246–249], the immediately most relevant, in terms of clinical development, include the tyrosine kinase inhibitors IM, nilotinib, dasatinib and PKC412. During in vitro experiments involving both cell lines and primary cells from patients with SM, IM inhibited wild-type KIT and KIT mutants with trans-membrane (F522C) and juxta-membrane (V559G and V560G) but not kinase (D816V or D814V) domain mutations [222, 250, 251]. Similarly, not all juxta-membrane mutations are sensitive to IM (e.g. V559I) [252]. Consistent with these in vitro observation, IM therapy is ineffective in SM associated with KITD816V [253] whereas others have shown activity in MCD associated with the trans-membrane KITF522C mutation [222]. In contrast to the experience from my own institution, Droogendijk et al. treated 11 patients with D816V-positive SM with 400 mg/day of IM and found some ‘clinical improvement’ in some patients [254].
Nilotinib is more potent than IM in its in vitro anti-BCR-ABL activity but is similarly ineffective against KITD816V [255]. Other studies have, however, shown a more promising activity of nilo-tinib against kinase domain KIT mutations [256]. In contrast, dasatinib has shown potent anti-KIT activity in mast cell and leukaemia cell lines with different KIT mutants including those with D816V [257, 258]. In the largest study of dasatinib therapy in SM [259], the drug was given at a starting dose of 70 mg PO bid to 33 SM patients: 18 indolent, 9 aggressive and 6 with associated non-mast cell myeloid neoplasm. Two (6%) patients, both of whom were D816V-negative, achieved complete remission. Nine (27%) patients experienced symptomatic improvement. The results of another case report series of four patients with SM treated with dasatinib were equally unimpressive [260].
PKC412 has shown in vitro activity against transformed and primary cells with kinase domain KIT mutants (D816Y and D816V) [261, 262]. In a preliminary report, transient improvement in liver function test abnormalities, peripheral blood mast cell percentage and plasma histamine levels were seen in a patient with SM treated with PKC412 [263]. Similarly, oral PKC412 (100 mg bid) was administered to 15 SM patients (60% with detectable KITD816V) with evidence of clinical activity in 11 (73%) patients including increase in haemoglobin (n= 1) and reduction of ascites (n= 2) or pleural effusion (n = 2). In four patients, the bone marrow mast cell burden was reported to have decreased from 50–60% to 10–15% range.
PDGFR mutations
Both platelet-derived growth factor receptors a (PDGFRA located on chromosome 4q12) and p (PDGFRB located on chromosome 5q31-q32) are involved in MPN-relevant activating mutations. Clinical phenotype in both instances includes prominent blood eosinophilia and excellent response to IM therapy.
PDGFRA mutations
In regards to PDGFRA mutations, the most intensively studied has been the FIP1L1-PDGFRA, a karyotypically occult del(4)(q12), that was described in 2003 as an imatinib-sensitive activating mutation [12]. Subsequent studies have demonstrated the stem cell origin of the particular mutation [264, 265] and functional studies have demonstrated transforming properties in cell lines and the induction of MPD in mice [266, 267]. Cloning of the FIP1L1-PDGFRA fusion gene identified a novel molecular mechanism for generating this constitutively active fusion tyrosine kinase, wherein a ∼800 kb interstitial deletion within 4q12 fuses the 5’ portion of FIP1L1 to the 3’ portion of PDGFRA [12]. Molecular studies have shown that the breakpoint in FIP1L1 is relatively promiscuous, whereas the PDGFRA breakpoint is restricted to exon 12 that encodes part of the protein-protein interaction module with two fully conserved tryptophans (WW domain) containing the JM region with resultant disruption of its autoinhibitory activity [268]. Further biochemical analysis has shown that, in contrast with most tyrosine kinase fusions associated with human cancers, the FIP1L1 encoded sequences are dispensable for transformation, and there is no requirement for a dimerization motif; disruption of the autoinhibitory juxtamembrane motif as an invariant consequence of disruption of exon 12 is the basis for constitutive activation of PDGFRA kinase activity [269].
FIP1L1-PDGFRA occurs in a very small subset of patients who present with the phenotypic features of either SM or HES but the presence of the mutation reliably predicts complete haematologic and molecular response to imatinib therapy [67, 69, 270]. FIP1L1-PDGFRA mutation (T674I) that is homologous to the resistance-inducing, ‘gatekeeper’ T315I mutation in BCR-ABL has been described [12, 271, 272] and in vitro salvage with other kinase inhibitors including PKC412 [266] and sorafenib [273] has been demonstrated whereas such activity has not been conclusively shown for nilotinib [274, 275].
PDGFRA activation associated with CEL has also been described with karyotypically apparent fusion mutations including KIF5B-PDGFRA, t(4: 10)(q12; p11) [276], BCR-PDGFRA, t(4; 22)(q12; q11) [277] and CDK5RAP2-PDGFRA, ins(9; 4)(q33; q12q25) [278]. In the former instance, the breakpoints involved exon 3 of the kinesin family member 5B and exon 12 of PDGFRA resulting in an in-frame fusion. The patient achieved complete haematological and molecular remission with imatinib therapy [276]. BCR-PDGFRA represents an in-frame fusion with BCR breakpoints in intron 7/exon 12/exon 1/exon 17 and PDGFRAbreakpoint in exon 12/exon 13 and is also sensitive to imatinib therapy [277, 279, 280]. CDK5RAP2-PDGFRA also represents an imatinib-sensitive in-frame fusion involving exon 13 of CDK5RAP2 and intron 9/exon 12 of PDGFRA [278]. As is the case with FIP1L1-PDGFRA, currently known PDGFRA breakpoints are noted to be tightly clustered in the JM region, which once again highlights a key regulatory role for this domain.
PDGFRB mutations
The association between eosinophilic myeloid malignancies and PDGFRB rearrangement was first characterized and published in 1994 where fusion of the tyrosine kinase encoding region of PDGFRB to the ets- like gene, ETV6 (ETV6-PDGFRB, t(5; 12)(q33; p13) was demonstrated [11]. The fusion protein was transforming to cell lines and resulted in constitutive activation of PDGFRB signalling [281]. Since then, several other PDGFRB fusion transcripts with similar disease phenotypes have been described [282–290], cell line transformation [287–289] and MPD-induction in mice has been demonstrated [287], and imatinib therapy was effective when employed [23, 283, 284, 286, 290]. Additional evidence regarding the oncogenicity of activated PDGFRB comes from experiments with mice where either ETV6-PDGFRB or H4-PDGFRB induced lymphoblastic lymphoma [289, 291]. In most of these mutations, PDGFRB is fused to the N-terminal segment of a partner protein that encodes for one or more oligomerization domains.
FGFR1 mutations
There are at least four members of the FGFR protein family (FGFR1, FGFR2, FGFR3 and FGFR4) with additional variants resulting from alternative splicing. Structurally, FGFRs feature extracellular, trans-membrane, juxta-membrane and kinase domains. FGFRs are functionally important in embryonal development and tissue integrity and germline FGFR mutations result in various birth defects including achondroplasia/hypochondroplasia (FGFR3), Crouzon syndrome (FGFR2) and Kallmann syndrome (FGFR1). Somatic FGFR3 mutations have been described in benign skin tumours, urothelial cancer and multiple myeloma and FGFR2 mutations in endometrial carcinoma. In the context of myeloid malignancies, somatic FGFR1 mutations have been specifically associated with SCLL (also known as the 8p11 myeloproliferative syndrome).
SCLL is an extremely rare disease usually characterized by a mixed myeloid and lymphoid phenotype. The typical presentation is one of aggressive lymphoblastic lymphoma (usually T cell), eosinophilia, monocytosis and other blood and bone marrow evidence of clonal myeloproliferation. The disease rapidly progresses into acute leukaemia (usually myeloid). The hallmark of SCLL is a cytogenetic translocation between chromosome 8p11 and a spectrum of partner chromosomes as outlined in Table 2; these translocations result in fusion oncogenes that always involve exon 9 of FGFR1, located on 8p11 (Table 2) [292–307].
In SCLL, FGFR1 mutation is present in both myeloid and lymphoid lineage cells and some of the associated fusion genes have been shown transform cell lines [301, 308–310] and induce SCLL (ZNF198-FGFR1) [311] or CML (BCR-FGFR1) [310] like disease in mice. Consistent with this laboratory observation, some patients with BCR-FGFR1 mutation manifest a more indolent, compared to SCLL, disease phenotype [301]. The mechanism of FGFR1 activation in SCLL is similar, in most cases, to that seen with PDGFRB-associated CEL; the tyrosine kinase domain of FGFR1 is juxtaposed to a dimerization domain from the partner gene, although oligomerization motifs are not always identified in the partner gene [307].
SCLL is refractory to usual chemotherapy and some (e.g. PKC412) but not other (e.g. imatinib) kinase inhibitors have been shown to inhibit in vitro kinase activity as well as cell proliferation induced by ZNF198-FGFR1[301, 312]. Haematopoietic stem cell expression of ZNF198-FGFR1 in mice induces an MPN phenotype as well as activation of the downstream effector molecules PLC-7, STAT5 and phosphatidylinositol 3-kinase/AKT [312]. PKC412 (N-benzoyl-staurosporine) is a small molecule tyrosine kinase inhibitor with activity against FLT3, protein kinase C, CDC2 and receptors for PDGF, FGF and VEGF. The drug has been shown to inhibit ZNF198-FGFR1 kinase and downstream effector activity reduce proliferation of ZNF198-FGFR1 transformed cell lines and favourably affect survival of mice with ZNF198-FGFR1-induced MPN [312]. Whether or not the drug will work in human beings with SCLL is not currently clear although one PKC412-treated patient experienced an improvement in leukocytosis, lymphadenopathy and splenomegaly over 6 months before receiving allogeneic stem cell transplant [312].
References
- 1.Vardiman JW, Brunning RD, Harris NL. In: World Health Organization classification of tumors: tumours of the haematopoietic and lymphoid tissues. Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. International Agency for Research on Cancer (IARC) Press; 2001. pp. 17–44. [Google Scholar]
- 2.Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6:372–75. [PubMed] [Google Scholar]
- 3.Tefferi A. The history of myeloproliferative disorders: before and after Dameshek. Leukemia. 2008;22:3–13. doi: 10.1038/sj.leu.2404946. [DOI] [PubMed] [Google Scholar]
- 4.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 5.Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109. [PubMed] [Google Scholar]
- 6.Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science. 1960;132:1497. [Google Scholar]
- 7.Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myel-ogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci USA. 1990;87:6649–53. doi: 10.1073/pnas.87.17.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–30. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 9.Elefanty AG, Hariharan IK, Cory S. bcr-abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. EMBO J. 1990;9:1069–78. doi: 10.1002/j.1460-2075.1990.tb08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736–44. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5; 12) chromosomal translocation. Cell. 1994;77:307–16. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 12.Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, Tefferi A, Malone J, Alam R, Schrier SL, Schmid J, Rose M, Vandenberghe P, Verhoef G, Boogaerts M, Wlodarska I, Kantarjian H, Marynen P, Coutre SE, Stone R, Gilliland DG. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–14. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 13.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 14.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 15.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 16.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 17.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, Deangelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litzow MR, Gilliland DG, Tefferi A. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–6. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 20.Pardanani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinicopatho-logic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia. 2007;21:1960–3. doi: 10.1038/sj.leu.2404810. [DOI] [PubMed] [Google Scholar]
- 21.Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, Ferrari M, Gisslinger H, Kralovics R, Cremonesi L, Skoda R, Cazzola M. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111:1686–9. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 22.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 23.Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, Chase A, Chessells JM, Colombat M, Dearden CE, Dimitrijevic S, Mahon FX, Marin D, Nikolova Z, Olavarria E, Silberman S, Schultheis B, Cross NC, Goldman JM. Response to imatinib mesy-late in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347:481–7. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- 24.Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci USA. 1967;58:1468–71. doi: 10.1073/pnas.58.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myeloge-nous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–3. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 26.Heisterkamp N, Groffen J, Stephenson JR, Spurr NK, Goodfellow PN, Solomon E, Carritt B, Bodmer WF. Chromosomal localization of human cellular homologues of two viral oncogenes. Nature. 1982;299:747–9. doi: 10.1038/299747a0. [DOI] [PubMed] [Google Scholar]
- 27.de Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, Bootsma D, Spurr NK, Heisterkamp N, Groffen J, Stephenson JR. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765–7. doi: 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 28.Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–9. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- 29.Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature. 1985;315:758–61. doi: 10.1038/315758a0. [DOI] [PubMed] [Google Scholar]
- 30.Caracciolo D, Valtieri M, Venturelli D, Peschle C, Gewirtz AM, Calabretta B. Lineage-specific requirement of c-abl function in normal hematopoiesis. Science. 1989;245:1107–10. doi: 10.1126/science.2672339. [DOI] [PubMed] [Google Scholar]
- 31.Hernandez SE, Krishnaswami M, Miller AL, Koleske AJ. How do Abl family kinases regulate cell shape and movement? Trends Cell Biol. 2004;14:36–44. doi: 10.1016/j.tcb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida K, Miki Y. Enabling death by the Abl tyrosine kinase: mechanisms for nuclear shuttling of c-Abl in response to DNA damage. Cell Cycle. 2005;4:777–9. doi: 10.4161/cc.4.6.1746. [DOI] [PubMed] [Google Scholar]
- 33.Pendergast AM. Stress and death: breaking up the c-Abl/14–3-3 complex in apop-tosis. Nat Cell Biol. 2005;7:213–4. doi: 10.1038/ncb0305-213. [DOI] [PubMed] [Google Scholar]
- 34.Wei G, Li AG, Liu X. Insights into selective activation of p53 DNA binding by c-Abl. J Biol Chem. 2005;280:12271–8. doi: 10.1074/jbc.M409522200. [DOI] [PubMed] [Google Scholar]
- 35.Van Etten RA. Mechanisms of transformation by the BCR-ABL oncogene: new perspectives in the post-imatinib era. Leuk Res. 2004;28:S21–8. doi: 10.1016/j.leukres.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Bernards A, Rubin CM, Westbrook CA, Paskind M, Baltimore D. The first intron in the human c-abl gene is at least 200 kilobases long and is a target for translocations in chronic myelogenous leukemia. Mol Cell Biol. 1987;7:3231–6. doi: 10.1128/mcb.7.9.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melo JV. BCR-ABL gene variants. Baillieres Clin Haematol. 1997;10:203–22. doi: 10.1016/s0950-3536(97)80003-0. [DOI] [PubMed] [Google Scholar]
- 38.Karlic H, Grill R, Schlogl E. Minor BCR (m-bcr) rearrangements may appear in major BCR (M-bcr)-positive CML cases. Hematol Pathol. 1992;6:203–7. [PubMed] [Google Scholar]
- 39.Costello RT, Gabert J, Brunel V, Sainty D, Arnoulet C, Mozziconacci MJ, Camerlo J, Perret C, Gastaut JA, Bouabdallah R. Minor breakpoint cluster region (m-BCR) positive chronic myeloid leukaemia with an acute lymphoblastic leukaemia onset: a case report. Br J Haematol. 1995;91:428–30. doi: 10.1111/j.1365-2141.1995.tb05317.x. [DOI] [PubMed] [Google Scholar]
- 40.Saglio G, Guerrasio A, Rosso C, Zaccaria A, Tassinari A, Serra A, Rege-Cambrin G, Mazza U, Gavosto F. New type of Bcr/Abl junction in Philadelphia chromosome-positive chronic myelogenous leukemia. Blood. 1990;76:1819–24. [PubMed] [Google Scholar]
- 41.Tefferi A, Bren GD, Wagner KV, Schaid DJ, Ash RC, Thibodeau SN. The location of the Philadelphia chromosomal breakpoint site and prognosis in chronic granulocytic leukemia. Leukemia. 1990;4:839–42. [PubMed] [Google Scholar]
- 42.Stam K, Heisterkamp N, Grosveld G, de Klein A, Verma RS, Coleman M, Dosik H, Groffen J. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N Engl J Med. 1985;313:1429–33. doi: 10.1056/NEJM198512053132301. [DOI] [PubMed] [Google Scholar]
- 43.Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–4. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- 44.Konopka JB, Witte ON. Detection of c-abl tyrosine kinase activity in vitro permits direct comparison of normal and altered abl gene products. Mol Cell Biol. 1985;5:3116–23. doi: 10.1128/mcb.5.11.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl onco-gene products. Science. 1990;247:1079–82. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 46.Pendergast AM, Muller AJ, Havlik MH, Maru Y, Witte ON. BCR sequences essential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine-dependent manner. Cell. 1991;66:161–71. doi: 10.1016/0092-8674(91)90148-r. [DOI] [PubMed] [Google Scholar]
- 47.Sattler M, Salgia R, Okuda K, Uemura N, Durstin MA, Pisick E, Xu G, Li JL, Prasad KV, Griffin JD. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3’ kinase pathway. Oncogene. 1996;12:839–46. [PubMed] [Google Scholar]
- 48.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–83. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 49.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 50.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, Lydon NB. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-pheny-laminopyrimidine derivative. Cancer Res. 1996;56:100–4. [PubMed] [Google Scholar]
- 51.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Regenass U, Lydon NB. Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2- phenylaminopyrimidine class. Proc Natl Acad Sci USA. 1995;92:2558–62. doi: 10.1073/pnas.92.7.2558. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Okuda K, Weisberg E, Gilliland DG, Griffin JD. ARG tyrosine kinase activity is inhibited by STI571. Blood. 2001;97:2440–8. doi: 10.1182/blood.v97.8.2440. [DOI] [PubMed] [Google Scholar]
- 53.Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96:925–32. [PubMed] [Google Scholar]
- 54.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 55.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy, safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 56.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 57.Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, Talpaz M, Druker B, Goldman J, O’Brien SG, Russell N, Fischer T, Ottmann O, Cony-Makhoul P, Facon T, Stone R, Miller C, Tallman M, Brown R, Schuster M, Loughran T, Gratwohl A, Mandelli F, Saglio G, Lazzarino M, Russo D, Baccarani M, Morra E. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–52. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- 58.Talpaz M, Silver RT, Druker BJ, Goldman JM, Gambacorti-Passerini C, Guilhot F, Schiffer CA, Fischer T, Deininger MW, Lennard AL, Hochhaus A, Ottmann OG, Gratwohl A, Baccarani M, Stone R, Tura S, Mahon FX, Fernandes-Reese S, Gathmann I, Capdeville R, Kantarjian HM, Sawyers CL. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99:1928–37. doi: 10.1182/blood.v99.6.1928. [DOI] [PubMed] [Google Scholar]
- 59.Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW, Fischer T, O’Brien SG, Stone RM, Gambacorti-Passerini CB, Russell NH, Reiffers JJ, Shea TC, Chapuis B, Coutre S, Tura S, Morra E, Larson RA, Saven A, Peschel C, Gratwohl A, Mandelli F, Ben-Am M, Gathmann I, Capdeville R, Paquette RL, Druker BJ. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–9. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 60.Kantarjian HM, Cortes J, O’Brien S, Giles FJ, Albitar M, Rios MB, Shan J, Faderl S, Garcia-Manero G, Thomas DA, Resta D, Talpaz M. Imatinib mesylate (STI571) therapy for Philadelphia chromosome-positive chronic myelogenous leukemia in blast phase. Blood. 2002;99:3547–53. doi: 10.1182/blood.v99.10.3547. [DOI] [PubMed] [Google Scholar]
- 61.Kantarjian HM, O’Brien S, Cortes JE, Smith TL, Rios MB, Shan J, Yang Y, Giles FJ, Thomas DA, Faderl S, Garcia-Manero G, Jeha S, Wierda W, Issa JP, Kornblau SM, Keating M, Resta D, Capdeville R, Talpaz M. Treatment of philadelphia chromosome-positive, accelerated-phase chronic myelogenous leukemia with imatinib mesylate. Clin Cancer Res. 2002;8:2167–76. [PubMed] [Google Scholar]
- 62.Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, Tura S, Fischer T, Deininger MW, Schiffer CA, Baccarani M, Gratwohl A, Hochhaus A, Hoelzer D, Fernandes-Reese S, Gathmann I, Capdeville R, O’Brien SG. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100:1965–71. doi: 10.1182/blood-2001-12-0181. [DOI] [PubMed] [Google Scholar]
- 63.van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, Martens M, Webb A, Sciot R, Van Glabbeke M, Silberman S, Nielsen OS. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358:1421–3. doi: 10.1016/s0140-6736(01)06535-7. [DOI] [PubMed] [Google Scholar]
- 64.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stro-mal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 65.Gleich GJ, Leiferman KM, Pardanani A, Tefferi A, Butterfield JH. Treatment of hypereosinophilic syndrome with imatinib mesilate. Lancet. 2002;359:1577–8. doi: 10.1016/S0140-6736(02)08505-7. [DOI] [PubMed] [Google Scholar]
- 66.Pardanani A, Reeder T, Porrata LF, Li CY, Tazelaar HD, Baxter EJ, Witzig TE, Cross NC, Tefferi A. Imatinib therapy for hypere-osinophilic syndrome and other eosinophilic disorders. Blood. 2003;101:3391–7. doi: 10.1182/blood-2002-10-3103. [DOI] [PubMed] [Google Scholar]
- 67.Pardanani A, Elliott M, Reeder T, Li C-Y, Baxter EJ, Cross NCP, Tefferi A. Imatinib for systemic mast-cell disease. Lancet. 2003;362:535–6. doi: 10.1016/s0140-6736(03)14115-3. [DOI] [PubMed] [Google Scholar]
- 68.Pardanani A, Ketterling RP, Brockman SR, Flynn HC, Paternoster SF, Shearer BM, Reeder TL, Li CY, Cross NCP, Cools J, Gilliland DG, Dewald GW, Tefferi A. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mas-tocytosis associated with eosinophilia and predicts response to imatinib therapy. Blood. 2003;102:3093–6. doi: 10.1182/blood-2003-05-1627. [DOI] [PubMed] [Google Scholar]
- 69.Pardanani A, Brockman SR, Paternoster SF, Flynn HC, Ketterling RP, Lasho TL, Ho CL, Li CY, Dewald GW, Tefferi A. FIP1L1-PDGFRA fusion: prevalence and clinico-pathologic correlates in 89 consecutive patients with moderate to severe eosinophilia. Blood. 2004;104:3038–45. doi: 10.1182/blood-2004-03-0787. [DOI] [PubMed] [Google Scholar]
- 70.Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, Apperley J, Cervantes F, Cortes J, Deininger M, Gratwohl A, Guilhot F, Horowitz M, Hughes T, Kantarjian H, Larson R, Niederwieser D, Silver R, Hehlmann R. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108:1809–20. doi: 10.1182/blood-2006-02-005686. [DOI] [PubMed] [Google Scholar]
- 71.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 72.Azam M, Daley GQ. Anticipating clinical resistance to target-directed agents: the BCR-ABL paradigm. Mol Diagn Ther. 2006;10:67–76. doi: 10.1007/BF03256446. [DOI] [PubMed] [Google Scholar]
- 73.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8:1018–29. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 74.O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–9. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 75.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 76.Azam M, Nardi V, Shakespeare WC, Metcalf CA, 3rd, Bohacek RS, Wang Y, Sundaramoorthi R, Sliz P, Veach DR, Bornmann WG, Clarkson B, Dalgarno DC, Sawyer TK, Daley GQ. Activity of dual SRC-ABL inhibitors highlights the role of BCR/ABL kinase dynamics in drug resistance. Proc Natl Acad Sci USA. 2006;103:9244–9. doi: 10.1073/pnas.0600001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, Cortes J, Alland L, Ottmann OG. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–51. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 78.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, Blackwood-Chirchir MA, Iyer V, Chen TT, Huang F, Decillis AP, Sawyers CL. Dasatinib in ima-tinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 79.Druker BJ. Circumventing resistance to kinase-inhibitor therapy. N Engl J Med. 2006;354:2594–6. doi: 10.1056/NEJMe068073. [DOI] [PubMed] [Google Scholar]
- 80.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, Floyd M, Ford JM, Grotzfeld RM, Herrgard S, Insko DE, Mehta SA, Patel HK, Pao W, Sawyers CL, Varmus H, Zarrinkar PP, Lockhart DJ. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA. 2005;102:11011–6. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, Warmuth M. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on hedgehog pathway activation. Cancer Cell. 2008;14:238–49. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 82.Walz C, Cross NC, Van Etten RA, Reiter A. Comparison of mutated ABL1 and JAK2 as oncogenes and drug targets in myeloproliferative disorders. Leukemia. 2008;22:1320–34. doi: 10.1038/leu.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hannemann JR, Healy LE, Ridge SA, Wiedemann LM. The second ETV6 allele is not necessarily deleted in acute leukemias with a ETV6/ABL fusion. Genes Chromosomes Cancer. 1998;21:256–9. doi: 10.1002/(sici)1098-2264(199803)21:3<256::aid-gcc11>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 84.Papadopoulos P, Ridge SA, Boucher CA, Stocking C, Wiedemann LM. The novel activation of ABL by fusion to an ets-related gene, TEL. Cancer Res. 1995;55:34–8. [PubMed] [Google Scholar]
- 85.Van Limbergen H, Beverloo HB, van Drunen E, Janssens A, Hahlen K, Poppe B, Van Roy N, Marynen P, De Paepe A, Slater R, Speleman F. Molecular cytogenetic and clinical findings in ETV6/ABL1-positive leukemia. Genes Chromosomes Cancer. 2001;30:274–82. [PubMed] [Google Scholar]
- 86.Griesinger F, Janke A, Podleschny M, Bohlander SK. Identification of an ETV6-ABL2 fusion transcript in combination with an ETV6 point mutation in a T-cell acute lymphoblastic leukaemia cell line. Br J Haematol. 2002;119:454–8. doi: 10.1046/j.1365-2141.2002.03850.x. [DOI] [PubMed] [Google Scholar]
- 87.Golub TR, Goga A, Barker GF, Afar DE, McLaughlin J, Bohlander SK, Rowley JD, Witte ON, Gilliland DG. Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol Cell Biol. 1996;16:4107–16. doi: 10.1128/mcb.16.8.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O’Brien SG, Vieira SA, Connors S, Bown N, Chang J, Capdeville R, Melo JV. Transient response to imatinib mesylate (STI571) in a patient with the ETV6-ABL t(9; 12) translocation. Blood. 2002;99:3465–7. doi: 10.1182/blood.v99.9.3465. [DOI] [PubMed] [Google Scholar]
- 89.La Starza R, Trubia M, Testoni N, Ottaviani E, Belloni E, Crescenzi B, Martelli M, Flandrin G, Pelicci PG, Mecucci C. Clonal eosinophils are a morphologic hallmark of ETV6/ABL1 positive acute myeloid leukemia. Haematologica. 2002;87:789–94. [PubMed] [Google Scholar]
- 90.Andreasson P, Johansson B, Carlsson M, Jarlsfelt I, Fioretos T, Mitelman F, Hoglund M. BCR/ABL-negative chronic myeloid leukemia with ETV6/ABL fusion. Genes Chromosomes Cancer. 1997;20:299–304. [PubMed] [Google Scholar]
- 91.Barbouti A, Ahlgren T, Johansson B, Hoglund M, Lassen C, Turesson I, Mitelman F, Fioretos T. Clinical and genetic studies of ETV6/ABL1-positive chronic myeloid leukaemia in blast crisis treated with imatinib mesylate. Br J Haematol. 2003;122:85–93. doi: 10.1046/j.1365-2141.2003.04391.x. [DOI] [PubMed] [Google Scholar]
- 92.Keung YK, Beaty M, Steward W, Jackle B, Pettnati M. Chronic myelocytic leukemia with eosinophilia, t(9; 12)(q34; p13), and ETV6-ABL gene rearrangement: case report and review of the literature. Cancer Genet Cytogenet. 2002;138:139–42. doi: 10.1016/s0165-4608(02)00609-x. [DOI] [PubMed] [Google Scholar]
- 93.Million RP, Aster J, Gilliland DG, Van Etten RA. The Tel-Abl (ETV6-Abl) tyrosine kinase, product of complex (9; 12) translo-cations in human leukemia, induces distinct myeloproliferative disease in mice. Blood. 2002;99:4568–77. doi: 10.1182/blood-2001-12-0244. [DOI] [PubMed] [Google Scholar]
- 94.Wilks AF. The JAK kinases: not just another kinase drug discovery target. Semin Cell Dev Biol. 2008 doi: 10.1016/j.semcdb.2008.07.020. in press. [DOI] [PubMed] [Google Scholar]
- 95.Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zurcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057–65. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luo H, Rose P, Barber D, Hanratty WP, Lee S, Roberts TM, D’Andrea AD, Dearolf CR. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol Cell Biol. 1997;17:1562–71. doi: 10.1128/mcb.17.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–63. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 98.Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ, Yoo NJ, Lee SH. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14:3716–21. doi: 10.1158/1078-0432.CCR-07-4839. [DOI] [PubMed] [Google Scholar]
- 99.Tomasson MH, Xiang Z, Walgren R, Zhao Y, Kasai Y, Miner T, Ries RE, Lubman O, Fremont DH, McLellan MD, Payton JE, Westervelt P, DiPersio JF, Link DC, Walter MJ, Graubert TA, Watson M, Baty J, Heath S, Shannon WD, Nagarajan R, Bloomfield CD, Mardis ER, Wilson RK, Ley TJ. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood. 2008;111:4797–808. doi: 10.1182/blood-2007-09-113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, Ariola C, Fodale V, Clappier E, Paoloni F, Martinelli S, Fragale A, Sanchez M, Tavolaro S, Messina M, Cazzaniga G, Camera A, Pizzolo G, Tornesello A, Vignetti M, Battistini A, Cave H, Gelb BD, Renauld JC, Biondi A, Constantinescu SN, Foa R, Tartaglia M. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205:751–8. doi: 10.1084/jem.20072182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xiang Z, Zhao Y, Mitaksov V, Fremont DH, Kasai Y, Molitoris A, Ries RE, Miner TL, McLellan MD, DiPersio JF, Link DC, Payton JE, Graubert TA, Watson M, Shannon W, Heath SE, Nagarajan R, Mardis ER, Wilson RK, Ley TJ, Tomasson MH. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood. 2008;111:4809–12. doi: 10.1182/blood-2007-05-090308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walters DK, Mercher T, Gu TL, O’Hare T, Tyner JW, Loriaux M, Goss VL, Lee KA, Eide CA, Wong MJ, Stoffregen EP, McGreevey L, Nardone J, Moore SA, Crispino J, Boggon TJ, Heinrich MC, Deininger MW, Polakiewicz RD, Gilliland DG, Druker BJ. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10:65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 103.Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, Gilliland DG, Tefferi A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both 殿typical_ myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005;106:1207–9. doi: 10.1182/blood-2005-03-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Steensma DP, McClure RF, Karp JE, Tefferi A, Lasho TL, Powell HL, DeWald GW, Kaufmann SH. JAK2 V617F is a rare finding in de novoacute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia. 2006;20:971–8. doi: 10.1038/sj.leu.2404206. [DOI] [PubMed] [Google Scholar]
- 105.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, Cario G, Schrappe M, Stanulla M, Strehl S, Haas OA, Mann G, Binder V, Borkhardt A, Kempski H, Trka J, Bielorei B, Avigad S, Stark B, Smith O, Dastugue N, Bourquin JP, Tal NB, Green AR, Izraeli S. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet. 2008;372:1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 106.Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, Monpoux F, Van Rompaey L, Baens M, Van den Berghe H, Marynen P. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9; 12) in a lymphoid and t(9; 15; 12) in a myeloid leukemia. Blood. 1997;90:2535–40. [PubMed] [Google Scholar]
- 107.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–12. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 108.Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B, Berger U, Telford N, Aruliah S, Yin JA, Vanstraelen D, Barker HF, Taylor PC, O’Driscoll A, Benedetti F, Rudolph C, Kolb HJ, Hochhaus A, Hehlmann R, Chase A, Cross NC. The t(8; 9)(p22; p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65:2662–7. doi: 10.1158/0008-5472.CAN-04-4263. [DOI] [PubMed] [Google Scholar]
- 109.Murati A, Gelsi-Boyer V, Adelaide J, Perot C, Talmant P, Giraudier S, Lode L, Letessier A, Delaval B, Brunel V, Imbert M, Garand R, Xerri L, Birnbaum D, Mozziconacci MJ, Chaffanet M. PCM1-JAK2 fusion in myeloproliferative disorders and acute erythroid leukemia with t(8; 9) translocation. Leukemia. 2005;19:1692–6. doi: 10.1038/sj.leu.2403879. [DOI] [PubMed] [Google Scholar]
- 110.Bousquet M, Quelen C, De Mas V, Duchayne E, Roquefeuil B, Delsol G, Laurent G, Dastugue N, Brousset P. The t(8; 9)(p22; p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene. 2005;24:7248–52. doi: 10.1038/sj.onc.1208850. [DOI] [PubMed] [Google Scholar]
- 111.Adelaide J, Perot C, Gelsi-Boyer V, Pautas C, Murati A, Copie-Bergman C, Imbert M, Chaffanet M, Birnbaum D, Mozziconacci MJ. A t(8; 9) translocation with PCM1-JAK2 fusion in a patient with T-cell lymphoma. Leukemia. 2006;20:536–7. doi: 10.1038/sj.leu.2404104. [DOI] [PubMed] [Google Scholar]
- 112.Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, Wormann B, Haase D, Bohlander SK. A BCR-JAK2 fusion gene as the result of a t(9; 22)(p24; q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44:329–33. doi: 10.1002/gcc.20235. [DOI] [PubMed] [Google Scholar]
- 113.Poitras JL, Dal Cin P, Aster JC, DeAngelo DJ, Morton CC. Novel SSBP2-JAK2 fusion gene resulting from a t(5; 9)(q14.1; p24.1) in pre-B acute lymphocytic leukemia. Genes Chromosomes Cancer. 2008;47:884–9. doi: 10.1002/gcc.20585. [DOI] [PubMed] [Google Scholar]
- 114.Cirmena G, Aliano S, Fugazza G, Bruzzone R, Garuti A, Bocciardi R, Bacigalupo A, Ravazzolo R, Ballestrero A, Sessarego M. A BCR-JAK2 fusion gene as the result of a t(9; 22)(p24; q11) in a patient with acute myeloid leukemia. Cancer Genet Cytogenet. 2008;183:105–8. doi: 10.1016/j.cancergencyto.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 115.Carron C, Cormier F, Janin A, Lacronique V, Giovannini M, Daniel MT, Bernard O, Ghysdael J. TEL-JAK2 transgenic mice develop T-cell leukemia. Blood. 2000;95:3891–9. [PubMed] [Google Scholar]
- 116.dos Santos NR, Ghysdael J. A transgenic mouse model for TEL-JAK2-induced B-cell lymphoma/leukemia. Leukemia. 2006;20:182–5. doi: 10.1038/sj.leu.2404026. [DOI] [PubMed] [Google Scholar]
- 117.Kennedy JA, Barabe F, Patterson BJ, Bayani J, Squire JA, Barber DL, Dick JE. Expression of TEL-JAK2 in primary human hematopoietic cells drives erythropoietin-independent erythropoiesis and induces myelofibrosis in vivo. Proc Natl Acad Sci USA. 2006;103:16930–5. doi: 10.1073/pnas.0604902103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Poitras JL, Cin PD, Aster JC, Deangelo DJ, Morton CC. Novel SSBP2-JAK2 fusion gene resulting from a t(5; 9)(q14.1; p24.1) in pre-B acute lymphocytic leukemia. Genes Chromosomes Cancer. 2008;47:884–9. doi: 10.1002/gcc.20585. [DOI] [PubMed] [Google Scholar]
- 119.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, White H, Zoi C, Loukopoulos D, Terpos E, Vervessou EC, Schultheis B, Emig M, Ernst T, Lengfelder E, Hehlmann R, Hochhaus A, Oscier D, Silver RT, Reiter A, Cross NC. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–8. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 120.Melzner I, Weniger MA, Menz CK, Moller P. Absence of the JAK2 V617F activating mutation in classical Hodgkin lymphoma and primary mediastinal B-cell lymphoma. Leukemia. 2006;20:157–8. doi: 10.1038/sj.leu.2404036. [DOI] [PubMed] [Google Scholar]
- 121.Lee JW, Soung YH, Kim SY, Nam SW, Park WS, Lee JY, Yoo NJ, Lee SH. JAK2 V617F mutation is uncommon in non-Hodgkin lymphomas. Leuk Lymphoma. 2006;47:313–4. doi: 10.1080/10428190500271723. [DOI] [PubMed] [Google Scholar]
- 122.Sulong S, Case M, Minto L, Wilkins B, Hall A, Irving J. The V617F mutation in Jak2 is not found in childhood acute lym-phoblastic leukaemia. Br J Haematol. 2005;130:964–5. doi: 10.1111/j.1365-2141.2005.05697.x. [DOI] [PubMed] [Google Scholar]
- 123.Levine RL, Loriaux M, Huntly BJ, Loh ML, Beran M, Stoffregen E, Berger R, Clark JJ, Willis SG, Nguyen KT, Flores NJ, Estey E, Gattermann N, Armstrong S, Look AT, Griffin JD, Bernard OA, Heinrich MC, Gilliland DG, Druker B, Deininger MW. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377–9. doi: 10.1182/blood-2005-05-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee JW, Soung YH, Kim SY, Nam SW, Park WS, Lee JY, Yoo NJ, Lee SH. Absence of JAK2 V617F mutation in gastric cancers. Acta Oncol. 2006;45:222–3. doi: 10.1080/02841860500341223. [DOI] [PubMed] [Google Scholar]
- 125.Lee JW, Kim YG, Soung YH, Han KJ, Kim SY, Rhim HS, Min WS, Nam SW, Park WS, Lee JY, Yoo NJ, Lee SH. The JAK2 V617F mutation in de novoacute myelogenous leukemias. Oncogene. 2006;25:1434–6. doi: 10.1038/sj.onc.1209163. [DOI] [PubMed] [Google Scholar]
- 126.Scott LM, Campbell PJ, Baxter EJ, Todd T, Stephens P, Edkins S, Wooster R, Stratton MR, Futreal PA, Green AR. The V617F JAK2 mutation is uncommon in cancers and in myeloid malignancies other than the classic myeloproliferative disorders. Blood. 2005;106:2920–1. doi: 10.1182/blood-2005-05-2087. [DOI] [PubMed] [Google Scholar]
- 127.Tefferi A, Sirhan S, Lasho TL, Schwager SM, Li CY, Dingli D, Wolanskyj AP, Steensma DP, Mesa R, Gilliland DG. Concomitant neutrophil JAK2 mutation screening and PRV-1 expression analysis in myeloproliferative disorders and secondary polycythaemia. Br J Haematol. 2005;131:166–71. doi: 10.1111/j.1365-2141.2005.05743.x. [DOI] [PubMed] [Google Scholar]
- 128.Kralovics R, Teo SS, Buser AS, Brutsche M, Tiedt R, Tichelli A, Passamonti F, Pietra D, Cazzola M, Skoda RC. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood. 2005;106:3374–6. doi: 10.1182/blood-2005-05-1889. [DOI] [PubMed] [Google Scholar]
- 129.Renneville A, Quesnel B, Charpentier A, Terriou L, Crinquette A, Lai JL, Cossement C, Lionne-Huyghe P, Rose C, Bauters F, Preudhomme C. High occurrence of JAK2 V617 mutation in refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Leukemia. 2006;20:2067–70. doi: 10.1038/sj.leu.2404405. [DOI] [PubMed] [Google Scholar]
- 130.Verstovsek S, Silver RT, Cross NC, Tefferi A. JAK2V617F mutational frequency in polycythemia vera: 100%, >90%, less? Leukemia. 2006;20:2067. doi: 10.1038/sj.leu.2404379. [DOI] [PubMed] [Google Scholar]
- 131.Vizmanos JL, Ormazabal C, Larrayoz MJ, Cross NC, Calasanz MJ. JAK2 V617F mutation in classic chronic myeloproliferative diseases: a report on a series of 349 patients. Leukemia. 2006;20:534–5. doi: 10.1038/sj.leu.2404086. [DOI] [PubMed] [Google Scholar]
- 132.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–81. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lacout C, Pisani DF, Tulliez M, Moreau Gachelin F, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–60. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 134.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, Skoda RC. Ratio of mutant JAK2-V617F to wild type JAK2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 135.Shide K, Shimoda HK, Kumano T, Karube K, Kameda T, Takenaka K, Oku S, Abe H, Katayose KS, Kubuki Y, Kusumoto K, Hasuike S, Tahara Y, Nagata K, Matsuda T, Ohshima K, Harada M, Shimoda K. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia. 2008;22:87–95. doi: 10.1038/sj.leu.2405043. [DOI] [PubMed] [Google Scholar]
- 136.Kittur J, Knudson RA, Lasho TL, Finke CM, Gangat N, Wolanskyj AP, Li CY, Wu W, Ketterling RP, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia. Cancer. 2007;109:2279–84. doi: 10.1002/cncr.22663. [DOI] [PubMed] [Google Scholar]
- 137.Tefferi A, Strand JJ, Lasho TL, Knudson RA, Finke CM, Gangat N, Pardanani A, Hanson CA, Ketterling RP. Bone marrow JAK2V617F allele burden and clinical correlates in polycythemia vera. Leukemia. 2007;21:2074–5. doi: 10.1038/sj.leu.2404724. [DOI] [PubMed] [Google Scholar]
- 138.Tefferi A, Lasho TL, Huang J, Finke C, Hanson CA, Mesa RA, Pardanani A. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, predicts inferior overall and leukemia-free survival. Leukemia. 2008;22:756–61. doi: 10.1038/sj.leu.2405097. [DOI] [PubMed] [Google Scholar]
- 139.Vannucchi AM, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, Marchioli R, Marfisi RM, Finazzi G, Guerini V, Fabris F, Randi ML, De Stefano V, Caberlon S, Tafuri A, Ruggeri M, Specchia G, Liso V, Rossi E, Pogliani E, Gugliotta L, Bosi A, Barbui T. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–6. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 140.Dupont S, Masse A, James C, Teyssandier I, Lecluse Y, Larbret F, Ugo V, Saulnier P, Koscielny S, Le Couedic JP, Casadevall N, Vainchenker W, Delhommeau F. The JAK2 617V>F mutation triggers erythropoietin hypersensitiv-ity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood. 2007;110:1013–21. doi: 10.1182/blood-2006-10-054940. [DOI] [PubMed] [Google Scholar]
- 141.Kralovics R, Passamonti F, Buser AS, Soon-Siong T, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain of function mutation in Jak2 is frequently found in patients with myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 142.Plo I, Nakatake M, Malivert L, de Villartay JP, Giraudier S, Villeval JL, Wiesmuller L, Vainchenker W. JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood. 2008;112:1402–12. doi: 10.1182/blood-2008-01-134114. [DOI] [PubMed] [Google Scholar]
- 143.Lippert E, Boissinot M, Kralovics R, Girodon F, Dobo I, Praloran V, Boiret-Dupre N, Skoda RC, Hermouet S. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108:1865–7. doi: 10.1182/blood-2006-01-013540. [DOI] [PubMed] [Google Scholar]
- 144.Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F JAK2 mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006;108:2435–7. doi: 10.1182/blood-2006-04-018259. [DOI] [PubMed] [Google Scholar]
- 145.Jamieson CH, Gotlib J, Durocher JA, Chao MP, Mariappan MR, Lay M, Jones C, Zehnder JL, Lilleberg SL, Weissman IL. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci USA. 2006;103:6224–9. doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tefferi A, Lasho TL, Gilliland G. JAK2 mutations in myeloproliferative disorders. N Engl J Med. 2005;353:1416–7. doi: 10.1056/NEJMc051878. [DOI] [PubMed] [Google Scholar]
- 147.Delhommeau F, Dupont S, Tonetti C, Masse A, Godin I, Le Couedic JP, Debili N, Saulnier P, Casadevall N, Vainchenker W, Giraudier S. Evidence that the JAK2 G1849T (V617F) mutation occurs in a lymphomyeloid progenitor in polycythemia vera and idiopathic myelofibrosis. Blood. 2007;109:71–7. doi: 10.1182/blood-2006-03-007146. [DOI] [PubMed] [Google Scholar]
- 148.Lasho TL, Mesa R, Gilliland DG, Tefferi A. Mutation studies in CD3+, CD19+ and CD34+ cell fractions in myeloproliferative disorders with homozygous JAK2(V617F) in granulocytes. Br J Haematol. 2005;130:797–9. doi: 10.1111/j.1365-2141.2005.05682.x. [DOI] [PubMed] [Google Scholar]
- 149.Bellanne-Chantelot C, Chaumarel I, Labopin M, Bellanger F, Barbu V, De Toma C, Delhommeau F, Casadevall N, Vainchenker W, Thomas G, Najman A. Genetic and clinical implications of the Val617Phe JAK2 mutation in 72 families with myeloproliferative disorders. Blood. 2006;108:346–52. doi: 10.1182/blood-2005-12-4852. [DOI] [PubMed] [Google Scholar]
- 150.Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic cell lineages by the JAK2V617F mutation in polycythemia vera. Blood. 2006;108:3128–34. doi: 10.1182/blood-2006-04-017392. [DOI] [PubMed] [Google Scholar]
- 151.Pardanani A, Lasho TL, Finke C, Mesa RA, Hogan WJ, Ketterling RP, Gilliland DG, Tefferi A. Extending Jak2V617F and MplW515 mutation analysis to single hematopoietic colonies and B and T lymphocytes. Stem Cells. 2007;25:2358–62. doi: 10.1634/stemcells.2007-0175. [DOI] [PubMed] [Google Scholar]
- 152.Kralovics R, Teo SS, Li S, Theocharides A, Buser AS, Tichelli A, Skoda RC. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377–80. doi: 10.1182/blood-2005-11-009605. [DOI] [PubMed] [Google Scholar]
- 153.Nussenzveig RH, Swierczek SI, Jelinek J, Gaikwad A, Liu E, Verstovsek S, Prchal JF, Prchal JT. Polycythemia vera is not initiated by JAK2(V617F) mutation. Exp Hematol. 2007;35:32–8. doi: 10.1016/j.exphem.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 154.Theocharides A, Boissinot M, Girodon F, Garand R, Teo SS, Lippert E, Talmant P, Tichelli A, Hermouet S, Skoda RC. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110:375–9. doi: 10.1182/blood-2006-12-062125. [DOI] [PubMed] [Google Scholar]
- 155.Campbell PJ, Baxter EJ, Beer PA, Scott LM, Bench AJ, Huntly BJP, Erber WN, Kusec R, Larsen TS, Glraudler S, Le Bousse-Kerdlles M-C, Grlesshammer M, Reilly JT, Cheung BY, Harrison CN, Green AR. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108:3548–55. doi: 10.1182/blood-2005-12-013748. [DOI] [PubMed] [Google Scholar]
- 156.Pardananl A, Frldley BL, Lasho TL, Gllllland DG, Tefferi A. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood. 2008;111:2785–9. doi: 10.1182/blood-2007-06-095703. [DOI] [PubMed] [Google Scholar]
- 157.Schnlttger S, Bacher U, Kern W, Schroder M, Haferlach T, Schoch C. Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia. 2006;20:2195–7. doi: 10.1038/sj.leu.2404325. [DOI] [PubMed] [Google Scholar]
- 158.Grunebach F, Bross-Bach U, Kanz L, Brossart P. Detection of a new JAK2 D620E mutation in addition to V617F in a patient with polycythemia vera. Leukemia. 2006;20:2210–1. doi: 10.1038/sj.leu.2404419. [DOI] [PubMed] [Google Scholar]
- 159.Mallnge S, Ben-Abdelall R, Settegrana C, Radford-Welss I, Debre M, Beldjord K, Maclntyre EA, Vllleval JL, Valnchenker W, Berger R, Bernard OA, Delabesse E, Penard-Lacronlque V. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood. 2007;109:2202–4. doi: 10.1182/blood-2006-09-045963. [DOI] [PubMed] [Google Scholar]
- 160.Zhang SJ, LI JY, LI WD, Song JH, Xu W, Qiu HX. The investigation of JAK2 mutation in Chinese myeloproliferative diseases-identification of a novel C616Y point mutation in a PV patient. Int J Lab Hematol. 2007;29:71–2. doi: 10.1111/j.1365-2257.2006.00864.x. [DOI] [PubMed] [Google Scholar]
- 161.Wong CL, Ma ES, Wang CL, Lam HY, Ma SY. JAK2 V617F due to a novel TG –> CT mutation at nucleotides 1848–1849: diagnostic implication. Leukemia. 2007;21:1344–6. doi: 10.1038/sj.leu.2404654. [DOI] [PubMed] [Google Scholar]
- 162.Karow A, Waller C, Relmann C, Niemeyer CM, Kratz CP. JAK2 mutations other than V617F: a novel mutation and mini review. Leuk Res. 2008;32:365–6. doi: 10.1016/j.leukres.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 163.Kratz CP, Boll S, Kontny U, Schrappe M, Niemeyer CM, Stanulla M. Mutational screen reveals a novel JAK2 mutation, L611S, in a child with acute lymphoblastic leukemia. Leukemia. 2006;20:381–3. doi: 10.1038/sj.leu.2404060. [DOI] [PubMed] [Google Scholar]
- 164.Mercher T, Wernig G, Moore SA, Levine RL, Gu TL, Frohling S, Cullen D, Polakiewicz RD, Bernard OA, Boggon TJ, Lee BH, Gilliland DG. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 2006;108:2770–9. doi: 10.1182/blood-2006-04-014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Scott LM, Beer PA, Bench AJ, Erber WN, Green AR. Prevalance of JAK2 V617F and exon 12 mutations in polycythaemia vera. Br J Haematol. 2007;139:511–2. doi: 10.1111/j.1365-2141.2007.06806.x. [DOI] [PubMed] [Google Scholar]
- 167.Butcher CM, Hahn U, To LB, Gecz J, Wilkins EJ, Scott HS, Bardy PG, D’Andrea RJ. Two novel JAK2 exon 12 mutations in JAK2V617F-negative poly-cythaemia vera patients. Leukemia. 2008;22:870–3. doi: 10.1038/sj.leu.2404971. [DOI] [PubMed] [Google Scholar]
- 168.Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, Ferrari M, Gisslinger H, Kralovics R, Cremonesi L, Skoda R, Cazzola M. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111:1686–9. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 169.Percy MJ, Scott LM, Erber WN, Harrison CN, Reilly JT, Jones FG, Green AR, McMullin MF. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica. 2007;92:1607–14. doi: 10.3324/haematol.11643. [DOI] [PubMed] [Google Scholar]
- 170.Martinez-Aviles L, Besses C, Alvarez-Larran A, Cervantes F, Hernandez-Boluda JC, Bellosillo B. JAK2 exon 12 mutations in polycythemia vera or idiopathic erythrocytosis. Haematologica. 2007;92:1717–8. doi: 10.3324/haematol.12011. [DOI] [PubMed] [Google Scholar]
- 171.Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, Hanson CA, Barosi G, Verstovsek S, Birgegard G, Mesa R, Reilly JT, Gisslinger H, Vannucchi AM, Cervantes F, Finazzi G, Hoffman R, Gilliland DG, Bloomfield CD, Vardiman JW. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombo-cythemia, and primary myelofibrosis: rec-ommendations from an ad hoc international expert panel. Blood. 2007;110:1092–7. doi: 10.1182/blood-2007-04-083501. [DOI] [PubMed] [Google Scholar]
- 172.Li S, Kralovics R, De Libero G, Tichelli A, Skoda RC. Lineage distribution of JAK2 exon12 mutations and JAK2-V617F in patients with polycythemia vera. ASH Ann Meet Abstr. 2007;110:1527. [Google Scholar]
- 173.Duke VM, Gurunlian N, Yogashangari B, Colley T, McNamara C, Hoffbrand AV, Foroni L. Frequency of JAK2 exon 12 mutations in JAK2 exon 14 V617F-negative patients: high frequency in ET patients. ASH Ann Meet Abstr. 2007;110:2539. [Google Scholar]
- 174.Colaizzo D, Amitrano L, Tiscia GL, Grandone E, Guardascione MA, Margaglione M. A new JAK2 gene mutation in patients with polycythemia vera and splanchnic vein thrombosis. Blood. 2007;110:2768–9. doi: 10.1182/blood-2007-05-092502. [DOI] [PubMed] [Google Scholar]
- 175.Lok S, Kaushansky K, Holly RD, Kuijper JL, Lofton-Day CE, Oort PJ, Grant FJ, Heipel MD, Burkhead SK, Kramer JM. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature. 1994;369:565–8. doi: 10.1038/369565a0. [DOI] [PubMed] [Google Scholar]
- 176.Kaushansky K. Lineage-specific hemato-poietic growth factors. N Engl J Med. 2006;354:2034–45. doi: 10.1056/NEJMra052706. [DOI] [PubMed] [Google Scholar]
- 177.Abkowitz JL, Chen J. Studies of c-Mpl function distinguish the replication of hematopoietic stem cells from the expansion of differentiating clones. Blood. 2007;109:5186–90. doi: 10.1182/blood-2006-08-044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Le Coniat M, Souyri M, Vigon I, Wendling F, Tambourin P, Berger R. The human homolog of the myeloproliferative virus maps to chromosome band 1p34. Hum Genet. 1989;83:194–6. doi: 10.1007/BF00286717. [DOI] [PubMed] [Google Scholar]
- 179.Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, Tsuboi K, Nitta M, Miyazaki H, Iida S, Ueda R. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoi-etin. Blood. 2004;103:4198–200. doi: 10.1182/blood-2003-10-3471. [DOI] [PubMed] [Google Scholar]
- 180.Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA, Hara T. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proc Natl Acad Sci USA. 1999;96:3132–6. doi: 10.1073/pnas.96.6.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A, Krukemeier S, Eilers M, Strauss G, Welte K. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytope-nia. Blood. 2001;97:139–46. doi: 10.1182/blood.v97.1.139. [DOI] [PubMed] [Google Scholar]
- 182.Germeshausen M, Ballmaier M, Welte K. MPL mutations in 23 patients suffering from congenital amegakaryocytic throm-bocytopenia: the type of mutation predicts the course of the disease. Hum Mutat. 2006;27:296. doi: 10.1002/humu.9415. [DOI] [PubMed] [Google Scholar]
- 183.Moliterno AR, Williams DM, Gutierrez-Alamillo LI, Salvatori R, Ingersoll RG, Spivak JL. Mpl Baltimore: a thrombopoi-etin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci USA. 2004;101:11444–7. doi: 10.1073/pnas.0404241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pardanani AD, Levine RL, Lasho TL, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litzow MR, Gilliland DG, Tefferi A. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–6. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 185.Schnittger S, Haferlach C, Beelen DW, Bojko P, Dengler R, Diestelrath A, Eckart MJ, Eckert R, Fries S, Kochling G, Laubenstein HP, Planker M, Pihusch R, Kern W, Haferlach T. Detection of three different MPLW515 mutations in 10.1% of all JAK2 V617 unmutated ET and 9.3% of all JAK2 V617F unmutated OMF: a study of 387 patients. ASH Ann Meet Abstr. 2007;110:2546. [Google Scholar]
- 186.Beer P, Campbell P, Erber W, Scott L, Bench A, Bareford D, Bridget W, Wheatley K, Buck G, Harrison C, Green A. Clinical significance of MPL mutations in essential thrombocythemia: analysis of the PT-1 cohort. ASH Ann Meet Abstr. 2007;110:677. [Google Scholar]
- 187.Vannucchi AM, Antonioli E, Pancrazzi A, Guglielmelli P, Di Lollo S, Alterini R, Guerini V, Baroni G, Rambaldi A, Bosi A, Barbui T. The clinical phenotype of patients with essential thrombocythemia harboring MPL 515W>L/K mutation. ASH Annu Meet Abstr. 2007;110:678. [Google Scholar]
- 188.Guglielmelli P, Pancrazzi A, Bergamaschi G, Rosti V, Villani L, Antonioli E, Bosi A, Barosi G, Vannucchi AM. Anaemia characterises patients with myelofibrosis harbouring Mpl mutation. Br J Haematol. 2007;137:244–7. doi: 10.1111/j.1365-2141.2007.06565.x. [DOI] [PubMed] [Google Scholar]
- 189.Szpurka H, Gondek LP, Mohan SR, Hsi ED, Theil KS, Maciejewski JP. UPD1p indicates the presence of MPL W515L mutation in RARS-T, a mechanism analogous to UPD9p and JAK2 V617F mutation. Leukemia. 2008 doi: 10.1038/leu.2008.249. in press. [DOI] [PubMed] [Google Scholar]
- 190.Pardanani A, Lasho TL, Finke C, Mesa RA, Hogan WJ, Ketterling RP, Gilliland DG, Tefferi A. Extending Jak2V617F and Mpl515 mutation analysis to single myeloid colonies and T and B lymphocytes. Stem Cells. 2007;25:2358–62. doi: 10.1634/stemcells.2007-0175. [DOI] [PubMed] [Google Scholar]
- 191.Chaligne R, James C, Tonetti C, Besancenot R, Le Couedic JP, Fava F, Mazurier F, Godin I, Maloum K, Larbret F, Lecluse Y, Vainchenker W, Giraudier S. Evidence for MPL W515L/K mutations in hematopoietic stem cells in primitive myelofibrosis. Blood. 2007;110:3735–43. doi: 10.1182/blood-2007-05-089003. [DOI] [PubMed] [Google Scholar]
- 192.Pardanani A, Lasho TL, Finke C, Markovic SN, Tefferi A. Demonstration of MPLW515K, but not JAK2V617F, in in vitro expanded CD4+ T lymphocytes. Leukemia. 2007;21:2206–7. doi: 10.1038/sj.leu.2404749. [DOI] [PubMed] [Google Scholar]
- 193.Lasho TL, Pardanani A, McClure RF, Mesa RA, Levine RL, Gary Gilliland D, Tefferi A. Concurrent MPL515 and JAK2V617F mutations in myelofibrosis: chronology of clonal emergence and changes in mutant allele burden over time. Br J Haematol. 2006;135:683–7. doi: 10.1111/j.1365-2141.2006.06348.x. [DOI] [PubMed] [Google Scholar]
- 194.Tefferi A, Lasho TL, Schwager SM, Strand JS, Elliott M, Mesa R, Li CY, Wadleigh M, Lee SJ, Gilliland DG. The clinical phenotype of wild-type, heterozygous, and homozygous JAK2V617F in polycythemia vera. Cancer. 2006;106:631–5. doi: 10.1002/cncr.21645. [DOI] [PubMed] [Google Scholar]
- 195.Vannucchi AM, Antonioli E, Guglielmelli P, Longo G, Pancrazzi A, Ponziani V, Bogani C, Ferrini PR, Rambaldi A, Guerini V, Bosi A, Barbui T. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21:1952–9. doi: 10.1038/sj.leu.2404854. [DOI] [PubMed] [Google Scholar]
- 196.Wolanskyj AP, Lasho TL, Schwager SM, McClure RF, Wadleigh M, Lee SJ, Gary Gilliland D, Tefferi A. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol. 2005;131:208–13. doi: 10.1111/j.1365-2141.2005.05764.x. [DOI] [PubMed] [Google Scholar]
- 197.Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, Ponziani V, Longo G, Bosi A, Vannucchi AM. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia. 2005;19:1847–9. doi: 10.1038/sj.leu.2403902. [DOI] [PubMed] [Google Scholar]
- 198.Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, Duffy A, Boyd EM, Bench AJ, Scott MA, Vassiliou GS, Milligan DW, Smith SR, Erber WN, Bareford D, Wilkins BS, Reilly JT, Harrison CN, Green AR. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005;366:1945–53. doi: 10.1016/S0140-6736(05)67785-9. [DOI] [PubMed] [Google Scholar]
- 199.Tefferi A, Lasho TL, Schwager SM, Steensma DP, Mesa RA, Li CY, Wadleigh M, Gary Gilliland D. The JAK2 tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: lineage specificity and clinical correlates. Br J Haematol. 2005;131:320–8. doi: 10.1111/j.1365-2141.2005.05776.x. [DOI] [PubMed] [Google Scholar]
- 200.Barosi G, Bergamaschi G, Marchetti M, Vannucchi AM, Guglielmelli P, Antonioli E, Massa M, Rosti V, Campanelli R, Villani L, Viarengo G, Gattoni E, Gerli G, Specchia G, Tinelli C, Rambaldi A, Barbui T. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007;110:4030–6. doi: 10.1182/blood-2007-07-099184. [DOI] [PubMed] [Google Scholar]
- 201.Patel RK, Lea NC, Heneghan MA, Westwood NB, Milojkovic D, Thanigaikumar M, Yallop D, Arya R, Pagliuca A, Gaken J, Wendon J, Heaton ND, Mufti GJ. Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd-Chiari Syndrome. Gastroenterology. 2006;130:2031–8. doi: 10.1053/j.gastro.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 202.Campbell PJ, Griesshammer M, Dohner K, Dohner H, Kusec R, Hasselbalch HC, Larsen TS, Pallisgaard N, Giraudier S, Le Bousse-Kerdiles MC, Desterke C, Guerton B, Dupriez B, Bordessoule D, Fenaux P, Kiladjian JJ, Viallard JF, Briere J, Harrison CN, Green AR, Reilly JT. V617F mutation in JAK2 is associated with poorer survival in idiopathic myelofibrosis. Blood. 2006;107:2098–100. doi: 10.1182/blood-2005-08-3395. [DOI] [PubMed] [Google Scholar]
- 203.Tefferi A, Lasho TL, Huang J, Finke C, Mesa RA, Li CY, Wu W, Hanson CA, Pardanani A. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22:756–61. doi: 10.1038/sj.leu.2405097. [DOI] [PubMed] [Google Scholar]
- 204.Pardanani A. JAK2 inhibitor therapy in myeloproliferative disorders: rationale, preclinical studies and ongoing clinical trials. Leukemia. 2008;22:23–30. doi: 10.1038/sj.leu.2404948. [DOI] [PubMed] [Google Scholar]
- 205.Lasho TL, Tefferi A, Hood JD, Verstovsek S, Gilliland DG, Pardanani A. TG101348, a JAK2-selective antagonist, inhibits primary hematopoietic cells derived from myeloproliferative disorder patients with JAK2V617F, MPLW515K or JAK2 exon 12 mutations as well as mutation negative patients. Leukemia. 2008;22:1790–2. doi: 10.1038/leu.2008.56. [DOI] [PubMed] [Google Scholar]
- 206.Verstovsek S, Pardanani AD, Shah NP, Sokol L, Wadleigh M, Gilliland DG, List AF, Tefferi A, Kantarjian HM, Bui LA, Clary DO. A phase I study of XL019, a selective JAK2 inhibitor, in patients with primary myelofibrosis and post-polycythemia vera/essential thrombocythemia myelofibrosis. ASH Ann Meet Abstr. 2007;110:553. [Google Scholar]
- 207.Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, Angeles T, Emerson SG, Carroll M, Ruggeri B, Dobrzanski P. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663–71. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Goh KC, Ong WC, Hu C, Hentze H, Liang AL, Stunkel W, Tan YC, Sangthongpitag K, Goh SK, Sieh S, Bonday ZQ, William AD, Lee A, Llanchard S, Liu TC, Sattler M, Griffin JD, Dymock BW, Kantharaj E, Wood JM. SB1518: a potent and orally active JAK2 inhibitor for the treatment of myeloproliferative disorders. Blood. 2007;110:165a–6a. [Google Scholar]
- 209.Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, Finke C, Mak CC, Mesa R, Zhu H, Soll R, Gilliland DG, Tefferi A. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21:1658–68. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 210.Lasho TL, Finke C, Hood JD, Soll R, Noronha G, Mesa RA, Jamieson C, Verstovsek S, Cortes J, Kantarjian H, Gilliland DG, Tefferi A, Pardanani A. Primary cell experiments with TG101348, a JAK2-selective inhibitor, in the presence of myeloproliferative disorder-associated JAK2V617F, MPLW515L/K, and JAK2 exon 12 mutations. Leukemia. 2008;22:1790–2. doi: 10.1038/leu.2008.56. [DOI] [PubMed] [Google Scholar]
- 211.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, Mak CC, Noronha G, Martin M, Ko YD, Lee BH, Soll R, Tefferi A, Hood JD, Gilliland DG. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–20. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 212.Geron I, Abrahamsson A, Barroga C, Kavelerchik E, Gotlib JR, Hood JD, Durocher J, Mak CC, Noronha G, Soll R, Tefferi A, Kaushansky K, Jamieson CHM. Selective inhibition of JAK2 driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008;13:321–30. doi: 10.1016/j.ccr.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 213.Verstovsek S, Kantarjian H, Pardanani A, Thomas D, Cortes J, Mesa R, Redman J, Staschen C-M, Fridman J, Vaddi K, Tefferi A. INCB018424, an oral, selective JAK2 inhibitor, shows significant clinical activity in a phase I/II study in patients with primary myelofibrosis (PMF) and post polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF) ASH Annu Meet Abstr. 2007;110:558. [Google Scholar]
- 214.Verstovsek S, Tefferi A, Kornblau S, Thomas D, Cortes J, Ravandi-Kashani F, Garcia-Manero G, Kantarjian H. Phase II study of CEP701, an orally available JAK2 inhibitor, in patients with primary myelofibrosis and post polycythemia vera/essential thrombocythemia myelofi-brosis. ASH Annu Meet Abstr. 2007;110:3543. [Google Scholar]
- 215.Tsujimura T, Furitsu T, Morimoto M, Isozaki K, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y. Ligand-inde-pendent activation of c-kit receptor tyrosine kinase in a murine mastocytoma cell line P-815 generated by a point mutation. Blood. 1994;83:2619–26. [PubMed] [Google Scholar]
- 216.Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD. Identification of a point mutation in the catalytic domain of the pro-tooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA. 1995;92:10560–4. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yanagihori H, Oyama N, Nakamura K, Kaneko F. c-kit Mutations in patients with childhood-onset mastocytosis and geno-type-phenotype correlation. J Mol Diagn. 2005;7:252–7. doi: 10.1016/S1525-1578(10)60552-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Buttner C, Henz BM, Welker P, Sepp NT, Grabbe J. Identification of activating c-kit mutations in adult-, but not in childhood-onset indolent mastocytosis: a possible explanation for divergent clinical behavior. J Invest Dermatol. 1998;111:1227–31. doi: 10.1046/j.1523-1747.1998.00414.x. [DOI] [PubMed] [Google Scholar]
- 219.Longley BJ, Jr, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA. 1999;96:1609–14. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312–4. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 221.Pignon JM, Giraudier S, Duquesnoy P, Jouault H, Imbert M, Vainchenker W, Vernant JP, Tulliez M. A new c-kit mutation in a case of aggressive mast cell disease. Br J Haematol. 1997;96:374–6. doi: 10.1046/j.1365-2141.1997.d01-2042.x. [DOI] [PubMed] [Google Scholar]
- 222.Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103:3222–5. doi: 10.1182/blood-2003-11-3816. [DOI] [PubMed] [Google Scholar]
- 223.Garcia-Montero AC, Jara-Acevedo M, Teodosio C, Sanchez ML, Nunez R, Prados A, Aldanondo I, Sanchez L, Dominguez M, Botana LM, Sanchez-Jimenez F, Sotlar K, Almeida J, Escribano L, Orfao A. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108:2366–72. doi: 10.1182/blood-2006-04-015545. [DOI] [PubMed] [Google Scholar]
- 224.Kahler C, Didlaukat S, Feller AC, Merz H. Sensitive and reliable detection of Kit point mutation Asp 816 to Val in pathological material. Diagn Pathol. 2007;2:37. doi: 10.1186/1746-1596-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ma Y, Longley BJ, Wang X, Blount JL, Langley K, Caughey GH. Clustering of activating mutations in c-KIT's juxtamembrane coding region in canine mast cell neoplasms. J Invest Dermatol. 1999;112:165–70. doi: 10.1046/j.1523-1747.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- 226.Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. Human protooncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–51. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.d’Auriol L, Mattei MG, Andre C, Galibert F. Localization of the human c-kit pro-tooncogene on the q11-q12 region of chromosome 4. Hum Genet. 1988;78:374–6. doi: 10.1007/BF00291740. [DOI] [PubMed] [Google Scholar]
- 228.Roskoski R., Jr Structure and regulation of Kit protein-tyrosine kinase–the stem cell factor receptor. Biochem Biophys Res Commun. 2005;338:1307–15. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 229.Geissler EN, Liao M, Brook JD, Martin FH, Zsebo KM, Housman DE, Galli SJ. Stem cell factor (SCF), a novel hematopoietic growth factor and ligand for c-kit tyrosine kinase receptor, maps on human chromosome 12 between 12q14.3 and 12qter. Somat Cell Mol Genet. 1991;17:207–14. doi: 10.1007/BF01232978. [DOI] [PubMed] [Google Scholar]
- 230.Martin FH, Suggs SV, Langley KE, Lu HS, Ting J, Okino KH, Morris CF, McNiece IK, Jacobsen FW, Mendiaz EA. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63:203–11. doi: 10.1016/0092-8674(90)90301-t. [DOI] [PubMed] [Google Scholar]
- 231.Mathew S, Murty VV, Hunziker W, Chaganti RS. Subregional mapping of 13 single-copy genes on the long arm of chromosome 12 by fluorescence in situ hybridization. Genomics. 1992;14:775–9. doi: 10.1016/s0888-7543(05)80184-3. [DOI] [PubMed] [Google Scholar]
- 232.Miettinen M, Lasota J. KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol. 2005;13:205–20. doi: 10.1097/01.pai.0000173054.83414.22. [DOI] [PubMed] [Google Scholar]
- 233.Ronnstrand L. Signal transduction viathe stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004;61:2535–48. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- 234.Akin C, Kirshenbaum AS, Semere T, Worobec AS, Scott LM, Metcalfe DD. Analysis of the surface expression of c-kit and occurrence of the c-kit Asp816Val activating mutation in T cells, B cells, and myelomonocytic cells in patients with mas-tocytosis. Exp Hematol. 2000;28:140–7. doi: 10.1016/s0301-472x(99)00145-9. [DOI] [PubMed] [Google Scholar]
- 235.Yavuz AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100:661–5. doi: 10.1182/blood-2002-01-0203. [DOI] [PubMed] [Google Scholar]
- 236.Taylor ML, Sehgal D, Raffeld M, Obiakor H, Akin C, Mage RG, Metcalfe DD. Demonstration that mast cells, T cells, and B cells bearing the activating kit mutation D816V occur in clusters within the marrow of patients with mastocytosis. J Mol Diagn. 2004;6:335–42. doi: 10.1016/S1525-1578(10)60529-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Piao X, Bernstein A. A point mutation in the catalytic domain of c-kit induces growth factor independence, tumorigenic-ity, and differentiation of mast cells. Blood. 1996;87:3117–23. [PubMed] [Google Scholar]
- 238.Kitayama H, Tsujimura T, Matsumura I, Oritani K, Ikeda H, Ishikawa J, Okabe M, Suzuki M, Yamamura K, Matsuzawa Y, Kitamura Y, Kanakura Y. Neoplastic transformation of normal hematopoietic cells by constitutively activating mutations of c-kit receptor tyrosine kinase. Blood. 1996;88:995–1004. [PubMed] [Google Scholar]
- 239.Zappulla JP, Dubreuil P, Desbois S, Letard S, Hamouda NB, Daeron M, Delsol G, Arock M, Liblau RS. Mastocytosis in mice expressing human Kit receptor with the activating Asp816Val mutation. J Exp Med. 2005;202:1635–41. doi: 10.1084/jem.20050807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ferrao PT, Gonda TJ, Ashman LK. Constitutively active mutant D816VKit induces megakayocyte and mast cell differentiation of early haemopoietic cells from murine foetal liver. Leuk Res. 2003;27:547–55. doi: 10.1016/s0145-2126(02)00272-2. [DOI] [PubMed] [Google Scholar]
- 241.Taylor ML, Dastych J, Sehgal D, Sundstrom M, Nilsson G, Akin C, Mage RG, Metcalfe DD. The Kit-activating mutation D816V enhances stem cell factor– dependent chemotaxis. Blood. 2001;98:1195–9. doi: 10.1182/blood.v98.4.1195. [DOI] [PubMed] [Google Scholar]
- 242.Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC, Besmer P. Differential roles of PI3-kinase and Kit tyrosine 821 in Kit receptor-mediated proliferation, survival and cell adhesion in mast cells. EMBO J. 1995;14:473–83. doi: 10.1002/j.1460-2075.1995.tb07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Timokhina I, Kissel H, Stella G, Besmer P. Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. EMBO J. 1998;17:6250–62. doi: 10.1093/emboj/17.21.6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jelacic T, Linnekin D. PKCdelta plays opposite roles in growth mediated by wild-type Kit and an oncogenic Kit mutant. Blood. 2005;105:1923–9. doi: 10.1182/blood-2004-04-1450. [DOI] [PubMed] [Google Scholar]
- 245.Lahortiga I, Akin C, Cools J, Wilson TM, Mentens N, Arthur DC, Maric I, Noel P, Kocabas C, Marynen P, Lessin LS, Wlodarska I, Robyn J, Metcalfe DD. Activity of imatinib in systemic mastocyto-sis with chronic basophilic leukemia and a PRKG2-PDGFRB fusion. Haematologica. 2008;93:49–56. doi: 10.3324/haematol.11836. [DOI] [PubMed] [Google Scholar]
- 246.Tanaka A, Konno M, Muto S, Kambe N, Morii E, Nakahata T, Itai A, Matsuda H. A novel NF-kappaB inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-kit receptors. Blood. 2005;105:2324–31. doi: 10.1182/blood-2004-08-3247. [DOI] [PubMed] [Google Scholar]
- 247.Gabillot-Carre M, Lepelletier Y, Humbert M, de Sepuvelda P, Hamouda NB, Zappulla JP, Liblau R, Ribadeau-Dumas A, Machavoine F, Letard S, Baude C, Hermant A, Yang Y, Vargaftig J, Bodemer C, Morelon E, Lortholary O, Recher C, Laurent G, Dy M, Arock M, Dubreuil P, Hermine O. Rapamycin inhibits growth and survival of D816V-mutated c-kit mast cells. Blood. 2006;108:1065–72. doi: 10.1182/blood-2005-06-2433. [DOI] [PubMed] [Google Scholar]
- 248.Pan J, Quintas-Cardama A, Kantarjian HM, Akin C, Manshouri T, Lamb P, Cortes JE, Tefferi A, Giles FJ, Verstovsek S. EXEL-0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivoin human mast cells expressing the KIT D816V mutation. Blood. 2007;109:315–22. doi: 10.1182/blood-2006-04-013805. [DOI] [PubMed] [Google Scholar]
- 249.Pan J, Quintas-Cardama A, Manshouri T, Cortes J, Kantarjian H, Verstovsek S. Sensitivity of human cells bearing onco-genic mutant kit isoforms to the novel tyrosine kinase inhibitor INNO-406. Cancer Sci. 2007;98:1223–5. doi: 10.1111/j.1349-7006.2007.00516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zermati Y, De Sepulveda P, Feger F, Letard S, Kersual J, Casteran N, Gorochov G, Dy M, Ribadeau Dumas A, Dorgham K, Parizot C, Bieche Y, Vidaud M, Lortholary O, Arock M, Hermine O, Dubreuil P. Effect of tyrosine kinase inhibitor STI571 on the kinase activity of wild-type and various mutated c-kit recep-tors found in mast cell neoplasms. Oncogene. 2003;22:660–4. doi: 10.1038/sj.onc.1206120. [DOI] [PubMed] [Google Scholar]
- 251.Akin C, Brockow K, D’Ambrosio C, Kirshenbaum AS, Ma Y, Longley BJ, Metcalfe DD. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp Hematol. 2003;31:686–92. doi: 10.1016/s0301-472x(03)00112-7. [DOI] [PubMed] [Google Scholar]
- 252.Nakagomi N, Hirota S. Juxtamembrane-type c-kit gene mutation found in aggressive systemic mastocytosis induces ima-tinib-resistant constitutive KIT activation. Lab Invest. 2007;87:365–71. doi: 10.1038/labinvest.3700524. [DOI] [PubMed] [Google Scholar]
- 253.Pardanani A, Elliott M, Reeder T, Li C Y, Baxter EJ, Cross NC, Tefferi A. Imatinib for systemic mast-cell disease. Lancet. 2003;362:535–6. doi: 10.1016/s0140-6736(03)14115-3. [DOI] [PubMed] [Google Scholar]
- 254.Droogendijk HJ, Kluin-Nelemans HJ, van Doormaal JJ, Oranje AP, van de Loosdrecht AA, van Daele PL. Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial. Cancer. 2006;107:345–51. doi: 10.1002/cncr.21996. [DOI] [PubMed] [Google Scholar]
- 255.Verstovsek S, Akin C, Manshouri T, Quintas-Cardama A, Huynh L, Manley P, Tefferi A, Cortes J, Giles FJ, Kantarjian H. Effects of AMN107, a novel aminopy-rimidine tyrosine kinase inhibitor, on human mast cells bearing wild-type or mutated codon 816 c-kit. Leuk Res. 2006;30:1365–70. doi: 10.1016/j.leukres.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 256.von Bubnoff N, Gorantla SH, Kancha RK, Lordick F, Peschel C, Duyster J. The systemic mastocytosis-specific activating cKit mutation D816V can be inhibited by the tyrosine kinase inhibitor AMN107. Leukemia. 2005;19:1670–1. doi: 10.1038/sj.leu.2403887. [DOI] [PubMed] [Google Scholar]
- 257.Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, Bokemeyer C, Deininger MW, Druker BJ, Heinrich MC. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–81. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 258.Shah NP, Lee FY, Luo R, Jiang Y, Donker M, Akin C. Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood. 2006;108:286–91. doi: 10.1182/blood-2005-10-3969. [DOI] [PubMed] [Google Scholar]
- 259.Verstovsek S, Tefferi A, Jorge C, O’Brien S, Garcia-Manero G, Pardanani A, Akin C, Faderl S, Thomas D, Kantarjian H. Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mas-tocytosis. Clin Cancer Res. 2008;14:3906. doi: 10.1158/1078-0432.CCR-08-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Purtill D, Cooney J, Sinniah R, Carnley B, Cull G, Augustson B, Cannell P. Dasatinib therapy for systemic mastocyto-sis: four cases. Eur J Haematol. 2008;80:456–8. doi: 10.1111/j.1600-0609.2008.01048.x. [DOI] [PubMed] [Google Scholar]
- 261.Growney JD, Clark JJ, Adelsperger J, Stone R, Fabbro D, Griffin JD, Gilliland DG. Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood. 2005;106:721–4. doi: 10.1182/blood-2004-12-4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gleixner KV, Mayerhofer M, Aichberger KJ, Derdak S, Sonneck K, Bohm A, Gruze A, Samorapoompichit P, Manley PW, Fabbro D, Pickl WF, Sillaber C, Valent P. PKC412 inhibits in vitro growth of neo-plastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006;107:752–9. doi: 10.1182/blood-2005-07-3022. [DOI] [PubMed] [Google Scholar]
- 263.Gotlib J, Berube C, Growney JD, Chen CC, George TI, Williams C, Kajiguchi T, Ruan J, Lilleberg SL, Durocher JA, Lichy JH, Wang Y, Cohen PS, Arber DA, Heinrich MC, Neckers L, Galli SJ, Gilliland DG, Coutre SE. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood. 2005;106:2865–70. doi: 10.1182/blood-2005-04-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Robyn J, Lemery S, McCoy JP, Kubofcik J, Kim YJ, Pack S, Nutman TB, Dunbar C, Klion AD. Multilineage involvement of the fusion gene in patients with FIP1L1/ PDGFRA-positive hypereosinophilic syndrome. Br J Haematol. 2006;132:286–92. doi: 10.1111/j.1365-2141.2005.05863.x. [DOI] [PubMed] [Google Scholar]
- 265.Tefferi A, Lasho TL, Brockman SR, Elliott MA, Dispenzieri A, Pardanani A. FIP1L1-PDGFRA and c-kit D816V mutation-based clonality studies in systemic mast cell disease associated with eosinophilia. Haematologica. 2004;89:871–3. [PubMed] [Google Scholar]
- 266.Cools J, Stover EH, Boulton CL, Gotlib J, Legare RD, Amaral SM, Curley DP, Duclos N, Rowan R, Kutok JL, Lee BH, Williams IR, Coutre SE, Stone RM, DeAngelo DJ, Marynen P, Manley PW, Meyer T, Fabbro D, Neuberg D, Weisberg E, Griffin JD, Gilliland DG. PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRalpha-induced myeloproliferative disease. Cancer Cell. 2003;3:459–69. doi: 10.1016/s1535-6108(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 267.Griffin JH, Leung J, Bruner RJ, Caligiuri MA, Briesewitz R. Discovery of a fusion kinase in EOL-1 cells and idiopathic hyper-eosinophilic syndrome. Proc Natl Acad Sci USA. 2003;100:7830–5. doi: 10.1073/pnas.0932698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Roche-Lestienne C, Lepers S, Soenen-Cornu V, Kahn JE, Lai JL, Hachulla E, Drupt F, Demarty AL, Roumier AS, Gardembas M, Dib M, Philippe N, Cambier N, Barete S, Libersa C, Bletry O, Hatron PY, Quesnel B, Rose C, Maloum K, Blanchet O, Fenaux P, Prin L, Preudhomme C. Molecular characterization of the idiopathic hypereosinophilic syndrome (HES) in 35 French patients with normal conventional cytogenetics. Leukemia. 2005;19:792–8. doi: 10.1038/sj.leu.2403722. [DOI] [PubMed] [Google Scholar]
- 269.Stover EH, Chen J, Folens C, Lee BH, Mentens N, Marynen P, Williams IR, Gilliland DG, Cools J. Activation of FIP1L1-PDGFRalpha requires disruption of the juxtamembrane domain of PDGFRalpha and is FIP1L1-independent. Proc Natl Acad Sci USA. 2006;103:8078–83. doi: 10.1073/pnas.0601192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Klion AD, Robyn J, Akin C, Noel P, Brown M, Law M, Metcalfe DD, Dunbar C, Nutman TB. Molecular remission and reversal of myelofibrosis in response to imatinib mesylate treatment in patients with the myeloproliferative variant of hypereosinophilic syndrome. Blood. 2004;103:473–8. doi: 10.1182/blood-2003-08-2798. [DOI] [PubMed] [Google Scholar]
- 271.von Bubnoff N, Sandherr M, Schlimok G, Andreesen R, Peschel C, Duyster J. Myeloid blast crisis evolving during ima-tinib treatment of an FIP1L1-PDGFR alpha-positive chronic myeloproliferative disease with prominent eosinophilia. Leukemia. 2005;19:286–7. doi: 10.1038/sj.leu.2403600. [DOI] [PubMed] [Google Scholar]
- 272.Ohnishi H, Kandabashi K, Maeda Y, Kawamura M, Watanabe T. Chronic eosinophilic leukaemia with FIP1L1-PDGFRA fusion and T674I mutation that evolved from Langerhans cell histiocytosis with eosinophilia after chemotherapy. Br J Haematol. 2006;134:547–9. doi: 10.1111/j.1365-2141.2006.06221.x. [DOI] [PubMed] [Google Scholar]
- 273.Lierman E, Folens C, Stover EH, Mentens N, Van Miegroet H, Scheers W, Boogaerts M, Vandenberghe P, Marynen P, Cools J. Sorafenib (BAY43–9006) is a potent inhibitor of FIP1L1-PDGFR{alpha} and the imatinib resistant FIP1L1-PDGFR{alpha} T674I mutant. Blood. 2006;108:1374–6. doi: 10.1182/blood-2006-02-004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Stover EH, Chen J, Lee BH, Cools J, McDowell E, Adelsperger J, Cullen D, Coburn A, Moore SA, Okabe R, Fabbro D, Manley PW, Griffin JD, Gilliland DG. The small molecule tyrosine kinase inhibitor AMN107 inhibits TEL-PDGFRbeta and FIP1L1-PDGFRalpha in vitro and in vivo. Blood. 2005;106:3206–13. doi: 10.1182/blood-2005-05-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.von Bubnoff N, Gorantla SP, Thone S, Peschel C, Duyster J. The FIP1L1-PDGFRA T674I mutation can be inhibited by the tyrosine kinase inhibitor AMN107 (nilotinib) Blood. 2006;107:4970–1. doi: 10.1182/blood-2006-01-0285. author reply 4972. [DOI] [PubMed] [Google Scholar]
- 276.Score J, Curtis C, Waghorn K, Stalder M, Jotterand M, Grand FH, Cross NC. Identification of a novel imatinib responsive KIF5B-PDGFRA fusion gene following screening for PDGFRA overexpression in patients with hypereosinophilia. Leukemia. 2006;20:827–32. doi: 10.1038/sj.leu.2404154. [DOI] [PubMed] [Google Scholar]
- 277.Baxter EJ, Hochhaus A, Bolufer P, Reiter A, Fernandez JM, Senent L, Cervera J, Moscardo F, Sanz MA, Cross NC. The t(4; 22)(q12; q11) in atypical chronic myeloid leukaemia fuses BCR to PDGFRA. Hum Mol Genet. 2002;11:1391–7. doi: 10.1093/hmg/11.12.1391. [DOI] [PubMed] [Google Scholar]
- 278.Walz C, Curtis C, Schnittger S, Schultheis B, Metzgeroth G, Schoch C, Lengfelder E, Erben P, Muller MC, Haferlach T, Hochhaus A, Hehlmann R, Cross NC, Reiter A. Transient response to imatinib in a chronic eosinophilic leukemia associated with ins(9; 4)(q33; q12q25) and a CDK5RAP2-PDGFRA fusion gene. Genes Chromosomes Cancer. 2006;45:950–6. doi: 10.1002/gcc.20359. [DOI] [PubMed] [Google Scholar]
- 279.Trempat P, Villalva C, Laurent G, Armstrong F, Delsol G, Dastugue N, Brousset P. Chronic myeloproliferative disorders with rearrangement of the platelet-derived growth factor alpha receptor: a new clinical target for STI571/Glivec. Oncogene. 2003;22:5702–6. doi: 10.1038/sj.onc.1206543. [DOI] [PubMed] [Google Scholar]
- 280.Safley AM, Sebastian S, Collins TS, Tirado CA, Stenzel TT, Gong JZ, Goodman BK. Molecular and cytogenetic characterization of a novel translocation t(4; 22) involving the breakpoint cluster region and platelet-derived growth factor receptor-alpha genes in a patient with atypical chronic myeloid leukemia. Genes Chromosomes Cancer. 2004;40:44–50. doi: 10.1002/gcc.20014. [DOI] [PubMed] [Google Scholar]
- 281.Carroll M, Tomasson MH, Barker GF, Golub TR, Gilliland DG. The TEL/platelet-derived growth factor beta receptor (PDGF beta R) fusion in chronic myelomonocytic leukemia is a transforming protein that self-associates and activates PDGF beta R kinase-dependent signaling pathways. Proc Natl Acad Sci USA. 1996;93:14845–50. doi: 10.1073/pnas.93.25.14845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Abe A, Emi N, Tanimoto M, Terasaki H, Marunouchi T, Saito H. Fusion of the platelet-derived growth factor receptor beta to a novel gene CEV14 in acute myel-ogenous leukemia after clonal evolution. Blood. 1997;90:4271–7. [PubMed] [Google Scholar]
- 283.Levine RL, Wadleigh M, Sternberg DW, Wlodarska I, Galinsky I, Stone RM, DeAngelo DJ, Gilliland DG, Cools J. KIAA1509 is a novel PDGFRB fusion partner in imatinib-responsive myeloprolifera-tive disease associated with a t(5; 14)(q33; q32) Leukemia. 2005;19:27–30. doi: 10.1038/sj.leu.2403548. [DOI] [PubMed] [Google Scholar]
- 284.Vizmanos JL, Novo FJ, Roman JP, Baxter EJ, Lahortiga I, Larrayoz MJ, Odero MD, Giraldo P, Calasanz MJ, Cross NC. NIN, a gene encoding a CEP110-like centrosomal protein, is fused to PDGFRB in a patient with a t(5; 14)(q33; q24) and an imatinib-responsive myeloproliferative disorder. Cancer Res. 2004;64:2673–6. doi: 10.1158/0008-5472.can-04-0144. [DOI] [PubMed] [Google Scholar]
- 285.Morerio C, Acquila M, Rosanda C, Rapella A, Dufour C, Locatelli F, Maserati E, Pasquali F, Panarello C. HCMOGT-1 is a novel fusion partner to PDGFRB in juvenile myelomonocytic leukemia with t(5; 17)(q33; p11.2) Cancer Res. 2004;64:2649–51. doi: 10.1158/0008-5472.can-03-4026. [DOI] [PubMed] [Google Scholar]
- 286.Wilkinson K, Velloso ER, Lopes LF, Lee C, Aster JC, Shipp MA, Aguiar RC. Cloning of the t(1; 5)(q23; q33) in a myeloproliferative disorder associated with eosinophilia: involvement of PDGFRB and response to imatinib. Blood. 2003;102:4187–90. doi: 10.1182/blood-2003-04-1150. [DOI] [PubMed] [Google Scholar]
- 287.Magnusson MK, Meade KE, Brown KE, Arthur DC, Krueger LA, Barrett AJ, Dunbar CE. Rabaptin-5 is a novel fusion partner to platelet-derived growth factor beta receptor in chronic myelomonocytic leukemia. Blood. 2001;98:2518–25. doi: 10.1182/blood.v98.8.2518. [DOI] [PubMed] [Google Scholar]
- 288.Ross TS, Bernard OA, Berger R, Gilliland DG. Fusion of Huntingtin interacting protein 1 to platelet-derived growth factor beta receptor (PDGFbetaR) in chronic myelomonocytic leukemia with t(5; 7)(q33; q11.2) Blood. 1998;91:4419–26. [PubMed] [Google Scholar]
- 289.Schwaller J, Anastasiadou E, Cain D, Kutok J, Wojiski S, Williams IR, LaStarza R, Crescenzi B, Sternberg DW, Andreasson P, Schiavo R, Siena S, Mecucci C, Gilliland DG. H4(D10S170), a gene frequently rearranged in papillary thyroid carcinoma, is fused to the platelet-derived growth factor receptor beta gene in atypical chronic myeloid leukemia with t(5; 10)(q33; q22) Blood. 2001;97:3910–8. doi: 10.1182/blood.v97.12.3910. [DOI] [PubMed] [Google Scholar]
- 290.Grand FH, Burgstaller S, Kuhr T, Baxter EJ, Webersinke G, Thaler J, Chase AJ, Cross NC. p53-Binding protein 1 is fused to the platelet-derived growth factor receptor beta in a patient with a t(5; 15)(q33; q22) and an imatinib-responsive eosinophilic myeloproliferative disorder. Cancer Res. 2004;64:7216–9. doi: 10.1158/0008-5472.CAN-04-2005. [DOI] [PubMed] [Google Scholar]
- 291.Tomasson MH, Williams IR, Hasserjian R, Udomsakdi C, McGrath SM, Schwaller J, Druker B, Gilliland DG. TEL/PDGFbetaR induces hematologic malignancies in mice that respond to a specific tyrosine kinase inhibitor. Blood. 1999;93:1707–14. [PubMed] [Google Scholar]
- 292.Xiao S, Nalabolu SR, Aster JC, Ma J, Abruzzo L, Jaffe ES, Stone R, Weissman SM, Hudson TJ, Fletcher JA. FGFR1 is fused with a novel zinc-finger gene, ZNF198, in the t(8; 13) leukaemia/ lymphoma syndrome. Nat Genet. 1998;18:84–7. doi: 10.1038/ng0198-84. [DOI] [PubMed] [Google Scholar]
- 293.Smedley D, Hamoudi R, Clark J, Warren W, Abdul-Rauf M, Somers G, Venter D, Fagan K, Cooper C, Shipley J. The t(8; 13)(p11; q11–12) rearrangement associated with an atypical myeloproliferative disorder fuses the fibroblast growth factor receptor 1 gene to a novel gene RAMP. Hum Mol Genet. 1998;7:637–42. doi: 10.1093/hmg/7.4.637. [DOI] [PubMed] [Google Scholar]
- 294.Popovici C, Adelaide J, Ollendorff V, Chaffanet M, Guasch G, Jacrot M, Leroux D, Birnbaum D, Pebusque MJ. Fibroblast growth factor receptor 1 is fused to FIM in stem-cell myeloproliferative disorder with t(8; 13) Proc Natl Acad Sci USA. 1998;95:5712–7. doi: 10.1073/pnas.95.10.5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Still IH, Chernova O, Hurd D, Stone RM, Cowell JK. Molecular characterization of the t(8; 13)(p11; q12) translocation associated with an atypical myeloproliferative disorder: evidence for three discrete loci involved in myeloid leukemias on 8p11. Blood. 1997;90:3136–41. [PubMed] [Google Scholar]
- 296.Reiter A, Sohal J, Kulkarni S, Chase A, Macdonald DH, Aguiar RC, Goncalves C, Hernandez JM, Jennings BA, Goldman JM, Cross NC. Consistent fusion of ZNF198 to the fibroblast growth factor receptor-1 in the t(8; 13)(p11; q12) myeloproliferative syndrome. Blood. 1998;92:1735–42. [PubMed] [Google Scholar]
- 297.Chaffanet M, Popovici C, Leroux D, Jacrot M, Adelaide J, Dastugue N, Gregoire MJ, Hagemeijer A, Lafage-Pochitaloff M, Birnbaum D, Pebusque MJ. t(6; 8), t(8; 9) and t(8; 13) transloca-tions associated with stem cell myelopro-liferative disorders have close or identical breakpoints in chromosome region 8p11–12. Oncogene. 1998;16:945–9. doi: 10.1038/sj.onc.1201601. [DOI] [PubMed] [Google Scholar]
- 298.Popovici C, Zhang B, Gregoire MJ, Jonveaux P, Lafage-Pochitaloff M, Birnbaum D, Pebusque MJ. The t(6; 8)(q27; p11) translocation in a stem cell myeloproliferative disorder fuses a novel gene, FOP, to fibroblast growth factor receptor 1. Blood. 1999;93:1381–9. [PubMed] [Google Scholar]
- 299.Mugneret F, Chaffanet M, Maynadie M, Guasch G, Favre B, Casasnovas O, Birnbaum D, Pebusque MJ. The 8p12 myeloproliferative disorder. t(8; 19) (p12; q13.3): a novel translocation involving the FGFR1 gene. Br J Haematol. 2000;111:647–9. doi: 10.1046/j.1365-2141.2000.02355.x. [DOI] [PubMed] [Google Scholar]
- 300.Sohal J, Chase A, Mould S, Corcoran M, Oscier D, Iqbal S, Parker S, Welborn J, Harris RI, Martinelli G, Montefusco V, Sinclair P, Wilkins BS, van den Berg H, Vanstraelen D, Goldman JM, Cross NC. Identification of four new translocations involving FGFR1 in myeloid disorders. Genes Chromosomes Cancer. 2001;32:155–63. doi: 10.1002/gcc.1177. [DOI] [PubMed] [Google Scholar]
- 301.Demiroglu A, Steer EJ, Heath C, Taylor K, Bentley M, Allen SL, Koduru P, Brody JP, Hawson G, Rodwell R, Doody ML, Carnicero F, Reiter A, Goldman JM, Melo JV, Cross NC. The t(8; 22) in chronic myeloid leukemia fuses BCR to FGFR1: transforming activity and specific inhibition of FGFR1 fusion proteins. Blood. 2001;98:3778–83. doi: 10.1182/blood.v98.13.3778. [DOI] [PubMed] [Google Scholar]
- 302.Fioretos T, Panagopoulos I, Lassen C, Swedin A, Billstrom R, Isaksson M, Strombeck B, Olofsson T, Mitelman F, Johansson B. Fusion of the BCR and the fibroblast growth factor receptor-1 (FGFR1) genes as a result of t(8; 22)(p11; q11) in a myeloproliferative disorder: the first fusion gene involving BCR but not ABL. Genes Chromosomes Cancer. 2001;32:302–10. doi: 10.1002/gcc.1195. [DOI] [PubMed] [Google Scholar]
- 303.Guasch G, Popovici C, Mugneret F, Chaffanet M, Pontarotti P, Birnbaum D, Pebusque MJ. Endogenous retroviral sequence is fused to FGFR1 kinase in the 8p12 stem-cell myeloproliferative disorder with t(8; 19)(p12; q13.3) Blood. 2003;101:286–8. doi: 10.1182/blood-2002-02-0577. [DOI] [PubMed] [Google Scholar]
- 304.Pini M, Gottardi E, Scaravaglio P, Giugliano E, Libener R, Baraldi A, Muzio A, Cornaglia E, Saglio G, Levis A. A fourth case of BCR-FGFR1 positive CML-like disease with t(8; 22) translocation showing an extensive deletion on the derivative chromosome 8p. Hematol J. 2002;3:315–6. doi: 10.1038/sj.thj.6200201. [DOI] [PubMed] [Google Scholar]
- 305.Grand EK, Grand FH, Chase AJ, Ross FM, Corcoran MM, Oscier DG, Cross NC. Identification of a novel gene, FGFR1OP2, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer. 2004;40:78–83. doi: 10.1002/gcc.20023. [DOI] [PubMed] [Google Scholar]
- 306.Belloni E, Trubia M, Gasparini P, Micucci C, Tapinassi C, Confalonieri S, Nuciforo P, Martino B, Lo-Coco F, Pelicci PG, Di Fiore PP. 8p11 myeloproliferative syndrome with a novel t(7; 8) translocation leading to fusion of the FGFR1 and TIF1genes. Genes Chromosomes Cancer. 2005;42:320–5. doi: 10.1002/gcc.20144. [DOI] [PubMed] [Google Scholar]
- 307.Hidalgo-Curtis C, Chase A, Drachenberg M, Roberts MW, Finkelstein JZ, Mould S, Oscier D, Cross NC, Grand FH. The t(1; 9)(p34; q34) and t(8; 12)(p11; q15) fuse pre-mRNA processing proteins SFPQ (PSF) and CPSF6 to ABL and FGFR1. Genes Chromosomes Cancer. 2008;47:379–85. doi: 10.1002/gcc.20541. [DOI] [PubMed] [Google Scholar]
- 308.Ollendorff V, Guasch G, Isnardon D, Galindo R, Birnbaum D, Pebusque MJ. Characterization of FIM-FGFR1, the fusion product of the myeloproliferative disorder-associated t(8; 13) translocation. J Biol Chem. 1999;274:26922–30. doi: 10.1074/jbc.274.38.26922. [DOI] [PubMed] [Google Scholar]
- 309.Guasch G, Ollendorff V, Borg JP, Birnbaum D, Pebusque MJ. 8p12 stem cell myeloproliferative disorder: the FOP-fibroblast growth factor receptor 1 fusion protein of the t(6; 8) translocation induces cell survival mediated by mitogen-acti-vated protein kinase and phosphatidylinos-itol 3-kinase/Akt/mTOR pathways. Mol Cell Biol. 2001;21:8129–42. doi: 10.1128/MCB.21.23.8129-8142.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Roumiantsev S, Krause DS, Neumann CA, Dimitri CA, Asiedu F, Cross NC, Van Etten RA. Distinct stem cell myeloprolifer-ative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell. 2004;5:287–98. doi: 10.1016/s1535-6108(04)00053-4. [DOI] [PubMed] [Google Scholar]
- 311.Guasch G, Delaval B, Arnoulet C, Xie MJ, Xerri L, Sainty D, Birnbaum D, Pebusque MJ. FOP-FGFR1 tyrosine kinase, the product of a t(6; 8) translocation, induces a fatal myeloproliferative disease in mice. Blood. 2004;103:309–12. doi: 10.1182/blood-2003-05-1690. [DOI] [PubMed] [Google Scholar]
- 312.Chen J, Deangelo DJ, Kutok JL, Williams IR, Lee BH, Wadleigh M, Duclos N, Cohen S, Adelsperger J, Okabe R, Coburn A, Galinsky I, Huntly B, Cohen PS, Meyer T, Fabbro D, Roesel J, Banerji L, Griffin JD, Xiao S, Fletcher JA, Stone RM, Gilliland DG. PKC412 inhibits the zinc finger 198-fibroblast growth factor receptor 1 fusion tyrosine kinase and is active in treatment of stem cell myeloproliferative disorder. Proc Natl Acad Sci USA. 2004;101:14479–84. doi: 10.1073/pnas.0404438101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Vardiman JW, Brunning RD, Arber DA, Le Beau MM, Porwit A, Tefferi A, Bloomfield CD, Thiele J. In: WHO classification of tumours of haematopoietic and lymphoid tissues. 4. Swerdlow SH, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. International Agency for Research on Cancer; 2008. pp. 18–30. [Google Scholar]