

Abstract
Minerval is an oleic acid synthetic analogue that impairs lung cancer (A549) cell proliferation upon modulation of the plasma membrane lipid structure and subsequent regulation of protein kinase C localization and activity. However, this mechanism does not fully explain the regression of tumours induced by this drug in animal models of cancer. Here we show that Minerval also induced apoptosis in Jurkat T-lymphoblastic leukaemia and other cancer cells. Minerval inhibited proliferation of Jurkat cells, concomitant with a decrease of cyclin D3 and cdk2 (cyclin-dependent kinase2). In addition, the changes that induced on Jurkat cell membrane organization caused clustering (capping) of the death receptor Fas (CD95), caspase-8 activation and initiation of the extrinsic apoptosis pathway, which finally resulted in programmed cell death. The present results suggest that the intrinsic pathway (associated with caspase-9 function) was activated downstream by caspase-8. In a xenograft model of human leukaemia, Minerval also inhibited tumour progression and induced tumour cell death. Studies carried out in a wide variety of cancer cell types demonstrated that apoptosis was the main molecular mechanism triggered by Minerval. This is the first report on the pro-apoptotic activity of Minerval, and in part explains the effectiveness of this non-toxic anticancer drug and its wide spectrum against different types of cancer.
Keywords: membrane-lipid therapy, leukaemia treatment, anti-cancer drug, regulation of cell signalling
Introduction
Minerval (2-hydroxyoleic acid) is a synthetic oleic acid (OA) derivative that binds to the plasma membrane and alters lipid organization [1], triggering a series of events that impairs lung cancer (A549) cell proliferation [2, 3]. OA and structural analogues (e.g. Minerval) modulate the plasma membrane lipid structure by increasing its propensity to form nonlamellar (hexagonal HII) phases [4, 5]. This modulation of the membrane lipid structure influences the localization and activity of amphitropic membrane proteins involved in cell signalling, such as G proteins and protein kinase C [6–10]. This antiproliferative effect of Minerval is not accompanied by apoptosis in A549 lung cancer cells. The present study was designed to investigate the pharmacological effectiveness of this drug in a number of cancer cell lines and the mechanism of action triggered by Minerval in these cells. In this context, it was found that this drug induced apoptosis in most cell lines studied, whereas it did not significantly affect normal fibroblasts. Moreover, it also impaired tumour progression and induction of cancer cell death in an animal model of leukaemia without apparent toxicity.
Programmed cell death or apoptosis can be triggered by external signals propagated within the cell either by receptors in the plasma membrane (extrinsic pathway), or by signals generated in the mitochondria (intrinsic pathway). In both pathways, the events that provoke apoptosis involve the activation of previously dormant cysteine-proteases called caspases. The first caspases activated by such cell death signals, the ‘initiator caspases’, are specific to the apoptotic pathway used. Thus, caspase-8 is associated with the ‘extrinsic’ membrane death receptor pathway and caspase-9 with the ‘intrinsic’ mitochondrial pathway [11]. Through proteolysis, these proteins activate ‘effector caspases’ (e.g. caspase-3, -6 and -7), which are also known as ‘executioner’ caspases because their activity results in the widespread cleavage of a variety of target proteins [12]. Here we showed that Minerval markedly induced apoptosis, preferentially through the ‘extrinsic’ membrane (caspase-8-mediated) death receptor pathway, upon membrane lipid re-organization and subsequent Fas receptor capping in the plasma membrane of Jurkat cells. In contrast, OA had a modest influence on proliferation and apoptosis, which justifies its preventive but not therapeutic activity.
The development of Minerval was based on the discovery that anthracyclines were able to exert anti-tumour activity by the sole interaction with the plasma membrane [13]. Regulation of cell signals through changes in membrane lipid structure (membrane-lipid therapy) is an approach that has been recently suggested as an alternative for treatment of cancer [14]. In the search of molecules capable of regulating membrane lipid structure, we found that OA was the most active compound [15]. For this reason, we designed Minerval, because alpha-hydroxy derivatives of fatty acids exhibit a lesser degradation or biological use [16]. In addition, this drug does not cause cellular or general toxicity (reference [2] and formal preclinical toxicological studies not shown here), which along with its oral administration and high efficacy provide evidence for the initiation of clinical trials in humans, which will probably start during this year.
Materials and methods
Cell lines and culture
The various cancer cell lines used in this study were obtained from the European Collection of Cell Cultures and cultured at 37°C and 5% CO2 in DMEM (M220 and HT-29) or RPMI 1640 (the rest of lines, except MDA-MB-231 cells) media supplemented with 10 mM Hepes, pH 7.4, 2 mM glutamine, 2 g/l bicarbonate, 1 g/l glucose, 10% (v/v) foetal bovine serum, 100 units/ml penicillin, 0.1 mg/ml streptomycin and 0.25 μg/ml Amphotericin B. MDA-MB-231 breast cancer cells were incubated in L-15 Leibowitz medium supplemented with 15% foetal bovine serum and the other substances above indicated. Media and other culture reagents were obtained from Sigma-Aldrich (Madrid, Spain).
Cell treatments, cell proliferation and caspase activity determinations
Cells were plated at a density of 1 × 105 cells in 24-well plates, incubated for 24 hrs, and then exposed to various concentrations of either Minerval or OA for another 24, 48 or 72 hrs in the above-mentioned culture media. At the end of the treatment, the cells were counted using an automated cell counter (Advia 120, Bayer Diagnostics, Leverkusen, Germany). In addition to cell count, cell proliferation was further determined using the MTT (methylthiazolyl diphenyl tetrazolium bromide) method. Both techniques produced similar results. Caspase activity was determined using the caspase detection kit (Calbiochem, San Diego, CA, USA), which is based on the nontoxic and irreversible binding of the caspase inhibitor, VAD-FMK, conjugated to the fluorescent moiety FITC (FITC-VAD-FMK). The increase in fluorescence because of caspase activation in apoptotic cells was determined by flow cytometry on a Coulter Epics XL flow cytometer (Beckman Coulter, Inc., Fullerton, CA, USA) using the FL-1 channel (excitation at 488 nm and emission at 510–550 nm).
Cell DNA content analysis
The cellular DNA content was determined by staining cells with ethidium bromide as indicated below and then performing single-cell fluorescence flow cytometry measurements. Jurkat cells were incubated for 0–96 hrs in the presence or absence (control) of 50 μM Minerval (time dependence) or for 72 hrs in the presence of 0–100 μM Minerval (concentration dependence). In another series of experiments, cells were incubated for 72 hrs in the presence of 50 μM Minerval and in the presence or absence of 20 μM of zIETD-fmk (Calbiochem) to inhibit caspase-8 activity, or 20 μM of zLEHD-fmk to inhibit caspase-9 activity. Then, cells were washed twice with phosphate-buffered saline (PBS: 137 mM NaCl, 2.7 mM potassium chloride, 12 mM dibasic sodium phosphate, 1.38 mM monobasic potassium phosphate at pH 7.4), resuspended in 500 μl of methanol and submitted to vortex shaking. The cells were subsequently incubated at room temperature for 1 hr, and then for 30 min. with 100 μg/ml ethidium bromide and 100 μg/ml RNAse A (Sigma-Aldrich) in PBS. Single-cell ethidium bromide fluorescence was measured on a Coulter Epics XL flow cytometer. The cell cycle was evaluated in populations, using EXPO 32 flow cytometry software (Beckman Coulter, Inc.). Gates were set to differentiate between G0/G1, S and G2+M phases, apoptotic hypodiploid cells formed a peak with less DNA than that which corresponded to cells in the G0/G1 phase (sub-G1 peak).
Electrophoresis, immunobloting and protein quantification
Cells were cultured in 6-well plates similarly as indicated above. After incubation in the presence or absence of Minerval or OA at the indicated concentrations and times (see the Results section), 150 μl of protein extraction buffer (10 mM Tris·HCl buffer at pH 7.4, containing 50 mM NaCl, 1 mM MgCl2, 2 mM EDTA, 1% SDS, 5 mM iodoacetamide, 1 mM PMSF) was added to each well and suspensions from two wells containing cells submitted to identical treatment were pooled. Cell suspensions were submitted to ultrasounds for 10 sec. at 50 W using a Braun Labsonic U sonicator and 30 μl aliquots were removed for protein quantification. The remaining suspension (about 270 μl) was mixed with 30 μl of 10 × electrophoresis loading buffer (120 mM Tris·HCl at pH 6.8, 4% SDS, 50% glycerol, 0.1% bromophenol blue, 10%β-mercaptoethanol) and boiled for 3 min. Proteins were separated by electrophoresis (SDS-PAGE) on 8% polyacrylamide (15-well and 1.5 mm thick) gels and then transferred onto nitrocellulose membranes (Schleicher & Schüell, Kent, UK). After immunoblotting, the nitrocellulose membranes were blocked for 1 hr at room temperature in PBS containing 5% non-fat dry milk, 0.5% bovine serum albumin and 0.1% Tween 20 (blocking solution). The membranes were then incubated overnight at 4°C in blocking solution containing one of the following primary antibodies: anti-poly(ADP-ribose) polymerase (anti-PARP; 1:1000); anti-cyclin D3 (1:1,000) or anti-cyclin-dependent kinase2 (anti-cdk2) (1:1,000; BD Transduction Laboratories, Heidelberg, Germany). On the other hand, caspase-8 and -9 (procaspases) and their cleavage products (activated caspases) were detected in mice tumours by immunoblotting using specific antibodies (Cell Signaling, 1:1,000 dilution), similarly as described in reference [6].
After removing the primary antibody, the membranes were washed three times for 10 min. with PBS and incubated for 1 hr at room temperature in fresh blocking solution with horseradish peroxidase-linked goat anti-mouse IgG (1:2,000 dilution; Amersham Pharmacia, Buckinghamshire, UK). Immunoreactivity was detected with the Enhanced Chemiluminescence Western Blot Detection system (ECL; Amersham Pharmacia), followed by exposure to ECL hyperfilm (Amersham Pharmacia). The α-tubulin content in the samples was determined by the same procedure. The films were scanned at a resolution of 400 dpi for quantification, and the concentration measured for a given protein was divided by the α-tubulin content, measured in the same sample.
Gene expression studies by microarray analyses
Total RNA was extracted from either Jurkat or A549 cells incubated in the absence or presence of Minerval (100 μM for 24 hrs) using the RNeasy midi kit (Qiagen). About 50–100 μg of total RNA was obtained from 0.5–1 × 108 cells and was processed following the manufacturer’s directions. The CNIO OncoChip [17] was used to identify differentially expressed genes (i.e. Minerval-treated versus untreated). Fluorescent cDNA (30 μg) was generated from total RNA samples and labelled using either Cy3-dUTP (treated samples) or Cy5-dUTP (untreated samples). Upon hybridization of fluorescent cDNAs to the OncoChip, these microarrays were scanned with the Agilent G2565BA scanner and images were subsequently quantified using the Agilent Technologies Feature Extraction software (Agilent Technologies, Santa Clara, CA, USA). After raw data filtering and normalization with ‘Lowess’ routine [18, 19] genes were defined as significantly up- or down-regulated if the ratio was either higher than 2 or lower than 0.5 (i.e. 2-fold or more difference).
Confocal microscopy
Jurkat cells were cultured as indicated above in the presence or absence of Minerval (50 μM, 6 hrs), washed with TBS buffer (137 mM NaCl, 2.7 mM KCl, 25 mM Tris·HCl, pH 7.4) and incubated for 10 min at 4°C with Vybrant Alexa Fluor 594 (Lipid Raft labelling kit, Molecular Probes, Paisley, UK) in fresh TBS buffer, which uses the affinity of cholera toxin B subunit for ganglioside GM1 to label membrane rafts (excitation at 542 nm and detection at 610–650 nm). Then, cells were fixed with 4% paraformaldehyde for 15 min at 4°C and incubated with 5% normal horse serum in TBS buffer for 1 hr at room temperature. Immediately, they were incubated overnight in the presence of monoclonal anti-Fas (1:1,00; Santa Cruz) at 4°C in TBS buffer supplemented with 2% horse serum. Finally, cells were washed with TBS buffer, incubated with the secondary antibody (Alexa Fluor, Paisley, UK, 488-labelled goat anti-mouse IgG at 1:200 dilution, Molecular Probes; excitation at 488 nm and detection at 510–550 nm) and mounted on glass slides using Glycerol Gelatin, Sigma). Images were acquired using a Leica TCS SP2 spectral confocal microscope with 630× optical magnification and 8× digital magnification (approximately 5,000× total magnification), and analysed with the software provided by the manufacturer.
Animal model of cancer and treatments
The protocols used in this study were revised and approved by the Ethics Committee of the University of the Balearic Islands. A xenograft model of human lymphoblastic leukaemia was established by s.c. injection of tissue culture-derived Jurkat cells (5 × 106 cells/animal) on the left dorsal surface of 6-week-old nude [Crl:Nu(Ico)-Fox1] male mice. The tumour volume (cubic millimetres) was calculated as w2×l/2, where w is the width and l is the length of the tumour, as measured with a digital calliper. On day 10 after implantation, tumours reached an average size of about 60 mm3 (100% tumour size). The animals were randomized into two groups of eight animals, so that the average tumour volumes were approximately the same for each group at the onset of treatment. Animals were treated for 21 days with Minerval (600 mg/kg daily, p.o.) or vehicle (2 ml/kg of 50% glycerol daily, p.o.), being the tumour volume determined in both groups. Finally, animals were killed and tumours were dissected for histological analysis. Briefly, tumours were fixed overnight in 10% paraformaldehyde, dehydrated and embedded in paraffin. Then, serial tissue sections were either stained with hematoxilin and eosyn or subjected to terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL), using the TumorTACS In situ Apoptosis Detection kit (R&D Systems, Inc., Minneapolis, MN, USA) to determine the extent of apoptosis in Minerval-treated and control animals. One small fragment from each tumour was removed to determine procaspase-8 and -9 activation (cleavage) by immunoblotting techniques, as described above.
Results
Effects of Minerval on Jurkat cell proliferation
We studied the effects of Minerval on Jurkat (human T-lymphoblast leukaemia) cells because treatments with this molecule increase the survival rate of mice infected with murine leukaemia cells (84% and 0% survival for mice treated and untreated with Minerval, respectively) [2] and the molecular mechanisms involved in this anti-cancer action have not been studied yet. The effects of Minerval and the structurally related fatty acid, OA, on the growth of Jurkat cells in culture were assessed (25, 50 and 75 μM for Minerval and 25, 50 and 100 μM for OA). Minerval impaired Jurkat cell growth in a time- and concentration-dependent manner, with an IC50 of ∼40 μM (48–72 hrs, Fig. 1A). In contrast, Jurkat cell growth was only modestly affected by similar concentrations of OA (Fig. 1B). Cell viability was also determined using the MTT method and presented similar results. Thus, Minerval induced a marked and significant reduction of cell viability (Fig. 1C), whereas OA only caused a modest inhibition (Fig. 1D). This impairment of Jurkat (leukaemia) cell growth by Minerval was in agreement with previous results showing induction of the cell cycle arrest in A549 (lung cancer) cells. Inhibition of Jurkat cell proliferation was associated with marked decreases in cyclin D3 and cdk2 with respect to α-tubulin (Fig. 1E). Hence, the molecular mechanism of the anti-tumourigenic effects of Minerval in Jurkat cells appeared to be in part similar to that (anti-proliferative) described in A549 cells [2]. In addition, it was also demonstrated that the modest chemical difference between Minerval and OA (one oxygen atom) had a marked effect on the pharmacological activity of these fatty acids.
Fig 1.
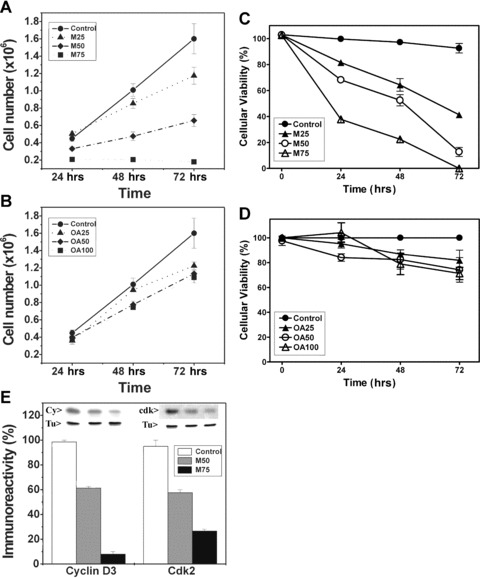
(A) The effects of 25 μM, 50 μM, and 75 μM Minerval (M25, M50 and M75, respectively) and (B) 25 μM, 50 μM, and 100 μM OA (OA25, OA50 and OA100, respectively) on Jurkat cell number. Cell proliferation was also measured using the MTT method in Jurkat cells treated with the same concentrations of Mineval (C) and OA (D). (E) The effects of 50 μM (M50) and 75 μM (M75) Minerval (48 hrs) on the cellular accumulation of cyclin D3 (Cy) and cdk2 (cdk) as determined by immunoblotting. Tu, tubulin.
Minerval induces apoptosis in Jurkat cells
In previous studies, it was shown that apoptosis was not involved in the anti-cancer effects of Minerval on human lung adenocarcinoma (A549) cells [2]. In contrast, Minerval did induce apoptosis in human leukaemia cells. As determined by flow cytometry of ethidium bromide stained cells, Minerval treatments induced DNA breakage (increases in sub-G1 peaks, Fig. 2A). When the sub-G1 peak was quantified it became clear that the apoptotic effect of Minerval was both time- and concentration-dependent. After a 72-hr exposure to 100 μM Minerval a 20-fold increase in the proportion of apoptotic cells was observed (from 3.4 ± 0.4% to 69.5 ± 4.9% in the absence and presence of Minerval, respectively). This time- and concentration-dependent effect of Minerval was most probably responsible for the reversion of leukaemia in animal models [2], because greater concentrations of this drug induced total depletion of cancer cells in cultures (data not shown). Minerval also induced a marked and concentration-dependent increase in the proteolytic cleavage of PARP, a molecular marker of apoptosis (Fig. 2B).
Fig 2.
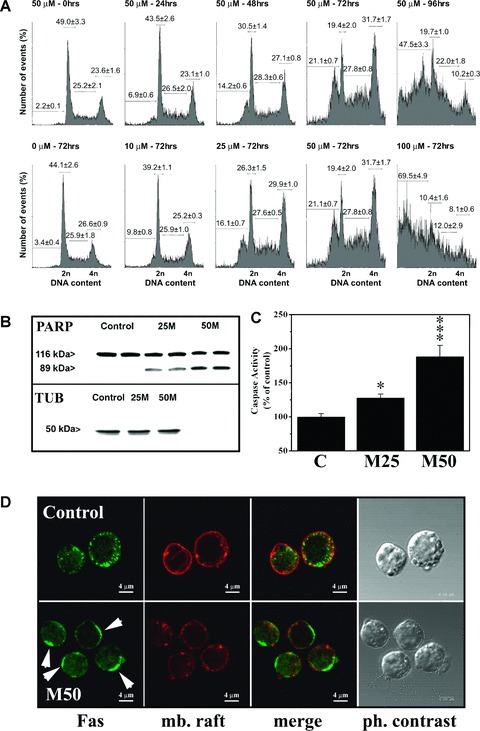
Assessment of the cell cycle and induction of apoptosis. (A) Time-dependence (upper row) and concentration-dependence (lower row) of induction of apoptosis by Minerval in Jurkat cells. DNA degradation was determined by flow cytometry in cells incubated at the times and concentrations indicated over each panel. The percentage of cells undergoing apoptosis corresponds to the sub-G1 peak (DNA content between 0 and 2 n). (B) Proteolytic cleavage of PARP in Jurkat cells cultured for 72 hrs in the presence of 25 and 50 μM of Minerval (M25 and M50, respectively). Untreated (control) cells showed no significant fragmentation of PARP. TUB, tubulin. (C) Caspase activity in Jurkat cells cultured for 48 hrs in the presence (M25, 25 μM; M50, 50 μM) or absence (control, C) of Minerval (*P < 0.05; ***P < 0.001). (D) Labelling of Fas receptor and GM1 (membrane rafts, mb. raft) in Jurkat cells cultured for 48 hrs in the presence (M50, 50 μM) or absence (Control) of Minerval. Capping of Fas receptors (arrows) and membrane raft disorganization upon treatment with Minerval were observed using confocal microscopy (bar = 4 μm). For further details see text.
On the other hand, the presence of Minerval (50 μM, 6 hrs) induced changes in membrane structure that induced membrane raft disorganization and capping of Fas receptor in Jurkat cell membranes (Fig. 2D). Various studies have shown that membrane lipid reorganization may induce clustering and activation of death receptors in the absence of ligand (see in the discussion section). Because Fas receptor activation promotes assembly of death-inducing signalling complex (DISC) and caspase-8 activation, we assessed caspase activity in Jurkat cells. Minerval also induced caspase activation, as determined by flow cytometry using the fluorescent caspase ligand FITC-VAD-FMK (100.0 ± 4.98% and 188.2 ± 16.82% in control and Minerval-treated cells, Fig. 2C). At greater concentrations of this drug or longer times, caspase activity could possibly be higher, although the lesser number of viable cells made it difficult to determine caspase activity using this technique. These observations taken together further support the conclusion that Minerval induced apoptosis in Jurkat cells. In addition, we observed that Jurkat cells underwent important morphological changes following exposure to Minerval with significant cell degeneration, including the formation of apoptotic bodies, membrane blebs, mitochondrial swelling, nuclear degeneration, etc. (Fig. 3).
Fig 3.
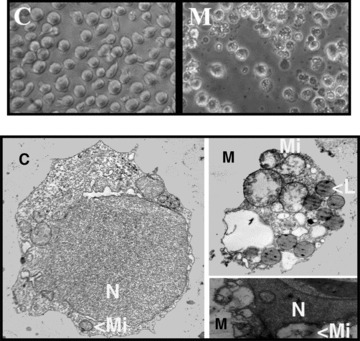
Upper panels: phase contrast microscopy (original magnification × 400) of Jurkat cells cultured for 72 hrs in the presence (M) or absence (C) of 50 μM Minerval. Minerval-treated cells show membrane blebbing and budding and cellular degeneration which was further assessed by electron microscopy. Lower panels: transmission electron microscopy of Jurkat cells incubated in the absence (Control, C, left, original magnification × 5,000) or presence of Minerval (M, right, original magnification × 8,000). The right TEM panels show in detail two apoptotic bodies also observed by phase contrast microscopy in the presence of Minerval. N, nucleus; Mi, mitochondria; L, lipid vesicles.
Caspase-8 mediates the effects of Minerval on Jurkat cells
Effector caspases can be activated by two general pathways: the ‘extrinsic’ membrane death receptor pathway that is associated with caspase-8 activation; and the ‘intrinsic’ mitocondrial pathway that is associated with caspase-9 activation. As an approximation to determine which of these pathways might be involved in Minerval-induced apoptosis, we determined the effects of inhibition of caspase-8 (using the inhibitor zIETD-fmk) and caspase-9 (using the inhibitor zLEHD-fmk) on the apoptosis of Jurkat cells induced by 50 μM Minerval during 72 hrs. The inhibition of caspase-8 activity (the extrinsic pathway) significantly reduced the occurrence of apoptotic events in Jurkat cells treated with Minerval (Fig. 4). In the presence of caspase-8 inhibitor, the proportion of Jurkat cells that underwent apoptosis decreased from 21.1 ± 0.7% (Minerval alone) to 6.4 ± 0.1% (Minerval plus zIETD-fmk) of the total cell population (Fig. 4). In contrast, caspase-9 inhibitor offered only modest protection to the apoptosis induced by Minerval (from 21.1 ± 0.7% to 14.7 ± 1.2% in the absence or presence of zLEHD-fmk, respectively: Fig. 4). When measured as the percentage of hypodiploid events, the inhibition of caspase-8 reduced the degree of apoptosis over basal values by about 78% whereas caspase-9 inhibition produced only a 33% decrease. Simultaneous inhibition of both caspases did not produce any synergism in the protection against apoptosis offered to Jurkat cell. In the presence of both inhibitors, the proportion of cells that underwent cell death was similar to that seen when caspase-8 alone was inhibited (data not shown). Moreover, neither the caspase-8 inhibitor nor the caspase-9 inhibitor significantly affected the cells in the absence of Minerval. These data suggest that caspase-8 is involved in induction of apoptosis in Jurkat cells and is upstream with respect to caspase-9 in the induction of apoptosis upon Minerval treatments.
Fig 4.
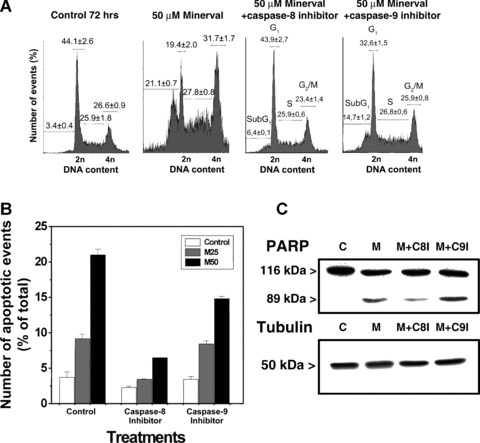
Effect of caspase-8 and caspase-9 inhibition on Minerval-induced apoptosis. (A) DNA content in Minerval-treated cells (50 μM) in the presence of caspase-8 (left) or caspase-9 (right) inhibitors. Basal apoptosis in the presence of 50 μM Minerval was 21.1% of the cells in culture. (B) Jurkat cells were cultured for 72 hrs in the presence or absence (open bars) of 25 (M25, grey bars) or 50 μM (M50, black bars) of Minerval, and in the presence or absence (control) of caspase-8 and caspase-9 inhibitors. C, PARP fragmentation in untreated Jurkat cells (control, C) and in cells treated with 50 μM Minerval in the presence or absence (M) of the caspase-8 inhibitor (M+C8I) or the caspase-9 inhibitor (M+9I). Immunoreactive α-tubulin bands from cells treated as above are also shown.
In agreement with flow cytometry data, caspase-8 inhibition induced a marked reduction of fragmentation of PARP after 72 hrs in the presence of 50 μM Minerval (81% decrease). In contrast, inhibition of caspase-9 activity did not significantly reduce the fragmentation of PARP (Fig. 4C). These results suggest that Minerval induced apoptosis mainly through the extrinsic (caspase-8-dependent) pathway.
Minerval on a xenograft model of human leukaemia
Minerval treatments markedly and significantly inhibited tumour growth in nude mice infected with Jurkat cells. Tumours from control (untreated) mice increased 431 ± 24% between days 10 and 31 after tumour implantation, whereas the average increase in tumour volume of Minerval-treated animals was 24 ± 16% (three out of eight animals showing tumour reductions) (Fig. 5). On the other hand, cytohistochemical analysis of the tumours dissected from these animals revealed that most cells were dead, containing pycnotic nuclei (hematoxilin-eosyn stainig), or apoptotic (TUNEL), in animals treated with Minerval. In turn, in tumours from untreated animals, most cancer cells did not show signs of apoptosis or cell death (Fig. 5). Similarly to Jurkat cells from in vitro cultures, we observed caspase-8 and caspase-9 activation (the former at a greater extent than the latter) in these tumours, as determined by the presence of immunoreactive bands corresponding to proteolytic caspase products (active caspases). The greater immunoreactivity of proteolysis fragments from caspase-8 also suggests that activation of the extrinsic pathway occurs upstream with respect to the intrinsic pathway (Fig. 5).
Fig 5.
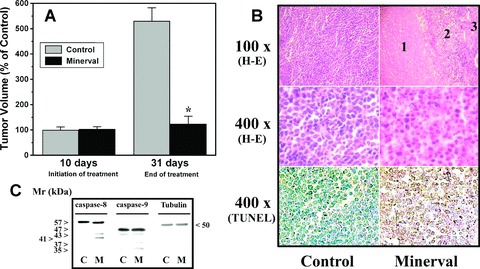
Effect of Minerval treatments on tumour growth in mice. (A) Nude mice were infected with Jurkat cells by s.c. injection on the dorsal surface. Ten days after implantation, tumours were measured and then animals were treated with vehicle (Control, grey bars) or Minerval (black bars) for another 21 consecutive days. The bars correspond to the mean ± SEM values of tumour volume at day 10 (before treatment) and 31 (end of treatment) post-implantation. (B) Histopathological analysis of tumours dissected from vehicle- (Control, left) and Minerval-treated (right) animals. Tumours from control animals showed a homogeneous histological organization, being most cells alive. In tumours from animals treated with Minerval (which were smaller than those from vehicle-treated animals), the greatest region corresponded to that of dead cells (1), followed by a transition area (2) containing dead cells and cells with pycnotic nuclei and a small region (3) in which most cells were alive. The lower micrographs of this panel show that most cells in Minerval-treated animals underwent apoptosis (TUNEL). (C) Immunoblotting detection of caspase-8, caspase-9 and tubulin in tumours from untreated (Control) and Minerval-treated (M) mice.
Molecular mechanisms of the anti-cancer action of Minerval in different cancer cell lines
We also studied the molecular mechanisms involved in the pharmacological action of Minerval in several cancer cell lines and non-tumour fibroblasts (Table 1). For this purpose, cell viability was determined, cell DNA content was quantified and determined the proportion of cells in the various phases of the cell cycle (G0/G1, S and G2/M) or in apoptosis (sub-G1 peaks) in the absence or presence of Minerval (25–400 μM) at different incubation times (0–144 hrs). Apoptosis was further assessed by quantification of PARP degradation in the absence or presence of Minerval. The mechanism of action of Minerval was assigned to induction of the cell cycle arrest (anti-proliferative, P) when the number of cells was reduced, they accumulated in the G0/G1 phase and no PARP degradation was observed, whereas degradation of this enzyme along with the presence of a sub-G1 peak (produced by DNA degradation) was associated with induction of apoptosis (A). In seven cancer cell lines (two lymphoblastic leukaemia, pancreas, prostate, breast, cervix, and colon), Minerval appeared to trigger only apoptosis (Table 1). In another three cell lines (lung, glioma, and neuroblastoma), Minerval only exerted anti-proliferative effects. In other two cell lines (prostate and myeloblastic leukaemia), Minerval induced anti-proliferative effects at lower concentrations and pro-apoptotic effects at higher concentrations. The assignment of the mechanism of action in these cell lines was relative to the type of studies carried out in them, which have been listed above. In contrast, in IMR90 (non-tumour fibroblast) cells Minerval did not induce important reductions in the number of viable cells and apoptosis was not observed (Table 1). In fact, the IC50 values for Minerval against most cancer cell lines was in the range of 30–100 μM, whereas for IMR90 cells IC50 was greater than 5,000 μM.
Table 1.
Effect of Minerval on cancer cell growth and mechanism of action
| Cell line | Cancer type | Mechanism of action* | Antitumoural effect† |
|---|---|---|---|
| PC3 | Prostate | PA | +++ |
| LNcaP | Prostate | A | +++ |
| MDA-MB-231 | Breast | A | ++ |
| M220 | Pancreas | A | ++ |
| L-1210 | Lymphoblastic leukaemia | A | +++ |
| Jurkat | Lymphoblastic leukaemia | A | +++ |
| HL-60 | Myeloblastic leukaemia | PA | +++ |
| HeLa | Cervix | A | +++ |
| HT-29 | Colon | A | ++ |
| C-6 | Brain-glioma‡ | P | +++ |
| SH-SY5Y | Brain-neuroblastoma | P | + |
| A549 | Lung | P | +++ |
| IMR90 | Non-tumour fibroblasts | None | −§ |
A, apoptosis induction; P, proliferation inhibition. PA, proliferation inhibition at lower concentrations and apoptosis induction at higher concentrations. Mechanisms based on the studies listed in the Results section.
+, cell growth inhibition; ++, cell growth arrest; +++, complete killing of tumour cells (data at 200 μM).
All cell lines listed are derived from human cancers except for C-6 rat glioma cells.
No effects were observed; IC50 > 5,000 μM.
Minerval and analogues (e.g. OA) are known to bind to membrane lipids and regulate membrane lipid structure, which results in changes of the localization and activity of important signalling peripheral proteins, such as G proteins and protein kinase C (PKC) [1, 2, 15], which eventually modulate gene expression. We studied this modulation in one cell line in which Minerval induced cell cycle arrest (A549 cell) and another cell line in which induced apoptosis (Jurkat cells). In this context, we found that Minerval treatments increased the transcriptional rate of several genes associated with the onset of apoptosis in Jurkat cells, most of them unchanged in A549 cells (Table 2). In contrast, the number of antiproliferative genes induced in A549 cells was greater than that induced in Jurkat cells (Table 2). Moreover, the regulatory effects of Minerval in these cell lines appeared to show differences that would justify the anti-cancer mechanisms induced in one or the other cell line.
Table 2.
Effect of Minerval on gene expression in Jurkat and A549 cells
| Gene symbol | Gene/protein name | Jurkat cells* | A549 cells* | Cellular effects† |
|---|---|---|---|---|
| CASP6 | Caspase 6 | + | = | A |
| DATF1 | Death associated transcription factor 1 | + | = | A |
| MADD | MAP-kinase activating death domain | + | = | A |
| PDCD5 | Programmed cell death 5 | + | = | A |
| PRG1 | Proteoglycan 1 | + | = | A |
| STK4 | Serine/threonine kinase 4 | + | = | A |
| TIAL1 | TIA1 CG-associated RNA binding protein-like 1 | + | = | A |
| TNFSF8 | TNF (ligand) superfamily, member 8 | + | = | A |
| TP53I3 | Tumour protein p53 inducible protein 3 | + | = | A |
| DUSP5 | Dual specificity phosphatase 5 | + | + | D / Pi |
| BCL2L11 | BCL2-like 11 (apoptosis facilitator) | = | + | A |
| BID | BH3 interacting domain death agonist | = | + | A |
| EPHA2 | EphA2 | = | + | D |
| HDAC5 | Histone deacetylase 5 | = | + | D / Pi |
| LIF | Leukaemia inhibitory (differentiation) factor | = | + | D |
| MYADM | Myeloid-associated differentiation marker | = | + | D |
| PPP1R15A | Protein phosphatase 1, inhibitory subunit 15A | = | + | A / Pi |
| RAI3 | Retinoic acid induced 3 | = | + | D / Pi |
| RIS1 | Ras induced senescence 1 | = | + | D |
| S100A2 | S100 calcium binding protein A2 | = | + | D / Pi |
| ABL1 | V-abl Abelson leukaemia oncogene homolog 1 | – | = | Pa |
| CCND1 | Cyclin D1 (PRAD1) | – | = | Pa |
| CDC25A y B | Cell division cycle 25A y B | – | = | Pa |
| CDK10 | Cyclin-dependent kinase 10 | – | = | Pa |
| CDK6 | Cyclin-dependent kinase 6 | – | = | Pa |
| CLK-2 | CDC-like kinase 2 | – | = | Pa |
| CRSP7 | Cofactor for Sp1 activation, subunit 7 | – | = | Pa |
| E2F2 | E2F transcription factor 2 | – | = | Pa |
| FOLR1 | Folate receptor 1 | – | = | Pa |
| MAP3K11 | MAP kinase kinase kinase 11 | – | = | Pa |
| RBL1 | Retinoblastoma-like 1 (p107) | – | = | Pa |
| CDCA8 | Cell division cycle associated 8 | – | – | Pa |
| MAP3K1 | Mitogen-activated protein kinase kinase kinase 1 | – | – | A / D / Pa |
| RBBP6 | Retinoblastoma binding protein 6 | – | – | A / D / Pa |
| CCNA2 | Cyclin A2 | = | – | D / Pa |
| CCNG1 | Cyclin G1 | = | – | Pa |
| ECT2 | Epithelial cell transforming sequence 2 oncogene | = | – | Pa |
| EIF5A y B | Eukaryotic translation initiation factor 5A y B | = | – | Pa |
| GSPT1 | G1 to S phase transition 1 | = | – | Pa |
Significant (P < 0.05) and marked (over 2-fold) increase (+) or decrease (−), or no significant changes (=), with respect to untreated cells.
Pathway(s) regulated by the gene: A, Apoptosis; P, cell proliferation inhibition (Pi) or activation (Pa); D, cell differentiation.
Discussion
The present study shows that Minerval turned on the apoptotic machinery in most cancer cell lines studied (9 out of 12 cell lines). In turn, inhibition of cell proliferation (i.e. exit from the cell cycle) was the sole mechanism triggered by this drug in three cell lines (Table 1). In contrast, Minerval did not induce anti-proliferative, pro-apoptotic, or any kind of cytotoxic effects in non-tumour cells (IMR90 fibroblasts). These results explain in part the activity of this drug against various types of cancer. In addition, these data further indicate a lack of cytotoxic effects in normal tissues, which in fact has been reported in previous histopathological studies in various tissues from rats treated with high doses of Minerval [2] and in formal and independent preclinical studies (data not shown). Evasion of apoptosis is one of the hallmarks of cancer cells and one of the aims of novel therapies designed against cancer. The present study shows how membrane lipid signals can differentially trigger cancer cell death without compromising health in animals. Because most chemotherapy agents show important side effects, the existence of a molecule with high pharmacological activity and low toxicity might be crucial for treatment of primary leukaemia and haematological cancers resulting from treatments of other tumours with genotoxic drugs. Moreover, the difference in IC50 values between normal cells (> 5,000 μM) and cancer cells (30–100 μM) indicates that the therapeutic window for this drug will fall far below the maximum tolerated dose (or minimum lethal dose), dislike most anticancer drugs currently used. These facts suggest that Minerval might be used as a first-line treatment or in combinatory therapies for leukaemia and other types of cancer.
The first target of this synthetic analogue of OA is the plasma membrane, in which it binds to membrane lipids and regulates lipid structure and peripheral signalling protein localization and activity [2]. In human lung cancer (A549) cells, this is followed by knockdown of certain cyclins and ckds and hypophosphorylation of the retinoblastoma protein (pRb), which remains bound to E2F-1, inhibiting the transcriptional activation of many genes involved in cell growth [3].
Minerval is structurally related to OA, and both compounds are capable of modulating the structure of the membrane, inducing the formation of nonlamellar membrane structures (hexagonal, HII phases) [1, 4]. Nonlamellar-phase propensity has been shown to regulate the interaction of signalling proteins with membranes, such as G proteins and PKC [2, 7–9], which are fundamental in the control of cell proliferation and other important cell functions [20, 21]. As such, the effects of Minerval and structurally related fatty acids (e.g. OA) have been associated with their influence on the activity of these signalling proteins induced by the alterations in membrane lipid structure [4, 5, 15]. The influence on membranes of these ‘boomerang-shaped’ lipids is founded on their structural properties, because the ‘rod-shaped’ fatty acids, elaidic (trans isomer of OA) and stearic (saturated analogue of OA) acids, did not exert the same effects on membrane organization or on the activity of peripheral proteins [1, 2, 4, 5, 15].
The plasma membrane is a meeting point for lipids and proteins and the scenario in which extracellular signals activate receptor proteins and messages are propagated into the cell by complex transduction systems. We have previously shown how membrane lipid structure regulates cell signalling [7, 9, 15]. Moreover, regulation of membrane lipid structure has proven to be a good strategy to treat a number of human pathologies [14]. In this context, the plasma membrane is involved in the mode of action of several anti-tumour drugs, such as anthracyclines, hexamethylene bisacetamide, and Minerval [1, 2, 8, 15, 22, 23]. In addition, certain anti-tumour compounds exhibit molecular structures similar to those of membrane lipids and may interact with cell membranes: edelfosine [Et-18-OCH3 (1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine)] and miltefosine [HePC (hexadecyl phosphocholine)] have an important hydrophobic moiety with long hydrocarbon chains (18 and 16 C atoms, respectively), and a polar region comprised a phosphate group and a choline moiety. On the other hand, the anti-cancer drug NEO6002 results from the covalent binding of gemcitabine to cardiolipin, a phospholipid typical of mitochondrial membranes, and it appears to be less toxic and more effective than gemcitabine alone [24]. The lipid modification of gemcitabine induces the membrane-mediated internalization of the compound, which is not blocked by nucleoside transporter inhibitors that affect the internalization of gemcitabine. Another type of lipid-interacting compound is propofol-DHA, which combines a well-known anaesthetic (propofol) with a polyunsaturated fatty acid (docosahexaenoic acid, DHA). The resulting compound induced apoptosis in MDA-MB-231 breast cancer cells [25] and so did Minerval in the same cell line (this work). Indeed, the pro-apoptotic properties of the lipid ceramide have already been attributed in part to its capacity to induce nonlamellar membrane lipid phases [26], a characteristic also displayed by Minerval [1, 2]. These studies and the present work indicate that the modification of membrane lipid structure can trigger apoptosis in cancer cells. In this line, cyclooxygenase-2 inhibition induces increases of arachidonic acid and ceramide accumulation, altering the membrane structural properties and clustering of the TRAIL receptor DR5 [27]. Similarly, edelfosine and other alkylphosphocholine anti-cancer compounds induce apoptosis in cancer cells through a mechanism that involves alteration of membrane lipid rafts [28]. Here, we showed that the regulation of membrane lipid structure promoted by Minerval induced important changes in the distribution of membrane rafts that may induce capping of the death receptor Fas (CD95). This clustering could promote DISC assembly and activation of caspase-8.
Additional mechanisms for induction of apoptosis may also be triggered by Minerval. Thus, although repression of cyclin D3 impairs proliferation of A549 cells [29], a decrease in cyclin D3 is associated with the induction of apoptosis in T-lymphocytes [30]. Similarly, cdk2 repression and down-regulation has anti-proliferative effects in A549 cells [2] and pro-apototic effects in Jurkat cells [31]. Therefore, through common molecular mechanisms, Minerval has a differential anti-cancer activity against A549 cells, Jurkat cells, and other cancer cells. The critical role of cdk2 in cell proliferation has been well established, raising interest in the potential to use cdk2 inhibitors in the treatment of cancer [31, 32]. However, Minerval is not strictly an inhibitor of cdk2 but the resulting down-regulation of this kinase produces a similar reduction of the enzyme’s activity. In the present study, we observed that Minerval induced a marked down-regulation of cyclin D3 and cdk2. These key events might be involved in the induction of apoptosis by Minerval in Jurkat cells and it would justify the fact that in certain cell lines this drug induces cell cycle arrest. In this context, cdk inhibition enhances TRAIL-induced formation of a cell death signalling complex and early processing of procaspase-8 in breast cancer cells [33], which also appeared to undergo apoptosis after Minerval treatments (Table 1). In this work, we showed that Minerval regulated more genes related to apoptosis in Jurkat cells than in A549 cells, whereas in the latter the number of anti-proliferative genes significantly regulated was greater (Table 2). Thus, the mechanism by which Minerval exerts its anti-cancer activity in different tumour cells depends on the balance of genes whose expression is regulated.
Minerval-induced apoptosis was reversed by inhibition of caspase-8 activity (about 78% reduction in the number of apoptotic cells), indicating that Minerval triggered the ‘extrinsic’ apoptosis (death-receptor-associated) pathway. In contrast, caspase-9 inhibition only led to modest reductions in apoptosis. In addition, simultaneous caspase-8 and caspase-9 inhibition did not augment the protection from apoptosis offered by inhibiting caspase-8 alone. Thus, activation of the ‘intrinsic’ pathway could be initiated as a downstream event of the ‘extrinsic’ pathway [11, 33]. Moreover, in an animal model of human cancer, we observed that Minerval inhibited tumour growth and induced caspase-8 activation and induction of apoptosis. These results further support the potential use of this drug for treatment of human leukaemia. In this context, a recent study shows that caspases can interact with substrates and inhibitors other than those indicated to be specific for each isozyme, so that the present results should be considered from this perspective [34]. However, both the caspase-8 substrate IETD and its related inhibitor zIETD-fmk do not show affinity for caspase-9 [34], the intrinsic pathway initiator caspase, which supports the hypothesis of activation of the extrinsic apoptosis pathway by Minerval.
OA is a natural nutrient that is metabolised in the mitochondria to obtain energy through β-oxidation. Our data show that Minerval (2-hydroxy-OA) induced apoptosis of Jurkat T-lymphoblastic leukaemia cells in a concentration- and time-dependent manner. In contrast, OA only had a modest effect on the proliferation of these cells at higher concentrations (≥ 250 μM; data not shown). Therefore, the additional oxygen atom on Minerval increases the pharmacological potency of this drug when compared to the natural free fatty acid, OA. In addition, this modification did not change the safety of the fatty acid, because doses up to 3 g/kg daily for 2 weeks did not produce toxicity in rats [2]. In line with this, epidemiological and experimental studies have shown that the intake of free fatty acids reduces the incidence of several types of cancer [35–41]. Moreover, fatty acids with a lesser rate of metabolic degradation show a greater potency against tumour growth [35–41]. Therefore, the reduced clearance of Minerval from the cell would directly enhance the effects of OA on membrane structures and cell signalling.
Protein–lipid interactions play a crucial role in signal propagation [42, 43] and Minerval can regulate membrane physical properties involved in such interactions [1, 2]. This constitutes a new therapeutic approach (membrane-lipid therapy), based on regulation of membrane lipid structure and subsequent modification of cell signalling [14]. In this study, we showed that apoptosis appeared to be the preferential mechanism of action induced by Minerval in most cancer cell lines studied. Differences in the expression of certain genes might be involved in the induction of other anti-cancer mechanisms, such as cell cycle arrest. In this context, apoptosis has been pointed out as a promising therapeutic approach for treatment of cancer and other pathologies [44], as far as this process does not affect other healthy cells, which is the case of Minerval. Thus, the present study explains in part the potent anti-tumour action of this compound without apparent side effects or toxicity, although further studies are required to fully understand the molecular bases of its pharmacological effects.
Acknowledgments
This work was supported in part by Grants BFU2007-61071 from the Ministerio de Educación y Ciencia (Spain), PCTIB-2005GC4-07 and PRIB2004-10131 from the Govern Balear and by ‘The Marathon Foundation’.
References
- 1.Barceló F, Prades J, Funari SS, et al. The hypotensive drug 2-hydroxyoleic acid modifies the structural properties of model membranes. Mol Membr Biol. 2004;21:261–8. doi: 10.1080/09687680410001716835. [DOI] [PubMed] [Google Scholar]
- 2.Martínez J, Vögler O, Casas J, et al. Membrane structure modulation, protein kinase Calpha activation, and anticancer activity of Minerval. Mol Pharmacol. 2005;67:531–40. doi: 10.1124/mol.104.000778. [DOI] [PubMed] [Google Scholar]
- 3.Martínez J, Gutiérrez A, Casas J, et al. The repression of E2F-1 is critical for the activity of Minerval against cancer. J Pharmacol Exp Ther. 2005;315:466–74. doi: 10.1124/jpet.105.088716. [DOI] [PubMed] [Google Scholar]
- 4.Funari SS, Barcelo F, Escribá PV. Effects of oleic acid and its congeners, elaidic and stearic acids, on the structural properties of phosphatidylethanolamine membranes. J Lipid Res. 2003;44:567–75. doi: 10.1194/jlr.M200356-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Prades J, Funari SS, Escribá PV, et al. Effects of unsaturated fatty acids and triacylglycerols on phosphatidylethanola-mine membrane structure. J Lipid Res. 2003;44:1720–7. doi: 10.1194/jlr.M300092-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Alemany R, Vögler O, Terés S, et al. Antihypertensive action of 2-hydroxyoleic acid in SHRs via modulation of the protein Kinase A pathway and Rho kinase. J Lipid Res. 2006;47:1762–70. doi: 10.1194/jlr.M500520-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Vögler O, Casas J, Capó D, et al. The Gβγ dimer drives the interaction of heterotrimeric Gi proteins with nonlamellar membrane structures. J Biol Chem. 2004;279:36540–5. doi: 10.1074/jbc.M402061200. [DOI] [PubMed] [Google Scholar]
- 8.Escribá PV, Sastre M, García-Sevilla JA. Disruption of cellular signaling pathways by daunomycin through destabilization of nonlamellar membrane structures. Proc Natl Acad Sci USA. 1995;92:7595–9. doi: 10.1073/pnas.92.16.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Escribá PV, Ozaita A, Ribas C, et al. Role of lipid polymorphism in G protein-membrane interactions: nonlamellar-prone phospholipids and peripheral protein binding to membranes. Proc Natl Acad Sci USA. 1997;94:11375–80. doi: 10.1073/pnas.94.21.11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giorgione J, Epand RM, Buda C, et al. Role of phospholipids containing docosahexaenoyl chains in modulating the activity of protein kinase C. Proc Natl Acad Sci USA. 1995;92:9767–70. doi: 10.1073/pnas.92.21.9767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basu A, Castle VP, Bouziane M, et al. Crosstalk between extrinsic and intrinsic cell death pathways in pancreatic cancer: synergistic action of estrogen metabolite and ligands of death receptor family. Cancer Res. 2006;66:4309–18. doi: 10.1158/0008-5472.CAN-05-2657. [DOI] [PubMed] [Google Scholar]
- 12.Haupt S, Berger M, Goldberg Z, et al. Apoptosis – the p53 network. J Cell Sci. 2003;116:4077–85. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- 13.Tritton TR, Yee G. The anticancer agent adriamycin can be actively cytotoxic without entering cells. Science. 1982;217:248–50. doi: 10.1126/science.7089561. [DOI] [PubMed] [Google Scholar]
- 14.Escribá PV. Membrane-lipid therapy: a new approach in molecular medicine. Trends Mol Med. 2006;12:34–43. doi: 10.1016/j.molmed.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Yang Q, Alemany R, Casas J, et al. Influence of the membrane lipid structure on signal processing via G protein-coupled receptors. Mol Pharmacol. 2005;68:210–7. doi: 10.1124/mol.105.011692. [DOI] [PubMed] [Google Scholar]
- 16.Galbiati F, Guzzi F, Magee AI, et al. Chemical inhibition of myristoylation of the G protein Gi1 alpha by 2-hydroxymyristate does not interfere with palmitoylation or membrane association. Evidence that palmioylation, but not myristoylation, regulates membrane attachment. Biochem J. 1996;313:717–20. doi: 10.1042/bj3130717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tracey L, Villuendas R, Ortiz P, et al. Identification of genes involved in resistance to interferon-alpha in cutaneous T-cell lymphoma. Am J Pathol. 2002;161:1825–37. doi: 10.1016/s0002-9440(10)64459-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smyth GK, Speed TP. Normalization of cDNA microarray data. Methods. 2003;31:265–73. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- 19.Yang YH, Dudoit S, Luu P, et al. Normalization of cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furui T, LaPushin R, Mao M, et al. Overexpression of edg-2/vzg-1 induces apoptosis and anoikis in ovarian cancer cells in a lysophosphatidic acid-independent manner. Clin Cancer Res. 1999;5:4308–18. [PubMed] [Google Scholar]
- 21.Wang Q, Worland PJ, Clark JL, et al. Apoptosis in 7-hydroxystaurosporine-treated T lymphoblasts correlates with activation of cyclin-dependent kinases 1 and 2. Cell Growth Differ. 1995;6:927–36. [PubMed] [Google Scholar]
- 22.Escribá PV, Ferrer-Montiel AV, Ferragut JA, et al. Role of membrane lipids in the interaction of daunomycin with plasma membranes from tumor cells: implications in drug-resistance phenomena. Biochemistry. 1990;29:7275–82. doi: 10.1021/bi00483a017. [DOI] [PubMed] [Google Scholar]
- 23.Escribá PV, Morales P, Smith A. Membrane phospholipid reorganization differentially regulates metallothionein and heme oxygenase by heme-hemopexin. DNA Cell Biol. 2002;21:355–64. doi: 10.1089/104454902753759762. [DOI] [PubMed] [Google Scholar]
- 24.Chen P, Chien PY, Khan AR, et al. In-vitro and in-vivo anti-cancer activity of a novel gemcitabine-cardiolipin conjugate. Anticancer Drugs. 2006;17:53–61. doi: 10.1097/01.cad.0000185182.80227.48. [DOI] [PubMed] [Google Scholar]
- 25.Siddiqui RA, Zerouga M, Wu M, et al. Anticancer properties of propofol-docosahexaenoate and propofol-eicosapentaenoate on breast cancer cells. Breast Cancer Res. 2005;7:R645–54. doi: 10.1186/bcr1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krönke M. Biophysics of ceramide signaling: interaction with proteins and phase transition of membrane. Chem Phys Lipids. 1999;101:109–21. doi: 10.1016/s0009-3084(99)00059-6. [DOI] [PubMed] [Google Scholar]
- 27.Martin S, Phillips DC, Szekely-Szucs K, et al. Cyclooxygenase-2 inhibition sensitizes human colon carcinoma cells to TRAIL-induced apoptosis through clustering of DR5 and concentrating death-inducing signaling complex components into ceramide-enriched caveolae. Cancer Res. 2005;65:11447–58. doi: 10.1158/0008-5472.CAN-05-1494. [DOI] [PubMed] [Google Scholar]
- 28.Zaremberg V, Gajate C, Cacharro LM, et al. Cytotoxicity of an anti-cancer lysophospholipid through selective modification of lipid raft composition. J Biol Chem. 2005;280:38047–58. doi: 10.1074/jbc.M502849200. [DOI] [PubMed] [Google Scholar]
- 29.Agathanggelou A, Bièche I, Ahmed-Choudhury J, et al. Identification of novel gene expression targets for the Ras association domain family 1 (RASSF1A) tumor suppressor gene in non-small cell lung cancer and neuroblastoma. Cancer Res. 2003;63:5344–51. [PMC free article] [PubMed] [Google Scholar]
- 30.Fimognari C, Nüsse M, Berti F, et al. Cyclin D3 and p53 mediate sulforaphane-induced cell cycle delay and apoptosis in non-transformed human T lymphocytes. Cell Mol Life Sci. 2002;59:2004–12. doi: 10.1007/PL00012523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu C, Rahmani M, Dai Y, et al. The lethal effects of cyclin-dependent inhibitors in human leukemia cells proceed through phosphatidylinositol 3-kinase/Akt-dependent process. Cancer Res. 2003;63:1822–33. [PubMed] [Google Scholar]
- 32.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 33.Palacios C, Yerbes R, Lopez-Rivas A. Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res. 2006;66:8858–69. doi: 10.1158/0008-5472.CAN-06-0808. [DOI] [PubMed] [Google Scholar]
- 34.McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322–331. doi: 10.1038/sj.cdd.4402260. [DOI] [PubMed] [Google Scholar]
- 35.Abdi-Dezfuli F, Froyland L, Thorsen T, et al. Eicosapentaenoic acid and sulphur substituted fatty acid analogues inhibit the proliferation of human breast cancer cells in culture. Breast Cancer Res Treat. 1997;45:229–39. doi: 10.1023/a:1005818917479. [DOI] [PubMed] [Google Scholar]
- 36.Akihisa T, Tokuda H, Ogata M, et al. Cancer chemopreventive effects of polyunsaturated fatty acids. Cancer Lett. 2004;205:9–13. doi: 10.1016/S0304-3835(03)00284-2. [DOI] [PubMed] [Google Scholar]
- 37.Begin ME, Ells G. Effects of C18 fatty acids on breast carcinoma cells in culture. Anticancer Res. 1987;7:215–7. [PubMed] [Google Scholar]
- 38.Fortes C, Forastiere F, Farchi S, et al. The protective effect of the Mediterranean diet on lung cancer. Nutr Cancer. 2003;46:30–7. doi: 10.1207/S15327914NC4601_04. [DOI] [PubMed] [Google Scholar]
- 39.Llor X, Pons E, Roca A, et al. The effects of fish oil, olive oil, oleic acid and linoleic acid on colorectal neoplastic processes. Clin Nutr. 2003;22:71–9. doi: 10.1054/clnu.2002.0627. [DOI] [PubMed] [Google Scholar]
- 40.Martin-Moreno JM, Willett WC, Gorgojo L, et al. Dietary fat, olive oil intake and breast cancer risk. Int J Cancer. 1994;58:774–80. doi: 10.1002/ijc.2910580604. [DOI] [PubMed] [Google Scholar]
- 41.Willett WC, Stampfer MJ, Colditz GA, et al. Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. N Engl J Med. 1990;323:1664–72. doi: 10.1056/NEJM199012133232404. [DOI] [PubMed] [Google Scholar]
- 42.Escribá PV, Wedegaertner PB, Goñi FM, et al. Lipid-protein interactions in GPCR-associated signaling. BBA Biomembranes. 2007;1768:836–52. doi: 10.1016/j.bbamem.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Escribá PV, González-Ros JM, Goñi FM, et al. Membranes: a meeting point for lipids, proteins and therapies. J Cell Mol Med. 2008;12:829–75. doi: 10.1111/j.1582-4934.2008.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lockshin RA, Zakeri Z. Cell death in health and disease. J Cell Mol Med. 2007;11:1214–24. doi: 10.1111/j.1582-4934.2007.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]