

Abstract
The pituitary gonadotropin follicle-stimulating hormone (FSH) interacts with its membrane-bound receptor to produce biologic effects. Traditional functions of FSH include follicular development and estradiol production in females, and the regulation of Sertoli cell action and spermatogenesis in males. Knockout mice for both the ligand (Fshb) and the receptor (Fshr) serve as models for FSH deficiency, while Fshb and Fshr transgenic mice manifest FSH excess. In addition, inactivating mutations of both human orthologs (FSHB and FSHR) have been characterized in a small number of patients, with phenotypic effects of the ligand disruption being more profound than those of its receptor. Activating human FSHR mutants have also been described in both sexes, leading to a phenotype of normal testis function (male) or spontaneous ovarian hyperstimulation syndrome (females). As determined from human and mouse models, FSH is essential for normal puberty and fertility in females, particularly for ovarian follicular development beyond the antral stage. In males, FSH is necessary for normal spermatogenesis, but there are differences in human and mouse models. The FSHB mutations in humans result in azoospermia; while FSHR mutations in humans and knockouts of both the ligand and the receptor in mice affect testicular function but do not result in absolute infertility. Available evidence also indicates that FSH may also be necessary for normal androgen synthesis in males and females.
Keywords: Follicle-stimulating hormone (FSH), FSH receptor, isolated FSH deficiency, FSHB gene, FSHR gene, FSHB knockout mouse, FSHB transgenic mouse, FSHR knockout mouse, FSHB mutations, FSHR mutations, ovarian hyperstimulation syndrome
Introduction
The coordination of the hypothalamic–pituitary–gonadal (HPG) axis is regulated by gonadotropin-releasing hormone (GnRH) action. Intriguingly, GnRH neurons originate outside the brain and migrate alongside olfactory nerves to reach the hypothalamus.1,2 The GnRH neurons from the arcuate nucleus within the hypothalamus secrete GnRH in a pulsatile manner into the hypophyseal portal system, where it acts on its cell surface receptor in the anterior pituitary.3 In turn, pituitary gonadotropes respond by increasing the production of the common α-subunit and specific β-subunits of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). These assembled α-β dimers of FSH and LH exert their principal function in the gonads, where gametogenesis, folliculogenesis, and steroidogenesis occur. The steroid hormones, estradiol (E2) and progesterone (P4) in females, and testosterone in males, feedback to both the hypothalamus and pituitary to modulate the production of GnRH and the gonadotropins. The recently characterized gonadotropin inhibitory hormone (GnIH), also known as RFRP-3, inhibits gonadotropin expression and serum levels and is likely to have an important regulatory function.4 The HPG axis is central to normal pubertal development and reproductive capacity, as the onset of puberty is accomplished through an activation of the GnRH pulse generator with FSH playing a vital role in reproductive maturation and function.3
The FSH ligand is produced principally from the anterior pituitary and targets its receptor found on the cell surface of granulosa cells of the ovary and Sertoli cells of the testes.5 The ovarian response to FSH induces aromatase, converting androgens to estrogens and inducing follicular maturation. In addition, the hormone inhibin B is produced by the granulosa cells and exerts negative feedback upon FSH. In the testes, FSH upregulates Sertoli cells and along with testosterone plays a key role in spermatogenesis.5 Mutations of both the FSHβ gene (FSHB) and its receptor (FSHR) have been reported in humans; and the corresponding knockouts (KOs) have been generated in mice. These recent discoveries have furthered the understanding of FSH signaling and metabolism, important for its effect upon sexual maturation and reproduction. The purpose of this review is to evaluate all of the existing literature on these very valuable “KOs of nature” in humans with mutations of FSHB and FSHR, as well as the corresponding KO and transgenic mice. Since activating FSHR mutations have also been described in humans, the phenotypic findings from excess FSH can be examined and compared with transgenic mouse overexpression models. Findings from these studies will then be incorporated into the understanding of FSH action in mammals as much has changed since we last summarized the literature more than a decade ago.5
The Human FSH Ligand
A dimeric pituitary glycoprotein hormone, FSH is composed of an α-subunit and a β-subunit, which are both expressed in the gonadotropes of the pituitary gland.6 The α-subunit is encoded by a single gene (CGA), which is common to FSH, LH, human chorionic gonadotropin (hCG), and thyroid-stimulating hormone (TSH).7 The α-subunit protein is noncovalently joined to the β-subunit to form a biologically active hormone, but it is the β-subunit that confers hormone specificity. The β-subunit is structurally similar to the members of the cysteine knot GF family.6
The 9.6 kb human CGA gene is localized to chromosome 6q14.3 and has 2 splice variants. Variant NM_001252383 represents the longer transcript and encodes the longer isoform, containing 5 exons, yielding a 147 amino acid protein that is N-linked glycosylated at positions 107 and 133 (Asn52 and Asn78 of the mature protein).6 The signal peptide is 24 amino acids and the mature protein 123 amino acids. The shorter variant NM_000735 encodes 116 amino acids and lacks an alternate in-frame exon 3 that the longer variant possesses. This protein has the same 24 amino acid signal peptide and a 92 amino acid mature protein. Tertiary structure of the α-subunit is provided by 5 disulfide bonds formed via 10 Cys residues, which are completely conserved across species.
The 4.26 kb human FSHB gene on chromosome 11p14.1 is composed of 3 exons (Figure 1), which encode a 129 amino acid FSH β-subunit (an 18 amino acid signal peptide and a 111 amino acid mature protein).8 The single FSHB gene possesses several unique characteristics compared to the other gonadotropin β-subunit genes.8 Alternative splicing of the first exon, which encodes the 5' untranslated regions, renders this small exon either 33 or 63 nucleotides in length.8 Exon 2 encodes the 18 amino acid signal peptide followed by the first 35 residues of the mature β-subunit, while the third exon encodes amino acids 54 to 129 and the 3′ untranslated region. Therefore, 2 different-sized transcripts (FSHB-NM_000510 and NM_001018080) are produced from alternative splicing of exon 1 and the presence of 2 different potential sites of polyadenylation; however, each results in the translation of the same FSHβ protein. The FSHβ-subunit possesses 2 N-linked glycosylation sites at Asn25 and Asn42 of the total protein (Asn7 and Asn24 of the mature protein) as well as 12 highly conserved Cys residues that form 6 disulfide bonds including Cys21-Cys69, Cys35-Cys84, Cys38-Cys122, Cys46-Cys100, Cys50-Cys102, and Cys105-Cys112 of the full-length protein.8
Figure 1.
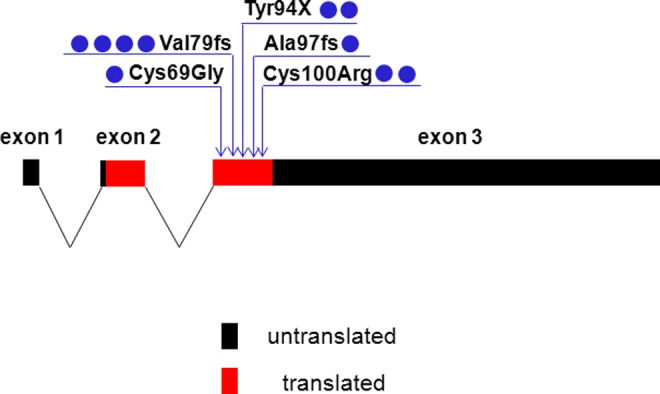
The structure of the human FSHB gene is shown with all known mutations, all of which reside within exon 3. The 3 exons are indicated as to whether they are translated or untranslated. The blue circles beside each mutation indicate the number of times the allele has been reported in a family with isolated FSH deficiency (note: there may be more than 1 affected member within the family). FSH indicates follicle-stimulating hormone. See online version for color reference.
Following transcription, translation, and glycosylation of individual α- and β-subunits, dimer formation occurs. The crystal structure of hCG was the first of the pituitary glycoprotein hormones to be reported9 but was subsequently accomplished for FSH6 and FSH complexing with its receptor.10 By lining up Cys residues from FSHβ and hCGβ of the mature protein, FSHβ Cys20 and Cys104 (Cys 38 and Cys122 of the total protein) form a “seatbelt” that wraps around and stabilizes the α-subunit.6,9 Only the mature FSH dimer binds the FSHR.
The Human FSHR
The human FSH receptor (FSHR) is a glycosylated, heptohelical G-protein-coupled receptor (GPCR) with an exceptionally long extracellular domain (ECD) constituting more than half of the full-length protein (residues 1-357), a 7 transmembrane domain (TMD1: 358-390, TMD2: 395-429, TMD3: 440-472, TMD4: 482-506, TMD5: 525-557, TMD6: 565-597, and TMD7: 605-642), 3 short intracellular loops (ICL1: 391-394, ICL2: 473-481, and ICL3: 558-564), and 3 extracellular loops (ECL1: 430-439, ECL2: 507-524, and ECL3: 598-604), and an intracellular tail (643-695; Figure 2).11–13 The receptor has 2 functional domains—an ECD and a serpentine domain that includes the transmembrane domain as well as extracellular and intracellular loops.
Figure 2.

The structure of the human FSHR protein is shown with the large extracellular domain (ECD) and the serpentine domain, which consists of 7 transmembrane domains (TMDs), 3 extracellular loops (ECLs); 3 intracellular loops (ICLs); and an intracytoplasmic tail carboxy-terminal (COOH) tail. Not all amino acids in the ECD are shown (including Tyr 335 in the hinge region) as no human mutations have been identified to affect it. Inactivating missense mutations are indicated by a black circle and white letter, corresponding to the location of the affected amino acid and mutant. Activating mutations are indicated by gray-shaded circles and a black letter, corresponding to the mutant amino acid residue. In both cases, the amino acid letter shown is the wild type. See online version for color reference.
Two transcript variants have been described in humans, a longer one (NM_000145) and a shorter one (NM_181446), which lacks an alternate in-frame exon 6 of the longer variant. The longer protein NP_000136 contains 695 amino acids in length and is encoded by a 192-kb gene on chromosome 2p16.3, which contains 10 exons. The first 9 exons range in size from 69 to 251 bp and encode the extracellular domain, while the last exon containing an open reading frame of 1234 bp encodes the entire transmembrane domain loops and intracellular domains of the receptor (Figure 2). The large ECD is made up of leucine-rich repeats (LRRs) and a horseshoe-shaped structure of α-helices and β-strands, conferring specificity and high-affinity binding.14 The rhodopsin-like domain comprising 7 hydrophobic transmembrane helices functions to activate the receptor which then use G proteins for signal transduction. The FSHR is principally expressed in granulosa cells in females and in Sertoli cells in males but has also recently been found in the endothelium of a wide variety of tumor blood vessels.14
In addition to the 2 human FSHR isoforms, FSHR isoforms have been described in other species. Yarney et al15 identified an alternatively spliced ovine FSHR isoform, which is identical to the full-length FSHR for most of its protein, but diverges in sequence beginning at residue 625 and results in a shorter protein by 25 amino acids. As further shown by these investigators,16 this FSHR isoform was shown to possess dominant negative qualities since co-transfection of the isoform in stably transfected HEK293 cells abrogated hormone response. Kraaij et al17 identified 2 FSHR splice variants in rat, but both messenger RNAs (mRNAs) encode truncated proteins, and when co-transfected with the WT FSHR, signal transduction was not affected. Therefore, no clear gonadal function could be attributed to these 2 isoforms. Similarly, Tena-Sempere et al18 identified 4 different mouse FSHR splice variants, but none were able to bind recombinant human FSH or elicit cyclic adenosine monophosphate (cAMP) or progesterone responses in vitro. More recently, Sairam et al19 characterized 4 ovine FSHR isoforms (R1-R4). R1 is the full-length FSHR comprised of 10 exons, while R2 has the 10 exons but has a different C-terminus (the dominant negative isoform mentioned above). R3 has exons 1-8, but is truncated, and appears to function as a cytokine/growth factor type receptor. R4 has only exons 1-4 and is a truncated receptor with uncertain function.19 Therefore, the complexity of the FSHR suggests diverse functions that will continue to be elucidated.
Follicle-Stimulating Hormone Signaling and Action
Dimeric FSH binds to the ectodomain of the FSHR in a handclasp fashion as β-strand residues of the receptor interact with the α- and β-subunits of the ligand.6 During this ligand binding, the FSHR dimerizes, which prompts changes as the 7-transmembrane domain then acts on a heterotrimeric Gs protein causing guanine nucleotide exchange and activation of adenylyl cyclase, which results in the production of protein kinase A.10,14 The resulting cAMP then initiates a signaling cascade culminating in steroid synthesis. In addition, there is evidence that FSH also activates other separate signaling cascades such as RAS,20 PI3K (likely via an SRC tyrosine kinase),20 and glycogen synthase kinase 3β (GSK3β).21
Recently, the crystal structure of FSH binding to the entire ectodomain of the receptor has been reported by Jiang et al.22 The ectodomain of the FSHR is comprised of 12 LRR with a hairpin loop between the 11th and 12th LRRs. This hairpin loop contains an important amino acid residue, a sulfated tyrosine at codon 335 (sTyr), that forms the hinge region (the hinge region was previously thought to be a separate structural unit).22 The convex surface formed by the LRR and the hinge region resembles the fingers and thumb, respectively, of a right hand; and the binding of FSH with its receptor is similar to a right hand clasping an American football. Unexpectedly, FSH binding to the entire ectodomain forms a trimeric arrangement, which suggested an additional binding site on the FSHR outside the primary region (termed an exosite).22 These findings suggest a 2-step process of FSH binding to its receptor. First, the quiescent state FSHR exposes LRR 1-8 to recruit FSH to the hormone-binding region. In the second step, FSH accepts the hinge region of the FSHR with sTyr, which now prepares the FSHR to enter the active state with Gs or β-arrestin.22
FSH functions to increase the differentiation and proliferation of granulosa cells and induces aromatase (CYP19A1) expression, converting androgens to estrogens, as well as FSHR and cyclin D2 (CCND2) expression.23 A variety of growth factors, notably activins and inhibin, as well as members of the TGFβ family are important in FSH action.23 In males, FSH is involved in Sertoli cell proliferation and regulation of spermatogenesis. Evidence from Fshb and Fshr KO mice and from humans with FSHB and FSHR gene mutations suggests additional roles for FSH function (see below).
Mutation of the Ligand
Females
The Fshb knockout mouse
Germline Fshb KO mice were generated using homologous recombination techniques.24 Although Fshb +/− heterozygotes had normal estrous cycles and fertility, homozygous (Fshb −/−) KO female mice demonstrated low serum FSH levels, a lack of estrous cyclicity, and sterility.24 Serum LH levels in the Fshb −/− mice were consistently elevated above baseline for all ages of development. Grossly, the uteri and ovaries of the Fshb −/− mice were markedly small, consistent with hypoestrogenism. Somewhat surprisingly, however, serum estradiol levels were normal in these Fshb −/− females, although it is notoriously difficult to discriminate low from low-normal estradiol by immunoassay.25 Microscopically, the ovaries contained mostly primordial and primary follicles, with only occasional small, degenerating, antral follicles. No preovulatory mature follicles or corpora lutea were identified in female Fshb −/− mice.24
These findings in Fshb −/− mice (Table 1) suggested that FSH is essential for estrous cyclicity and fertility in female mice. In addition, follicular development up to the primary and early antral stages in the ovary occurred in the absence of pituitary FSH.24 The observed phenotypic manifestations in female Fshb −/− mice were all reversible by the administration of exogenous gonadotropins.30
Table 1.
Phenotypea of Fshb 24 and Fshr 26,27 Knockout Homozygotes (−/−) Mice are Shown, as Well as the Overexpressing Fshb Transgenic Mice.28
| Fshb −/− | Fshr −/− | High Copy Overexpressing Fshb Tg Mouse | ||||
|---|---|---|---|---|---|---|
| Phenotype | Male | Female | Male | Female | Male | Female |
| FSH | Low | Low | High (3-fold) | High (15-fold) | Markedly increased | Markedly increased |
| LH | Normal | High | Normal26; elevated29 | Normal | Normal | |
| Testosterone | Normal | ND | 65%-85% reduction | Increased | Elevated | Elevated |
| Estradiol | Normalb | ND; normal estrous cycles | Elevated | |||
| Progesterone | Reduced 50% | |||||
| Fertility | Fertile | Sterile | Fertile (reduced) | Sterile | Infertile | Infertile |
| Litter size | ND | 0 | Normal (7.3 ±0.6 vs 8.1 ± 2.4 in controls | 0c | ||
| Ovarian follicles | Primordial mostly; few antral follicles; increased stroma | Primordial, primary, secondary; increased stroma | Hemorrhagic, cystic ovaries; no corpora lutea | |||
| Uterus | Small | Small | ||||
| Testes size | Decreased 50% | – | Decreased 20%-30% | – | Normal | |
| Sperm concentration | Decreased 75% | – | Decreased 35% | – | 75% increase | |
| Sperm motility | Decreased 40% | – | Decreased 25% | – | ||
| Sperm morphology | ND | – | Decreased 30% | – | ||
| Sperm function | ND | – | Multiple parameters affected (see text) | – | ND | – |
| Other | Intratesticular testosterone and Leydig cell numberb normal; Sertoli cell and germ cell number decreased 30%-39% | Steroid enzyme and cyclin expression abnormal | Intratesticular testosterone and Leydig cell numberb normal at first, but both reduced in adulthood; Sertoli cell and germ cell number decreased 30%-39% | Increased adiposity, weight, and skeletal abnormalities | Seminal vesicle size doubled; histology of testes normal | No estrous cycles; Progesterone elevated; enlarged cystic kidneys and die 6-13 weeks of life from bladder obstruction |
Abbreviation: ND, not determined.
a Only the high copy overexpressing mice are shown, and low copy expression mice were normal.
b Estradiol was measured as normal, although the phenotype indicates low levels.
Burns et al31 used northern blot and in situ hybridization methods to more precisely characterize the alterations in ovarian gene expression in Fshb −/− mice. Ovarian Fshr expression doubled, but Lhr was 5-fold less that of WT and heterozygotes. Serum/glucocorticoid-inducible kinase (Sgk), which is implicated in transitioning granulosa cells from a proliferative to a terminally differentiated luteal state, was reduced more than 5-fold. Steroid enzyme machinery was also affected as expected. Without FSH, both P450 side chain cleavage (Cyp11a) and aromatase (Cyp19) were decreased by about 6-fold, while 17-hydroxylase (Cyp17) expression was doubled (and found to be localized to the theca).31 Expression of inhibin subunits revealed a 50% reduction in the α-subunit (Inha), and no evidence of expression of either of the β-subunits of inhibin (Inhba or Inhbb). Follistatin (Fst) expression did not change. Cell cycle genes were also altered: cyclin D2 (Ccnd2), which is important for granulosa cell proliferation, was decreased by 30%; whereas Cdk2 and Cdk6 (associated with granulosa cell cycle withdrawal, differentiation, and luteinization) were increased by 50% and 20%, respectively. Inhibitors of the cyclins were all reduced—p27Kip1 (Cdkn1b; 3-fold), p21Cip1 (Cdkn1a; 5-fold), and p15Ink4b (Cdkn2b; >10-fold).31 The authors concluded that cyclin D2 loss could account for some of the defects in granulosa cell function, but the mechanism is different from cycle withdrawal at the time of luteinization.31
Human FSHB mutations
Only 5 different FSHB mutations have been found in humans (Table 2, Figure 1)— 6 females (Table 3) and 3 males (Table 4). The first patient with an FSHB mutation was described by Matthews et al36 in 1993. She was a 27-year-old female who presented with primary amenorrhea, low serum FSH, and high LH. At the age of 13, she had no breast development but was subsequently treated with estrogen. She later conceived with exogenous gonadotropin therapy. Upon DNA sequencing, the proband was found to possess a homozygous 2 base pair deletion at codon 61 (Val61X) of the mature protein. These investigators did not perform functional studies in vitro, but this frameshift mutation was predicted to alter amino acids 61 to 86, and then produce a stop codon, resulting in a truncated FSHβ protein lacking amino acids 87 to 111.36 It should be noted that for most of the described human FSHB mutations, the nomenclature only took into account the mature protein. However, in the now accepted mutation nomenclature, the 18 amino acid signal peptide should really be numbered as codon 1 as it represents the first in-frame ATG.41 In Table 2, the reported nomenclature of described FSHB mutations is shown, along with their corrected nomenclature. Therefore, this mutation is more correctly indicated as Val79fs or Val79fsX105, as it alters amino acids 79 to 104 and produces a stop codon truncating amino acids 105 to 129.
Table 2.
Reported Inactivating FSHB Mutations in Human Patients (Males and Females) for Which a Functional Effect Has Been Demonstrated.a
| # | Author | FSHB Mutation | Exon | Cells for In Vitro Analysis | Functional Effect In Vitro | ||
|---|---|---|---|---|---|---|---|
| Old Nomenclature | New Nomenclature | ||||||
| Nucleotide | Protein | ||||||
| 1 | Layman 199732 | p.Val61X | c.236-237delTG | p.Val79fs or p.Val79fsX105 | 3 | CHO | Absent I and B-FSH |
| 2 | Layman 199732 | p.Cys51Gly | c.205T>G | p.Cys69Gly | 3 | CHO | Absent I and B-FSH |
| 3 | Layman 200233 | p.Tyr76X | c.282C>A | p.Tyr94X | 3 | CHO | Absent I and B-FSH |
| 4 | Clark 200334 | p.Cys82Arg | c.298T>C | p.Cys100Arg | 3 | CHO | Absent I and B-FSH |
| 5 | Kottler 201035 | p.Ala79fs108X | c.289delG | p.Ala97fs or p.Ala97fsX127 | 3 | CHO | Absent I and B-FSH |
Abbreviations: B, bioactive; CHO, Chinese hamster ovary cells; I, immunoactive.
aNote that p.Val79fsX105 may also be indicated as p.Val79fs and p.Ala97fsX127 may be indicated as p.Ala97fs.
Table 3.
The FSHB Mutations in Human Females.a
| # | Author | FSHB Mutation | Age | Race | Breast | Menses | Ht | Wt | BMI | FSH (1-10 mIU/mL) | LH (1-9 mIU/mL) | E2 (30-300 pg/mL) | InB (20-220 pg/mL) | Ultrasound | Histology | Other | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basal | Peak | Basal | Peak | |||||||||||||||
| 1 | Matthews 199336 | p.Val79fsX105 | 18 | Italian | T1 | 10 Am | 176 | 73 | 23.6 | < 0.1 | < 0.1 | 28 | 104 | 27 | U | ND | ND | After 15 days of rFSH, InhB and E2 levels rose just above detection; later FSH treatment resulted in pregnancy |
| 2 | Layman 199732 | p.Val79fsX105 and p.Cys69Gly | 15.5 | USA | T1 | 10 Am | 155 | ND | ND | 0.6 | 0.8 | 21-33 | 170 | < 25 | ND | Multicystic ovaries—many small antral follicles | ND | ND; LH pulses similar to PCOS |
| 3 | Matthews 199737 | p.Val79fsX105 | 17 | Israeli | “Poor” | 10 Am | 163 | ND | ND | < 3 | ND | 50-90 | ND | < 10 | ND | ND | Ovarian biopsy shows primordial follicles | Laparoscopy: small uterus and ovaries |
| 4 | Layman 200233 | p.Tyr94X | 32 | Brazil | T2-3 | 10 Am | 156 | ND | ND | < 0.01 | < 0.01 | 51 | 159 | 12 | ND | ND | ND | ND |
| 5 | Berger 200538 | p.Tyr94X | 16 | Brazil | T3 | 10 Am | 158 | 48 | 19.2 | < 1 | < 1 | 30 | 98 | < 13 | ND | Small uterus (7 cc volume) normal ovaries | ND | ND |
| 6 | Kottler 201035 | p.Ala97fsX127 | 29 | Spanish | T2 | 10 Am | 165 | ND | ND | < 0.05 | < 0.05 | 49 | 170 | 7 | U | Small uterus; Ovaries 1 and 1.6 mL volume (6.6 ± 1.9): few follicles <3 mm | ND | Treatment with rFSH resulted in hyperstimulation |
Abbreviations: FSH, follicle-stimulating hormone; LH, luteinizing hormone; Am, amenorrhea; BMI, body mass index; E2, estradiol; ND, not determined; Ht, height in cm; InB, inhibin B; Ov Vol, ovarian volume; T, Tanner stage (T1, prepubertal and T5, adult); U, undetectable; Wt, weight in kg.
aAll are homozygous except case 2 who has compound heterozygous mutations. All were 46, XX and had normal pituitary imaging.
Table 4.
The FSHB Mutations in Males.
| Pt. | Author | FSHB Mutation | Age | Nationality | Puberty | Testes (15-25 cc) | Gynecomastia | Ht | Wt | BMI | Sperm | FSH (1-10 mIU/mL) | LH (1-9 mIU/mL) | Testosterone (270-1100 ng/dL) | Testicular Biopsy | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basal | Peak | Basal | Peak | ||||||||||||||
| 1 | Lindstedt 199839 | p.Cys100Arg | 28 | Serbian | Normal | 3 and 6 | No | 171 | 181 | 62 | Azoospermia | U | U | 12 | 41 | 246 | Leydig cells present but spermatogenic arrest |
| 2 | Phillip 199840 | p.Val79 fs or p.Val79fsX105 | 18 | Israeli | Absent | 1 and 2 | No | 178 | 59 | 19 | Azoospermia | < 0.5 | < 0.5 | 24 | 75 | 130 | ND |
| 3 | Layman 200233 | p.Tyr94X | 30 | Brazilian | Normal | 12 and 12 | No | 170 | N | ND | Azoospermia | 0.5 | 1 | 36 | 160 | 582 | Leydig cell hyperplasia; small seminiferous tubules with germinal cell aplasia, peritubular fibrosis, and few Sertoli cells |
Abbreviations: BMI, body mass index; Ht, height in cm; ND, not determined; T, testosterone; U, undetectable; Wt, weight in kg.
aAll were homozygous. Normal endocrine values are indicated.
Layman et al32 reported the first patient with an FSHB mutation that was confirmed by in vitro analysis. The proband was a 15 11/12 year-old female who presented with absent breast development, primary amenorrhea, low FSH, undetectable estradiol, elevated LH levels, and no pituitary tumor by MRI.32 She demonstrated compound heterozygosity for 2 different FSHB gene mutations, 1 being the previously described Val61X (Val79fs or Val79fsX105) inherited from her mother, while the other was a Cys51Gly (Cys69Gly, new nomenclature) missense mutation inherited from her father. When mutants versus controls were separately transfected into Chinese hamster ovary cells, both the Val79fs and the Cys69Gly proteins resulted in unmeasurable immunoreactive and bioactive FSH levels in the cellular media compared with controls.32 These in vitro data confirmed the phenotypic findings of the patient and indicated that FSH is necessary for pubertal development and fertility in females. There were 5 heterozygotes within the family who were fertile, therefore suggesting that only biallelic FSHB mutations impair reproductive function.
Since these 2 patients were described, there have only been 4 more females with FSHB mutations reported (cases 3-6 in Table 3).33,37,38 Rather than completely lacking breast development as was present in the first 2 patients, these females all had partial breast development. Nevertheless, all 6 described females share certain phenotypic findings—primary amenorrhea, low serum FSH, low serum estradiol, and elevated LH (Table 3). When GnRH stimulation was performed in 5 of these patients, no FSH rise from baseline was observed, whereas there was a marked rise in serum LH in all 5 patients who were studied. These findings could be predicted since the absence of estradiol negative feedback should elicit an increase in GnRH secretion and a subsequent but appropriate rise in LH. However, no corresponding rise in FSH occurs because of the FSHB mutation, which likely interferes with dimer formation and subsequent secretion.32 As shown in Tables 2 and 3, only 5 different mutations have been reported in these 6 females—5 as homozygotes and 1 as a compound heterozygote. Two are frameshift; 1 is a stop mutation; and 1 is a missense involving the highly conserved Cys residue. All 4 reported FSHB mutations in females resulted in unmeasurable immuno and bioactive FSH in vitro, regardless of the clinical severity of the mutations (ie, the presence or absence of breast development).32,33
Several interesting features of the patient with the compound heterozygous Val61X/Cys51Gly (Val79fx/Cys69Gly, new nomenclature) mutations deserve further mention.32 Although serum LH levels were elevated, this patient had a normal testosterone and no evidence of hirsutism.32,42,43 These findings were unexpected, considering that women with polycystic ovary syndrome (PCOS) demonstrate elevated LH levels and elevated androgens.44 This suggested that perhaps FSH was necessary for the LH-induced theca cells to make androgens. In order to test this hypothesis, serial gonadotropin levels were obtained for 11 hours at 10-minute intervals. Consistent with the baseline levels, serum FSH was unmeasurable, while the mean LH level was elevated (49 mIU/mL) with 9 LH pulses and an elevated amplitude (46 mIU/mL), as is shown in Figure 3. 42 This pulsatile pattern of LH closely resembles that seen in women with PCOS.44
Figure 3.

The LH pulses are shown for a female with an FSHB mutation (patient #2 in Table 3) who had serum LH levels drawn every 10 minutes overnight. Note the LH pulses indicated by arrowheads, which are similar to women with PCOS. Modified from Barnes et al.42 LH indicates luteinizing hormone; PCOS, polycystic ovary syndrome.
This FSH-deficient female was further studied by Barnes et al.42,43 By ultrasound, she had multiple small antral follicles, similar to a PCO appearing ovary except there was no dense stroma.42 She was given dexamethasone to suppress the adrenal contribution of androgens for 4 days and then 5000 IU hCG was administered (endocrine studies were done at baseline and 24 hours later). Following a washout period of 1 month, baseline studies were obtained, and then she received 300 IU recombinant FSH (rFSH), and then hCG again 24 hours later.43 Androgens increased somewhat in response to hCG alone, but there was a more dramatic rise in testosterone (which more than tripled) after rFSH followed by hCG. These data add further support to the hypothesis that LH-induced androgen production in the theca requires the presence of FSH for a normal physiologic response, perhaps by inducing inhibin, LH receptors, or growth factors.43 In primates, there is a positive correlation between expression of the FSHR and the androgen receptor in healthy, growing follicles.43
Similar studies were carried out in the Brazilian female with the p.Tyr76X (p.Tyr94X, new nomenclature) mutation by Lofrano-Porto et al.45 By overnight sampling, she had 8 LH pulses/8 hours similar to the other patient. However, she had no response to hCG, rFSH, or hCG + rFSH, which could be an age-related effect since she was 41 at the time of the studies.45
Males
The Fshb knockout mouse
FSH-deficient Fshb −/− male mice had normal sexual development (Table 1); and serum levels of LH and testosterone were similar to control Fshb +/+ males. It is likely that testosterone’s negative feedback in these Fshb −/− mice accounts for the quantitative difference in serum LH values between male and female Fshb −/− mice. In contrast to female Fshb −/− mice that are infertile, the male Fshb −/− mice are fertile despite a decrease in testicular size and a 75% reduction in sperm. Microscopic examination of the testes reveals a relative diminution in all testicular elements, but progression through spermatogenesis as well as Leydig cell numbers appears to be normal. Unfortunately, however, the litter sizes were not provided. It seems possible that reduced litters or prolonged intervals between pregnancies would be observed because of the oligospermia, but this has not been reported. The combination of small testes and fertility in Fshb −/− male mice suggests that FSH is necessary for testicular volume but may not be absolutely essential for spermatogenesis and normal fertility in mice.
Further support that these observed effects of Fshb −/− mice are specifically caused by FSH deficiency comes from rescue of the Fshb −/− mice of both sexes by the introduction of the human FSHB transgene,30 which is selectively expressed in the pituitary. In both females and males, all of the phenotypic effects are corrected, indicating that they are caused by isolated FSH deficiency.30
Since the original description of the Fshb −/− mice, Wreford et al46 examined testicular function in more detail. Testicular weights were decreased about 60%, and Sertoli cell number and germ cell numbers were decreased 30% to 39%. The germ cell attrition was more pronounced for later stages of spermatogenesis, as manifested by a 46% reduction in spermatogonia and 60% decrease in round spermatids. The investigators suggested that the testicular function in Fshb −/− male mice is due to the lack of FSH’s action on Sertoli cell proliferation and the ability of Sertoli cells to nurture germ cells.46
Baker et al47 studied Fshb −/− male mice to determine the effects of absent FSH upon testicular function over time. Diminished testicular volume was apparent as early as 20 days of life and was about one-third the normal size in adults. Reflecting normal serum LH levels in Fshb −/− male mice, Leydig cell number increased appropriately at days 1, 5, 20, and adult compared with control. Corresponding intratesticular testosterone was normal at 60 and 180 days. Leydig cell morphology was normal, but the reserve of the testis was impaired since hCG administration elicited an intratesticular response that approximated two-thirds of the WT animals.47 Disordered testicular steroidogenesis was observed in adult Fshb −/− males, however, as 17β-HSD type 3 (Hsd17b3) and StAR (Star) were increased. There were no changes in P450scc (Cyp11a1), 3β-HSD type 6 (Hsd3b6), and P450c17 (Cyp17a1).47 Collectively, these findings indicate impairment of both endocrine and exocrine functions of the testes in the Fshb −/− male mice.
Human FSHB Mutations
Only 3 males with FSHB mutations have been described in humans (Table 4, Figure 1). Based upon the findings in Fshb −/− mouse, the expected phenotype would be that of normal puberty (since LH-induced testosterone production should occur), but there could be varying degrees of spermatogenic failure. The first male described by Lindstedt et al39 with an FSHB mutation had exactly this presentation, with normal virilization and pubertal development although the testes were small (3 and 6 cc volume with normal being ∼15-25 cc). His testosterone level was in the lower range of normal; LH was mildly elevated, but FSH was undetectable (Table 4). He demonstrated homozygosity for a Cys82Arg (p.Cys100Arg in new nomenclature) missense mutation, which was predicted to interfere with dimer formation but was not studied in vitro. Subsequently, Clark et al34 demonstrated that this mutation resulted in absent immunoactive and bioactive FSH (Table 2).
Phillip et al40 reported the second FSH-deficient male that exhibited a more severe, and perhaps unexpected, phenotypic presentation. He had no evidence of pubertal development with very small testes (1 and 2 cc), a low testosterone (130 ng/dL; normal: 270-1070), elevated LH, and low FSH. He demonstrated homozygosity for the Val61X (p.Val79fs, new nomenclature),40 previously shown to be completely devoid of immuno- and bioactivity.32 The third male with an FSHB mutation characterized by Layman et al33 had a presentation similar to the first reported male. He had normal puberty, infertility, and azoospermia. His testicular biopsy demonstrated Leydig cell hyperplasia and sparse, small seminiferous tubules with germinal cell aplasia, peritubular fibrosis, and scant Sertoli cells.33
All 3 men with FSHB mutations had in common the finding of azoospermia despite testicular size ranging from completely prepubertal to nearly normal (Table 4). Interestingly, none had gynecomastia or cryptorchidism. Also, in all 3 males, FSH was low and did not respond to exogenous GnRH administration. Similar to females with FSHB mutations, and unlike male Fshb −/− mice, baseline LH levels were elevated and displayed an exaggerated LH response to GnRH. Inhibin B was low in the first 2 males, reflecting Sertoli cell dysfunction48 (unfortunately, inhibin B was not studied in the third male).33 Although only 2 had testicular biopsies, Leydig cells were present in 1 patient and numerous in the other, yet variable seminiferous tubule dysfunction was noted—germinal aplasia to spermatogenic arrest (Table 4).
It is intriguing that at least one of the males had low serum testosterone. Leydig cells do not express the FSHR, so direct evidence of FSH upon Leydig cells has not been demonstrated. However, several studies identified by Phillip et al40 indicated that FSH may have an indirect role upon Leydig cell testosterone production. In fact, supernatants of Sertoli cells incubated with FSH have been reported to stimulate testosterone secretion by Leydig cells and testicular explants from rats,49,50 hamsters,51 and humans.29 Therefore, human FSHB mutations in males clearly affect fertility by causing azoospermia and possibly could play a role in pubertal development. These mutations appear to be rare in eugonadal males with infertility, since none of 65 males studied, including 13 with nonobstructive azoospermia and 52 with oligozoospermia, had FSHB mutations (Clark AD and Layman LC, unpublished).
Inactivating Mutations of the Receptor
Females
The FSHR knockout mouse
Two different groups of investigators have knocked out the Fshr gene in mice.26,27 Similar to female Fshb −/− mice, female Fshr −/− mice, first described by Dierich et al,27 demonstrated sterility (Table 1). As expected, serum FSH levels were markedly elevated (15-fold) in female Fshr −/− mice versus WT Fshr +/+ mice.27 However, serum LH levels were reportedly normal by these investigators,27 which is in contrast to Abel et al26 who found that serum LH was markedly elevated. Female Fshr −/− mice had a small uterus and impaired follicular maturation with primordial, primary, and secondary follicles but absent mature follicles.26,27 Similar to the Fshb −/− mice, stromal hypertrophy was present.26 When Fshr −/− mice were studied further, uterine, ovarian, and vaginal weights were decreased compared with WT and heterozygotes.52 As expected, serum estradiol and progesterone were markedly decreased, but interestingly, serum testosterone was elevated (Table 1).52 There was also no change in gene expression of aromatase (Cyp19a1), estrogen receptor-α (Esr1), estrogen receptor-β (Esr2) or their encoded proteins, but there was a 60% reduction of protein expression for both the A and B isoforms of progesterone receptor.52 In addition, the expression of the FSH-responsive gene cyclin D2 (Ccnd2) was unaltered in Fshr −/− females.27
When Abel et al26 generated Fshr −/− mice, overall, they reported similar findings. In addition, they found that the vagina was imperforate and that both ovarian and serum inhibins (A and B) were undetectable in Fshr −/− mice versus heterozygote and wild-type animals. A notable finding by Dierich et al27 was that the pituitaries of female (but not male) Fshr −/− mice were enlarged, as both thyrotropes and gonadotropes were increased. Abel et al26 found that pituitary content of FSH, but not LH, was significantly elevated in Fshr −/− females. When these mice were treated with estradiol, serum LH was suppressed to the level of controls but FSH was only reduced by 50%. It has also been shown that transplantation of an ovary from WT animals into an Fshr −/− mouse can correct hypoestrogenism and hyperandrogenemia, although it cannot rescue the effects of the FSHR loss in host granulosa cells.53
Danilovich et al52 characterized additional features of Fshr KO mice involving body weight, adiposity, and the skeleton. Body weights of Fshr −/− mice were significantly (20%) higher than WT, and there was a doubling of their adipose tissue (abdominal, inguinal, and retroperitoneal fat pads) versus matched age Fshr +/+ mice.52 Beginning about 4 to 5 months of age, Fshr −/− females demonstrated a stooped posture kyphosis that was evident on X-ray; and the weight of the right femur was reduced in these females compared with wild-type mice.52 These skeletal changes are analogous to those in aging human females.
Interestingly, heterozygous female Fshr +/− mice also had phenotypic findings that were intermediate between Fshr −/− and Fshr +/+ mice. Fshr +/− mice had a reduction in litter size by 50% and pups/litter by 43%, as well as a longer time between pregnancies.52 Body weight and weights of the reproductive organs did not differ in heterozygotes versus wild-type mice. Serum estradiol was not different, but progesterone was decreased and testosterone increased compared with Fshr +/+ mice.52
Human inactivating FSHR mutations
Although inactivating mutations in the FSHR gene were predicted to cause 46, XX ovarian failure, no mutations were initially identified in a group of unrelated North American patients.54 The first reported FSHR gene mutations were identified in Finnish kindreds with 46, XX hypergonadotropic hypogonadism (mutation 1 in Table 5 and case 1 in Table 6, Figure 2). Aittomaki et al55 investigated all church records in Finland for families having women with primary amenorrhea under age 20, elevated gonadotropins, and a normal karyotype. Using linkage analysis of multiplex families, strong linkage at chromosome 2p21 was observed and the FSHR gene was found to be in linkage disequilibrium with this phenotype. DNA sequencing revealed a homozygous p.Ala189Val missense mutation in about one-third of all the Finnish ovarian failure patients studied. This mutation was shown to markedly reduce the binding capacity of the receptor in MSC-1 Sertoli cells, as well as to dramatically impair cAMP production in response to recombinant FSH (Table 5).55
Table 5.
Reported Inactivating FSHR Mutations in Human Patients (Males and Females) Supported by Functional In Vitro Analysis.
| Inactivating FSHR Mutations | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| # | Author | FSHR Mutation | Exon | Protein Region | In Vitro Analysis | |||||
| Nucleotide | Protein | Cells | Receptor Binding Capacity | Receptor Binding Affinity | Receptor Targeting | Cyclic AMP Response to FSH | ||||
| 1 | Aittomaki 199555 | c.566C>T | p.Ala189Val | 7 | ECD | MSC-1 | Marked reduction | Normal | ND | Negligible vs WT |
| 2 | Beau 199856 | c.479T>C | p.Ile160Thr | 6 | ECD | COS-7 | ND | Marked reduction | None seen on cell surface | 10% of WT |
| 3 | Beau 199856 | c.1717C>T | p.Arg573Cys | 10 | TMD6 | COS-7 | ND | Normal | Normal | 30% of WT |
| 4 | Touraine 199957 | c.1801C>G | p.Leu601Val | 10 | ECL3 | COS-7 | ND | Normal | Normal | 12% of WT |
| 5 | Touraine 199957 | c.671A>T | p.Asp224 Val | 9 | ECD | COS-7 | ND | Barely detectable | Impaired (intracellular) | 4% of WT |
| 6 | Doherty 200258 | c.1255G>A | p.Ala419Thr | 10 | TMD2 | COS-7 | Normal | Normal | Normal | Absent cAMP |
| 7 | Allen 200359 | c.1043C>G | p.Pro348Arg | 10 | ECD | COS-7 | Normal | No different vs empty vector | ND | Absent cAMP |
| 8 | Meduri 200360 | c.1555C>A | p.Pro519Thr | 10 | ECL2 | COS-7 | ND | Minimal | Impaired (intracellular) | Absent cAMP |
| 9 | Kuechler 201061 | c.1760C>A | p.Pro587His | 10 | TMD6 | COS-7 | ND, but predicted to cause a kink in TMD6 helix | ND | ND | Absent cAMP |
| 10 | Kuechler 201061a | ND precisely, Minimal deletion of 163 kb; Maximal deletion of 178 kb | Min del: p.Leu223-Asn695del; Max del: p.Leu175-Asn695del | Min del (9,10); Max del (7-10) | Part ECD; all TMD, ECL, ICL, and ICD | ND | ND | ND | ND | ND |
Abbreviations: ECD, extracellular domain; ECL, extracellular loop; ICL, intracellular loop; ND, not determined; TMD, transmembrane domain; WT, wild type; AMP, adenosine monophosphate.
a Kuechler et al found that the deletion was due to a 46, XX, t(2;8)(p16.3or21;p23.1), which was apparently balanced prior to chromosomal microarray analysis. The precise deletion breakpoints have not been determined. However, the minimal deletion would be 163 kb (chr2: 48 895 903-49 058 614) and the maximal deletion would be 178 kb (chr2: 48 887 046-49 065 417) based on the HG18 version. With the maximal deletion, the proximal breakpoint is within intron 6 of FSHR and so exons 7-10 would be deleted, encoding a truncated protein with the first 174 amino acids of full-length FSHR. In this case, almost of half of the ECD, all 7 TMD1-7, ICL1-3, and ECL1-3, and the intracellular domain would be gone. With the minimal deletion, the proximal breakpoint would be within intron 8 of FSHR and therefore exons 9 and 10 would be deleted, encoding a truncated protein with the first 222 amino acids of full-length FSHR. In this case, part of the ECD, all 7 TMD1-7, ICL1-3, and ECL1-3, and the intracellular domain (ICD) would be removed. The full mutation for case 10 is annotated as Chr2: g.(48 887 046-48 895 903)_(49 058 614-49 065 417)del (NCBI 36/hg18).
Table 6.
Human-Inactivating FSHR Mutations in Females Supported by Functional Analysis.a
| # | Author | FSHR Mutation | Age | Race | Breast | Menses | Ht | Wt | BMI | FSH < 10 | LH < 10 | E2 (pg/mL) | InB | US Ov Vol (cc) 6.6+0.19cc | Histology | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Aittomaki 199555,62 Vaskivuo 200263+ | p.Ala189Val HMZ | <20 | Finn | Absent to normal | 10 Am | 160.5 (150-172) | ND | ND | 61 (42-176) | 40 (17-119) | n = 12 16 (range: 8-81) | <10 (n=3)+ | n = 12; 0.7 (0.3-4.8); 6 of 12 had follicles seen | N = 9; primordial and primary; 2/9 with preantral; 1/9 with mature | 2 Females treated with FSH or FSH and hCG and no endocrine response; minimal GATA-4 or aromatase protein expression+ |
| 2 | Beau 199856 | p.Ile160Thr/p.Arg573Cys | 30 | Arm | “N” | 20 Am | 156 | 56 | 23 | 108 | 81 | 20-40 | 50 | ROV: 8.4 cc; LOV: 3.4 cc many 3-5 mm antral follicles | Primary, primordial, secondary follicles | FSHR protein expression normal but minimal LHR vs control |
| 3 | Touraine 199957 | p.Asp224Val p.Leu601Val | 19 | Cauc | “N” | 10 Am | 174 | 73 | 24.1 | 63 | 26 | 11, 22 | 30 | Ovaries stated “normal” with 10-12 follicles of 3 mm/ ovary | Primordial,, primary, secondary follicles seen; 3 antral follicles | Protein expression of zp450SCC; weak staining of 3BHSD; no controls shown; stimulated with 5625 IU rFSH without any response |
| 4 | Doherty 200258 | p.Ala419Thr/ p.Ala189Val | 17 | Finn | “N” | 10 Am | 163 | 51 | 19.2 | 91, 78 | 39, 33 | 16 | ND | “Small ovaries” | ND | ND |
| 5 | Allen 200359 | p.Pro348Arg (1 allele from Dad); 2nd allele unknown | 17 | ? | T3 | 10 Am | 160.6 | 50.8 | 19.6 | 105 | 36 | 21 | ND | ND | Streaks/ laparoscopy | MRI confirmed presence of follicles |
| 6 | Meduri 200360 | p.Pro519Thr HMZ | 26 | Cauc | ? | ? | 172 | 54 | 18.3 | 67 | 21 | < 10 | < 10 | 0.22 cc ovaries | Primordial and intermediary follicles (few primary follicles) | No follicles after 10,200 IU rFSH administration; c-Kit and PCNA labeling (no controls); no staining with SCC, Cyp17, Arom, or SF1 |
| 7 | Kuechler 201061 | p.Pro587His/del by translocation 46, XX, t(2;8) (p16.3or21;p23.1 | 17 | Ger | T3 | 10 Am | 159 | 48 | 19 | 106, 122 | 48, 47 | 13, 11 | ND | ND | Many primordial follicles, but no secondary or tertiary follicles | Laparoscopy—ovaries normal but small uterus |
Abbreviations: FSH, follicle-stimulating hormone; LH, luteinizing hormone; Am, amenorrhea; Arm, Armenian; Cauc, Caucasian; Finn, Finnish; Ger, German; LOV, left ovary; N, normal; ND, not determined; ROV, right ovary.
a Reports 2-7 are single cases, whereas report 1 consists of 22 of 75 women screened from 13 different Finnish families had the same homozygous inactivating FSHR mutation. Breast development is by Tanner staging: T1 prepubertal; T5 adult). Height (Ht) and weight (Wt) are measured in cm and kg, respectively. LH and FSH are in mIU/mL; Inhibin B (InB) is in pg/mL; ovarian volume (Ov Vol) is in cc. In case 6, spontaneous breast development and menses could not be determined because she was taking combined estrogen–progestin contraceptive pills at her first visit (this is indicated by a question mark). To convert pg/mL to pmol/L, multiply by 3.7. Normal follicular phase estradiol levels ∼ 20 to 200 pg/mL.
All Finnish women with this FSHR mutation had primary amenorrhea, but half had normal thelarche while the other half had impaired breast development.62 Ultrasound of the ovaries demonstrated follicles of varying sizes (Table 6). On histologic evaluation, only primordial follicles were seen in some patients, but in others, preantral or antral follicles were identified. In 1 patient, even a mature follicle was identified. Corpora lutea were not mentioned in the 9 women with FSHR gene mutations, suggesting that they did not ovulate, at least in the recent past. Stature of women with the FSHR gene mutations was less (160.5 cm) compared with ovarian failure patients without the mutation (165.8 cm). Although only a small sample size was studied, women with FSHR gene mutations (6 of 12) were more likely to have follicles on ultrasound than for those without mutations (1 of 11).62
Similar to patients with FSHB mutations, the inheritance of inactivating FSHR gene mutations was autosomal recessive. Although the carrier frequency of this mutation was common in Finland, the allele was not found in 35 unrelated US women with 46, XX ovarian failure.64 In a much larger international study, Jiang et al65 showed that the prevalence of FSHR gene mutations was exceedingly low in other ethnic populations—only 1 of 1200 from Switzerland, 0 of 1100 from Denmark, and 0 of 540 from Singapore were heterozygous carriers of the Finnish missense FSHR allele.
The second FSHR mutation was identified in an Armenian woman with secondary amenorrhea and hypergonadotropic hypogonadism.56 She had compound heterozygous FSHR missense mutations p.Ile160Thr/p.Arg573Cys (Tables 5 and 6). The Ile160Thr mutation in the ECD resulted in dramatic functional effects: severely impaired binding of iodinated FSH in COS-7 cells and a 90% reduction of cAMP production in response to exogenous FSH (Table 5).56 The other Arg573Cys allele in the third intracellular loop demonstrated normal binding affinity, but exhibited ∼30% of the cAMP production upon FSH stimulation compared with the wild type (Table 5). The affected proband had normal breast development, but menstruated only 3 times in a year-and-a-half, and then became amenorrheic. Serum gonadotropins were elevated and estradiols were low/low normal (20-40 pg/mL), and she had a 46, XX karyotype (Table 6).56 On transvaginal ultrasound, the ovaries were normal in size and contained numerous, small (3-5 mm in diameter) antral follicles (Table 6). The authors suggested that the compound heterozygous FSHR gene mutations in this patient produced a partially deficient state.56 However, it is also true that women with the Finnish missense mutation displayed evidence of hypofunction rather than null since half of these women manifested breast development; and follicular maturation (antral and mature follicles) was observed in some patients.62
After these 2 reports, 5 additional females with FSHR mutations, backed by in vitro and conformational studies, have been described (mutations 4-10 in Table 5 and cases 3-7 in Table 6).57 –61 Of the 5, 4 had primary amenorrhea and hypergonadotropic hypogonadism (Table 6). It is also likely that the other patient also did as well; however, she had been treated with oral contraceptives at age 17 and then had a normal exam about 10 years later. When examining these 10 inactivating FSHR mutations, it can be seen that 9 were missense (Table 5, Figure 2). The ECD was the most common site involved (4 of 10), but mutations were also found in the TMDs (n = 3) and ECLs (n = 2)—none were located in the ICLs. One mutation (case #10 in Table 5) involved a deletion that was not completely mapped at the genomic level but would be annotated as chr2: g.(48 887 046-48 895 903)-(49 058 614-49 065 417)del (NCBI 36/hg18) at the genomic level. The minimal deletion would be 163 kb, involving exons 9 and 10, while the maximal deletion would consist of a 178 kb deletion, affecting exons 7 to 10. In both cases, all 7 TMD1-7, ICL1-3, and ECL1-3, and the intracellular domain (ICD) would be removed. The difference between the 2 would be in how much of the ECD would be deleted (see Table 5 legend for details). When mutations occurred in the ECD, ligand binding was affected in 3 of 4 (cases 1, 2, 5, and 7 in Table 5) but binding was only affected in 1 of the 6 mutations outside the ECD (the ECL2 mutation). Of the 6 mutations, 3 analyzed impaired receptor targeting, but all 9 mutations studied (regardless of the location) impaired FSH signaling manifested as a reduction in cAMP.
All of the patients with the Finnish p.Ala189Val FSHR mutations were homozygous (Table 6). Of the remaining 6 patients, 3 had compound heterozygous missense mutations,57–58 1 had a homozygous missense mutation,60 1 had only 1 allele identified suggesting a possible unidentified mutation or deletion,59 and 1 had a missense mutation on 1 allele and a deletion on the other allele caused by an autosomal translocation61 (Table 6).The phenotype is relatively severe in patients with FSHR mutations in that all described patients except one56 had primary amenorrhea with elevated gonadotropins (Table 6). That patient, case #2 in Table 6 had secondary amenorrhea. These findings indicate that inactivating FSHR mutations in humans clearly cause the phenotype of hypergonadotropic hypogonadism.
Males
The Fshr knockout mouse
First reported by Dierich et al,27 the male Fshr −/− mouse exhibited oligospermia with reduced fertility and a ∼3-fold elevation of serum FSH, normal LH, and decreased testosterone levels (Table 1).27 This reduction in testosterone ranged from 65% to85% that of wild-type mice and was unlike male Fshb KO mice, which had normal testosterone levels (Table 1).27,66 However, Abel et al26 found elevations of both LH and FSH in the serum of Fshr −/− mice, but the pituitary content of either gonadotropin was not different from wild-type animals. These investigators also found that intratesticular inhibins A and B were not different in Fshr −/− mice compared to wild type or heterozygotes (although inhibin A was lower than inhibin B in all groups).26
Fshr −/− mice had small testes and seminiferous tubules and were initially reported by Dierich et al27 to have normal accessory structures (similar to male Fshb −/− mice). Subsequent studies by this same group quantified the reduction in testes weight by 50%, the seminal vesicle by 23%, and the epididymis by 30%.66 In addition, the tubal and luminal diameter of the seminiferous tubules and tubal diameter of the caput epididymis was reduced.66 These morphologic changes were likely related to the drastic reduction in serum testosterone.66
Sperm concentration, motility, and morphology were all reduced by 20% to 35% in Fshr −/− mice.27 In a subsequent, in-depth analysis by Krishnamurthy et al,66 there was no disruption or alteration of any of the steps of spermatogenesis, but 2 stages of spermatogenesis (VII and XI) were quantified. At 3 months of age, flow cytometric analysis indicated a 28% increase in spermatogonia and testicular somatic cells (2C fraction) and a 25% reduction in elongated spermatids in Fshr −/− mice.66 There was no difference in BrdU incorporation of S-phase cells indicating no increase in actively dividing spermatogonia. The expression of c-kit ligand (Kit), which stimulates DNA synthesis in type A spermatogonial cells, was doubled compared with WT. However, the expression of the FSH-responsive gene cAMP responsive element modulator (Crem), which is necessary for spermiogenesis, was unaltered.27 Elongated spermatids had reduced nuclear compaction, and there was evidence for inadequate condensation of sperm chromatin and poor sperm quality.66 Despite these abnormalities in sperm, litter size of Fshr −/− males was not different from wild-type or heterozygous mice as reported by Abel et al,26 although Dierich et al27 found that matings between heterozygous males and females resulted in a 35% to 50% litter size. Overall, the fertility of Fshr −/− male mice was more impaired than that of Fshb −/− mice and was thought to be due to the reduced testosterone levels in Fshr −/− mice.27
Interestingly, Fshb −/− and Fshr −/− males both have small testes, but Fshb −/− males have normal serum testosterone and LH levels, while Fshr −/− males have reduced testosterone and elevated LH levels. Baker et al47 compared Leydig cell function in these 2 KOs. Although Fshb −/− males had normal intratesticular testosterone and Leydig cells over time, Fshr −/− males had a reduction in intratesticular testosterone at 60 and 180 days (about one-third of the controls) and a reduction in Leydig cells at 20 (30% of controls) and 60 days (60% of controls). Similar to Fshb −/− males, Fshr −/− males demonstrated an impaired intratesticular testosterone response to hCG. There was a reduction of P450scc (Cyp11a1), 3βHSD6 (Hsd3b6), and StAR (Star) expression, but no change in P450c17 (Cyp17a1) or 17βHSD3 (Hsd17b3).47 They hypothesized that constitutively active FSHR in Fshb −/− male mice could regulate Leydig cell function and explain the different phenotypes. When the WT Fshr was transfected into a Leydig cell line, cAMP and progesterone were both upregulated 4-fold. These authors suggested that FSH signaling is not needed for the fetal population of Leydig cells (starting ∼day 5-10), as evidenced by normal Leydig cell number for both Fshb −/− and Fshr −/− at days 1 and 5.47 However, the adult population of Leydig cells was diminished by day 20 and 60 in Fshr −/− males (and not Fshb −/− males), but the constitutive action of the WT FSHR in Fshb −/− mice permitted sufficient signaling to effect normal function. Since only Sertoli cells (and not Leydig cells) express the FSHR, this is likely a permissive action of FSH upon Leydig cell function.47
Human inactivating FSHR mutations
Tapanainen et al67 reported the phenotypic findings of 5 men homozygous for the Finnish p.Ala189Val FSHR gene mutation who were identified from studies of ovarian failure families described above (Table 7, Figure 2). They all had normal puberty, with normal to small testes ranging from 4 to 15 cc (normal: 15-25 cc) and normal testosterone levels. All 5 had elevated FSH levels, with normal to elevated LH levels. In all, 2 males each had 2 children, but all 5 had abnormal sperm parameters with concentrations ranging from < 0.1 million/mL to 42 million/mL (normal >20 million/mL). Another 151 Finnish men with moderate-to-severe spermatogenic failure were studied for FSHR gene mutations but no homozygotes were identified. These authors suggested that FSH function may not be essential for spermatogenesis since fertility occurred in some men despite oligospermia.67 No other inactivating FSHR mutations have been described in men (http://omim.org/entry/136435).
Table 7.
Inactivating FSHR Mutations in Males.a
| Author | FSHR Mutation | Pt. | Age | Testes (15-25 cc) | Sperm | FSH (1-10.5 mIU/mL) | LH (1-8.4 mIU/mL) | Testosterone (8.2-34.6 nmol/L) | Inhibin B (76-447 ng/L) | Fertility | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tapanainen 1997 67 | p.Ala189Val | Sperm Conc. | Volume (mL) | % Normal | ||||||||
| 1 | 29 | 8.6 and 6 | 42 × 106 | 1.5 | 16% | 40 | 11 | 14.7 | 53 | Not tested | ||
| 2 | 42 | 8 and 8 | <1.0 × 106 | 2.5 | 16% | 21 | 16 | 26.2 | 54 | Not tested | ||
| 3 | 45 (30)b | 4 and 4 | <0.1 × 106 | 4.8 | Low | 24 | 16 | 14.5 | <15 | Infertile | ||
| 4 | 47 | 15 and 13.8 | 5.6 × 106 | 3 | Low | 13 | 6 | 8.8 | 33 | 2 children | ||
| 5 | 55 | 13.5 and 15.8 | <0.1 × 106 | 3.3 | Low | 15 | 4 | 15.8 | 62 | 2 children | ||
Abbreviations: FSH, follicle-stimulating hormone; LH, luteinizing hormone.
a All were homozygous and stated to be normally masculinized.
b Semen analysis was done at the stated age for all except patient #3 who was of age 30 when the semen analysis was performed. Height, weight, presence of gynecomastia, and sperm motility were not mentioned in the publication. To convert ng/dL to nmol/L multiply by 0.03.
Activating Mutations of the Receptor
Overexpression of FSH Using Transgenic Mice
In order to study how a gain of FSH function affects reproduction, transgenic mice were generated that ectopically produce FSH.28 In these experiments, metallothionein 1 promoter-driven expression of Cga in some mice and Fshb in other mice was accomplished. These overexpressing mice were then crossed to produce mice that could form dimeric FSH. Some strains of mice—the weak FSH expressers—had no phenotypic effects but had mildly elevated FSH levels (FSH = 48 mIU/mL in males; and 116 mIU/mL in females). In contrast, the high copy FSH expressers did manifest recognizable reproductive abnormalities. Male transgenic mice were infertile, despite having normal sized testes and increased sperm production (by ∼75%). In these mice, FSH levels were markedly elevated (150 000 mIU/mL), as were serum testosterone levels (18-fold increase compared to wild type). No obvious testicular defects were noted on histologic evaluation, but the seminal vesicle size was more than doubled. The infertility in these transgenic mice was thought to be due to either high testosterone levels or altered reproductive behavior. There is precedence for this as it is well known that the administration of exogenous testosterone to human males may act as a contraceptive agent by suppressing LH to decrease sperm concentration.68
In contrast, female high copy FSH expressers had markedly elevated FSH levels (360 000 mIU/mL), elevated estradiol, progesterone, and testosterone levels and demonstrated hemorrhagic, cystic ovaries similar to human ovarian hyperstimulation syndrome. Of interest, these female transgenic mice were infertile but usually died because of extreme bladder and kidney enlargement with obstruction.28
Activating Human FSHR Mutations
Males
The first activating FSHR mutation was reported by Gromoll et al69 who characterized a p.Asp567Gly missense mutation in a hypophysectomized man who fathered children solely with testosterone replacement (Figure 2). The patient had undetectable serum FSH and LH, though his testicular volume was normal and semen analysis parameters were essentially normal. In vitro analysis of the mutant FSHR revealed cAMP production that was increased by approximately 1.5-fold without ligand.69 These investigators70 then performed additional innovative studies involving the hypogonadal (hpg) mouse, which is gonadotropin deficient secondary to a partial deletion of Gnrh1.71 They created the p.Asp567Gly transgene and directed its expression using the rat androgen binding protein (Abp) gene promoter into the Sertoli cells of hpg mice. The insertion of the transgene resulted in a 2-fold-increased testicular weight, which contained mature Sertoli cells and premeiotic germ cells (absent in controls). Isolated Sertoli cells had a 2-fold increase in basal cAMP production and 1.5-fold increase in FSH-stimulated cAMP levels. Intriguingly, this transgene also resulted in increased testicular production of testosterone, suggesting a Sertoli-cell-mediated paracrine effect upon Leydig cells of the hpg mouse.70 These findings provide strong support for constitutive activation of mutant Asp563, a conserved residue that is also susceptible to mutation in the luteinizing hormone receptor (LHR) and TSHR.70 It is interesting that transgenic mouse models have reproduced the clinical phenotype of this missense FSHR mutation.70,78
Females
Similar to the phenotype of the overexpressing FSH mouse, human females with FSHR mutations manifest spontaneous ovarian hyperstimulation syndrome (sOHSS), previously only thought to be an iatrogenically induced condition in infertile females receiving exogenous gonadotropins for infertility treatment (Table 8). A hyperresponsiveness to gonadotropins in these patients results in excess follicular maturation, supranormal estradiol levels, with resultant ovarian enlargement, ascites, and hydrothorax if severe, as well as an increased risk for congestive heart failure, oliguria, and thrombotic events.79 These phenotypic findings are thought to be at least partially due to enhanced vascular endothelial growth factor action as well as alterations in cytokines and renin.79 Spontaneous OHSS is generally thought to be rare.
The first 2 descriptions of affected females with activating FSHR mutations were reported in back-to-back publications in 2003 (Tables 8 and 9, Figure 2).72,73 Vasseur et al72 described a Moroccan family in which 3 females manifested sOHSS. The proband was a 25-year-old woman who developed sOHSS in 4 of her 5 pregnancies, 1 of which was severe OHSS with hydrothorax and ascites. Two of her sisters had multiple occurrences of sOHSS during some, but not all of their pregnancies, while a third sister gave no such history. These investigators found that all 3 affected sisters demonstrated heterozygosity for a p.Thr449Ile missense mutation in TMD3 of the FSHR gene which was absent in the unaffected sister (Tables 8 and 9).
Table 8.
Reported Activating FSHR Mutations in Human Patients (Males and Females) Supported by Functional In Vitro Analysis.a
| # | Author | FSHR Mutation | Exon | Protein Region | In Vitro Analysis | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Protein | Binding Capacity | Binding Affinity | Receptor Targeting | Cyclic AMP | Constitutively Active and Other | ||||
| 1 | Gromoll 199669; Haywood 200270 | c.1700A>G | p.Asp567Gly | 10 | TMD6 | ND | ND | ND | Increased 1.5 fold over WT in COS; Tg mouse Sertoli cells of hpg mice increased 2-fold basally and 1.5 fold after FSH | Yes; 2-fold increase in testes weights; increased testosterone in absence of FSH/LH |
| 2 | Vasseur 20037,2 | c.1346C>T | p.Thr449Ile | 10 | TMD3 | Normal | Normal | Normal; No direct hCG binding; no displacement of labeled FSH to hCG | Normal basally and after FSH vs WT; >20-fold increase to hCG; No effect by TSH | No |
| 3 | Smits 200373; Olatunbosun 199674 | c.1699G>A | p.Asp567Asn | 10 | TMD6 | ND | Normal | Decreased cell surface expression | 3-fold increase basally without hormone; Increase similarly to WT with FSH; increase in response to hCG (1/1000 as sensitive as LHR); 3-fold increase to TSH | Yes |
| 4 | Montanelli 20047,5 | c.1455A>G | p.Thr449Ala | 10 | TMD3 | ND | ND | Mutant had 2-fold increased receptor expression | 2.5-fold constitutive increase in cAMP; response to rFSH similar to WT; increase in response to hCG (1/1000 as sensitive as LHR); increase to TSH by 2-fold | Yes |
| 5 | De Leener 20067,6 | c.1634T>C | p.Ile545Thr | 10 | TMD5 | ND | Markedly decreased | 23% reduction in cell surface expression | 30% increase in basal cAMP (no hormone);2-fold increase in cAMP with hCG or TSH | Yes |
| 6 | De Leener 20087,7 | c.383C>A | p.Ser128Tyr | 5 | ECD | ND | ND | 64% reduction of WT FSHR expression | No difference in cAMP without hormone; not sensitive to TSH; decreased specificity of the mutant FSHR toward hCG with an increase in sensitivity and affinity toward hCG | No |
Abbreviations: ECD, extracellular domain; ECL, extracellular loop; ICL, intracellular loop; ND, not determined; TMD, transmembrane domain; WT, wild type; hCG, human chorionic gonadotropin; TSH, thyroid-stimulating hormone; cAMP, cyclic adenosine monophosphate; rFSH, recombinant follicle-stimulating hormone.
a Case 1 is a male, whereas cases 2 to 6 are females. Hpg, hypogonadal mouse, which has gonadotropin deficiency secondary to a deletion in Gnrh1. All experiments were performed using COS-7 cells unless otherwise noted.
Table 9.
The Phenotypes of the Females With Activating FSHR Mutations Supported by Functional Analysis.
| # | Author | FSHR Mutation | Age | Race | Pregnancies | Pregnancy Outcome | Other | Family History |
|---|---|---|---|---|---|---|---|---|
| 1 | Vasseur 200372 | p.Thr449Ile | 25 | Moroccan | 4/5 pregnancies affected | Two affected sisters: 1).4/7 pregnancies and always began in 1st TM; 2) 2/4 pregnancies affected; One unaffected sister had 2 normal pregnancies | ||
| 1. Ascites and bilateral ovarian masses after TAB at 8 weeks | TAB | |||||||
| 2. SAB at 6 weeks—no OHSS | SAB | |||||||
| 3. Diagnosed 8 weeks and lasted all pregnancy | 2450 g female SGA | |||||||
| 4. TAB for OHSS at 9 weeks | TAB | |||||||
| 5. 8 weeks; 14 cm ovaries, which stayed large all pregnancy | Delivered normal female | hCG normal, FSH appropriately suppressed; estradiol high only in first TM; normal free T; inhibin B markedly elevated, but declined with hCG drop | ||||||
| 2 | Smits 200373 | p.Asp567Asn | 40 | Canadian | 4/4 pregnancies affected | |||
| 1. Age 24, multicystic, enlarged ovaries, acne; theca lutein cysts | Delivered term healthy 2635 g female | Normal ovarian and adrenal hormones | ||||||
| 2. Age 24, multicystic, enlarged ovaries | Death in utero 41.5 weeks | No fetal anomalies; ovaries normal 12 weeks postpartum; polycystic appearing ovaries | ||||||
| 3. Age 29, at 9 weeks, hydrothorax, ascites, bilateral multicystic ovaries | SAB at 10.5 weeks | Multiple paracenteses yielded 11 L of ascites; normal TSH, hCG, Hct; ovaries regressed to normal size 8 weeks after D&C | ||||||
| 4. Age 31, infertility evaluation showed PCO appearing ovaries | Infertility evaluation showed PCO appearing ovaries; CD#3 FSH = 7mIU/mL; LH = 18 mIU/mL | |||||||
| 3 | Montanelli 200475; Di Carlo 199780 | p.Thr449Ala | 24 | Italian | 1. Age 24, pain and enlarged multicystic ovaries at 10 weeks; uncertain if ascites | Delivered term healthy 3050 g female | ||
| 2. Age 26, pain and dyspnea at 10 weeks; 11X10X9 cm ROV; 13X11X10 LOV, ascites, hemoconcentration (severe) | Delivered a term healthy 3220 gram male | 25 days to resolution | Affected sister with severe OHSS who delivered 3470 g male | |||||
| 4 | De Leener 200676; Chae 200181 | p.Ile545Thr | 35 | Korean | 1. Normal pregnancy | Healthy male | ||
| 2. 12 weeks, she had severe OHSS 15 × 11 × 11 cm ROV; 12 × 9 × 12 cm LOV—both ovaries multilobulated 7 cystic; ascites was managed with 1000 mL of 5% dextrose/sodium;100 mL of 20% albumin and 500 mL of dextran | Healthy 3045 gram female at 41 4/7 weeks | Symptoms regressed in 2nd and 3rd TM and resolved postpartum; 22 1/7 weeks ROV was 9 × 4 × 5 cm; LOV was 8 × 4 × 6 cm, and small amount of ascites | ||||||
| 5 | De Leener 200877; Cepni 200682 | p.Ser128Tyr | 21 | Turkish | Presented with lower abdominal pain, distention, nausea, shortness of breath, and anuria for 24 hours; 11 week IUP with massive ascites and bilateral multilobulated cystic ovaries both 12 × 9 cm. Hemoconcentration, low sodium; high potassium; slightly elevated serum hepatic transaminases | Normal term vaginal delivery of male | Treated with normal saline (1 L/d) and 20% albumin (200 mL/day); felt better within 2 days and discharged on 10th day; Ovaries were normal in 3 months. | Unremarkable |
Abbreviations: ND, not determined; LOV, left ovary; ROV, right ovary; SAB, spontaneous abortion; TAB, therapeutic abortion; TM, trimester; OHSS, ovarian hyperstimulation syndrome; hCG, human chorionic gonadotropin; TSH, thyroid-stimulating hormone; FSH, follicle-stimulating hormone.
When this mutant FSHR was transfected into COS-7 cells, cell surface expression was normal and there was no constitutive activation basally (without ligand), and there was no difference in cAMP production after FSH administration compared to WT. However, the Thr449Ile mutant demonstrated an increased cAMP response to hCG. The wild-type FSHR had little or no activation of cAMP to increasing doses of hCG, but there was a profound increase (>20-fold) in maximal cAMP production of the mutant receptor compared to wild type. This occurred despite the observation that I125-labeled hCG did not bind to the mutant receptor; and there was no displacement of I125-labeled FSH by hCG. Additionally, TSH had no effect upon this receptor (Table 8). Therefore, this mutant receptor was not constitutively active, but exhibited a loss of binding specificity as it bound hCG in addition to FSH.72 It is interesting that although the ECD is responsible for ligand binding, activation of this mutant receptor by hCG without binding suggests an interplay between the 2 regions of the receptor resulting in decreased binding specificity.72
The other activating FSHR mutation was reported by Smits et al73 who found a heterozygous p.Asp567Asn in a 40-year-old woman with sOHSS in each of her 4 prior pregnancies (Table 8). This affected residue was located within the sixth transmembrane domain (TMD6) and interestingly was the same wild-type amino acid affected as in the hypophysectomized man (although it was a different mutation). Functional studies revealed that despite a 31% decrease in cell surface expression, the mutant exhibited a 3-fold basal increase in cAMP production relative to the wild type. Binding affinities were similar to the WT with increasing doses of exogenous FSH, as were the maximal responses for the mutant and wild-type receptors. However, there was an enhanced response of the mutant receptor, but not WT, to hCG. This response to hCG was only about 1 of 1000 as sensitive as the cAMP response of the LHR to hCG. Unlike p.Thr449Ile, the p.Asp567Asn mutant displayed constitutive activity, and an increased sensitivity to TSH (Table 8).73
Since these 2 studies, 3 additional cases were reported, each of whom had heterozygous activating FSHR mutations (Tables 8 and 9, Figure 2).75–77 The p.Thr449Ala in TMD3 described by Montanelli et al75 was constitutively active and displayed increased sensitivity to hCG and TSH (Table 8). Montanelli et al83 further studied each the reported activating FSHR mutations (p.Thr449Ile, p.Asp567Asn, and p.Thr449Ala) and showed that these 3 mutations affect the interhelical bonds between transmembrane helices which are known to keep the receptor in an inactive state.
In a patient with first trimester sOHSS and normal hCG levels, in 2006 De Leener et al76 characterized a p.Ile545Thr in TMD5 with a modest (30%) increase in basal cAMP activity, which was further increased by hCG or TSH (case 5 in Table 8). The sOHSS patients with reported FSHR mutations to this point all had normal hCG levels during pregnancy. These investigators76 studied 7 additional sOHSS patients with elevated hCG (n = 4) or TSH (n = 3) levels in pregnancy but did not find FSHR mutations, suggesting a natural stimulation of the WT receptor to high concentrations of the hormones. These studies indicate that not all patients with sOHSS have activating FSHR mutations or that they were not identified using current methodology. Whether these patients might possess mutations in regulatory regions of the receptor or have intragenic deletions or rearrangements not detectable by PCR-based DNA sequencing remains to be determined.
All reported activating FSHR mutations were localized to the transmembrane regions of the FSHR until 2008 when De Leener et al77 discovered the first mutation in the ligand binding domain—a heterozygous p.Ser128Tyr missense mutation (Tables 8 and 9, Figure 2). Functional analysis of the p.Ser128Tyr mutation resulted in a 64% reduction in cell surface expression, and there was no constitutive activation. However, the mutant receptor had increased affinity and sensitivity to hCG but not to TSH (Table 8).
When the identified heterozygous, activating FSHR mutations in 5 females with sOHSS are examined, it can be seen that 4 are in the serpentine region of the receptor and only 1 is in the ECD (Figure 2, cases 2-6 in Table 8). In fact 4 of the 6 activating FSHR mutations (when the 1 male is also included) occur in either the p.Asp567 (changing it to either a Gly or Asn) or a Thr449 (changing it to either an Ile or Ala). In both cases mutation of p.Asp567 resulted in a constitutively active protein, while Thr449Ala was constitutively active but Thr449Ile was not (Table 8). Overall, 2 of the 5 activating mutations in females (1 in TMD3 and 1 in the ECD) were not constitutively active. However, the common feature is that all 5 activating FSHR mutations, causing OHSS have reduced specificity such that they respond to the increasing hCG levels that occur during normal pregnancy. Perhaps this would be expected in the ascertainment of a phenotype exacerbated in pregnancy. A total of 3 also had an increased response to TSH further, demonstrating the decreased specificity of the mutant receptor.
When the phenotypes of the affected sOHSS women are examined, 2 of the 5 patients had other affected members, indicating autosomal dominant inheritance, while 3 were sporadic (Table 9). Mutations seem to affect different ethnicities. Interestingly, 2 of the patients described had 4 of 5 and 4 of 4 pregnancies affected with sOHSS, but 2 of 5 had some pregnancies that were not affected with OHSS. Therefore, the presence of an activating FSHR mutation did not preclude the possibility of an unaffected pregnancy. Overall, in these 5 patients, sOHSS occurred in 12 of 14 pregnancies with varying pregnancy outcomes. All 5 patients had at least 1 healthy live-born child during the cycle of hyperstimulation. Of the 12 pregnancies complicated by sOHSS, 1 resulted in a spontaneous abortion, 2 were electively aborted, 1 resulted in a death in utero at 41½ weeks, and 8 resulted in healthy live-born children—3 males and 5 females (Table 9).
Human FSHB and FSHR Polymorphisms
Single-nucleotide polymorphisms (SNPs) have been identified in the FSHR and FSHB genes, and there have been a number of studies attempting to associate them to fertility potential. Simoni et al84 studied men with infertility and did not find any causative FSHR mutations but did characterize 2 SNPs: p.Asn680Ser (rs6166) and p.Thr307Ala (rs6165). These missense variants had no functional effects in vitro; and the prevalence did not differ in fertile versus infertile men.84 Since this study, a number of investigators have conducted association studies of FSHR polymorphisms, with often contradictory results (reviewed elsewhere85 ,86). Perez Mayorga et al87 first found that the Ser680 allele is associated with higher serum FSH levels and the need for additional exogenous FSH in cycles of controlled ovarian hyperstimulation. In a review and pooling of data from 4 publications regarding Asn680Ser (the best studied of the FSHR SNPs), Moron and Ruiz86 concluded that the FSHR genotype is associated with response to exogenous gonadotropin therapy. The 3 studies showing association with the FSHR genotype enrolled patients in Spain,88 Greece,89 and the Ukraine,90 while no association was seen in the Netherlands population.91 Although promising, the clinical utility of the better studied FSHR SNPs, as well as FSHB SNPs, requires further investigation.
Immunoneutralization of FSH—Effects Upon Reproductive Phenotype
A number of investigators have shown that continuous bioneutralization of endogenous FSH may affect reproduction in males and females from a variety of species including rat, monkey, and human. Either passive or active immunization in fertile adult male monkeys has been shown to result in oligospermia and infertility.92,93 Moudgal et al93 showed that active immunization with ovine FSH (oFSH) isolated from sheep pituitaries elicited an antigenic response in adult male bonnet monkeys, but it did not affect the health, libido, or testosterone levels of the animals. However, there was a marked decrease (75%-100%) in the number of spermatozoa in seminal ejaculates by 150 days. These monkeys also manifested infertility, which was reversed when booster injections were discontinued.93 Similarly, blockade of FSH receptor in male monkeys by receptor antibody formation also resulted in testicular dysfunction and infertility.94 Moudgal et al95 injected human males with oFSH, and they produced specific antibodies capable of binding and neutralizing FSH bioactivity for 1 complete sperm cycle (∼65 days). Seminal plasma transferrin, a marker of Sertoli cell and seminiferous tubular function, was markedly reduced (30%-90%) and sperm counts were reduced (30%-74%) in some patients.95
To examine the spermatogenic dysfunction in more detail, Meachem et al96 used immunoneutralization with a polyclonal rat FSH antibody in male rats and demonstrated a significant reduction in the number of type A and B spermatogonia, pachytene spermatocytes, round spermatids, and elongated spermatids after 8.5 days of treatment. Krishnamurthy et al97 studied bonnet monkeys and normal, healthy fertile men that were actively immunized with oFSH and showed that sperm exhibited defective chromatin packaging and had a reduction in acrosomal glycoprotein content. Interestingly, these alterations in sperm quality occurred well in advance of decreased sperm counts in the ejaculate.97
Most of these studies have involved males. However, Ferro et al98 conjugated a variety of FSHβ peptides to tetanus toxoid, which were used to immunize female rats. All animals showed reduced estradiol levels and had disrupted estrous cycles. Similarly, Butterstein et al99 generated antibodies against human FSHβ that were administered to mature Sprague-Dawley rats. Continuous exposure to immunoneutralizing antibodies disrupted estrous cycles in all animals, but only animals with titers high enough to completely block binding of FSH to its receptor demonstrated infertility.99 Collectively, these findings provide additional support that FSH is essential for gonadal function in murine and primate species.
Synthesis of Human and Mouse Findings
Inactivating Mutations
Females
A small number of inactivating mutations in both the FSHB and the FSHR genes in humans have been described. The effect of mutations of the FSHB gene in human females is very similar to that described in Fshb −/− KO mice. These women had irreversibly delayed puberty and infertility with low serum FSH levels and elevated LH levels. Fshb −/− mice also were sterile with low FSH and elevated LH. Estradiol levels were low in the human patients; and although reportedly normal, they were likely low in the Fshb −/− mice, particularly given the appearance of the reproductive organs. Histologically, these mice had predominantly primordial follicles, although some degenerating antral follicles were present. Human females with FSHB mutations demonstrated primordial and small antral follicles. However, both mice and humans responded to exogenous FSH, indicating that even with endogenous depletion of the FSH ligand, the ovaries have the capacity to activate FSH signaling (except in the 1 patient who was older).
The phenotypes of women with inactivating FSHR mutations and the Fshr −/− KO mice were quite similar. The mice were sterile, while the women all had primary amenorrhea (with or without breast development). Both humans and mice had elevated serum gonadotropin levels. Most women with FSHR mutations had low or undetectable estradiol levels. The female Fshr −/− KO mice had low estradiol and progesterone, but elevated testosterone (not present in the humans with FSHR mutations); and these mice had no vaginal openings (indicating no puberty). The mice demonstrated primordial, primary, and secondary follicles, but antral follicles were not observed. Rarely, humans with FSHR mutations also had further follicular development with at least 1 of the Finnish patients having mature preovulatory follicles.
Women with inactivating FSHB or FSHR gene mutations possess mostly primordial follicles but may also have antral follicles. Therefore, FSH does not appear to be necessary for follicular development up to the small antral follicle stage but seems to be required for further follicular development. Since women with FSHB gene mutations have elevated LH levels but do not have hyperandrogenism, FSH may have some requisite function for ovarian thecal androgen production possibly by stimulating production of CYP17, LHR, inhibin, or other growth factors. These findings suggest that FSH is important for normal pubertal development and fertility.
Males
The phenotypic effects of males (human and mice) deficient in FSH are somewhat disparate, particularly for the ligand. Male Fshb −/− KO mice demonstrated smaller testes with a 75% reduction in sperm concentration and 40% reduction in motility, but they were fertile. Serum FSH was low, but LH was normal as was testosterone. All 3 human males with FSHB mutations had low FSH, elevated LH, and azoospermia. Interestingly, 2 males had small testes, but one of them also had low testosterone, suggesting that FSH deficiency could play some role in androgen production in the male.
Mutations in the receptor, whether in the human or mouse, had no demonstrable effect upon fertility in males. Fshr −/− KO male mice had a somewhat decreased testes size and a modest reduction in sperm concentration, motility, and morphology. As expected, serum FSH was elevated, but perhaps unexpectedly, serum testosterone was reduced by 65%. There have only been 5 human males with FSHR mutations—all ascertained in Finnish families with the p.Ala189Val mutation found in women with ovarian failure. They all had elevated serum FSH, normal to elevated LH, and normal testosterone levels. Their ages and sperm parameters were quite variable, but 2 of the 3 attempting fertility had children.
These findings in males suggest that perhaps there are species-specific differences between mice and men. In humans, mutations in the ligand gene are more severe than those of the receptor, while the opposite is true for the mouse. The endocrine function of human FSHB mutants may be more adversely affected than the human FSHR or either of the mouse genes since 1 of the 3 males with an FSHB mutation had low testosterone and delayed puberty. In the mouse, the Fshb −/− mouse may have a less severe phenotype because of the constitutively active FSHR, which may result in some FSH function without the presence of the ligand. However, both the Fshb −/− and Fshr −/− mice have decreased germ cell and Sertoli cell numbers (30%-40%). These studies also suggest that FSH is necessary in both males and females to augment LH-induced synthesis of androgens.
Inactivating FSHR and FSHB gene mutations in humans are both inherited as autosomal recessive traits, and they produce a somewhat similar phenotype in that they result in hypogonadism. However, both gonadotropins are elevated in patients with FSHR mutations (hypergonadotropic hypogonadism), while patients with FSHB mutations manifest reduced FSH levels (isolated FSH deficiency) and elevated (or normal) LH levels. Women with FSHR or FSHB mutations may have completely absent pubertal development or partial development. The difference lies in the reproductive potential of these patients with treatment as women with FSHB mutations have normal ovarian function and an excellent chance to conceive with exogenous gonadotropins; while women with FSHR mutations have impaired ovarian function and a poorer prognosis, as do other women with ovarian failure from any cause.
Activating FSHR Mutations
The creation of transgenic mice overexpressing human Fshb produced variable phenotypes depending upon the level of expression and the sex of the animal. With low Fshb expression, elevated FSH protein levels were observed similar to humans with gonadal failure, but the phenotype was normal in both male and female mice. However, with high Fshb expression, females demonstrated infertility and had enormously elevated levels of FSH with hemorrhagic ovarian cysts similar to iatrogenic human OHSS. Subsequently, human females with activating FSHR mutations were shown to have the same phenotype as the mouse. Although they are usually constitutively active, activating human FSHR mutations causing sOHSS have uniformly altered the receptor such that it responds to hCG, which is why ovarian enlargement is restricted to pregnancy. This sOHSS is inherited as an autosomal dominant trait. Therefore, hypofunction of both the ligand and the receptor are inherited as autosomal recessive disorders, while hyperfunction of the receptor is autosomal dominant, similar to the genetics of human LHR mutations. Lastly, when the single activating FSHR mutation in a human male was inserted into Sertoli cells of the hpg mouse, testosterone production increased. These data, in addition to that discussed above, provide further support that FSH action is necessary for LH-dependent androgen production in males as well as females.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Portions of this work were supported by the NICHD PHS-HD33004 to LCL.
References
- 1. Tobet SA, Schwarting GA. Minireview: recent progress in gonadotropin-releasing hormone neuronal migration. Endocrinology. 2006;147(3):1159–1165. [DOI] [PubMed] [Google Scholar]
- 2. Wierman ME, Pawlowski JE, Allen MP, Xu M, Linseman DA, Nielsen-Preiss S. Molecular mechanisms of gonadotropin-releasing hormone neuronal migration. Trends Endocrinol Metab. 2004;15(3):96–102. [DOI] [PubMed] [Google Scholar]
- 3. Plant TM. Hypothalamic control of the pituitary-gonadal axis in higher primates: key advances over the last two decades. J Neuroendocrinol. 2008;20(6):719–726. [DOI] [PubMed] [Google Scholar]
- 4. Bentley GE, Ubuka T, McGuire NL, et al. Gonadotrophin-inhibitory hormone: a multifunctional neuropeptide. J Neuroendocrinol. 2009;21(4):276–281. [DOI] [PubMed] [Google Scholar]
- 5. Layman LC, McDonough PG. Mutations of the follicle stimulating hormone-beta and its receptor in human and mouse: phenotype/genotype. Mol Cell Endocrinol. 2000;161(1-2):9–17. [DOI] [PubMed] [Google Scholar]
- 6. Fox KM, Dias JA, Van Roey P. Three-dimensional structure of human follicle-stimulating hormone. Mol Endocrinol. 2001;15(3):378–389. [DOI] [PubMed] [Google Scholar]
- 7. Fiddes JC, Goodman HM. The gene encoding the common alpha subunit of the four human glycoprotein genes. J Appl Genet. 1981;1(1):3–18. [PubMed] [Google Scholar]
- 8. Jameson JL, Becker CB, Lindell CM, Habener JF. Human follicle-stimulating hormone β-subunit gene encodes multiple messenger ribonucleic acids. Mol Endocrinol. 1988;2(9):806–815. [DOI] [PubMed] [Google Scholar]
- 9. Lapthorn AJ, Harris DC, Littlejohn A, et al. Crystal structure of human chorionic gonadotropin. Nature. 1994;369:455–461. [DOI] [PubMed] [Google Scholar]
- 10. Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature. 2005;433:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gromoll J, Pekel E, Nieschlag E. The structure and organization of the human follicle-stimulating hormone receptor (FSHR) gene. Genomics. 1996;35(2):308–311. [DOI] [PubMed] [Google Scholar]
- 12. Huhtaniemi IT, Aittomaki K. Mutations of follicle-stimulating hormone and its receptor; effects on gonadal function. Eur J Endocrinol. 1998;138(5):473–481. [DOI] [PubMed] [Google Scholar]
- 13. Kelton CA, Cheng SVY, Nugent NP, et al. The cloning of the human follcile-stimulating hormone receptor and its expression in COS-7, CHO, and Y-1 cells. Mol Cell Endocrinol. 1992;89(1-2):141–151. [DOI] [PubMed] [Google Scholar]
- 14. Meduri G, Bachelot A, Cocca MP, et al. Molecular pathology of the FSH receptor: new insights into FSH physiology. Mol Cell Endocrinol. 2008;282(1-2):130–142. [DOI] [PubMed] [Google Scholar]
- 15. Yarney TA, Jiang L, Khan H, MacDonald EA, Laird DW, Sairam MR. Molecular cloning, structure, and expression of a testicular follitropin receptor with selective alteration in the carboxy terminus that affects signaling function. Mol Reprod Dev. 1997;48(4):458–470. [DOI] [PubMed] [Google Scholar]
- 16. Sairam MR, Jiang LG, Yarney TA, Khan H. Follitropin signal transduction: alternative splicing of the FSH receptor gene produces a dominant negative form of receptor which inhibits hormone action. Biochem Biophys Res Commun. 1996;226(3):717–722. [DOI] [PubMed] [Google Scholar]
- 17. Kraaij R, Verhoef-Post M, Grootegoed JA, Themmen AP. Alternative splicing of follicle-stimulating hormone receptor pre-mRNA: cloning and characterization of two alternatively spliced mRNA transcripts. J Endocrinol 1998;158(1):127–136. [DOI] [PubMed] [Google Scholar]
- 18. Tena-Sempere M, Manna PR, Huhtaniemi I. Molecular cloning of the mouse follicle-stimulating hormone receptor complementary deoxyribonucleic acid: functional expression of alternatively spliced variants and receptor inactivation by a C566 T transition in exon 7 of the coding sequence. Biol Reprod 1999;60(6):1515–1527. [DOI] [PubMed] [Google Scholar]
- 19. Sairam MR, Babu PS. The tale of follitropin receptor diversity: a recipe for fine tuning gonadal responses? Mol Cell Endocrinol. 2007;260-262:163–171. [DOI] [PubMed] [Google Scholar]
- 20. Wayne CM, Fan HY, Cheng X, Richards JS. Follicle-stimulating hormone induces multiple signaling cascades: evidence that activation of Rous sarcoma oncogene, RAS, and the epidermal growth factor receptor are critical for granulosa cell differentiation. Mol Endocrinol. 2007;21:1940–1957. [DOI] [PubMed] [Google Scholar]
- 21. Gonzalez-Robayna IJ, Falender AE, Ochsner S, Firestone GL, Richards JS. Follicle-stimulating hormone (FSH) stimulates phosphorylation and activation of protein kinase B (PKB/Akt) and serum and glucocorticoid-induced kinase (Sgk): evidence for A kinase-independent signaling by FSH in granulosa cells. Mol Endocrinol. 2000;14:1283–1300. [DOI] [PubMed] [Google Scholar]
- 22. Jiang X, Liu H, Chen X, et al. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc Natl Acad Sci USA. 2012;109(31):12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richards JS, Pangas SA. The ovary: basic biology and clinical implications. J Clin Invest. 2010;120(4):963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle-stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15(2):201–204. [DOI] [PubMed] [Google Scholar]
- 25. Rinaldi S, Dechaud H, Biessy C, et al. Reliability and validity of commercially available, direct radioimmunoassays for measurement of blood androgens and estrogens in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2001;10(7):757–765. [PubMed] [Google Scholar]
- 26. Abel MH, Wootton AN, Wilkins V, Huhtaniemi I, Knight PG, Charlton HM. The effect of a null mutation in the follicle-stimulating hormone receptor gene on mouse reproduction. Endocrinology 2000;141(5):1795–1803. [DOI] [PubMed] [Google Scholar]
- 27. Dierich A, Sairam MR, Monaco L, et al. Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc Natl Acad Sci USA. 1998;95(23):13612–13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar TR, Palapattu G, Wang P, et al. Transgenic models to study gonadotropin function: the role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol Endocrinol. 1999;13(6):851–865. [DOI] [PubMed] [Google Scholar]
- 29. Rivarola MA, Belgorosky A, Berensztein E, de Davila MT. Human prepubertal testicular cells in culture: steroidogenic capacity, paracrine and hormone control. J Steroid Biochem Mol Biol. 1995;53(1-6):119–125. [DOI] [PubMed] [Google Scholar]
- 30. Kumar TR, Low MJ, Matzuk MM. Genetic rescue of follicle-stimulating hormone β-deficient mice. Endocrinol. 1998;139(7):3289–3295. [DOI] [PubMed] [Google Scholar]
- 31. Burns KH, Yan C, Kumar TR, Matzuk MM. Analysis of ovarian gene expression in follicle-stimulating hormone beta knockout mice. Endocrinology. 2001;142(7):2742–2751. [DOI] [PubMed] [Google Scholar]
- 32. Layman LC, Lee EJ, Peak DB, et al. Delayed puberty and hypogonadism caused by mutations in the follicle-stimulating hormone beta-subunit gene. N Engl J Med. 1997;337(9):607–611. [DOI] [PubMed] [Google Scholar]
- 33. Layman LC, Porto AL, Xie J, et al. FSH beta gene mutations in a female with partial breast development and a male sibling with normal puberty and azoospermia. J Clin Endocrinol Metab. 2002;87(8):3702–3707. [DOI] [PubMed] [Google Scholar]
- 34. Clark AD, Layman LC. Analysis of the Cys82Arg mutation in follicle-stimulating hormone beta (FSHbeta) using a novel FSH expression vector. Fertil Steril. 2003;79(2):379–385. [DOI] [PubMed] [Google Scholar]
- 35. Kottler ML, Chou YY, Chabre O, et al. A new FSHbeta mutation in a 29-year-old woman with primary amenorrhea and isolated FSH deficiency: functional characterization and ovarian response to human recombinant FSH. Eur J Endocrinol. 2010;162(3):633–641. [DOI] [PubMed] [Google Scholar]
- 36. Matthews CH, Borgato S, Beck-Peccoz P, et al. Primary amenorrhea and infertility due to a mutation in the β-subunit of follicle-stimulating hormone. Nat Genet. 1993;5(1):83–86. [DOI] [PubMed] [Google Scholar]
- 37. Matthews C, Chatterjee VK. Isolated deficiency of follicle-stimulating hormone re-revisited. N Engl J Med. 1997;337(9):642. [DOI] [PubMed] [Google Scholar]
- 38. Berger K, Souza H, Brito VN, d'Alva CB, Mendonca BB, Latronico AC. Clinical and hormonal features of selective follicle-stimulating hormone (FSH) deficiency due to FSH beta-subunit gene mutations in both sexes. Fertil Steril. 2005;83(2):466–470. [DOI] [PubMed] [Google Scholar]
- 39. Lindstedt G, Nystrom E, Matthews C, Ernest I, Janson PO, Chatterjee K. Follitropin (FSH) deficiency in an infertile male due to FSHbeta gene mutation. A syndrome of normal puberty and virilization but underdeveloped testicles with azoospermia, low FSH but high lutropin and normal serum testosterone concentrations. Clin Chem Lab Med. 1998;36(8):663–665. [DOI] [PubMed] [Google Scholar]
- 40. Phillip M, Arbelle JE, Segev Y, Parvari R. Male hypogonadism due to a mutation in the gene for the b-subunit of follicle stimulating hormone. N Engl J Med. 1998;338(24):1729–1732. [DOI] [PubMed] [Google Scholar]
- 41. Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat. 1998;11(1):1–3. [DOI] [PubMed] [Google Scholar]
- 42. Barnes RB, Namnoum A, Rosenfield RL, Layman LC. The role of LH and FSH in ovarian androgen secretion and ovarian follicular development: clinical studies in a patient with isolated FSH deficiency and multicystic ovaries. Hum Reprod. 2002;17(1):88–91. [DOI] [PubMed] [Google Scholar]
- 43. Barnes RB, Namnoum A, Rosenfield RL, Layman LC. Effects of follicle-stimulating hormone on ovarian androgen production in a woman with isolated follicle-stimulating hormone deficiency. N Engl J Med. 2000;343(16):1197–1198. [DOI] [PubMed] [Google Scholar]
- 44. Azziz R, Carmina E, Dewailly D, et al. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91(11):4237–4245. [DOI] [PubMed] [Google Scholar]
- 45. Lofrano-Porto A, Casulari LA, Nascimento PP, et al. Effects of follicle-stimulating hormone and human chorionic gonadotropin on gonadal steroidogenesis in two siblings with a follicle-stimulating hormone beta subunit mutation. Fertil Steril. 2008;90(4):1169–1174. [DOI] [PubMed] [Google Scholar]
- 46. Wreford NG, Rajendra Kumar T, Matzuk MM, de Kretser DM. Analysis of the testicular phenotype of the follicle-stimulating hormone beta-subunit knockout and the activin type II receptor knockout mice by stereological analysis. Endocrinology. 2001;142(7):2916–2920. [DOI] [PubMed] [Google Scholar]
- 47. Baker PJ, Pakarinen P, Huhtaniemi IT, et al. Failure of normal Leydig cell development in follicle-stimulating hormone (FSH) receptor-deficient mice, but not FSHbeta-deficient mice: role for constitutive FSH receptor activity. Endocrinology. 2003;144(1):138–145. [DOI] [PubMed] [Google Scholar]
- 48. Anawalt BD, Bebb RA, Matsumoto AM, et al. Serum inhibin B levels reflect Sertoli cell function in normal men and men with testicular dysfunction. J Clin Endocrinol Metab. 1996;81(9):3341–3345. [DOI] [PubMed] [Google Scholar]
- 49. Wu N, Murono EP. A Sertoli cell-secreted paracrine factor(s) stimulates proliferation and inhibits steroidogenesis of rat Leydig cells. Mol Cell Endocrinol 1994;106(1-2):99–109. [DOI] [PubMed] [Google Scholar]
- 50. Lecerf L, Rouiller-Fabre V, Levacher C, Gautier C, Saez JM, Habert R. Stimulatory effect of follicle-stimulating hormone on basal and luteinizing hormone-stimulated testosterone secretions by the fetal rat testis in vitro. Endocrinology. 1993;133(5):2313–2318. [DOI] [PubMed] [Google Scholar]
- 51. Sinha Hikim AP, Chandrashekar V, Bartke A, Russell LD. Sentinels of Leydig cell structural and functional changes in golden hamsters in early testicular regression and recrudescence. Int J Androl. 1993;16(5):324–342. [DOI] [PubMed] [Google Scholar]
- 52. Danilovich N, Babu PS, Xing W, Gerdes M, Krishnamurthy H, Sairam MR. Estrogen deficiency, obesity, and skeletal abnormalities in follicle-stimulating hormone receptor knockout (FORKO) female mice. Endocrinology 2000;141(11):4295–4308. [DOI] [PubMed] [Google Scholar]
- 53. Tiwari-Pandey R, Yang Y, Aravindakshan J, Sairam MR. Normalization of hormonal imbalances, ovarian follicular dynamics and metabolic effects in follitrophin receptor knockout mice. Mol Hum Reprod. 2007;13(5):287–297. [DOI] [PubMed] [Google Scholar]
- 54. Whitney EA, Layman LC, Chan PJ, Lee A, Peak DB, McDonough PG. The follicle-stimulating hormone receptor gene is polymorphic in premature ovarian failure and normal controls. Fertil Steril. 1995;64(2):518–524. [DOI] [PubMed] [Google Scholar]
- 55. Aittomaki K, Lucena JL, Pakarinen P, et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82(6):959–968. [DOI] [PubMed] [Google Scholar]
- 56. Beau I, Touraine P, Meduri G, et al. A novel phenotype related to partial loss of function mutations of the follicle stimulating hormone receptor. J Clin Invest. 1998;102(7):1352–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Touraine P, Beau I, Gougeon A, et al. New natural inactivating mutations of the follicle-stimulating hormone receptor: correlations between receptor function and phenotype. Mol Endocrinol. 1999;13(11):1844–1854. [DOI] [PubMed] [Google Scholar]
- 58. Doherty E, Pakarinen P, Tiitinen A, et al. A Novel mutation in the FSH receptor inhibiting signal transduction and causing primary ovarian failure. J Clin Endocrinol Metab. 2002;87(3):1151–1155. [DOI] [PubMed] [Google Scholar]
- 59. Allen LA, Achermann JC, Pakarinen P, et al. A novel loss of function mutation in exon 10 of the FSH receptor gene causing hypergonadotrophic hypogonadism: clinical and molecular characteristics. Hum Reprod. 2003;18(2):251–256. [DOI] [PubMed] [Google Scholar]
- 60. Meduri G, Touraine P, Beau I, et al. Delayed puberty and primary amenorrhea associated with a novel mutation of the human follicle-stimulating hormone receptor: clinical, histological, and molecular studies. J Clin Endocrinol Metab. 2003;88(8):3491–3498. [DOI] [PubMed] [Google Scholar]
- 61. Kuechler A, Hauffa BP, Koninger A, et al. An unbalanced translocation unmasks a recessive mutation in the follicle-stimulating hormone receptor (FSHR) gene and causes FSH resistance. Eur J Hum Genet. 2010;18(6):656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aittomaki K, Herva R, Stenman U-H, et al. Clinical features of primary ovarian failure caused by a point mutation in the follicle stimulating hormone receptor gene. J Clin Endocrinol Metab. 1996;81(10):3722–3726. [DOI] [PubMed] [Google Scholar]
- 63. Vaskivuo TE, Aittomaki K, Anttonen M, et al. Effects of follicle-stimulating hormone (FSH) and human chorionic gonadotropin in individuals with an inactivating mutation of the FSH receptor. Fertil Steril. 2002;78(1):108–113. [DOI] [PubMed] [Google Scholar]
- 64. Layman LC, Amde S, Cohen DP, Jin M, Xie J. The Finnish follicle stimulating hormone receptor (FSHR) gene mutation in women with 46, XX ovarian failure is rare in the United States. Fertil Steril. 1998;69(2):300–302. [DOI] [PubMed] [Google Scholar]
- 65. Jiang M, Aittomaki K, Nilsson C, et al. The frequency of an inactivating point mutation (566C-->T) of the human follicle-stimulating hormone receptor gene in four populations using allele-specific hybridization and time-resolved fluorometry. J Clin Endocrinol Metab. 1998;83(12):4338–4343. [DOI] [PubMed] [Google Scholar]
- 66. Krishnamurthy H, Danilovich N, Morales CR, Sairam MR. Qualitative and quantitative decline in spermatogenesis of the follicle-stimulating hormone receptor knockout (FORKO) mouse. Biol Reprod. 2000;62(5):1146–1159. [DOI] [PubMed] [Google Scholar]
- 67. Tapanainen JS, Aittomaki K, Min J, Vaskivuo T, Huhtaniemi IT. Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nat Genet. 1997;15(2):205–206. [DOI] [PubMed] [Google Scholar]
- 68. Nieschlag E, Vorona E, Wenk M, Hemker AK, Kamischke A, Zitzmann M. Hormonal male contraception in men with normal and subnormal semen parameters. Int J Androl. 2011;34(6 pt 1):556–567. [DOI] [PubMed] [Google Scholar]
- 69. Gromoll J, Simoni M, Nieschlag E. An activating mutation of the follicle stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J Clin Endocrinol Metab. 1996;81(4):1367–1370. [DOI] [PubMed] [Google Scholar]
- 70. Haywood M, Tymchenko N, Spaliviero J, et al. An activated human follicle-stimulating hormone (FSH) receptor stimulates FSH-like activity in gonadotropin-deficient transgenic mice. Mol Endocrinol. 2002;16(11):2582–2591. [DOI] [PubMed] [Google Scholar]
- 71. Mason AJ, Hayflick JS, Zoeller RT, et al. A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science. 1986;234(4782):1366–1371. [DOI] [PubMed] [Google Scholar]
- 72. Vasseur C, Rodien P, Beau I, et al. A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. N Engl J Med. 2003;349(8):753–759. [DOI] [PubMed] [Google Scholar]
- 73. Smits G, Olatunbosun O, Delbaere A, Pierson R, Vassart G, Costagliola S. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. N Engl J Med. 2003;349(8):760–766. [DOI] [PubMed] [Google Scholar]
- 74. Olatunbosun OA, Gilliland B, Brydon LA, Chizen DR, Pierson RA. Spontaneous ovarian hyperstimulation syndrome in four consecutive pregnancies. Clin Exp Obstet Gynecol. 1996;23(3):127–132. [PubMed] [Google Scholar]
- 75. Montanelli L, Delbaere A, Di Carlo C, et al. A mutation in the follicle-stimulating hormone receptor as a cause of familial spontaneous ovarian hyperstimulation syndrome. J Clin Endocrinol Metab. 2004;89(4):1255–1258. [PubMed] [Google Scholar]
- 76. De Leener A, Montanelli L, Van Durme J, et al. Presence and absence of follicle-stimulating hormone receptor mutations provide some insights into spontaneous ovarian hyperstimulation syndrome physiopathology. J Clin Endocrinol Metab. 2006;91(2):555–562. [DOI] [PubMed] [Google Scholar]
- 77. De Leener A, Caltabiano G, Erkan S, et al. Identification of the first germline mutation in the extracellular domain of the follitropin receptor responsible for spontaneous ovarian hyperstimulation syndrome. Hum Mutat. 2008;29(1):91–98. [DOI] [PubMed] [Google Scholar]
- 78. Allan CM, Garcia A, Spaliviero J, Jimenez M, Handelsman DJ. Maintenance of spermatogenesis by the activated human (Asp567Gly) FSH receptor during testicular regression due to hormonal withdrawal. Biol Reprod. 2006;74(5):938–944. [DOI] [PubMed] [Google Scholar]
- 79. Grossman LC, Michalakis KG, Browne H, Payson MD, Segars JH. The pathophysiology of ovarian hyperstimulation syndrome: an unrecognized compartment syndrome. Fertil Steril. 2010;94(4):1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Di Carlo C, Bruno P, Cirillo D, Morgera R, Pellicano M, Nappi C. Increased concentrations of renin, aldosterone and Ca125 in a case of spontaneous, recurrent, familial, severe ovarian hyperstimulation syndrome. Hum Reprod. 1997;12(10):2115–2117. [DOI] [PubMed] [Google Scholar]
- 81. Chae HD, Park EJ, Kim SH, Kim CH, Kang BM, Chang YS. Ovarian hyperstimulation syndrome complicating a spontaneous singleton pregnancy: a case report. J Assist Reprod Genet. 2001;18(2):120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cepni I, Erkan S, Ocal P, Ozturk E. Spontaneous ovarian hyperstimulation syndrome presenting with acute abdomen. J Postgrad Med. 2006;52(2):154–155. [PubMed] [Google Scholar]
- 83. Montanelli L, Van Durme JJ, Smits G, et al. Modulation of ligand selectivity associated with activation of the transmembrane region of the human follitropin receptor. Mol Endocrinol. 2004;18(8):2061–2073. [DOI] [PubMed] [Google Scholar]
- 84. Simoni M, Gromoll J, Hoppner W, et al. Mutational analysis of the follicle-stimulating hormone (FSH) receptor in normal and infertile men: identification and characterization of two discrete FSH receptor isoforms. J Clin Endocrinol Metab. 1999;84(2):751–755. [DOI] [PubMed] [Google Scholar]
- 85. Laan M, Grigorova M, Huhtaniemi IT. Pharmacogenetics of follicle-stimulating hormone action. Curr Opin Endocrinol Diabetes Obes. 2012;19(3):220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Moron FJ, Ruiz A. Pharmacogenetics of controlled ovarian hyperstimulation: time to corroborate the clinical utility of FSH receptor genetic markers. Pharmacogenomics. 2010;11(11):1613–1618. [DOI] [PubMed] [Google Scholar]
- 87. Perez Mayorga M, Gromoll J, Behre HM, Gassner C, Nieschlag E, Simoni M. Ovarian response to follicle-stimulating hormone (FSH) stimulation depends on the FSH receptor genotype. J Clin Endocrinol Metab. 2000;85(9):3365–3369. [DOI] [PubMed] [Google Scholar]
- 88. de Castro F, Moron FJ, Montoro L, et al. Human controlled ovarian hyperstimulation outcome is a polygenic trait. Pharmacogenetics. 2004;14(5):285–293. [DOI] [PubMed] [Google Scholar]
- 89. Loutradis D, Patsoula E, Minas V, et al. FSH receptor gene polymorphisms have a role for different ovarian response to stimulation in patients entering IVF/ICSI-ET programs. J Assist Reprod Genet. 2006;23(4):177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Livshyts G, Podlesnaja S, Kravchenko S, Sudoma I, Livshits L. A distribution of two SNPs in exon 10 of the FSHR gene among the women with a diminished ovarian reserve in Ukraine. J Assist Reprod Genet. 2009;26(1):29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Klinkert ER, te Velde ER, Weima S, et al. FSH receptor genotype is associated with pregnancy but not with ovarian response in IVF. Reprod Biomed Online. 2006;13(5):687–695. [DOI] [PubMed] [Google Scholar]
- 92. Murty GS, Rani CS, Moudgal NR, Prasad MR. Effect of passive immunization with specific antiserum to FSH on the spermatogenic process and fertility of adult male bonnet monkeys (Macaca radiata). J Reprod Fertil Suppl. 1979;(26):147–163. [PubMed] [Google Scholar]
- 93. Moudgal NR, Ravindranath N, Murthy GS, Dighe RR, Aravindan GR, Martin F. Long-term contraceptive efficacy of vaccine of ovine follicle-stimulating hormone in male bonnet monkeys (Macaca radiata). J Reprod Fertil 1992;96(1):91–102. [DOI] [PubMed] [Google Scholar]
- 94. Moudgal NR, Sairam MR, Krishnamurthy HN, Sridhar S, Krishnamurthy H, Khan H. Immunization of male bonnet monkeys (M. radiata) with a recombinant FSH receptor preparation affects testicular function and fertility. Endocrinology. 1997;138(7):3065–3068. [DOI] [PubMed] [Google Scholar]
- 95. Moudgal NR, Murthy GS, Prasanna Kumar KM, et al. Responsiveness of human male volunteers to immunization with ovine follicle stimulating hormone vaccine: results of a pilot study. Hum Reprod. 1997;12(3):457–463. [DOI] [PubMed] [Google Scholar]
- 96. Meachem SJ, McLachlan RI, Stanton PG, Robertson DM, Wreford NG. FSH immunoneutralization acutely impairs spermatogonial development in normal adult rats. J Androl. 1999;20(6):756–762; discussion 755. [PubMed] [Google Scholar]
- 97. Krishnamurthy H, Kumar KM, Joshi CV, Krishnamurthy HN, Moudgal RN, Sairam MR. Alterations in sperm characteristics of follicle-stimulating hormone (FSH)-immunized men are similar to those of FSH-deprived infertile male bonnet monkeys. J Androl. 2000;21(2):316–327. [PubMed] [Google Scholar]
- 98. Ferro VA, Stimson WH. Fertility-disrupting potential of synthetic peptides derived from the beta-subunit of follicle-stimulating hormone. Am J Reprod Immunol. 1998;40(3):187–197. [DOI] [PubMed] [Google Scholar]
- 99. Butterstein GM, Sachar D, Dias JA. Immunoneutralization of heterodimeric FSH using FSH beta subunit as the immunogen. Am J Reprod Immunol. 1993;29(1):48–55. [DOI] [PubMed] [Google Scholar]