

Abstract
Autism spectrum disorder (ASD) is a highly heritable complex neurodevelopmental condition characterized by impairments in social interaction and communication and restricted and repetitive behaviors. Although roles for both de novo and familial genetic variation have been documented, the underlying disease mechanisms remain poorly elucidated. In this study, we defined and explored distinct etiologies of genetic variants that affect genes regulated by Fragile-X mental retardation protein (FMRP), thought to play a key role in neuroplasticity and neuronal translation, in ASD-affected individuals. In particular, we developed the Trend test, a pathway-association test that is able to robustly detect multiple-hit etiologies and is more powerful than existing approaches. Exploiting detailed spatiotemporal maps of gene expression within the human brain, we identified four discrete FMRP-target subpopulations that exhibit distinct functional biases and contribute to ASD via different types of genetic variation. We also demonstrated that FMRP target genes are more likely than other genes with similar expression patterns to contribute to disease. We developed the hypothesis that FMRP targets contribute to ASD via two distinct etiologies: (1) ultra-rare and highly penetrant single disruptions of embryonically upregulated FMRP targets (“single-hit etiology”) or (2) the combination of multiple less penetrant disruptions of nonembryonic, synaptic FMRP targets (“multiple-hit etiology”). The Trend test provides rigorous support for a multiple-hit genetic etiology in a subset of autism cases and is easily extendible to combining information from multiple types of genetic variation (i.e., copy-number and exome variants), increasing its value to next-generation sequencing approaches.
Introduction
Autism spectrum disorder (ASD [MIM 209850]) is a neurological early-onset condition characterized by restricted interests, repetitive behavior, and impairments in social communication; it affects ∼1% of the population. The etiology of ASD still remains to be elucidated, although twin and family studies have shown that ASD is highly heritable1 at around 80%.2 Linkage, association, and copy-number variant (CNV) studies have identified a role for both de novo and familial variation.3–5 The list of ASD candidate genes is steadily increasing, but no single locus accounts for >1% of cases.6
Although ASD is predominantly a complex disorder, approximately 5% of cases are due to a high comorbidity with the monogenic Fragile-X syndrome (FXS [MIM 300624]), the most common single-gene defect associated with ASD.7 FXS is caused by a loss of function of Fragile-X mental retardation protein (FMRP),8 a neuronal and gonadal protein with key roles in neuroplasticity and neuronal translation.9 Under normal function, FMRP has been reported to negatively regulate translation by stalling ribosomal translocation across the mRNA of 842 genes (herein termed “FMRP targets”), including a significantly large number of previously identified ASD candidates genes.10 A recent exome sequencing study by Iossifov et al.11 found that genes affected by exonic de novo nonsense, splice-site, and frameshift mutations identified in ASD probands are significantly enriched in FMRP targets; the authors replicated the enrichment in an analogous ASD candidate gene set compiled by three other exome sequencing studies.12–14 However, the role of FMRP regulation has yet to be fully elucidated; genes targeted by FMRP play diverse roles in both embryonic and adult neurogenesis and in synapse structure and function.15,16 Thus, the causal relationship between the dysregulation of the 842 FMRP targets in FXS and the general pathology of ASD is unclear: whether all FMRP target genes causally contribute to ASD and whether those that do act through common or diverse mechanisms remain unknown. Other previous pathway analyses of ASD have separately implicated synaptic genes17–21 and chromatin modifiers,12 as well as embryonic transcription regulators.22 These separate studies have focused on particular types of genetic variation, whereas a holistic model of genetic causality in ASD within which these different types of variation can be interpreted has not yet been determined.
We hypothesized that different classes of sequence variants contribute to ASD to varying extents. Thus, we considered the role played by FMRP targets in ASD across the spectrum of genetic variation by exploiting data from SNPs, copy-number variants (CNVs), and disrupting single-gene de novo mutations (Table S1, available online). Crucially, whereas de novo gene disruptions might be singularly sufficient to cause ASD, variants inherited from unaffected parents are necessarily of incomplete penetrance. In particular, the presence of multiple CNVs in many individuals with ASD23,24 suggests that inherited variation might lead to ASD through cumulative effects of deleterious variants affecting a relevant biological pathway (a “multiple-hit” disease mechanism). This hypothesis is also supported by the observed influence of background variation on phenotypes in model organisms25 and could explain the phenotypic heterogeneity observed for variants associated with a range of neuropsychiatric phenotypes.24
In this systematic analysis of FMRP targets in ASD, we developed and explored the hypothesis that different groups of FMRP targets contribute to ASD in distinct ways; we did this most notably by distinguishing those genes affected by highly penetrant, de novo gene disruptions that tend to contribute via a single-hit etiology from those genes affected by less penetrant, inherited variants that tend to contribute via a multiple-hit etiology. First, we showed that comprehensive expression data from human brains reveal subgroups (modules) of FMRP targets with distinct spatiotemporal expression patterns and biological roles. Second, we examined the contribution of individual FMRP target modules to the overlap with ASD candidate genes on the basis of de novo single-gene disruptions. Third, we developed an approach to conducting gene-set analysis on the basis of gene disruptions found in CNV case-control data sets. Importantly, our approach explicitly considers the contribution of multiple gene disruptions in each individual and is demonstrably more powerful than approaches applied previously. Using this approach, we then examined the disruptions of FMRP target modules in rare CNVs and considered the association between SNPs located in FMRP targets and ASD diagnosis. Finally, we investigated whether the association between FMRP targets and ASD can be extended to genes with brain expression patterns similar to those of specific FMRP target subgroups and demonstrated that FMRP target genes are more likely to cause disease.
Material and Methods
Ethics Statement
Institutional-review-board approval and informed consent were given for all data sets in previously published papers.
De Novo Single-Gene Disruptions in ASD Data
Lists of genes disrupted by de novo nonsense, frameshift, or splice-site point mutations in autism probands were obtained from Iossifov et al.11 (59 genes; referred to as “I-exomes”) and three other recent studies by Sanders et al.,12 O’Roak et al.,13 and Neale et al. 14 (65 genes combined from all three; referred to as “SON-exomes”) (Table S1, available online). A list of genes disrupted by breakpoints of balanced chromosomal abnormalities (BCAs) observed in individuals with ASD was obtained from Talkowski et al.26 (32 genes; referred to as “T-BCAs”) (Table S1).
National Blood Service CNV Data
The CNV data for the National Blood Service (NBS) cohort were provided by the Wellcome Trust Case Control Consortium 1 (WTCCC1,27 09/02/09 release) after quality control, and CNVs were called from an Affymetrix 500k chip with SWArray (1,343 individuals). We only considered deletions ≥ 1 kb, with at least three probes, and with a confidence score ≥ 0.2 (false-discovery rate [FDR] was estimated at 22% by the WTCCC1; note that we only used this data set for simulations and therefore did not require very high call accuracy).
ASD CNV Data
All rare deletion CNVs that passed quality control were taken from Sanders et al.;28 we compared 872 probands to their matched unaffected siblings and compared 1,124 probands to their 2,248 parents (Table S1). Moreover, all rare deletion CNVs from ASD probands and unaffected controls passing quality controls from an Autism Genome Project (AGP) study29 were provided by the AGP Consortium (Table S1). In the AGP set, individuals with CNVs were subdivided into two groups on the basis of whether they met the strict criteria for ASD diagnosis (autism was diagnosed according to both the Autism Diagnostic Observation Schedule30 and the Autism Diagnostic Interview, Revised31); to consider only “strict autism” cases in our analysis, we did not include individuals without CNVs. Following the original definitions, Sanders et al. classified a CNV as rare if ≤50% of its length overlapped regions present at >1% frequency in the Database of Genomic Variants of March 2010;28 for the AGP CNV data set, a CNV was classified as rare if it was present in <1% of the AGP total sample.29
ASD SNP Data
We obtained access to an autism family-based genotype data set (“CHOP_cleaned”) with partial quality control from the Autism Genetic Resource Exchange (AGRE). A subset of the data was utilized in a genome-wide association study (GWAS) by Wang et al.,32 and we tried to match their filtering steps as closely as possible (see Appendix A). The final set consisted of 472,487 autosomal SNPs, 1,334 cases, and 1,764 controls (Table S1). From the transmission disequilibrium test for all autosomal SNPs, the genomic control was calculated as 1.04, matching the value from Wang et al.;32 the p values for the most associated SNPs were also similar to those calculated by Wang et al.32 (Table S2).
FMRP Targets
Brain Specificity
We obtained expression levels for 17,226 human genes with Ensembl gene IDs33,34 from the GNF2 expression atlas35 and further considered 4 fetal and 31 adult tissues (Table S3). For each gene G, we calculated the ratio of expression in the brain to the median of all tissues separately for fetal and adult tissues and obtained the brain-specificity ratios BrainSpecfetal(G) and BrainSpecadult(G), respectively. We used logistic regression to determine whether BrainSpecadult or being an FMRP target (indicator variable) had significant effects on the probability that a gene would be found as disrupted in ASD in I-exomes, SON-exomes, and T-BCAs from above, as well as in a combined list of the three. The analysis was repeated for BrainSpecfetal instead of BrainSpecadult.
Coding-Sequence Length
We obtained the coding-sequence (CDS) length of all protein-coding genes with Ensembl gene IDs from Ensembl mart (downloaded June 17, 2013). Each gene was assigned the maximum CDS length of its transcripts.
Coexpression and Temporal Specificity
Normalized gene expression data determined by RNA sequencing and representing 16 human brain regions were obtained via BrainSpan (downloaded May 8, 2012; 41 individuals). A total of 14,886 genes with Ensembl gene IDs and at least one read per kilobase per million (RPKM) in ≥95% of the samples were kept; these included 832 of the 842 reported FMRP targets.10 We used the R package WGCNA (weighted correlation network analysis)36 according to the procedure (including parameterization) recommended by the authors to cluster the 832 FMRP targets according to their expression patterns.
Six developmental stages were defined as suggested by the experimental proceedings of BrainSpan. For each gene, the expression level at a developmental stage was calculated as the median expression level across all samples (individuals and brain areas) from that time period. We noticed that the median expression during the stage of “childhood” was generally lower for all human genes, and we accounted for this by increasing median expression by a constant factor (see Appendix A).
Functional Enrichment
A total of 7,271 GO_BP_FAT (Gene Ontology [GO] Biological Processes) and 1,031 GO_CC_FAT (GO Cellular Compartment) gene sets (“pathways”) were downloaded from the Database for Annotation, Visualization, and Integrated Discovery (DAVID)37 (on June 15, 2012). To reduce multiple-testing and uninformative results,38 we further considered only 262 pathways that contained ≥250 genes.
Insights into a gene’s function can also be gained by consideration of the phenotypes that result from the disruption of that gene’s unique ortholog in a model organism.39,40 We proceeded as did Shaikh et al.41 In brief, we obtained the phenotypes exhibited by mouse models possessing a targeted disruption of a protein-coding gene from Mouse Genomics Informatics (MGI; downloaded November 16, 2011). The annotations consist of 30 general (overarching) phenotypes with finer terms in multiple hierarchical levels.42 Using 1:1 orthology between mouse and human genes defined by MGI, we mapped all mouse phenotypes to the human ortholog genes to obtain mouse phenotype assignments for 6,401 human genes. For each phenotype, the assigned human genes were referred to as a “pathway.” To reduce uninformative results, we only considered subphenotypes assigned to at least 1% of the genes of the overarching phenotype.38
Haploinsufficiency
Huang et al.43 reported the probability of haploinsufficiency for 12,218 genes with Ensembl gene IDs. We defined haploinsufficient (“HIS”) genes by applying a probability cutoff of ≥0.5 (3,362 genes). We confirmed pairwise differences in the distribution of HIS probabilities between gene sets (Figure S1).
Testing Differences between Gene Lists
For functional annotations and HIS genes, enrichment among a gene list was tested with a one-sided hypergeometric test, using a 5% Benjamini-Hochberg FDR for each pathway list. GO and MGI enrichments among FMRP modules were tested with all FMRP targets as the background; otherwise, the background consisted of all annotated genes. Differences in annotations between two gene lists were tested with a two-sided Fisher’s test. Differences in distributions of haploinsufficiency probabilities and brain specificity (separately for fetal and adult tissues) between two gene lists were tested with a two-sided Mann-Whitney U-test in R (v.2.13.0).
Variation in the Human Population
Single-nucleotide variants discovered by exome sequencing were downloaded from the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (ESP5400 release). Gene symbols were mapped to Ensembl gene IDs; only exonic variants in protein-coding genes found in individuals of European ancestry were considered (yielding variants for 17,846 genes; 279 genes in FMRP module 1 and 235 in module 2). For each gene, we calculated the proportion of variants that had a minor allele frequency (MAF) < 0.02% (“ultra-rare”), 0.02%–1% (“rare-not-ultra-rare”), or >1% (“common”). For each of the frequency categories, the pairwise differences in distribution of proportions between gene sets was calculated with a Mann-Whitney U-test in R and corrected for multiple testing at 5% FDR. In the second step, we repeated all calculations by considering only those variants classified as missense mutations, splice-site mutations, or mutations introducing or removing stop codons. Finally, we repeated the calculations for variants assigned a PolyPhen44 score of “probably damaging” or “possibly damaging” (contained in the downloaded data).
Methods for CNV Pathway Analysis
Trend and Fisher’s Tests for CNVs
For each individual, CNV coordinates in NCBI36 were compared to gene-transcript locations from Ensembl mart 54.33,34 Conservatively, a gene was defined as “hit” if a CNV overlapped at least one exon for every transcript of that gene. A pathway was defined as “hit” if at least one gene from the pathway was hit by CNVs in that individual. A pathway was defined as “hit h times” in an individual if h genes in that pathway were hit by CNVs in that individual. A Fisher’s test statistic was obtained from a two-sided exact Fisher’s test on the numbers of cases and controls with and without hits. We developed the Trend test as a Cochran-Armitage test for trend in the number of cases and controls with h hits as h increased (after pooling numbers when there were too few observations for large h, see Appendix A); we weighted observations for h hits by (see Appendix A).
Matching Random Gene Sets to a Pathway
It is crucial to account for differences in CNV burden between cases and controls to avoid false-positive pathway associations.45 We chose to account for differences in general CNV burden between cases and controls through randomizations and obtained an empirical Trend test p value for a pathway from random gene sets matched for the number of genes and the gene lengths. To match by gene length, we divided all human protein-coding genes (21,219 downloaded from Ensembl mart 54) by their gene length into 100 bins, b(1), ..., b(100), of equal size. If a pathway P had p(1), ... p(100) genes from the respective bins, we obtained a random gene set by sampling p(i) genes without replacement from bins i = 1, ..., 100.
Test for Risk Conferred by Multiple Hits
We noted that a significant association from the Trend test could also result from a single-hit disease model and would therefore not be sufficient to show a multiple-hit model of disease. Consequently, we developed the following testing procedure for pathways with significant results from the Trend test and the occurrence of multiple hits in several individuals. For a given case-control data set and a pathway P of interest, we calculated the numbers of cases and controls with h hits, h = 1, ..., m (after pooling hits with too few observations, see Appendix A). We then defined the sequence (R′ (h))h = 1,...,m as for h = 1, ..., m. A linear model was fitted to (R′ (h))h = 1,...,m on the basis of an intercept and the number of hits h. If the regression coefficient C for the number of hits was significant at single-test level, we obtained a one-sided empirical p value by calculating the proportion of 10,000 random pathways matched to P for which the coefficient C(rand) for the number of hits was also significant at single-test level and at least as large as C.
ASD De Novo Single-Gene Mutation Enrichment in FMRP Targets
We tested the enrichment of all FMRP targets and the four FMRP modules in the genes found disrupted in I-exomes, SON-exomes, and T-BCAs (Table S1) by using the right-tailed hypergeometric test and a conservative estimate of 15,000 genes as the background. Multiple testing for FMRP modules was corrected at 5% FDR for each data set. We also considered the pairwise overlap of I-exomes, SON-exomes, and T-BCAs: no overlap existed between T-BCAs and I-exomes, and no FMRP target was in both I-exomes and SON-exomes. To account for the overlap of T-BCAs and SON-exomes, we also performed an enrichment test for T-BCAs in which the genes found in SON-exomes were excluded. We also applied logistic regression to account for CDS length and (1) adult-brain specificity and (2) fetal-brain specificity when testing the enrichment of all FMRP targets. Additionally, we applied logistic regression to account for CDS length when analyzing the association of FMRP modules (because neither brain-specificity index was significantly associated with I-exomes, SON-exomes, or T-BCAs, we did not include brain specificity in this analysis).
ASD Trend-Test CNV Pathway Analysis Using FMRP Modules
We used the Trend test to examine the role of all FMRP targets and modules 1–4 in rare deletion CNVs in ASD. ASD probands and their unaffected siblings from Sanders et al.28 were used as a discovery cohort; the expanded set of probands and their parents were used for further validation. AGP “strict autism” cases and controls were used as a final replication sample.29 Empirical p values were obtained with 10,000 random gene sets matched to a pathway of interest as described above. Multiple testing for FMRP modules was corrected at 5% FDR for each data set.
ASD SNP Pathway Analysis Using FMRP Modules
On the basis of the AGRE SNP data set, for FMRP modules 1–4, we assigned each autosomal SNP to a module if it was located between the gene start and end based on GRCh37 (Ensembl). For each module, we then carried out a set-based association test in Plink to obtain empirical p values for the average chi-square statistic for each module from 100,000 permutations of case-control status. To account for linkage disequilibrium, we only took into account SNPs considered independent under an r2 threshold of 0.5; as the only significant module, module 2 was also tested with r2 thresholds 0.2 and 0.8. Multiple testing for FMRP modules was corrected at 5% FDR.
Extension of FMRP Modules
We aimed to find non-FMRP target genes with expression patterns similar to FMRP modules. Using BrainSpan data and the WGCNA R package “eigengene” function, we obtained a representative brain expression pattern, the “eigengene,” for each FMRP module. We then calculated the correlation between each FMRP target and the eigengene of its module. Subsequently, we obtained a set of 938 “module-1-like” genes as those non-FMRP target genes whose expression pattern in BrainSpan was at least as correlated with the FMRP module 1 eigengene as that of 50% of module 1 genes. Analogously, we obtained 339 module-2-like genes.
Functional annotations from MGI and HIS probabilities were obtained for the module-like genes as above for the FMRP modules. Enrichments of module-like genes among genes disrupted by de novo mutations in ASD were tested with the joint list of I-exomes, SON-exomes, and T-BCAs. For an FMRP module, Fisher’s test was used to test for a difference between the numbers of module genes and module-like genes disrupted by de novo mutations in ASD. Similarly, for rare deletion CNVs, the association between the module-like gene sets and ASD was calculated with the Trend test as for the FMRP modules.
Results
FMRP Targets in ASD De Novo Single-Gene Disruptions
A role for FMRP targets in ASD has recently been proposed by Iossifov et al.11 on the basis of a significant enrichment among genes disrupted by deleterious de novo point mutations in ASD probands. To initially confirm, before refining, the role played by FMRP targets in ASD, we first obtained two lists of genes identified from exome studies as disrupted by particularly deleterious de novo mutations (nonsense, frameshift, and splice-site variants) within ASD probands: more specifically, the I-exomes from Iossifov et al.11 and the SON-exomes from Sanders et al.,14 O’Roak et al.,12 and Neale et al.13 (see Material and Methods, Table S1). We confirmed the enrichment of FMRP targets among both I-exomes (4.5-fold, p = 6.07 × 10−7) and SON-exomes (3.6-fold, p = 5.29 × 10−5). We additionally considered the T-BCAs from Talkowski et al.26 (see Material and Methods, Table S1) and again found an enrichment in FMRP targets (4.4-fold, p = 0.0003).
Because ASD is a neurodevelopmental disorder and FMRP has been suggested to play a prominent role in the brain,10 we also used logistic regression to test whether FMRP targets are enriched among the ASD de novo single-gene disruptions while accounting for relative brain expression levels (see Material and Methods, Table S4). Moreover, we simultaneously accounted for the CDS length of genes, given that FMRP targets tend to be long genes and such genes can be more often mutated by chance. We indeed found that CDS length significantly influenced the probability of a gene’s being disrupted by de novo single-point mutations in ASD. By contrast, on the basis of tissue expression from the GNF2 human atlas (see Material and Methods), relative expression levels in neither the fetal nor the adult brain were significantly associated. After accounting for both CDS length and relative gene expression levels in the adult brain, we found that being targeted by FMRP significantly increased the probability that a gene would occur in I-exomes (β = 1.84, p = 7.85 × 10−9), SON-exomes (β = 1.80, p = 8.23 × 10−8), T-BCAs (β = 2.06, p = 2.58 × 10−6), and their combination (β = 1.62, p = 9.61 × 10−13) (Table S4). We repeated the analysis by using fetal tissue expression with very similar results: being targeted by FMRP significantly increased the probability that a gene would occur in I-exomes (β = 1.81, p = 4.97 × 10−9), SON-exomes (β = 1.66, p = 6.31 × 10−7), T-BCAs (β = 1.86, p = 7.69 × 10−6), and their combination (β = 1.53, p = 1.79 × 10−12) (Table S4). Thus, we broadened the enrichment of FMRP targets in ASD de novo single-gene disruptions and rejected the hypothesis that high relative brain expression or high CDS length explains the implication of FMRP targets in ASD.
Four Modules of FMRP Targets
Given that many genes causally implicated in ASD are not FMRP targets, we asked whether those FMRP targets that might contribute to ASD are drawn generally from the set of all FMRP targets or whether subsets of FMRP targets with particular functions relevant to ASD etiology are preferentially disrupted. Given the role of FMRP in neuronal translational regulation, we examined whether the targets of FMRP regulation might form distinct subgroups on the basis of their differential regional and longitudinal expression within the human brain.
Clustering the 832 FMRP targets with available data within a weighted gene coexpression network constructed from highly detailed maps of gene expression in the brain (BrainSpan;46 see Material and Methods) revealed four robust gene modules with distinct spatiotemporal expression patterns and functional biases (Figure 1). In particular, the two largest modules, module 1 (287 genes) and module 2 (230 genes), showed differential temporal expression: whereas genes in module 1 tended to be specifically upregulated during fetal development, genes in module 2 were generally upregulated in adolescence and adulthood. The relatively small numbers of genes in modules 3 and 4 showed expression patterns with relatively more moderate temporal variation than observed for module 1 or 2 (Figure 1A). Genes in module 3 (130 genes) were generally more constantly expressed, whereas genes in module 4 (120 genes) tended to be upregulated during fetal development but were still relatively more highly expressed during other stages than genes in module 1.
Figure 1.
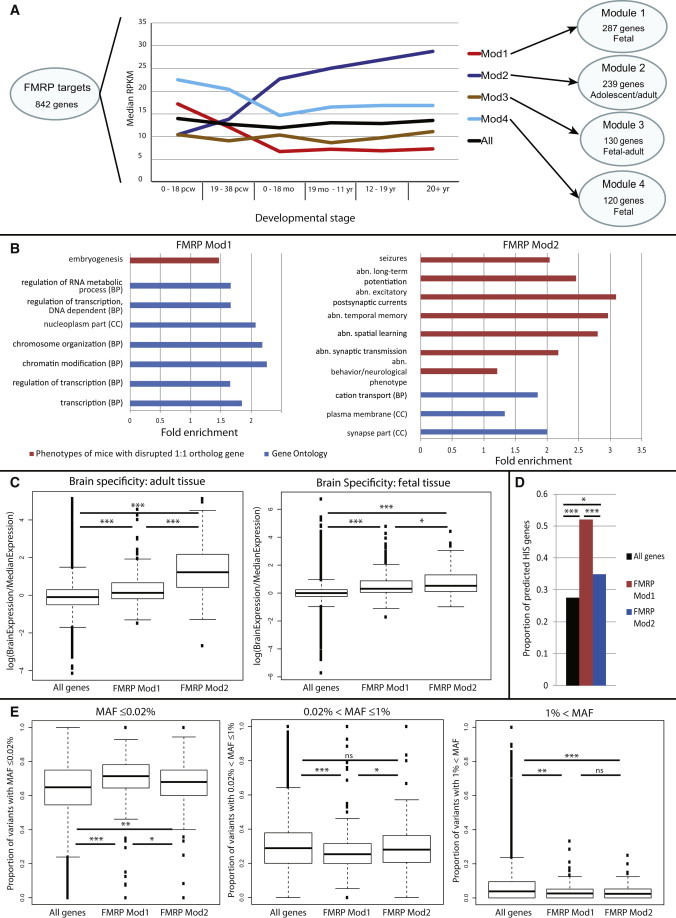
Subpopulations among FMRP Target Genes
(A) Brain temporal expression patterns across six developmental stages among the four identified subpopulations of FMRP targets (modules 1–4). Expression data from BrainSpan are shown (see Material and Methods). The following abbreviations are used: pcw, postconceptional weeks; mo, months; and yr, years.
(B) Top functional enrichments for FMRP modules 1 (Mod1) and 2 (Mod2). All enrichments shown are relative to all FMRP targets and significant at 5% FDR.
(C) Specificity of the expression of all FMRP target and module genes to the adult and fetal brain. Differences are significant at ∗p < 0.05 or ∗∗∗p < 0.0001. Error bars represent the most extreme point with a distance from the box ≤ 1.5-fold the interquartile range.
(D) Proportion of all FMRP target and module genes predicted to be HIS are enriched among FMRP modules 1 and 2. Differences are significant at ∗p < 0.05 or ∗∗∗p < 0.0001.
(E) Differences in the proportion of nonsynonymous ultra-rare (MAF < 0.02%), rare (0.02% ≤ MAF ≤ 1%), and common (MAF > 1%) variants between all FMRP targets and module genes. ns = not significant. ∗p < 0.05, ∗p < 0.001, ∗∗∗p < 0.0001. Error bars represent the most extreme point with a distance from the box ≤ 1.5-fold the interquartile range.
To ascribe function to these modules’ genes, we considered GO Biological Process and Cellular Component annotation terms,47 as well as the phenotypes that arise after the disruption of the 1:1 mouse ortholog of each of these genes (provided by MGI;42 see Material and Methods). Conservatively comparing modules to a background of all FMRP targets, we found that module 1 was significantly enriched with genes annotated with terms relating to embryogenesis, transcriptional regulation, and chromatin organization (Figure 1B and Table S5), whereas module 2 was significantly enriched with genes annotated by terms relating to synaptic function and seizures (Figure 1B and Tables S6 and S7). No significant functional enrichments were observed for the two smaller modules, modules 3 and 4. Given the opposing expression patterns and distinct functional biases of modules 1 and 2, as well as the absence of clear functional biases in modules 3 and 4, we took forward only modules 1 and 2 for further analyses as exemplars of the diversity of FMRP targets. Nonetheless, we considered and statistically accounted for the other modules throughout and return to considering them further in the Discussion. Together, the genes in modules 1 and 2 account for 517/842 (61%) of all FMRP targets.
Given the differing functional biases of FMRP module 1 and 2 genes, we compared the brain specificity of the genes within each of these modules by using data from the GNF2 gene expression atlas (Figure 1C). For both adult and fetal tissues, we found that genes within module 2 were more specific to the brain than those in module 1 (Mann-Whitney U-test, adult p < 2.2 × 10−16, fetal p = 0.007) and that both modules 1 and 2 had higher relative expression in the brain than did the background of all genes (Mann-Whitney U-test, for all comparisons p < 1 × 10−14). These findings are in good agreement with the functional biases and together infer a more general neural developmental role for module 1 genes and more specific synaptic roles for module 2 genes.
To examine the likely deleteriousness of mutations in module 1 and 2 genes, we first considered their probabilities of being HIS. On the basis of predictions of haploinsufficiency by Huang et al.,43 we found significant enrichments of predicted HIS genes in both module 1 (1.9-fold, p = 2.64 × 10−15) and module 2 (1.3-fold, p = 0.02; Figure 1D). Moreover, module 1 had a significantly higher proportion of predicted HIS genes than did module 2 (1.5-fold, Fisher’s exact test, p = 6 × 10−4). Confirming the likely deleteriousness of mutations in these genes, we found that genes from modules 1 and 2 had a significantly smaller proportion of common variation (MAF ≥ 1%) in the general population than did the background of all genes (Mann-Whitney U-test, p = 1.6 × 10−4 and p = 0.025, respectively; NHLBI Exome Variant Server data, see Material and Methods and Table S8). Notably, genes in module 1 had a significantly higher proportion of ultra-rare variants than did those in module 2 (MAF < 0.02%; Mann-Whitney U-test, p = 7.8 × 10−6), whereas genes in module 2 had a significantly higher proportion of rare-not-ultra-rare variants than did those in module 1 (0.02% ≤ MAF < 1%; Mann-Whitney U-test, p = 9.4 × 10−5). Our conclusions remained when we restricted our analysis to variants more likely to damage protein function (missense and splice-site mutations, change of stop codons) (Figure 1E and Table S8). The observations that variants in module 1 tended to be rarer than those in module 2 and that module 1 had a higher proportion of HIS genes than did module 2 indicate that module 1 genes were subject to stronger negative selection than module 2 genes.
Taken together, our findings infer that mutations disrupting the functioning of genes from both module 1 and module 2 are likely to be phenotypically consequential but that mutations affecting module 1 genes are likely to be more phenotypically consequential than those affecting module 2 genes.
ASD De Novo Single-Gene Disruptions Preferentially Affect Genes in the Embryonically Upregulated FMRP Module 1
Having characterized FMRP modules 1 and 2, we wanted to investigate to what extent they contribute to the enrichment of FMRP targets among genes disrupted by deleterious de novo point mutations in ASD probands. On the basis of the lists of variants from I-exomes, SON-exomes, and T-BCAs as above (Table S1), we found that the enrichment of FMRP targets was mainly due to highly significant enrichments of module 1 genes (I-exomes: 5.3-fold, p = 9 × 10−4; SON-exomes: 5.6-fold, p = 2 × 10−4; T-BCAs: 9.8-fold, p = 5.2 × 10−5; Figure 2 and Table S9). By contrast, FMRP module 2 was not significantly enriched with I-exomes (2.12-fold, p = 0.24) and had no overlap with SON-exomes or T-BCAs. As for all FMRP targets, we also used logistic regression to account for the CDS length of genes (see Material and Methods). We found that module 1 remained significantly associated with ASD de novo gene disruptions in all three lists of I-exomes, SON-exomes, and T-BCAs after CDS length had been accounted for (Table S10).
Figure 2.
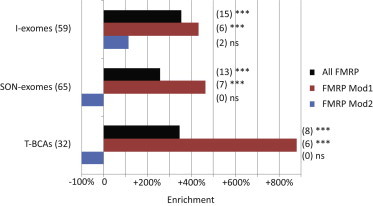
De Novo Single-Gene Disruptions in ASD Are Significantly Enriched in FMRP Targets, Particularly in Module 1 Genes
Gene numbers are in parentheses. ns = not significant at 5% FDR. ∗∗∗p < 0.0001.
For module 1, taken together with a high probability of haploinsufficiency and the rarity of variation in the general population as described above, the disruptions of genes in this more embryonically expressed module of FMRP targets provide strong evidence of these genes’ role in ASD via highly penetrant, single-gene de novo mutations.
CNV Pathway Analysis for Both Single- and Multiple-Hit Etiologies
To examine the disruption of FMRP target genes by rare deletion CNVs in ASD, we devised a test sensitive to the number of FRMP targets disrupted within each individual. In contrast to the de novo mutations considered above, the vast majority of the CNVs considered here are inherited28,29,48 and thus by themselves are unlikely to cause ASD. Nevertheless, such CNVs could contribute to the ASD phenotype through environmental interactions or, as reasoned in the Introduction, through the cumulative effects of multiple genetic variants. Currently, gene-set (“pathway”) analyses based on case-control CNV data have predominantly employed one of two approaches. Each of these approaches considers genes that are affected (“hit”) by CNVs by comparing either (1) the number of pathway genes that are hit in cases against the number of the same pathway genes hit in controls17 or (2) the proportion of cases with at least one hit in the pathway to the equivalent proportion of controls (“Fisher’s test”29).
The first approach does not take into account how many individuals contribute the hit genes, whereas the second method does not consider the number of genes hit in the pathway. To account for both the number of individuals with a pathway hit by CNVs and the number of times a pathway is hit within each individual, we devised the Trend test, which is suitable for both single- and multiple-hit disease etiologies. In brief, we tested whether the ratio of cases to controls with a number of pathway hits h varies as the number of hits h increases (see Material and Methods for further details). In a first step, we compared the performance of this Trend test to the currently applied Fisher’s test. Robustness and false-positive rates for the Trend test and Fisher’s test were investigated with extensive simulations for two data sets and five pathways of different sizes (see Appendix A). Although we found that type-I-error rates for both tests tended to be conservative, Fisher’s test was generally less sensitive than the Trend test (Figure 3). Notably, at each significance level, estimates of false-positive rates for both tests decreased with pathway size and became less robust (Figure 3).
Figure 3.
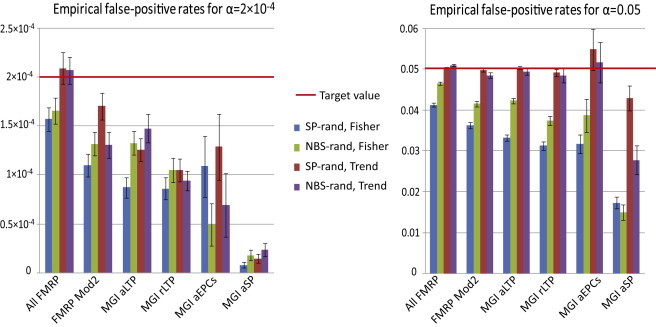
Trend and Fisher’s Test Comparison Using Empirical False-Positive Rates for Two Randomized Data Sets and Multiple Pathway Sizes
Simulations were performed for randomized case-control data sets on the basis of SandersParents (SP-rand) and NBS (NBS-rand) data sets. Random gene sets were matched to five pathways of different sizes: all FMRP targets (842 genes), FMRP module 2 (mod2) (239 genes), MGI abnormal long-term potentiation (aLTP) (187 genes), MGI reduced long-term potentiation (rLTP) (122 genes), MGI abnormal excitatory postsynaptic currents (aEPCs) (104 genes), and MGI abnormal synaptic plasticity (aSP) (42 genes). Error bars represent the SE for estimates from 100 random gene sets.
Comparing the power of the Trend test to that of Fisher’s test for a variety of scenarios, we found that even under a single-hit disease model, the Trend test was more powerful for the scenarios of interest (see Appendix A, Table S11). Whereas the power of Fisher’s test did not change under a multiple-hit model, the Trend test’s explicit modeling of this phenomenon resulted in a further increase in power.
Inherited Variation in ASD Is Associated with the Postnatally Upregulated FMRP Module 2
Using our Trend test and considering rare deletion CNVs and SNPs (Table S1), we asked whether FMRP targets and, separately, the target genes in module 1 or 2 play a role in inherited variation in ASD. In order to account for any variation in mutational burden between cases and controls, we deployed a method to calculate empirical p values by comparing results obtained from randomized pathways that we matched in both gene number and gene size (Figure S2; see Material and Methods).
First, we compared the gene disruptions caused by CNVs in 872 ASD probands to those disruptions in their matched unaffected siblings from Sanders et al.28 (Table 1A). We found that the probands had significantly more disruptions of all FMRP targets (Trend test p = 0.0011; empirical p = 0.0016) and of module 2 genes in particular (Trend test p = 0.0012; empirical p = 0.0017). We replicated this in the expanded set of 1,124 probands compared to their parents from Sanders et al.28 (FMRP targets: Trend test p = 0.0039, empirical p = 0.019; module 2: Trend test p = 8 × 10−4, empirical p = 0.0042; Table 1B). Finally, we replicated the result in an independent data set of 561 “strict autism” probands and 1,146 unrelated unaffected controls from the AGP29 (FMRP targets: Trend test p = 2.8 × 10−5, empirical p = 9 × 10−4; module 2: Trend test p = 0.0014, empirical p = 0.01; Table 1C). These results affirm with replication that rare deletion CNVs in ASD probands give rise to significantly more disruptions of FMRP targets, particularly genes in the postnatally upregulated module 2, than do rare deletion CNVs in their parents, their unaffected siblings, and the general control population.
Table 1.
The Trend Test Shows that Disruptions of FMRP Targets and Module 2 Genes, Particularly by Rare Deletion CNVs, Are Significantly Associated with ASD
| Gene Set | Trend Test p Value | Cases with Hit | Controls with Hit | Empirical p Value |
|---|---|---|---|---|
| (A) ASD Matched Probands versus Siblings (Sanders) | ||||
| Module 2a | 0.0011 | 23 | 6 | 0.0016b |
| All FMRP targetsa | 0.0012 | 54 | 29 | 0.0017b |
| Module 1 | 0.0066 | 23 | 8 | 0.0080b |
| (B) ASD Probands versus Parents (Sanders) | ||||
| Module 2a | 0.0008 | 26 | 21 | 0.0042b |
| All FMRP targetsa | 0.0039 | 68 | 89 | 0.0193b |
| Module 1 | 0.0514 | 29 | 36 | NA |
| (C) ASD Strict Cases versus Controls (AGP) | ||||
| Module 2a | 0.0014 | 8 | 2 | 0.0134b |
| All FMRP targetsa | 2.76 × 10−5 | 20 | 9 | 0.0009b |
| Module 1 | NA | NA | NA | NA |
A “hit” is defined as a rare deletion CNV overlapping a FMRP target or module gene such that at least one exon from every transcript is affected. Empirical p values were obtained from 10,000 random gene sets matched for gene number and length (see Material and Methods). Gene sets with significant results in (A) were validated in (B); likewise, results validated in (B) were tested for replication in (C). See Table S12 for modules 3 and 4. The following abbreviation is used: NA, not applicable.
The empirical p value for a gene set is significant for (A)–(C) at 5% FDR (Benjamini-Hochberg).
Significant at 5% FDR (Benjamini-Hochberg).
We sought to formally test whether the risk of ASD for an individual increases with the number of FMRP-target disruptions by rare deletion CNVs he or she possesses. Using regression and calculating empirical p values, which account for variation in mutational burden in cases (see Material and Methods; Figure 4), we found that an increased number of hits significantly increased the risk of ASD for all FMRP targets in the Sanders et al. proband-sibling cohort (empirical p = 0.02), the Sanders et al. proband-parent cohort (empirical p = 0.0027), and the AGP “strict ASD” proband-control cohort (empirical p < 1 × 10−4). Similarly, we found that multiple disruptions of FMRP module 2 genes significantly increased the risk of ASD in the Sanders et al. proband-parent cohort (empirical p = 0.01); however, the clearly visible trend in the Sanders et al. proband-sibling cohort was not significant (Figure 4), and no multiple hits in module 2 were observed in the AGP “strict ASD” proband-control cohort. Notably, the Sanders et al. proband-parent cohort was the largest of the three cohorts we examined, so a lack of power in the two smaller cohorts could explain why significance was not reached for them.
Figure 4.
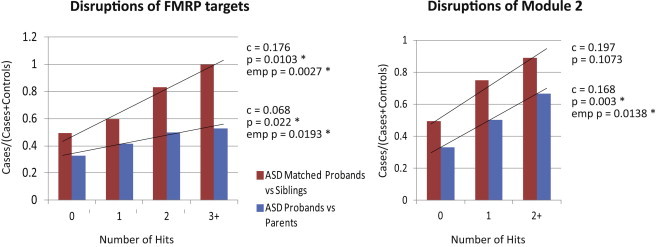
The Proportion of Autism Cases among All Individuals with Deletion CNVs Hitting an FMRP Target Gene Increases Dramatically with the Number of Hits
An individual was defined to have h hits in a gene set if the individual’s rare deletion CNVs overlapped h genes in the gene set (see Material and Methods). Using regression, we obtained the coefficient c for a linear increase in the proportion of cases among all individuals with an increasing number of hits and the associated p value. We then obtained empirical p values by comparing the coefficient c to corresponding coefficients calculated for 10,000 random gene sets (see Material and Methods). An asterisk indicates that a coefficient is significant for the number of hits versus risk increase on the basis of linear regression at a threshold of 0.05.
By definition, observations of rare deletion CNVs are infrequent, and thus large data sets are needed for investigating even those variants with large effects. Consequently, we also wanted to consider the role of FMRP targets in ASD on the basis of the more common inherited SNP variation. Although single SNPs are expected to have very small effect sizes, for FMRP modules 1 and 2, we could combine information across all autosomal SNPs located in each module’s genes. Based on a GWAS data set on ASD-affected families from AGRE (Table S1), SNP p values calculated with the transmission disequilibrium test were used in a set-based association test in Plink.49 Accounting for linkage disequilibrium of the SNPs at an r2 threshold of 0.5, we found that module 2 showed significant association at 5% FDR (p = 0.0062), whereas module 1 did not (p = 0.28; Table S13). We retested module 2 at r2 thresholds of 0.2 and 0.8 and again found significant association (p = 0.0022 and p = 0.0059, respectively).
We conclude that FMRP targets, and module 2 in particular, can be implicated in ASD via both rare deletion CNVs and SNPs. Notably, we replicated the risk increase conferred by multiple deletions of FMRP targets after correction for mutational burden in cases, providing statistically robust evidence of a multiple-hit disease etiology in at least a subset of ASD cases and illustrating the Trend test’s power to detect such etiologies.
FMRP Targets Are Associated with ASD More Than Other Genes with Similar Brain Expression Patterns
Having differentially implicated FMRP modules 1 and 2 in ASD, we investigated whether this association was either generalizable to genes with expression patterns similar to those of module 1 or 2 genes or else FMRP-target specific. To this end, we compiled a list of 938 non-FMRP target genes with expression patterns very similar to those of module 1 genes (see Material and Methods); this list was referred to as “module-1-like” genes. Analogously, 339 “module-2-like” genes were also compiled. We found that module-1-like genes were not significantly enriched among genes disrupted by deleterious de novo point mutations in ASD probands (I-exomes, SON-exomes, and T-BCAs joined: 1.38-fold, p = 0.15). Comparing directly, despite the much larger size of the module-1-like gene set and a 1.54-fold higher total CDS length of module-1-like genes, a deleterious de novo point mutation in an ASD proband is 4.27-fold more likely to disrupt a module 1 gene than a module-1-like gene (I-exomes, SON-exomes, and T-BCAs joined: Fisher’s test p = 7.33 × 10−5).
Unlike module 2 genes, module-2-like genes did not show a significant association with ASD in any of the three CNV data sets considered above in the Trend test (empirical p > 0.05 for all three data sets).
These results show that the association between ASD and both FMRP modules 1 and 2 does not apply to non-FMRP target genes with highly similar expression patterns in the human brain. We asked whether this might be due to functional differences between the FMRP modules and the module-like genes. Using functional annotations obtained from MGI as above and testing the overarching categories, we indeed found significant differences (Figure 5): compared to the corresponding module-like genes, both FMRP module 1 and 2 genes were significantly enriched in genes yielding a nervous system phenotype when disrupted in mice (module 1: 2.17-fold, Fishers’ p = 2.35 × 10−4; module 2: 2.56-fold, Fishers’ p = 3.6 × 10−4). Moreover, looking at the probability of haploinsufficiency, we found that both FMRP module 1 and 2 genes were significantly more likely to be HIS than the module-1-like or module-2-like genes, respectively (Figure 5; Wilcox p = 2.52 × 10−6 and p = 2.98 × 10−3, respectively). Thus, FMRP target genes from these two modules are both more sensitive to copy loss and more likely to yield nervous system phenotypes upon disruption than are non-FMRP target genes with similar expression profiles.
Figure 5.
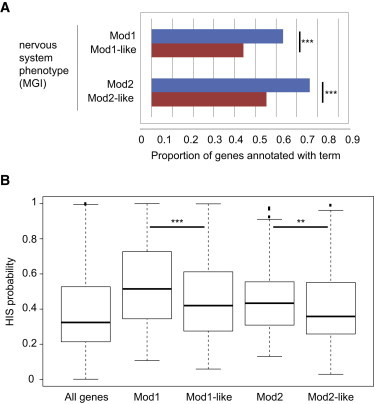
Differences between FMRP Modules and Module-like Genes
(A) Module 1 and 2 genes were significantly more often annotated to a nervous system phenotype than the corresponding module-like genes (Fisher’s test), despite their similar expression patterns. The only other significant difference is that module 2 genes were less often annotated to an immune system phenotype from MGI than module-2-like genes (0.4-fold, Fishers’ p = 1.59 × 10−3).
(B) Different distributions in predicted HIS probabilities (Mann-Whitney U-test). ∗∗p < 0.01, ∗∗∗p < 10−3. Error bars represent the most extreme point with a distance from the box ≤ 1.5-fold the interquartile range.
Discussion
In this work, we have dissected the role of FMRP target genes in ASD by identifying distinct subpopulations of FMRP targets and showing that these subpopulations are differentially affected by different classes of genetic variation. Specifically, we showed that (1) FMRP targets can be readily divided into subpopulations with distinct spatiotemporal expression patterns and functional biases, that (2) single-gene disruptions by de novo mutations in ASD are highly enriched in an embryonically expressed subgroup of FMRP targets, whereas (3) rare deletion CNVs and, separately, SNPs in ASD are associated with a subgroup of FMRP targets highly specific to the brain and upregulated in adolescence and adulthood, and finally that (4) FMRP targets within these two subgroups are more likely to yield nervous system phenotypes upon copy loss than are non-FMRP target genes with similar expression patterns. Importantly, we developed a powerful CNV association test that explicitly considers multiple-hit etiologies and used it to demonstrate that the risk of ASD increases with the number of disrupted FMRP target genes.
Our findings suggest that de novo mutations and inherited variation contribute to ASD through two different genetic etiologies: (1) single highly penetrant de novo mutations predominantly arising in genes encoding embryonic transcription factors and chromatin modifiers and (2) multiple, often inherited, pathway disruptions particularly enriched in synaptic genes. Our findings are well supported by the direct functional assessment of FMRP targets among disrupted genes in ASD (Tables S14–S16). Notably, de novo CNVs that affect multiple genes might also contribute to ASD via a multiple-hit mechanism resulting from the cumulative effects of multiple simultaneously copy-number-changed genes—for instance, region 16p11.2, with recurrent CNVs in autism,50 contains a total of four FMRP targets (MAZ [MIM 600999], SEZ6L2, TAOK2 [MIM 613199], and ALDOA [MIM 103850], see Table S14).
Taken together, our findings provide a framework that is based on the penetrance of a genetic variant and through which previous ASD pathway analyses, which have separately implicated synaptic genes17–20 and chromatin modifiers,12 can be unified. Etiologies that lie between these two extremes could be found, and it will be of interest to investigate to what extent less penetrant disruptions of synaptic genes can modify phenotypes caused by highly penetrant disruptions of embryonic transcription factors, for example. The FMRP targets implicated here can be used for prioritizing candidate genes for further study; in particular, only 19 out of the 105 FMRP targets found disrupted in ASD in this study are known ASD candidate genes (Table S14). Crucially, we note that although an enrichment of an FMRP target module among genes disrupted in ASD probands supports a causal role in ASD for biological processes represented by the module, it does not imply that each and every disrupted module gene is causal to the disorder. Moreover, there are types of genetic variation that we have not examined. Among them are de novo missense mutations within genes: these are also more common in ASD probands than in their siblings11,14 but less so than the highly deleterious de novo mutations examined in this study, and the biological consequences of missense mutations are less predictable. In addition, although we have extensively analyzed rare deletion CNVs, we initially did not consider rare duplication CNVs because the two original studies by Sanders et al.28 and the AGP29 found deletion CNVs to be more likely to contribute to ASD. In agreement with this, using the Trend test, we found an association between FMRP targets and ASD via rare duplication CNVs in the AGP cohort (empirical p = 0.018), but not in the two Sanders cohorts (empirical p > 0.05). Thus, the contribution of FMRP targets to ASD via additional types of genetic variation remains unclear.
Initially, we subdivided FMRP targets into four modules, but we observed distinct functional biases only for the two largest modules (modules 1 and 2; Figure 1B), as compared to the set of all FMRP targets, and thus we subsequently characterized only these in detail. From the analysis of gene disruptions in ASD, we found a significant enrichment of module 3 genes among the two lists of genes disrupted by damaging de novo mutations identified in recent exome sequencing studies (I-exomes and SON-exomes; Table S9). However, using a correction for gene length, we found that module 3 was significantly associated with ASD via de novo damaging mutations in all three lists of I-exomes, SON-exomes, and T-BCAs (Table S10). By contrast, applying the Trend test to rare deletion CNVs in ASD, we found that module 4 was significantly associated with ASD in only two of the three data sets we considered (Table S12). There was no other evidence of an association between ASD and module 3 or 4 (Tables S9, S12, and S13). The proportion of predicted HIS genes and the single-nucleotide variation in the general population for modules 3 and 4 imply that the deleteriousness of mutations in module 3 and 4 genes lies between that of modules 1 and 2 (Figures S1 and S3) and that there is a significant difference in haploinsufficiency probabilities between modules 2 and 3, but no significant difference between modules 1 and 3. Because module 3 genes are typically expressed throughout development, in agreement with the high probabilities of haploinsufficiency, they might play biological roles indispensable for embryonic stages onward, thus conforming closely to the two distinct genetic etiologies suggested above. Unfortunately, because of the relative small number of module 3 genes and the resulting lack of power, we could not determine this via functional enrichment analyses. As regulation by FMRP targeting is further elucidated, the functional interpretation of these smaller clusters of coexpressed genes might become clearer.51,52 The coexpression patterns of the 105 FMRP targets affected by the de novo single-gene mutations and rare deletion CNVs considered in this study confirm this modular pattern in that the genes from modules 3 and 4 link the more distinct clusters of disrupted genes from module 1 and, separately, module 2 (Figure S4; Appendix A). Notably, we found that non-FMRP target genes with brain expression patterns highly similar to those of FMRP module 1 and 2 genes did not show a strong association with ASD.
Our systemic insight into the role of particular subpopulations of FMRP targets in ASD is suggestive of etiological progression. It is plausible that a number of the earlier expressed module 1 FMRP targets are involved in establishing or maintaining the same neurological processes that the later expressed synaptically focused module 2 FMRP targets participate in. Unfortunately, the data are currently too sparse to establish whether module 2 gene expression is under the control of earlier expressed FMRP target genes. Nonetheless, the participation in shared processes would also explain how the disruption of different genes gives rise to a common phenotype. This participation would also suggest that the same process can be disrupted at different time points and have important therapeutic consequences for ASD: ASD-associated module 2 genes, often expressed at the synapse in adolescence and adulthood, might be far more therapeutically attractive targets for modulating this process than module 1 genes, often transcriptional regulators that are upregulated embryonically. Indeed, it has been shown recently that some symptoms in a mouse model of ASD can be rescued at a juvenile stage.53 The evidence that we found for a multiple-hit etiology in ASD is consistent with two different mechanisms of disease onset: (1) in an additive model, each disruption of relevant biological processes leads to an increase in ASD traits until ASD is diagnosed; and (2) in a threshold model, ASD emerges as soon as the mutational load affecting relevant biological processes exceeds a critical threshold. Further support for an additive model stems from the observation that unaffected parents in families with multiple affected children score higher on the broader autism phenotype assessment than do parents in families with only one affected child.54,55
The Trend test, which we developed to investigate multiple-hit contributions to ASD, is applicable to any complex genetic condition. From the results of our simulations, we provide the statistically founded recommendation to only consider gene sets with at least 100 genes for the initial stage of a CNV pathway analysis (Figure 3). For gene sets with a significant result in the initial stage, it is then possible to test smaller subsets in a second step. The Trend test will be particularly useful for highly polygenic disorders for which approaches that consider single genetic loci in isolation cannot readily uncover all causal variants (classically known as the “missing heritability”56). Moreover, the Trend test can readily combine information from multiple types of genetic variants (structural or nucleotide variation), increasing its potential use with the advent of next-generation sequencing techniques.
Acknowledgments
We thank the anonymous reviewers for their comments and suggestions. This work was supported by the Wellcome Trust (grants 093941/Z/10/Z to J.S. and 090532/Z/09/Z to the Wellcome Trust Centre for Human Genetics), Medical Research Council (funding to C.W), and the European Union’s Seventh Framework Programme project GENCODYS (grant 241995 to C.W.). We gratefully acknowledge the resources provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. The AGRE is a program of Autism Speaks and is supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to Clara M. Lajonchere (principal investigator). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A
Temporal Specificity of FMRP Targets
When investigating temporal specificity of FMRP target modules, we noticed that all FMRP targets had generally lower median RPKM measure C for the “child” stage. This was also the case for all genes contained in the BrainSpan data set and was potentially an experimental artifact. Consequently, we chose to increase C by a constant factor F(C) for each gene when looking at temporal specificity only and calculated it as follows.
For each gene g, let M(g,s) be the median expression in stage s. For each stage s, let
For all FRP targets, we calculated F(C) = 1.9773 (which is close to 1.9032, obtained when all genes in BrainSpan were used).
Trend and Fisher’s Tests for CNVs
The details for the Trend test are as follows. Define U and A as the numbers of unaffected and affected individuals, respectively, in the data set: N = A + U. Let m be the maximum number of hits for a pathway in any individual, let U(h) and A(h) be the number of unaffected and affected individuals, respectively, with exactly h hits in the pathway: h = 0, 1, ..., m. Also, let . We defined adjusted numbers U′(h) = U(h) and A′(h) = A(h) for h = 0, 1, ..., m′ − 1 and , . The Trend test statistic is
for which
where H′(h) = A′(h) + U′(h) for h = 0, 1, ..., m′. According to the results of Cochran and Armitage, , i.e., , follows a standard normal distribution.
Comparison of Trend Test to Fisher’s Test
To compare robustness and false-positive rates for the Trend test and Fisher’s test, we carried out simulations by creating randomized sets based on two different data sets: rare deletion CNVs in the parents of children with autism from Sanders et al.28 (SandersParents) and deletion CNVs in the NBS data set. To investigate the effect of varying gene numbers within pathways, we chose five example pathways representing a wide range of gene counts (Figure 3) and matched 100 random gene sets by gene number and gene length to each example pathway. The simulations consisted of 10,000 random splits of individuals from either the SandersParents or NBS data sets into cases and controls and were followed by the calculation of Trend and Fisher’s test statistics for the random gene sets; we then estimated the false-positive rates of the Trend and Fisher’s tests for the corresponding pathway size at significance levels 0.05 and 2 × 10−4.
To compare the power of the Fisher’s and Trend tests, we obtained association p values from both tests for a variety of scenarios: a data set (1) of either 800 cases and 800 controls or 600 cases and 600 controls; (2) with between 10 and 10% of controls possessing one hit; and (3) with a ratio of cases to controls with one hit equal to r in (1.5, 2, 2.5, 3, 3.5, 4). To avoid further assumptions about the increase in risk conferred by multiple hits, initially no individual was assumed to have more than one hit.
SNP Pathway Analysis
Following the additional quality-control steps of Wang et al.,32 we removed all monozygotic twins and sample duplicates (as detailed in the download information) and excluded individuals with a call rate < 0.95. We used multidimensional scaling in Plink49 (v.1.07) to identify individuals of European descent and matched the parameters of Wang et al.32 as closely as possible. We manually looked through 11 pairs of genotype duplicates and trisomy 21 cases from the paper by Wang et al.32 and excluded one case still found in the data set. Finally, we excluded SNPs with a MAF < 0.05 and Hardy-Weinberg equilibrium p < 0.001.
BrainSpan Gene Correlation Network
The BrainSpan gene expression data as described in the Material and Methods were used for building a gene correlation network. Genes with less than one RPKM in more than 95% of the samples were excluded. For all samples, we calculated Pearson’s correlation coefficient for all gene pairs. A network was built with genes as nodes and edges between two genes weighted with their correlation coefficient r. Considering only edges with weight r ≥ 0.5 gave 14,886 unique genes with at least one edge.
We calculated the number of links between the FMRP targets disrupted in ASD probands by de novo single-gene mutations and rare deletion CNVs (Table S14). We then obtained an empirical one-sided p value by comparing random gene sets as follows.
Of the FMRP targets disrupted in ASD probands by de novo single-gene mutations (in the lists of I-exomes, SON-exomes, and T-BCAs; see Material and Methodsand Results), 34 genes were in the correlation network; of the FMRP targets disrupted by rare deletion CNVs, but not by de novo single-gene mutations, 69 were in the correlation network. In each of 10,000 simulations performed, we
-
1.
chose 34 out of the 141 genes from I-exomes, SON-exomes, and T-BCAs (these were in the correlation network)
-
2.
chose 69 out of 1,246 genes disrupted in ASD probands by rare deletion CNVs in the data sets from AGP (strict ASD) and Sanders (see Material and Methods) (these were in the correlation network, but not in the 141 genes from I-exomes, SON-exomes, or T-BCAs)
-
3.
calculated the number of links between all 34 + 69 = 103 genes chosen in steps 1 and 2.
We found that there were more links between the disrupted FMRP targets than between any of the random gene sets (giving an empirical p value of 10−4; Figure S5). To confirm robustness of our results, we repeated the experiment with more stringent correlation thresholds for links in the network. For correlation thresholds 0.6, 0.7, 0.8, and 0.9, there were significantly more links between the disrupted FMRP targets than between random gene sets (empirical p values of 10−4, 2.9 × 10−3, 0.042, and 0.027, respectively; Figure S5).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Autism Genetic Resource Exchange (AGRE), https://research.agre.org/
BrainSpan: Atlas of the Developing Human Brain, http://www.brainspan.org/
Database for Annotation, Visualization, and Integrated Discovery (DAVID), http://david.abcc.ncifcrf.gov/
Mouse Genome Informatics (MGI), http://www.informatics.jax.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
The R Project for Statistical Computing, http://www.r-project.org/
References
- 1.Muhle R., Trentacoste S.V., Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- 2.Uher R. The role of genetic variation in the causation of mental illness: an evolution-informed framework. Mol. Psychiatry. 2009;14:1072–1082. doi: 10.1038/mp.2009.85. [DOI] [PubMed] [Google Scholar]
- 3.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 4.Klei L., Sanders S.J., Murtha M.T., Hus V., Lowe J.K., Willsey A.J., Moreno-De-Luca D., Yu T.W., Fombonne E., Geschwind D. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism. 2012;3:9. doi: 10.1186/2040-2392-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy D., Ronemus M., Yamrom B., Lee Y.H., Leotta A., Kendall J., Marks S., Lakshmi B., Pai D., Ye K. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 6.State M.W., Šestan N. Neuroscience. The emerging biology of autism spectrum disorders. Science. 2012;337:1301–1303. doi: 10.1126/science.1224989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLennan Y., Polussa J., Tassone F., Hagerman R. Fragile x syndrome. Curr. Genomics. 2011;12:216–224. doi: 10.2174/138920211795677886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budimirovic D.B., Kaufmann W.E. What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 2011;33:379–394. doi: 10.1159/000330213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soden M.E., Chen L. Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J. Neurosci. 2010;30:16910–16921. doi: 10.1523/JNEUROSCI.3660-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang T., Bray S.M., Warren S.T. New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 2012;22:256–263. doi: 10.1016/j.gde.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Callan M.A., Zarnescu D.C. Heads-up: new roles for the fragile X mental retardation protein in neural stem and progenitor cells. Genesis. 2011;49:424–440. doi: 10.1002/dvg.20745. [DOI] [PubMed] [Google Scholar]
- 17.Gai X., Xie H.M., Perin J.C., Takahashi N., Murphy K., Wenocur A.S., D’arcy M., O’Hara R.J., Goldmuntz E., Grice D.E. Rare structural variation of synapse and neurotransmission genes in autism. Mol. Psychiatry. 2012;17:402–411. doi: 10.1038/mp.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-David E., Shifman S. Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet. 2012;8:e1002556. doi: 10.1371/journal.pgen.1002556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skafidas E., Testa R., Zantomio D., Chana G., Everall I.P., Pantelis C. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol. Psychiatry. 2012 doi: 10.1038/mp.2012.126. Published online September 11, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noh H.J., Ponting C.P., Boulding H.C., Meader S., Betancur C., Buxbaum J.D., Pinto D., Marshall C.R., Lionel A.C., Scherer S.W., Webber C. Network topologies and convergent aetiologies arising from deletions and duplications observed in individuals with autism. PLoS Genet. 2013;9:e1003523. doi: 10.1371/journal.pgen.1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilman S.R., Iossifov I., Levy D., Ronemus M., Wigler M., Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ben-David E., Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol. Psychiatry. 2013;18:1054–1056. doi: 10.1038/mp.2012.148. [DOI] [PubMed] [Google Scholar]
- 23.Leblond C.S., Heinrich J., Delorme R., Proepper C., Betancur C., Huguet G., Konyukh M., Chaste P., Ey E., Rastam M. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Girirajan S., Rosenfeld J.A., Coe B.P., Parikh S., Friedman N., Goldstein A., Filipink R.A., McConnell J.S., Angle B., Meschino W.S. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012;367:1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doetschman T. Influence of Genetic Background on Genetically Engineered Mouse Phenotypes. In: Wurst W., Kühn R., editors. Gene Knockout Protocols. Humana Press; New York: 2009. pp. 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talkowski M.E., Rosenfeld J.A., Blumenthal I., Pillalamarri V., Chiang C., Heilbut A., Ernst C., Hanscom C., Rossin E., Lindgren A.M. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lord C., Risi S., Lambrecht L., Cook E.H., Jr., Leventhal B.L., DiLavore P.C., Pickles A., Rutter M. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000;30:205–223. [PubMed] [Google Scholar]
- 31.Lord C., Rutter M., Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 32.Wang K., Zhang H., Ma D., Bucan M., Glessner J.T., Abrahams B.S., Salyakina D., Imielinski M., Bradfield J.P., Sleiman P.M.A. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flicek P., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., Fitzgerald S. Ensembl 2012. Nucleic Acids Res. 2012;40(Database issue):D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 38.Webber C., Hehir-Kwa J.Y., Nguyen D.-Q., de Vries B.B.A., Veltman J.A., Ponting C.P. Forging links between human mental retardation-associated CNVs and mouse gene knockout models. PLoS Genet. 2009;5:e1000531. doi: 10.1371/journal.pgen.1000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeLorey T.M., Sahbaie P., Hashemi E., Li W.-W., Salehi A., Clark D.J. Somatosensory and sensorimotor consequences associated with the heterozygous disruption of the autism candidate gene, Gabrb3. Behav. Brain Res. 2011;216:36–45. doi: 10.1016/j.bbr.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Austin C.P., Battey J.F., Bradley A., Bucan M., Capecchi M., Collins F.S., Dove W.F., Duyk G., Dymecki S., Eppig J.T., The Comprehensive Knockout Mouse Project Consortium The knockout mouse project. Nat. Genet. 2004;36:921–924. doi: 10.1038/ng0904-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaikh T.H., Haldeman-Englert C., Geiger E.A., Ponting C.P., Webber C. Genes and biological processes commonly disrupted in rare and heterogeneous developmental delay syndromes. Hum. Mol. Genet. 2011;20:880–893. doi: 10.1093/hmg/ddq527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eppig J.T., Blake J.A., Bult C.J., Kadin J.A., Richardson J.E., Mouse Genome Database Group The Mouse Genome Database (MGD): comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res. 2012;40(Database issue):D881–D886. doi: 10.1093/nar/gkr974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang N., Lee I., Marcotte E.M., Hurles M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raychaudhuri S., Korn J.M., McCarroll S.A., Altshuler D., Sklar P., Purcell S., Daly M.J., International Schizophrenia Consortium Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet. 2010;6:e1001097. doi: 10.1371/journal.pgen.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M.M., Pletikos M., Meyer K.A., Sedmak G. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A.R., Green T., Autism Consortium Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 51.Ascano M., Jr., Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L., Langlois C., Munschauer M., Dewell S., Hafner M. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jayaseelan S., Tenenbaum S.A. Neurodevelopmental disorders: Signalling pathways of fragile X syndrome. Nature. 2012;492:359–360. doi: 10.1038/nature11764. [DOI] [PubMed] [Google Scholar]
- 53.Baudouin S.J., Gaudias J., Gerharz S., Hatstatt L., Zhou K., Punnakkal P., Tanaka K.F., Spooren W., Hen R., De Zeeuw C.I. Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science. 2012;338:128–132. doi: 10.1126/science.1224159. [DOI] [PubMed] [Google Scholar]
- 54.Gerdts J., Bernier R. The broader autism phenotype and its implications on the etiology and treatment of autism spectrum disorders. Autism Res. Treat. 2011;2011:545901. doi: 10.1155/2011/545901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernier R., Gerdts J., Munson J., Dawson G., Estes A. Evidence for broader autism phenotype characteristics in parents from multiple-incidence autism families. Autism Res. 2012;5:13–20. doi: 10.1002/aur.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.