

Abstract
Common variable immunodeficiency (CVID) is a heterogeneous disorder characterized by antibody deficiency, poor humoral response to antigens, and recurrent infections. To investigate the molecular cause of CVID, we carried out exome sequence analysis of a family diagnosed with CVID and identified a heterozygous frameshift mutation, c.2564delA (p.Lys855Serfs∗7), in NFKB2 affecting the C terminus of NF-κB2 (also known as p100/p52 or p100/p49). Subsequent screening of NFKB2 in 33 unrelated CVID-affected individuals uncovered a second heterozygous nonsense mutation, c.2557C>T (p.Arg853∗), in one simplex case. Affected individuals in both families presented with an unusual combination of childhood-onset hypogammaglobulinemia with recurrent infections, autoimmune features, and adrenal insufficiency. NF-κB2 is the principal protein involved in the noncanonical NF-κB pathway, is evolutionarily conserved, and functions in peripheral lymphoid organ development, B cell development, and antibody production. In addition, Nfkb2 mouse models demonstrate a CVID-like phenotype with hypogammaglobulinemia and poor humoral response to antigens. Immunoblot analysis and immunofluorescence microscopy of transformed B cells from affected individuals show that the NFKB2 mutations affect phosphorylation and proteasomal processing of p100 and, ultimately, p52 nuclear translocation. These findings describe germline mutations in NFKB2 and establish the noncanonical NF-κB signaling pathway as a genetic etiology for this primary immunodeficiency syndrome.
Introduction
Common variable immunodeficiency (CVID [MIM 607594]), which occurs in approximately 1:10,000 to 1:50,000 people,1–3 is a clinically and genetically heterogeneous disorder characterized by hypogammaglobulinemia with poor response to antigens. CVID is one of the most common primary immune deficiencies, with affected individuals frequently presenting with recurrent sinopulmonary infections as well as immune dysregulatory or autoimmune features. Approximately 10%–20% of cases are familial.4,5 The genetic defects responsible for CVID have been identified in less than 10%–15% of all cases4,6 and include mutations in CD197 (MIM 613493), MS4A18 (CD20 [MIM 613495]), CR29 (CD21 [MIM 614699]), ICOS10 (MIM 607594), TNFRSF13C11 (BAFFR [MIM 613494]), TNFRSF13B12,13 (TACI [MIM 240500]), PLCG214 (phospholipase Cγ2 [MIM 614878]), CD8115 (MIM 613496), LRBA16 (MIM 614700), and PRKCD17 (protein kinase Cδ [MIM 176977]). Causative variants in these genes typically affect peripheral or terminal B cell development, in contrast to more severe antibody deficiencies, such as X-linked agammaglobulinemia (MIM 300755), which affect early B cell development.18 Of individuals with CVID, 8%–10% have been identified with homozygous and heterozygous variants in TNFRSF13B, 1%–2% of individuals harbor variants in ICOS, and the remainder of the genes contribute to <1% of cases. The functional contribution of TNFRSF13B variants in the disease is less clear, because some variants are present in CVID subjects, normal controls, and healthy relatives.19,20 A genome-wide association study of CVID identified SNP associations as well as copy-number variants in several gene regions,21 providing additional evidence that both single-gene mutations and more complex genetic mechanisms contribute to the pathogenesis of CVID. This genetic heterogeneity may result in differences in the CVID phenotype, including age of onset, susceptibility to infection, autoimmunity, and malignancy. These differences highlight the importance of identifying additional molecular causes to predict disease onset, enable improved disease screening, and provide new targets for treatment.
The NF-κB signal transduction pathway is well known for its role in inflammatory and immune responses and includes the structurally homologous transcription factors NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel. The canonical pathway, which includes NF-κB1, primarily mediates broad inflammatory responses, whereas the noncanonical pathway, which includes NF-κB2, affects specific aspects of B cell maturation, peripheral lymphoid development, bone metabolism, and thymic development.22–24 In the nonactivated, resting state, NF-κB proteins are typically sequestered in the cytoplasm. Activation of either pathway results in proteasomal processing and translocation to the nucleus, where NF-κB1 or NF-κB2 bind their gene targets (see Figure S1 available online). As opposed to the broad function and numerous ligands of the canonical (NF-κB1) pathway that lead to the production of inflammatory cytokines, the noncanonical (NF-κB2) pathway has functional specificity. The primary noncanonical NF-κB2 ligands include lymphotoxin β, B cells activating factor (BAFF), CD40 ligand (CD40L), and receptor activator of NF-κB ligand (RANKL). BAFF receptor (BAFFR) deficiency is associated with a form of CVID. Somatic mutations that activate the noncanonical pathway/NF-κB2 have been associated with B cell lymphomas and other hematogenous malignancies.25–27 Together, these findings suggest that mutant proteins in the noncanonical pathway are excellent candidates for CVID pathogenesis.
We studied an index family affected by autosomal-dominant, early-onset hypogammaglobulinemia with variable autoimmune features as well as adrenal insufficiency. By using an exome-sequencing approach, we identified a heterozygous mutation in NFKB2 (MIM 164012), the gene that encodes NF-κB2, that affects the C terminus of the protein, which prompted screening for additional NFKB2 mutations in our CVID cohort. We identified a second NFKB2 mutation, also affecting the C terminus of NF-κB2, in an unrelated CVID-affected individual with the same features of hypogammaglobulinemia, autoimmunity, and adrenal insufficiency. The causal effect of these mutations is supported by our functional analysis and by Nfkb2 mouse models with similar CVID-like phenotypes. We demonstrate the effects that the mutations have upon NF-κB2 nuclear translocation and highlight the importance of the noncanonical pathway for the study of CVID.
Material and Methods
Subjects
Blood samples were obtained from individuals with CVID, healthy family members, and healthy control subjects. Informed consent was obtained from all individuals and the legal guardians of minors participating in studies approved by the University of Utah Institutional Review Board.
Clinical Laboratory Studies
Determination of lymphocyte subpopulations and immunoglobulin levels was carried out via standard laboratory techniques, including flow cytometry and nephelometry. Toll-like receptor (TLR) functional assays with ligands to TLR1–TLR8 were performed on peripheral blood mononuclear cells with supernatant measurement of interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNF-α), and IL-6 production measured by Luminex multi-analyte technology (ARUP Laboratories). Cortisol and adrenocorticotropic hormone (ACTH) levels were measured by standard assays for clinical analysis.
B Cell Immunophenotyping
Whole blood was stained with CD19 PE (Beckman Coulter, #6603846), CD27 APC (BD Bioscience, #655019), CD45 PerCP (BD Bioscience, #340334), and IgD FITC (Dako, #F0189) in BD TruCOUNT tubes (BD Bioscience, #340334). The data were acquired on a FACS Canto II flow cytometer and analyzed with FACSDiva software (BD Bioscience).
Exome Capture and Sequencing
Exome capture was performed on 3 μg of DNA samples with Roche Nimblegen SeqCap EZ Human Exome Library capture probes v.2.0, 44.1 Mb (A.II.1, P1, P2, and P3) or v.3, 65 Mb (P4) performed according to the manufacturer’s instructions followed by sequencing on the Illumina HiSeq 2000. Exome libraries from family A were sequenced on the HiSeq 2000 in individual lanes with paired-end v2.5 chemistry, and individual P4 was indexed and pooled with a second exome and sequenced in one lane of the HiSeq 2000 via paired-end, v.3 sequencing chemistry. High Density Cytoscan SNP arrays (Affymetrix) were run on family A to detect large structural changes. Fastq files from exome data were mapped and aligned to the reference genome (Genome Reference Consortium Human build 37 [GRCh37]) with Burrows-Wheeler Aligner (BWA).28 Alignments were subsequently refined by local realignment around indels, PCR duplicate removal, and recalibration of base quality scores via the Genome Analysis Toolkit software (GATK)29 and SAMTools.30 Variants were called and variant call format (VCF) files were generated with SAMTools, and GATK was used for variant annotation. The SNP and Variation Suite (SVS) software (Golden Helix) was used to filter the data sets for family A based on the assumption of a dominant inheritance pattern. The genomic position corresponding to c.2564delA (chr10: 104,161,900) was Sanger sequenced in the mother (P1), father (A.II.1), son (P3), maternal grandparents (A.I.1 and A.I.2), and unaffected child (A.III.3) by means of the primers shown in Table S1.
VAAST Analysis
Exome data from the four individuals in family A (A.II.1, P1, P2, and P3) and the affected individual from family B (P4) were analyzed with a version of the Variant Annotation, Analysis and Search Tool (VAAST),31 which annotates and prioritizes SNVs and indels. A dominant inheritance model and an expected disease frequency of 1:10,000 were assumed. Variant files from all three affected individuals in family A (P1, P2, and P3) were intersected, the father (A.II.1) was subtracted, and a union was performed between this variant file and the variant file from the affected individual in family B (P4). Genes were annotated and ranked by VAAST, and false positives were removed with VarBin, a tool that prioritizes variants based on a false variant likelihood prediction (J.D. Durtschi and R.L. Margraf, personal communication).
Genetic Screening of NFKB2 in a Collection of CVID Simplex Cases
Genetic screening of NFKB2 by long-range PCR and Illumina sequencing of a collection of 33 simplex CVID-affected individuals were performed as described in Durtschi and Margraf et al.32 Two primer sets with a 5′ Amino Modifier C6 block were used to span NFKB2 ± 1,000 bp to reduce the potential for allelic dropout. Primers were designed to amplify a 9,648 base pair amplicon and a 9,695 base pair amplicon (Table S1). CVID simplex samples were equimolarly pooled in two different pools (one with 16 samples and one with 17 samples) with 250 ng total DNA in each pool. DNA from the unaffected father in family A (A.I.1) was used as a reference and pooled to a total of 160 ng. The c.2557C>T variant was Sanger sequenced with the primers shown in Table S1.
Cell Culture
Peripheral blood mononuclear cells (PBMCs) and Epstein-Barr virus (EBV)-transformed B cell lines were maintained in complete medium with RPMI-1640 with 2 mM L-glutamine, 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in a humidified environment with 5% CO2.
Immunoblotting
EBV-B cell lines at 1 × 106 cells/ml were incubated with or without human CD40L (2 μg/ml) (Cell Signaling) for 4 hr in complete medium. Cell lysates were prepared in RIPA buffer with HALT protease inhibitor cocktail (Pierce) and benzonase (Sigma-Aldrich). A NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific) was used according to manufacturer’s instructions for isolation of nuclear and cytoplasmic cell fractions. To ensure equal loading, protein concentrations were assessed via Bradford protein assay (Bio-Rad). For protein identification, the following antibodies from Cell Signaling were used: NF-κB2 p100/p52 (#4882), phospho-NF-κB2p100 serine 866/870 (#4810), RelB (#4992), and IKKα (#2682). Anti-IKKα (phospho S176 + S180) antibody (Abcam, ab17943) was used. A histone deacetylase 1 antibody (HDAC1, H-51) (Santa Cruz Biotechnology, sc-7872) was used as a nuclear fraction loading control and a HRP-conjugated β-actin antibody (Abcam, ab20272) as a cytoplasmic or whole-cell lysate loading control.
Immunocytochemistry and Laser-Scanning Confocal Microscopy
EBV-B cells (1 × 106 cells/ml) were stimulated with CD40L at 2 μg/ml for 4 hr. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline, permeabilized with 0.3% Triton X-100, and blocked with 5% goat serum. Cells were immunostained with human-specific anti-NF-κB2 p100/p52 IgG (Cell Signaling, #3017). Biotin labeling was completed with Biotin-XX goat anti-rabbit IgG (Life Technologies, #B2770), followed by incubation with Streptavidin Alexa 488-conjugated streptavidin (Life Technologies, #S32354). Slides were counterstained with To-Pro-3, which stains the nucleus, and mounted with Prolong Gold antifade reagent (Molecular Probes) prior to imaging. All images were generated on an Olympus FV-1000-XY confocal microscope with a 60× oil immersion objective at 1× zoom with z spacing of 1 μm. Identical gain settings were used for image recording for all samples. Olympus Fluoview 1000 v.3.0 software (Olympus) was used for blinded scoring analysis of all images. Percent nuclear translocation of p52 was quantified with immunofluorescence confocal imaging of wild-type (WT) and mutant (CVID) cell lines by observing colocalization of the signal from anti-NF-κB2 p100/p52 IgG and To-Pro-3. A minimum of 300 cells per slide per cell line were counted in the control and affected groups, with 3 to 6 microscopy field images obtained per cell line.
Statistical Analysis
p values for confocal imaging analysis were calculated by a nonparametric Wilcoxon rank-sum (Mann-Whitney) test implemented in the STATA statistical software package (Stata Corp. LP: STATA Statistical Software: Release 11).
Results
Exome Sequencing and Genetic Screening Identifies Heterozygous NFKB2 Mutations in CVID-Affected Individuals
We studied a family affected by an autosomal-dominant form of childhood-onset CVID (family A; Figure 1). Clinical findings are summarized in Table 1, and case report and laboratory findings are summarized in Table 2. Affected individuals were diagnosed with CVID based on (1) the presence of markedly reduced levels of serum immunoglobulins, including low IgG levels, (2) poor response to antigens, and (3) a lack of criteria for diagnosis of other well-defined antibody deficiency syndromes.
Figure 1.
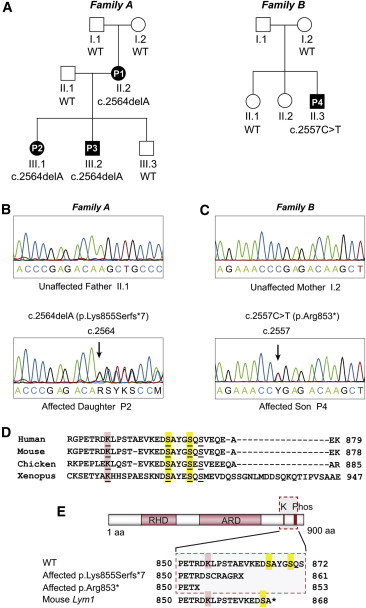
NFKB2 Mutations in CVID
(A) Pedigrees for two families affected by childhood-onset CVID with adrenal insufficiency. Mutation status of NFKB2 c.2564 for family A and NFKB2 c.2557 for family B are indicated beneath symbols for each individual (others were unavailable for testing). WT indicates wild-type.
(B) Sanger sequencing of two individuals in family A over the c.2564 position in NFKB2. Upper panel shows wild-type c.2564 position and lower panel shows the c.2564delA variant. Arrow points to c.2564delA variant.
(C) Sanger sequencing of two individuals in family B over the c.2557 position in NFKB2. Upper panel shows wild-type c.2557 position and lower panel shows the c.2557C>T variant. Arrow points to the c.2557C>T variant.
(D) Multiple sequence alignment of amino acid sequences in the C terminus of NF-κB2. The conserved lysine that acts as an acceptor for ubiquitin is underlined and highlighted in red. The two conserved serines required for phosphorylation are underlined and highlighted in yellow.
(E) Schematic of NF-κB2 with the Rel Homology Domain (RHD) and Ankyrin Repeat Domain (ARD) indicated by red boxes. The location of the conserved lysine (K) and two conserved serines (Phos) is indicated at top. The wild-type amino acid sequence is listed below the protein figure with the conserved lysine and serines marked with red and yellow boxes, respectively. The amino acid sequence for affected individuals in family A is shown second. The amino acid sequence of the affected individuals in family B is shown third, and the amino acid sequence for the mouse Lym1 mutation is shown fourth.
Table 1.
Clinical Features of Affected Individuals with Mutations in NFKB2
| Subject | A.II.2 (P1) | A.III.1 (P2) | A.III.2 (P3) | B.II.3 (P4) |
|---|---|---|---|---|
| Sex | female | female | male | male |
| Age at time of study | 36 yr | 13 yr | 11 yr | 24 yr |
| Age at onset of infections | childhood | 18 mo | 2 yr | <2 yr |
| Age at CVID diagnosis | 30 yr | 6 yr | 3 yr | 10 yr |
| Infections | VRIs, pneumonias, sinusitis | VRIs, otitis media | VRIs, pneumonias, sinusitis, otitis media | VRIs, pneumonias, sinusitis, otitis media |
| recurrent herpes labialis, giardia | recurrent herpes labialis, giardia | recurrent herpes labialis | ||
| Autoimmune features | none | alopecia totalis, trachyonychia, vitiligo | none | alopecia totalis, trachyonychia, psoriasiform dermatitis |
| Autoantibodies present | none | thyroid peroxidase Ab | glutamate decarboxylase Ab | thyroglobulin Ab, thyroid peroxidase Ab, (negative adrenal Ab) |
| Central adrenal insufficiency (ACTH deficiency) | + | + | + | + |
| Additional findings | asthma | asthma | asthma | asthma |
| Bell’s palsy | onychomycosis (Candida albicans culture positive) | type I Chiari malformation, otherwise normal brain MRI | interstitial lung disease with bronchial wall thickening | |
| type I Chiari malformation | ||||
| developmental delay | ||||
| aseptic meningitis | ||||
| pyloric stenosis | ||||
Abbreviations: plus sign (+), presence of; Ab, antibody, MRI, magnetic resonance imaging; VRIs, viral respiratory infections; yr, years.
Table 2.
Laboratory Findings in Affected Subjects with Mutations in NFKB2
| Subject | A.II.2 (P1)a | A.III.1 (P2)a | A.III.2 (P3)a | B.II.3 (P4)a |
|---|---|---|---|---|
| Lymphocyte Subsets | ||||
| Total lymphocyte count (lympocytes/μl) | 2,300 | 5,100 | 5,780 | 3,000 (1,500–7,000) |
| CD3+ | 2,189 (570–4,000) | 4,207∗ (1,000–2,200) | 5,374∗ (1,200–2,600) | 2,739∗ (677–2,383) |
| CD4+ | 1,512 (430–1,800) | 3,181∗ (400–2,100) | 3,409∗ (300–2,000) | 2,062∗ (425–1,509) |
| CD8+ | 672 (210–1,200) | 872 (330–920) | 1,849∗ (300–1,800) | 628 (169–955) |
| CD16+CD56+ | 47∗ (78–470) | 770∗ (70–480) | 462 (100–480) | 71∗ (101–678) |
| CD19+ | 65∗ (91–601) | 333 (200–600) | 244∗ (270–860) | 262 (99–527) |
| CD27−IgD+ naive B cells (% of CD19+) | 24.4%∗ (48.4%–79.7%)b | 89.5%∗ (51.3%–82.5%)b | 92.2%∗ (51.3%–82.5%)b | ND |
| CD27+IgD+ marginal zone/nonswitched memory B cells (% of CD19+) | 2.4%∗ (7.0%–23.8%)b | 1.7%∗ (4.6%–18.2%)b | 2.0%∗ (4.6%–18.2%)b | 0.7%∗ (1.7%–29.3%)c |
| CD27+IgD− switched memory B cells (% of CD19+) | 6.7%∗ (8.3%–27.8%)b | 5.0%∗ (8.7%–25.6%)b | 3.1%∗ (8.7%–25.6%)b | 1.9%∗ (2.3%–31.8%)c |
| Immunoglobulins | ||||
| IgG | 75∗ (639–1,349) | 148∗ (633–1,280) | 144∗ (441–1,135) | 269∗ (768–1,632) |
| IgM | 11∗ (56–352) | 12∗ (48–207) | 5∗ (47–200) | 13∗ (60–263) |
| IgA | <15∗ (70–312) | 8∗ (33–202) | 8∗ (22–159) | 16∗ (68–378) |
| IgE | ND | <2 | <2 | 0.2 |
| Vaccine Responses | ||||
| Tetanus IgG (IU/ml) | ND | ND | 0.80∗ (≥1.00)d,e | 1.61 (>0.10)d,e |
| Diphtheria IgG (IU/ml) | ND | ND | 0.05∗ (≥1.00)d,e | 0.20 (>0.10)d,e |
| Pneumococcal IgG (μg/ml) | ND | ND | ≥1.5 in 1 of 14 serotypes∗e | no response; <1.5 in all 14 serotypes∗e |
| Toll-like Receptor Function | ||||
| TLR 1-8 stimulation with measurement of IL-1β, IL-6, and TNF-α production | normal | normal | normal | normal |
| T Cell Spectratyping | ||||
| small monoclonal TCRγ monoclonal population with polyclonal background | normal | normal | ND | |
| Endocrine Studies | ||||
| Cortisol (μg/dl) | 1∗ (1–25) | <1.0∗ (1–23) | <1.1∗ (1–23) | <1.1∗ (1–23) |
| ACTH (pg/ml) | <5∗ (5–27) | 2–4∗ (5–46) | <2–2∗ (5–46) | 1–2∗ (6–55) |
| Failed ACTH stimulation test | ND | ND | + | + |
| Antithyroid antibodies | ND | thyroid peroxidase Ab 36.5∗ (0.0–3.9);d thyroglobulin Ab 6.5 (0.0–14.4)d | ND | thyroid peroxidase Ab 68.9∗ (0.0–2.0);d thyroglobulin Ab 14.3∗ (0.0–2.0)d |
| Antiadrenal antibodies | ND | ND | ND | negative |
| Other findings | mild transaminitis | |||
Asterisk (∗) indicates abnormal value; Abbreviations are as follows: plus sign (+), presence; Ab, antibody; ND, not determined; TCR, T cell receptor; TLR, toll-like receptor.
Normal values for age provided in parentheses.
Reference range obtained from Piatosa et al.33
Reference range obtained from Mayo Medical Laboratories.
Reference range obtained from ARUP Laboratories.
Obtained 1 month after immunization.
Individual P1 (A.II.2) is the biological mother of P2 (A.III.1), P3 (A.III.2), and A.III.3. She was diagnosed at 30 years of age with hypogammaglobulinemia at the time her son (P3) was diagnosed. She had a history of recurrent upper respiratory infections since childhood, including sinusitis and pneumonias. Individual P2 is a daughter of P1, born to nonconsanguineous parents of European descent, and is the full sibling of P3. She was evaluated at 6 years of age after the CVID diagnosis of P3. She had a history of recurrent infections, including multiple episodes of otitis media and asthma. She has had significant autoimmune features of alopecia areata, vitiligo, and trachyonychia (Figure S2). P2 had interim patchy hair growth after initiation of hydrocortisone replacement for adrenal insufficiency, which suggests an autoimmune mechanism of hair loss. Recent nail cultures demonstrated presence of Candida albicans. Individual P3 was evaluated at 3.5 years of age with a history of recurrent infections, asthma, and profound hypogammaglobulinemia (Table 1). He had approximately six episodes of pneumonia (several requiring hospitalization), recurrent otitis media, and asthma. After multiple episodes of hypoglycemia, he was found to have low ACTH and cortisol levels. After the administration of synthetic ACTH, his cortisol level remained low, which was consistent with central adrenal insufficiency at the hypothalamic or pituitary level. P1 and P2 were subsequently diagnosed with central adrenal insufficiency. Of note, both P3 and his healthy father (A.II.1) have type I Chiari malformations (MIM 118420). The maternal grandparents (A.I.1 and A.I.2) do not have a history of recurrent infections and have normal immunoglobulin levels and lymphocyte counts (not shown). The youngest sibling in this family (A.III.3) has not had recurrent infections and had normal immunoglobulin levels at 20 months of age.
The transmission pattern in family A suggested a Mendelian mode of inheritance, for which exome sequencing is ideally suited.34,35 Exome sequencing and genomic SNP array analysis, followed by heuristic filtering of four members of family A (A.II.1, P1, P2, and P3), showed that all three affected individuals have a heterozygous frameshift deletion (c.2564delA [p.Lys855Serfs∗7]; RefSeq accession number NM_001077494.2) in NFKB2, on chromosome 10q24 (Figure 1 and Tables S2 and S3). There were no large structural variants shared between the affected individuals in family A involving this gene or any of the known CVID causative genes (data not shown). Heuristic filtering was performed as described in Table S4.
An additional 33 CVID-affected individuals were tested for variants in the NFKB2 gene, and one person (P4; Figure 1, family B, B.II.3) with mutation c.2557C>T (p.Arg853∗; RefSeq NM_001077494.2) was identified (Table S5). Among these 33 CVID-affected individuals, this was the only one with known central adrenal insufficiency. CVID-affected individual P4 is unrelated to family A and is of northern European descent. He presented with hypogammaglobulinemia and recurrent upper respiratory infections including recurrent otitis media and pneumonia (Tables 1 and 2). He developed alopecia totalis at 10 years of age. Additional autoimmune features included trachyonychia (twenty-nail dystrophy) and psoriasiform dermatitis of the upper extremities confirmed by biopsy with presence of numerous, mainly small lymphocytes in the epidermis. He had a prolonged bout of chronic aseptic meningitis with pleomorphic lymphocytic cerebral spinal fluid at 16 years. He was later found to have central adrenal insufficiency characterized by low ACTH, low cortisol levels, intermittent hypoglycemia, and a failed ACTH stimulation test. He tested negative for anti-adrenal antibodies, which also supports a central underlying cause of adrenal insufficiency. The aseptic meningitis improved after initiation of glucocorticoid and mineralocorticoid replacement for his adrenal insufficiency, although it is not clear if the two diagnoses were related.
Both the c.2564delA and the c.2557C>T mutations are heterozygous, suggesting an autosomal-dominant mode of transmission in both families. The father (B.I.1) of P4 was unavailable for testing. Neither variant was found in dbSNP, Human Gene Mutation Database (HGMD),36 1000 Genomes,37 the Exome Variant Server database, or an internal database of 50 exomes unrelated to families A and B sequenced on the Illumina HiSeq 2000 instrument at ARUP Laboratories. Exome sequencing was then performed on P4. VAAST31 analysis with the exome data from family A alone demonstrated that NFKB2 ranked fourth genome-wide and met the criterion for genome-wide significance (p < 2.38 × 10−6) (Table S6). Addition of the exome-sequencing data from P4 into the VAAST analysis resulted in NFKB2 ranking highest, with genes ranked 2 and 3 predicted to be false positives by VarBin (Table S7). In total, four individuals affected with CVID and central adrenal insufficiency from two families (Figure 1) harbored heterozygous mutations in the same C-terminal region of NFKB2. In addition, none of the affected individuals harbored candidate causal variants in any of the genes previously associated with CVID.
B Cell Immunophenotyping Demonstrates Reduced Memory B Cells in the CVID-Affected Individuals
All individuals with NFKB2 mutations had low serum levels of IgA, IgG, and IgM (Table 2). In two of the four affected individuals, counts of CD19+ B cells were normal. B cell counts were markedly reduced in P1. Switched-memory B cells as defined by CD19+CD27+IgD− were low in all affected individuals tested, and marginal zone/nonswitched memory B cells as defined by CD19+CD27+IgD+ (Table 2 and Figure 2) were low in all affected individuals.
Figure 2.
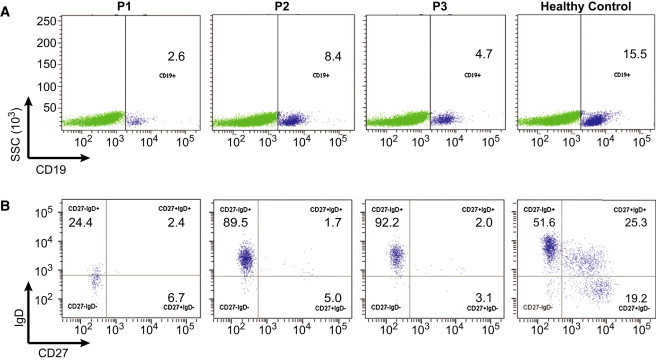
B Cell Immunophenotyping
(A) Dot plots demonstrate gating for CD19+ B cells.
(B) CD19+ B cells were then analyzed for CD27 and IgD expression.
Affected individuals (P1, P2, P3) demonstrate reduced absolute numbers and percent of switched-memory B cells (CD19+CD27+IgD−) as well as marginal zone/nonswitched memory B cells (CD19+CD27+IgD+) compared to a healthy control. In addition, P1 demonstrates markedly reduced CD19+ B cell numbers.
Substitutions p.Lys855Serfs∗7 and p.Arg853∗ Truncate NF-κB2
Full-length NF-κB2 is sequestered in the cytoplasm as the precursor p100 in a complex with RelB. Activation of NF-κB2 signaling requires a kinase cascade resulting in phosphorylation of p100 at two conserved C-terminal serines (Ser866, Ser870)38 by the IKKα kinase (Figure S1). This is followed by ubiquitination of a conserved lysine (Lys855), also located at the C terminus of p100.24 Ubiquitination acts as a signal for proteasomal processing of the p100 C terminus to generate p52, which, in its heterodimeric configuration with RelB, is then translocated into the nucleus where the active complex acts as a transcription factor.
In CVID family A, the NFKB2 c.2564delA variant is a frameshift deletion that changes the conserved Lys855 to a serine and introduces a premature stop codon at amino acid 861 (Figure 1). In family B, the NFKB2 c.2557C>T mutation introduces a premature stop codon at amino acid 853. We hypothesized that the location of the two substitutions not only would truncate NF-κB2 but also would prevent (1) phosphorylation at the critical serine sites, (2) ubiquitination, and (3) subsequent proteasomal processing into active p52. NF-κB2 is conserved through zebrafish, and the human protein shares 92% identity at the amino acid level with mouse NF-κB2. Murine Nfkb2 knockouts and an Nfkb2 mutant, c.2854A>T (Nfkb2Lym1/Lym1, nonsense mutation p.Tyr868∗), have been demonstrated to have a CVID-like phenotype.39–41 The heterozygous NFKB2 mutations in the two CVID families result in a C-terminal amino acid substitution less than 10 amino acids upstream from the murine Lym1 Nfkb2 mutation site. The Nfkb2Lym1/Lym1 mouse displays decreased serum immunoglobulins similar to the phenotype in the two CVID kindreds (Table S8).
To test the effect of the two NFKB2 mutations on protein size and stability, we isolated extracts from EBV-B cells derived from kindred controls (A.I.2 and A.II.1), an unrelated healthy pediatric control, and individuals with c.2564delA (P1, P2, and P3) and c.2557C>T (P4). Detection of NF-κB2 by immunoblotting demonstrated that the premature stop codon results in a truncated form of p100 at 94 kDa (Figures 3 and S3). Because the affected individuals are heterozygous for the respective mutations, both wild-type and mutant p100 are present in cells derived from the affected individuals.
Figure 3.
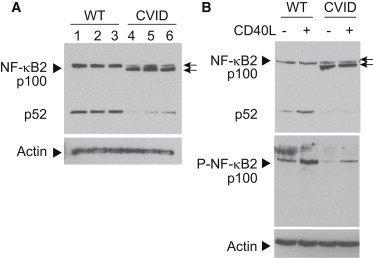
NF-κB2 Substitutions Lead to Protein Truncation and Phosphorylation Defects
(A) Immunoblot of wild-type and truncated mutant NF-κB2/p100 (arrows) from whole-cell lysates of EBV-B cells derived from unaffected (pediatric control, lane 1; A.I.2, lane 2; and A.II.1, lane 3) and affected (P2, lane 4; P3, lane 5; P4, lane 6) individuals are shown.
(B) Top panel shows immunoblot of wild-type and mutant NF-κB2 (arrows) from whole-cell lysates of EBV-B cells derived from unaffected (WT, A.II.1) and affected (CVID, P3) individuals. Cells were either treated (+) with CD40 ligand (CD40L) to stimulate the NF-κB2 pathway or left untreated (−) to compare the levels of phosphorylation in wild-type and mutant proteins. Bottom panel shows the same whole-cell lysates probed with an antibody that specifically recognizes the phosphorylated p100 protein (P-NF-κB2). Immunoblotting of actin was used as a loading control. Arrows point to full-length (top) and truncated (bottom) forms of NF-κB2. Arrowheads denote signals for protein being probed.
To test whether the truncated p100 mutant protein can be phosphorylated, we exposed cells to exogenous CD40 ligand (CD40L), a noncanonical NF-κB pathway agonist,42 and probed cell extracts with phospho-specific NF-κB2 antibody. The truncation removes the conserved phosphorylation sites required for activation of p100 to p52. This was confirmed in the mutant cell lines by the presence of phosphorylated signal on the full-length, wild-type p100 protein but not the truncated protein, as well as reduced p52 signal (Figures 3 and S3). Similar results were seen in all cell lines (data not shown). Therefore, the truncated p100 protein in the mutant cell lines cannot undergo C-terminal phosphorylation and ubiquitination required for proteosomal processing into p52.
NF-κB2 Substitutions Result in Reduced Nuclear Translocation of p52
We further investigated the effects of protein mutation (or truncation) on lymphocyte function by examining nuclear translocation of p52 and RelB, because p52:RelB dimers are the principal transcriptional activators of the noncanonical pathway. Mutant cell line immunoblots demonstrated decreased nuclear translocation of p52 and RelB compared to wild-type cell lines after upregulation of the noncanonical pathway with CD40L (Figure 4A, left). In order to phosphorylate p100, IKKα (CHUK [MIM 600664]; IκB kinase α) must first be phosphorylated by NIK (NF-κB-inducing kinase) (Figure S1), raising the possibility that NF-κB2 phosphorylation and nuclear translocation defects could result from an upstream abnormality in IKKα levels or function. Immunoblotting of total IKKα levels in the cytoplasmic fractions did not show a difference between mutant and wild-type cell lines (Figure 4A, middle). In addition, immunoblotting of phosphorylated IKKα showed that cytoplasmic phospho-IKKα levels in mutant cells were increased compared to wild-type cells, confirming that protein levels in upstream signaling molecules were not a limiting factor in reduced NF-κB2 translocation (Figure 4A, right). To determine whether the increase in phospho-IKKα protein levels in cells from CVID-affected individuals originated at the level of transcription, we performed transcriptome analyses of primary B cells stimulated with CD40L and saw no difference in CHUK expression (data not shown). Phospho-IKKα activates NF-κB2 at two C-terminal serine residues, both of which are missing in the CVID mutant proteins. This suggests that there is an accumulation of cytoplasmic phospho-IKKα resulting from a lack of NF-κB2 substrate rather than because of compensatory feedback mechanisms.
Figure 4.
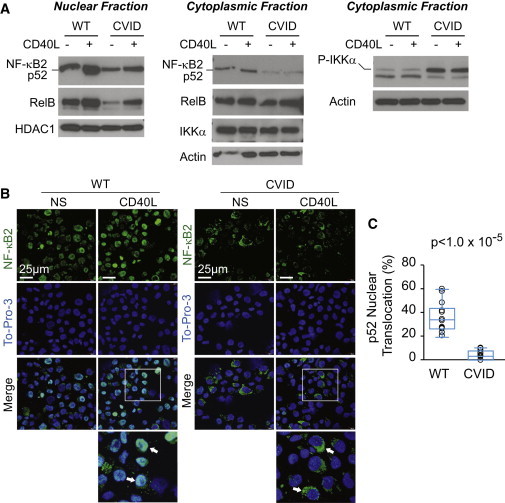
NF-κB2 Substitutions Affect Nuclear Localization of p52
(A) Nuclear (left) and cytoplasmic (middle and right) fractions of EBV-B cells derived from unaffected (WT, A.II.1) and affected (CVID, P2) individuals were subjected to immunoblotting with NF-κB2, RelB, HDAC1, IKKα, phospho-IKKα, and actin antibodies with (+) and without (−) induction with CD40 ligand (CD40L) for 4 hr. HDAC1 and actin were used as loading controls for the nuclear and cytoplasmic fractions, respectively.
(B) EBV-B cells derived from unaffected (WT, A.II.1) and affected (CVID, P3) individuals were stained with a monoclonal antibody against NF-κB2/p52 (green) and To-Pro-3 (blue) nuclear stain. Scale bars represent 25 μM. Cells were either stimulated with CD40 ligand for 4 hr (CD40L) or not stimulated (NS). Inset box in the merged CD40L images is zoomed in the image directly below. Arrows highlight WT cells with NF-κB2 nuclear colocalization, compared to those with reduced NF-κB2 nuclear colocalization in the CVID cells.
(C) Percent nuclear translocation of p52 was quantified via immunofluorescence confocal imaging of wild-type and mutant EBV-B cell lines after CD40L stimulation for 4 hr. A minimum of 300 cells per slide, per cell line were counted in the control (WT; pediatric control, A.I.2, and A.II.1) and affected (CVID; P2, P3, and P4) groups. Circles represent one microscopy field for each of three cell lines per group. n = 14 fields per group. Box plot denotes quartiles including median. Whiskers indicate min and max values. p value was calculated by a nonparametric Wilcoxon rank-sum test.
The immunoblot observations were supported by immunofluorescence microscopy in EBV-B cells from P3 (Figure 4B) and B cell lines derived from the three other CVID individuals. The EBV-B cells derived from an unaffected individual (WT; A.II.1) demonstrated increased NF-κB2 signal (green) colocalizing with the nuclear stain (To-Pro-3, blue) upon pathway stimulation by CD40L compared to unstimulated cells. In the mutant cell lines, there was a statistically significant reduced (p < 1.0 × 10−5), but not completely abolished, NF-κB2 signal colocalizing with nuclear signal upon CD40L stimulation compared to three control cell lines, suggestive of a functional haploinsufficiency resulting from the heterozygous NFKB2 mutations. Taken together, these results demonstrate that NF-κB2 nuclear translocation is reduced in the CVID-affected individuals.
Genetic Screening of Adrenal Insufficiency
Notably, all CVID-affected individuals in this study have central adrenal insufficiency, in which both ACTH and cortisol levels are low. This condition was identified after the diagnosis of CVID. Central adrenal insufficiency is an unusual feature among individuals with CVID. Genes associated with central or primary adrenal insufficiency, including PROP1 (MIM 601538), POU1F1 (MIM 173110), TBX19 (MIM 604614), POMC (MIM 176830), CRH (MIM 122560), HESX1 (MIM 601802), OTX2 (MIM 600037), LHX3 (MIM 600577), LHX4 (MIM 602146), SOX3 (MIM 313430), CRHR1 (MIM 122561), CRHR2 (MIM 602034), NR0B1 (MIM 300473), and AIRE (MIM 607358), were analyzed in our cohort for potential causative variants. Variants in these genes were considered if they met the following criteria: read depth > 8, minor allele frequency (MAF) < 5% in the 1000 Genomes Project database, location within 10 bp of an exon, and presence in affected individuals and absence in unaffected individuals. No variants met these criteria in family A. Exome sequencing data from individual P4 were analyzed separately and two synonymous variants were found in two different candidate genes (LHX3 and LHX4) for adrenal insufficiency (Table S9). Both LHX3 and LHX4 mutations have been associated with a combined pituitary hormone deficiency (MIM 221750 and 262700) involving decreased levels in multiple hormones generated by the pituitary gland as well as other syndromic features.43–45 This phenotype differs substantially from that of the described CVID-affected individuals and is therefore unlikely to be causing the central adrenal insufficiency seen in P4.
Discussion
In this study, we demonstrate that heterozygous mutations in NFKB2 cause a unique form of early-onset CVID that also presents with central adrenal insufficiency. Although CVID diagnoses occurred at 3 years of age or later, all affected individuals (P1, P2, P3, and P4) presented with recurrent infections during early childhood, have reduced serum immunoglobulin levels, and require immunoglobulin replacement therapy to prevent infections. Two of the four have significant autoimmune manifestations involving skin, hair, and nails, and all four have ACTH deficiency. The four individuals from two families harbor two distinct substitutions in the C terminus of NF-κB2. Both substitutions introduce a premature stop codon in the region of the protein that is required for posttranslational modifications, specifically phosphorylation and ubiquitination, leading to proteasomal processing. In both cases, this results in reduced protein activation and nuclear translocation.
Tucker et al. reported the Nfkb2 Lym1 mouse model, which was created by chemical mutagenesis in a forward genetic screen resulting in the nonsense mutation c.2854T>A (p.Tyr868∗) located at the C terminus of the protein in the same region as the described individuals’ mutations (Figure 1D and Table S8).40 The Nfkb2Lym1/+ heterozygous mice demonstrated an intermediate phenotype compared to the homozygous mutant, including slightly increased mortality compared to wild-type, absence of some peripheral lymph nodes, intermediate foci of lung and liver inflammatory infiltrates, and intermediate splenic architecture disruption. Nfkb2Lym1/Lym1 homozygous mice demonstrated reduced fertility, absence of all peripheral lymph nodes, 100% mortality at 250 days, severe inflammatory lesions in the lung and liver that worsened with age, and severely disorganized splenic architecture (see Table S10 for further comparisons). Although not reported in the heterozygous mutant mouse, the homozygous mutant had normal early B cell development in the bone marrow but decreased serum immunoglobulin levels and reduced peripheral B cell development, similar to the CVID phenotype seen in the affected individuals from families A and B.
In our study, we show that B cell lines from individuals with heterozygous NFKB2 mutations have defective processing of p100 to p52, and p52 translocation to the nucleus is reduced but not completely blocked upon stimulation of the noncanonical pathway (Figure 3). Processing of p52 in the heterozygous Nfkb2Lym1/+ mouse had similar defects, with reduced but not completely abrogated nuclear translocation (Table S10). This is in contrast to the homozygous Nfkb2Lym1/Lym1 mouse, which showed a complete absence of p52 upon stimulation of the noncanonical pathway. The Nfkb2Lym1/+ mouse model is consistent with our two CVID families, in which there appears to be a functional haploinsufficiency as a result of the heterozygous C-terminal NFKB2 mutations. Although we cannot absolutely exclude the possibility that expressed, truncated alleles inhibit the pathway, we believe that haploinsufficiency is the more likely mechanism. If the mutations in the CVID individuals were acting in a dominant-negative manner with respect to NF-κB2 nuclear translocation, we would expect that no or very little p52 from the wild-type copy of NF-κB2 would be translocated to the nucleus. Toll-like receptor assays in the individuals were normal compared to controls, suggesting normal canonical NF-κB signaling. Finally, Liang et al. demonstrated that cell lines with mutations in either of the critical C-terminal serine phosphorylation sites result in blocked p100 processing.46 Thus, we suspect that the leaky defect, in which some p52 is translocated to the nucleus in the families with heterozygous NFKB2 mutations, is a result of the presence of the wild-type p100, albeit in lower amounts compared to controls, which can be processed and ubiquitinated, and ultimately be translocated to the nucleus for transcriptional regulation. Our data suggest that CVID pathogenesis in humans with heterozygous NFKB2 mutations results from haploinsufficiency restricted to the noncanonical NF-κB pathway.
There are enough similarities to suggest that the Lym1 mouse provides an excellent model system for further study of the relationship between NFKB2 and CVID. The B cell maturational defects in the described individuals provide additional evidence to support the causality of the two NFKB2 mutations in our families. Mouse model knockouts and mutants in the noncanonical NF-κB pathway, including the Nfkb2 knockout mouse (Table S8),39,40 have aberrant splenic and lymph node architecture affecting germinal centers, which are critical for B cell differentiation, as well as reduced antigen-specific antibody responses. Individuals with CVID may have normal total B cell numbers, but the memory, class-switched, and marginal zone B cells are often reduced in those with increased complications of the disease, which are indicative of defects in peripheral and late B cell differentiation.47,48 All four of the CVID-affected individuals reported here have reduced antibody levels, poor response to immunizations, and reduced memory and marginal zone B cells.
The effects of NF-κB2 are not limited to lymphoid organ development and B cell maturation. The biological roles of the pathway include thymic development49 and T cell differentiation.24 The relationship of the noncanonical NF-κB signaling pathway to thymic development and autoimmune regulator (AIRE) expression has important implications for central tolerance50,51 and can provide a mechanism for development of autoimmunity in CVID. AIRE is a transcription factor required for expression of self-tissue antigens in the thymus, which removes autoreactive T cells, ensuring an immune competent T cell repertoire that is also self-tolerant. Reduced AIRE expression can cause self-reactive T cells to enter the circulation. This results in autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED or APS-1 [MIM 240300]),52 a syndrome with clinical heterogeneity characterized by mucocutaneous candidal infections as well as autoimmune organ disease, particularly involving the endocrine glands. The candidal infections may in part be due to autoantibodies against cytokines that are important for the immune response against Candida,53,54 and the nail dystrophy and alopecia seen in many of these individuals are additional autoimmune findings. The Lym1 mouse demonstrates reduced thymocyte AIRE expression.40 P2 has nail dystrophy, candidal nail infection, alopecia, and central adrenal insufficiency, bearing very striking similarities to APECED. P4 has nail dystrophy, alopecia, and central adrenal insufficiency. These observations may provide future directions for studying the autoimmunity seen in the 20% of CVID individuals with autoimmune features.55,56 Interestingly, none of our described CVID individuals has demonstrated autoimmune blood cytopenias, which is one of the more common autoimmune findings in CVID. Thus, specific gene mutations in CVID-affected individuals may have specific phenotypic consequences, and the mutations in NFKB2 may not result in autoimmune cytopenias, but may result in other autoimmune sequelae. By extension, mutations in other genes, known or yet to be identified as CVID causative, may have other autoimmune sequelae such as cytopenias, and remain to be investigated in further molecular detail.
A unique feature in our CVID cohort is the presence of central adrenal insufficiency. Although rare, individuals with hypogammaglobulinemia or CVID as well as ACTH deficiency have been reported without a known genetic etiology.57–59 In addition, NFKBIA (NF-κB inhibitor IκBα [MIM 164008]) is a critical gene in the canonical NF-κB pathway, with hypermorphic mutations known to cause autosomal-dominant ectodermal dysplasia with immunodeficiency (EDA-ID [MIM 612132]). A recent study described an individual with a heterozygous NFKBIA mutation who, in addition to the standard EDA-ID phenotype, also had a polyendocrinopathy with hypothyroidism and hypopituitarism.60 Adrenal insufficiency, whether central or primary, can result from congenital, autoimmune, infectious, or trauma-related causes. Adrenal function has not been assessed in the Lym1 or Nfkb2 knockout mouse models. The cause of the ACTH deficiency in the CVID-affected individuals reported here is unknown, but known genetic causes were ruled out through filtering of shared variants as well as screening of known genes associated with adrenal insufficiency. This suggests that the NFKB2 variants are causative for both the CVID and the central adrenal insufficiency (resulting from ACTH deficiency) phenotypes. The link between the noncanonical NF-κB pathway and adrenal or pituitary development and function has not previously been described but should be further investigated in mouse models. Based on what is known in APECED and Nfkb2 mouse models, we speculate that alterations in thymic AIRE expression may result in aberrant T cell-mediated self-tolerance of endocrine-related organs.
Adrenal insufficiency can cause severe hypoglycemia, seizures, electrolyte imbalances, and nonspecific symptoms such as anorexia, nausea, vomiting, abdominal pain, and weakness. The signs and symptoms of adrenal insufficiency may become apparent only during times of stress or illness. P4 had a prolonged bout of presumed aseptic meningitis with nausea and vomiting, which in part seemed to be corrected by addressing the adrenal deficiency through steroid supplementation. P3 developed hypoglycemia after an infection. In individuals with CVID, we suggest that clinicians have a high clinical suspicion and low screening threshold for adrenal insufficiency to reduce morbidity and mortality associated with this easily treated condition.
The genetic etiology of 85%–90% of CVID cases remains unknown. Here we report four individuals with highly penetrant NFKB2 variants in a collection of 37 CVID-affected individuals, making this gene an excellent candidate for diagnostic testing. The mainstay of CVID treatment is lifelong immune globulin replacement, but immune globulin does not treat the autoimmune features of the disease. With a better understanding of the molecular pathogenesis of CVID, drugs targeting specific pathways can potentially be developed to allow for immune reconstitution, addressing both the immune deficient as well as autoimmune components seen in affected individuals. In conclusion, our findings identify aberrant NF-κB2 function as a cause of CVID with adrenal insufficiency and describe two unique germline human mutations in NFKB2. Further studies of the NF-κB2 signaling pathway will increase our understanding of the genetic and molecular mechanisms underlying CVID pathogenesis that could eventually lead to improved therapeutics.
Acknowledgments
The authors would like to thank the families for their willingness to participate in this study; the Phenotyping Core in the Department of Pediatrics, Division of Medical Genetics at the University of Utah including study coordinators Megan Grandemange, Kyle Berg, Suzy Jones, and Ann Rutherford; the University of Utah School of Medicine Cell Imaging Facility; and Kalyan Mallempati, Mark Cody, C. Con Yost, and Eugene Ravkov for their assistance. We would also like to thank Diana Lim for assistance with figure generation. Confocal images were obtained at the University of Utah School of Medicine Cell Imaging Facility. DNA extractions and B cell transformation was performed by the University of Utah Center for Clinical & Translational Science (CCTS) Translational Technologies & Resources (TTR) core. This research was funded in part by an NIH 1R21AI094004-01 grant to K.V.V., A.K., and H.R.H.; NIH 5K12HD001410-10 Child Health Research Career Development Award (PI: E.B. Clark) from the University of Utah Department of Pediatrics to K.C.; and the Primary Children’s Medical Center Foundation Early Career Development Award to K.C. L.B.J. is supported by NIH grant GM59290 and the Utah Genome Project. A.S.W. and G.A.Z. are supported by NIH U54HL112311-01. G.A.Z. is also supported by NIH R37HL6044525. The views expressed in this article are those of the author (A.V.G.) and do not necessarily represent the views of the Department of Veterans Affairs.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
Human Gene Mutation Database, http://www.hgmd.org/
Human Genome Reference Consortium, http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
SAMtools, http://samtools.sourceforge.net/
Accession Numbers
The dbSNP accession numbers for the NFKB2 c.2564delA (p.Lys855Serfs∗7) and NFKB2 c.2557C>T (p.Arg853∗) sequences reported in this paper are rs397514331 and rs397514332, respectively.
References
- 1.Primary immunodeficiency diseases. Report of a WHO scientific group. Clin. Exp. Immunol. 1997;109(Suppl 1):1–28. [PubMed] [Google Scholar]
- 2.Oksenhendler E., Gérard L., Fieschi C., Malphettes M., Mouillot G., Jaussaud R., Viallard J.F., Gardembas M., Galicier L., Schleinitz N., DEFI Study Group Infections in 252 patients with common variable immunodeficiency. Clin. Infect. Dis. 2008;46:1547–1554. doi: 10.1086/587669. [DOI] [PubMed] [Google Scholar]
- 3.Geha R.S., Notarangelo L.D., Casanova J.L., Chapel H., Conley M.E., Fischer A., Hammarström L., Nonoyama S., Ochs H.D., Puck J.M., International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J. Allergy Clin. Immunol. 2007;120:776–794. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunningham-Rundles C., Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin. Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 5.Hammarström L., Vorechovsky I., Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID) Clin. Exp. Immunol. 2000;120:225–231. doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salzer U., Unger S., Warnatz K. Common variable immunodeficiency (CVID): exploring the multiple dimensions of a heterogeneous disease. Ann. N Y Acad. Sci. 2012;1250:41–49. doi: 10.1111/j.1749-6632.2011.06377.x. [DOI] [PubMed] [Google Scholar]
- 7.van Zelm M.C., Reisli I., van der Burg M., Castaño D., van Noesel C.J., van Tol M.J., Woellner C., Grimbacher B., Patiño P.J., van Dongen J.J., Franco J.L. An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 8.Kuijpers T.W., Bende R.J., Baars P.A., Grummels A., Derks I.A., Dolman K.M., Beaumont T., Tedder T.F., van Noesel C.J., Eldering E., van Lier R.A. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J. Clin. Invest. 2010;120:214–222. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiel J., Kimmig L., Salzer U., Grudzien M., Lebrecht D., Hagena T., Draeger R., Völxen N., Bergbreiter A., Jennings S. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J. Allergy Clin. Immunol. 2012;129:801–810. doi: 10.1016/j.jaci.2011.09.027. e6. [DOI] [PubMed] [Google Scholar]
- 10.Grimbacher B., Hutloff A., Schlesier M., Glocker E., Warnatz K., Dräger R., Eibel H., Fischer B., Schäffer A.A., Mages H.W. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat. Immunol. 2003;4:261–268. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 11.Warnatz K., Salzer U., Rizzi M., Fischer B., Gutenberger S., Böhm J., Kienzler A.K., Pan-Hammarström Q., Hammarström L., Rakhmanov M. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc. Natl. Acad. Sci. USA. 2009;106:13945–13950. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castigli E., Wilson S.A., Garibyan L., Rachid R., Bonilla F., Schneider L., Geha R.S. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat. Genet. 2005;37:829–834. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 13.Salzer U., Chapel H.M., Webster A.D., Pan-Hammarström Q., Schmitt-Graeff A., Schlesier M., Peter H.H., Rockstroh J.K., Schneider P., Schäffer A.A. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat. Genet. 2005;37:820–828. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 14.Ombrello M.J., Remmers E.F., Sun G., Freeman A.F., Datta S., Torabi-Parizi P., Subramanian N., Bunney T.D., Baxendale R.W., Martins M.S. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N. Engl. J. Med. 2012;366:330–338. doi: 10.1056/NEJMoa1102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Zelm M.C., Smet J., Adams B., Mascart F., Schandené L., Janssen F., Ferster A., Kuo C.C., Levy S., van Dongen J.J., van der Burg M. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J. Clin. Invest. 2010;120:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Herrera G., Tampella G., Pan-Hammarström Q., Herholz P., Trujillo-Vargas C.M., Phadwal K., Simon A.K., Moutschen M., Etzioni A., Mory A. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am. J. Hum. Genet. 2012;90:986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salzer E., Santos-Valente E., Klaver S., Ban S.A., Emminger W., Prengemann N.K., Garncarz W., Müllauer L., Kain R., Boztug H. B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase C δ. Blood. 2013;121:3112–3116. doi: 10.1182/blood-2012-10-460741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vetrie D., Vorechovský I., Sideras P., Holland J., Davies A., Flinter F., Hammarström L., Kinnon C., Levinsky R., Bobrow M. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 19.Pan-Hammarström Q., Salzer U., Du L., Björkander J., Cunningham-Rundles C., Nelson D.L., Bacchelli C., Gaspar H.B., Offer S., Behrens T.W. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat. Genet. 2007;39:429–430. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castigli E., Wilson S., Garibyan L., Rachid R., Bonilla F., Schneider L., Morra M., Curran J., Geha R. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat. Genet. 2007;39:430–431. doi: 10.1038/ng0407-430. [DOI] [PubMed] [Google Scholar]
- 21.Orange J.S., Glessner J.T., Resnick E., Sullivan K.E., Lucas M., Ferry B., Kim C.E., Hou C., Wang F., Chiavacci R. Genome-wide association identifies diverse causes of common variable immunodeficiency. J. Allergy Clin. Immunol. 2011;127:1360–1367. doi: 10.1016/j.jaci.2011.02.039. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonizzi G., Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Hayden M.S., Ghosh S. NF-κB in immunobiology. Cell Res. 2011;21:223–244. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fracchiolla N.S., Lombardi L., Salina M., Migliazza A., Baldini L., Berti E., Cro L., Polli E., Maiolo A.T., Neri A. Structural alterations of the NF-kappa B transcription factor lyt-10 in lymphoid malignancies. Oncogene. 1993;8:2839–2845. [PubMed] [Google Scholar]
- 26.Keats J.J., Fonseca R., Chesi M., Schop R., Baker A., Chng W.J., Van Wier S., Tiedemann R., Shi C.X., Sebag M. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shanmugam R., Gade P., Wilson-Weekes A., Sayar H., Suvannasankha A., Goswami C., Li L., Gupta S., Cardoso A.A., Al Baghdadi T. A noncanonical Flt3ITD/NF-κB signaling pathway represses DAPK1 in acute myeloid leukemia. Clin. Cancer Res. 2012;18:360–369. doi: 10.1158/1078-0432.CCR-10-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yandell M., Huff C., Hu H., Singleton M., Moore B., Xing J., Jorde L.B., Reese M.G. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011;21:1529–1542. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Margraf R.L., Durtschi J.D., Dames S., Pattison D.C., Stephens J.E., Voelkerding K.V. Variant identification in multi-sample pools by illumina genome analyzer sequencing. J. Biomol. Tech. 2011;22:74–84. [PMC free article] [PubMed] [Google Scholar]
- 33.Piątosa B., Wolska-Kuśnierz B., Pac M., Siewiera K., Gałkowska E., Bernatowska E. B cell subsets in healthy children: reference values for evaluation of B cell maturation process in peripheral blood. Cytometry B Clin. Cytom. 2010;78:372–381. doi: 10.1002/cyto.b.20536. [DOI] [PubMed] [Google Scholar]
- 34.Ku C.S., Naidoo N., Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum. Genet. 2011;129:351–370. doi: 10.1007/s00439-011-0964-2. [DOI] [PubMed] [Google Scholar]
- 35.Coonrod E.M., Durtschi J.D., Margraf R.L., Voelkerding K.V. Developing genome and exome sequencing for candidate gene identification in inherited disorders: an integrated technical and bioinformatics approach. Arch. Pathol. Lab. Med. 2013;137:415–433. doi: 10.5858/arpa.2012-0107-RA. [DOI] [PubMed] [Google Scholar]
- 36.Stenson P.D., Ball E.V., Mort M., Phillips A.D., Shiel J.A., Thomas N.S., Abeysinghe S., Krawczak M., Cooper D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 37.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao G., Harhaj E.W., Sun S.C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 39.Caamaño J.H., Rizzo C.A., Durham S.K., Barton D.S., Raventós-Suárez C., Snapper C.M., Bravo R. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J. Exp. Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tucker E., O’Donnell K., Fuchsberger M., Hilton A.A., Metcalf D., Greig K., Sims N.A., Quinn J.M., Alexander W.S., Hilton D.J. A novel mutation in the Nfkb2 gene generates an NF-kappa B2 “super repressor”. J. Immunol. 2007;179:7514–7522. doi: 10.4049/jimmunol.179.11.7514. [DOI] [PubMed] [Google Scholar]
- 41.Franzoso G., Carlson L., Poljak L., Shores E.W., Epstein S., Leonardi A., Grinberg A., Tran T., Scharton-Kersten T., Anver M. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J. Exp. Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coope H.J., Atkinson P.G., Huhse B., Belich M., Janzen J., Holman M.J., Klaus G.G., Johnston L.H., Ley S.C. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonfig W., Krude H., Schmidt H. A novel mutation of LHX3 is associated with combined pituitary hormone deficiency including ACTH deficiency, sensorineural hearing loss, and short neck-a case report and review of the literature. Eur. J. Pediatr. 2011;170:1017–1021. doi: 10.1007/s00431-011-1393-x. [DOI] [PubMed] [Google Scholar]
- 44.Machinis K., Pantel J., Netchine I., Léger J., Camand O.J., Sobrier M.L., Dastot-Le Moal F., Duquesnoy P., Abitbol M., Czernichow P., Amselem S. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am. J. Hum. Genet. 2001;69:961–968. doi: 10.1086/323764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfaeffle R.W., Hunter C.S., Savage J.J., Duran-Prado M., Mullen R.D., Neeb Z.P., Eiholzer U., Hesse V., Haddad N.G., Stobbe H.M. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J. Clin. Endocrinol. Metab. 2008;93:1062–1071. doi: 10.1210/jc.2007-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang C., Zhang M., Sun S.C. beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell. Signal. 2006;18:1309–1317. doi: 10.1016/j.cellsig.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 47.Warnatz K., Denz A., Dräger R., Braun M., Groth C., Wolff-Vorbeck G., Eibel H., Schlesier M., Peter H.H. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–1551. doi: 10.1182/blood.v99.5.1544. [DOI] [PubMed] [Google Scholar]
- 48.Piqueras B., Lavenu-Bombled C., Galicier L., Bergeron-van der Cruyssen F., Mouthon L., Chevret S., Debré P., Schmitt C., Oksenhendler E. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J. Clin. Immunol. 2003;23:385–400. doi: 10.1023/a:1025373601374. [DOI] [PubMed] [Google Scholar]
- 49.Boehm T., Scheu S., Pfeffer K., Bleul C.C. Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR. J. Exp. Med. 2003;198:757–769. doi: 10.1084/jem.20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akiyama T., Shimo Y., Yanai H., Qin J., Ohshima D., Maruyama Y., Asaumi Y., Kitazawa J., Takayanagi H., Penninger J.M. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. doi: 10.1016/j.immuni.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 51.Chin R.K., Lo J.C., Kim O., Blink S.E., Christiansen P.A., Peterson P., Wang Y., Ware C., Fu Y.X. Lymphotoxin pathway directs thymic Aire expression. Nat. Immunol. 2003;4:1121–1127. doi: 10.1038/ni982. [DOI] [PubMed] [Google Scholar]
- 52.Nagamine K., Peterson P., Scott H.S., Kudoh J., Minoshima S., Heino M., Krohn K.J., Lalioti M.D., Mullis P.E., Antonarakis S.E. Positional cloning of the APECED gene. Nat. Genet. 1997;17:393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 53.Kisand K., Bøe Wolff A.S., Podkrajsek K.T., Tserel L., Link M., Kisand K.V., Ersvaer E., Perheentupa J., Erichsen M.M., Bratanic N. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puel A., Döffinger R., Natividad A., Chrabieh M., Barcenas-Morales G., Picard C., Cobat A., Ouachée-Chardin M., Toulon A., Bustamante J. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010;207:291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chapel H., Lucas M., Lee M., Bjorkander J., Webster D., Grimbacher B., Fieschi C., Thon V., Abedi M.R., Hammarstrom L. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 56.Agarwal S., Cunningham-Rundles C. Autoimmunity in common variable immunodeficiency. Curr. Allergy Asthma Rep. 2009;9:347–352. doi: 10.1007/s11882-009-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tovo P.A., Lala R., Martino S., Pastorelli G., De Sanctis C. Isolated adrenocorticotropic hormone deficiency associated with common variable immunodeficiency. Eur. J. Pediatr. 1991;150:400–402. doi: 10.1007/BF02093717. [DOI] [PubMed] [Google Scholar]
- 58.Quentien M.H., Delemer B., Papadimitriou D.T., Souchon P.F., Jaussaud R., Pagnier A., Munzer M., Jullien N., Reynaud R., Galon-Faure N. Deficit in anterior pituitary function and variable immune deficiency (DAVID) in children presenting with adrenocorticotropin deficiency and severe infections. J. Clin. Endocrinol. Metab. 2012;97:E121–E128. doi: 10.1210/jc.2011-0407. [DOI] [PubMed] [Google Scholar]
- 59.Younes J.S., Secord E.A. Panhypopituitarism in a child with common variable immunodeficiency. Ann. Allergy Asthma Immunol. 2002;89:322–325. doi: 10.1016/S1081-1206(10)61963-1. [DOI] [PubMed] [Google Scholar]
- 60.Schimke L.F., Rieber N., Rylaarsdam S., Cabral-Marques O., Hubbard N., Puel A., Kallmann L., Sombke S.A., Notheis G., Schwarz H.P. A novel gain-of-function IKBA mutation underlies ectodermal dysplasia with immunodeficiency and polyendocrinopathy. J. Clin. Immunol. 2013;33:1088–1099. doi: 10.1007/s10875-013-9906-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.