

Abstract
Prions are unconventional infectious agents that are composed of misfolded aggregated prion protein. Prions replicate their conformation by template-assisted conversion of the endogenous prion protein PrP. Templated conversion of soluble proteins into protein aggregates is also a hallmark of other neurodegenerative diseases. Alzheimer's disease or Parkinson's disease are not considered infectious diseases, although aggregate pathology appears to progress in a stereotypical fashion reminiscent of the spreading behavior ofmammalian prions. While basic principles of prion formation have been studied extensively, it is still unclear what exactly drives PrP molecules into an infectious, self-templating conformation. In this review, we discuss crucial steps in the life cycle of prions that have been revealed in ex vivo models. Importantly, the persistent propagation of prions in mitotically active cells argues that cellular processes are in place that not only allow recruitment of cellular PrP into growing prion aggregates but also enable the multiplication of infectious seeds that are transmitted to daughter cells. Comparison of prions with other protein aggregates demonstrates that not all the characteristics of prions are equally shared by prion-like aggregates. Future experiments may reveal to which extent aggregation-prone proteins associated with other neurodegenerative diseases can copy the replication strategies of prions.
1. Prions—Infectious Agents Composed Predominately of Protein
Prion diseases or transmissible spongiform encephalopathies (TSEs) are invariably fatal neurodegenerative diseases that are associated with severe spongiform vacuolation and nerve cell loss [1]. Animal and human TSEs are infectious diseases that either naturally spread between individuals of the same species or have been accidentally transmitted through food contaminants, blood and medical products, or during surgery [1]. Animal prion diseases include scrapie in sheep and goats, bovine spongiform encephalopathy, and chronic wasting disease in elk and deer [1]. TSEs in humans also occur sporadically or are of familial origin. Human prion diseases manifest as Creutzfeldt-Jakob disease, fatal familial insomnia, Gerstmann-Sträussler-Scheinker syndrome and Kuru. Familial prion diseases are associated with dominantly inherited mutations in the coding region of the cellular prion protein. Many prion strains have also been successfully adapted to laboratory animals. Prion strains can be propagated in the same inbred mouse lines, where they produce phenotypically distinct neurological diseases [2]. Interestingly, prion strains selectively target specific brain regions, while leaving others unaffected [3, 4]. During the course of the disease, the cellular prion protein PrPC misfolds and aggregates in the affected brain areas, leading to accumulation of abnormal PrP (termed PrPSc) intracellularly or extracellularly. PrPSc purified from brains of diseased animals is closely associated with prion infectivity, arguing for a causal relation between the conformational state of the protein and its infectious properties [5]. According to the prion hypothesis [5] and compelling new evidence [6–8], prions constitute a class of proteinaceous infectious agents composed almost exclusively of protein without coding nucleic acid. For propagation, PrPSc acts as a template that catalyzes the conformational conversion of the cellular prion protein PrPC. The existence of prion strains that can propagate in the same inbred mouse lines has posed a conundrum to the prion hypothesis. Accumulating evidence now argues that prion strains are encoded by different PrP conformers capable of faithfully replicating their specific structure in the affected hosts. The strain-specific biological and biochemical signatures are likely enciphered by conformational variants of PrPSc [9–11].
The precursor of PrPSc, PrPC, is a natively folded protein, with an unstructured amino terminal domain. Mature PrPC is a glycosylphosphatidylinositol- (GPI-) anchored membrane protein that is abundantly expressed in the central nervous system but also in other tissues. The exact function of PrPC has not been elucidated, but several studies suggest it has a cytoprotective function in neurons [12]. PrPSc fundamentally differs from PrPC in its biophysical properties. PrPSc has a high β-sheet content, is insoluble in nonionic detergents, and its globular domain (approximately amino acid residues 89–230) is resistant to proteinase K (PK) [13]. Treatment of histological specimen or tissue lysates with PK is used routinely to identify prions in biological samples. It is important to note, however, that the exact PrP conformer with infectious properties has not been defined. Recent data argue that infectious PrP propagated in vivo and in cell culture can also be PK-sensitive, adding another layer of complexity to the characterization of infectious proteins [10, 14]. For simplicity, we will refer to infectious PrP molecules as PrPSc.
Propagation of prions is thought to occur through a process of nucleation-dependent polymerization, in which a seed of aggregated PrPSc templates the conformational conversion of its soluble homotypic isoform. The initial step of seed or oligomer formation is a slow and rate limiting process. In a subsequent elongation step, monomeric protein is recruited into the structurally ordered β-pleated fibrils, so-called amyloid. In a third step, secondary nucleation events such as filament fragmentation produce additional seeds that elongate and multiply [15]. Flow field-flow fractionation has recently been used to separate prions by size, demonstrating that particles composed of 14–28 monomers exhibit the highest infection properties in vivo [16]. Two lines of evidence argue that aggregate shearing is crucial for prion multiplication. First, aggregate fragmentation is an essential step in the so-called protein misfolding cyclic amplification (PMCA) developed by Saborio and colleagues [17]. In this assay, PrPSc present in brain homogenate serves as a template that is mixed with substrate PrPC present in normal, uninfected brain homogenate. Consecutive steps of incubation and sonication catalyze PrPSc growth and segregation, leading to an exponential increase of prion polymers with infectious properties. Second, protein aggregate fragmentation is crucial for the propagation of prion-like protein aggregates in yeast and filamentous fungi [18, 19]. Prions in lower eukaryotes are protein-only epigenetic elements of inheritance that replicate by a seeded polymerization/fragmentation process similar to mammalian prions. Interestingly, yeast prions are fragmented by chaperone Hsp104 in conjunction with additional chaperones [20, 21]. Hsp104 has no homologue in mammalian cells, and the in vivo mechanism of mammalian prion fragmentation is so far unknown.
2. PrPSc Formation in Cell Culture
The establishment of prion cell culture models has greatly enhanced our understanding of the cellular mechanisms of prion formation. However, even decades after the first successful prion infection ex vivo, many aspects of prion replication still remain elusive. The most puzzling observation is that the susceptibility of a given cell line can only be determined empirically. Most PrPC expressing cell lines are refractory to mammalian prion infection for unknown reasons [22, 23]. Prion strains also demonstrate an exquisite host cell tropism not only in vivo [3, 4] but also in tissue culture cells [24–29]. Prion propagation ex vivo is not restricted to neuronal cells, and also epithelial cells or fibroblast cell lines are permissive to certain strains. Many prion strains from various origins have never been successfully propagated in cell culture [30]. These observations suggest that so far unidentified strain and host cell specific factors control the replication of prion strains. Because of the usually low infection rates, most studies have been performed with previously established cell lines persistently infected with prion strains RML or 22L. In the following sections, we will briefly review basic findings on the uptake, the initiation of an infection, and the propagation of prions in permanent cell culture models.
Uptake of PrPSc is an early step of prion infection and is independent of PrPC expression [31]. PrPSc uptake is also observed in nonpermissive cell lines and thus not indicative of a productive infection [31, 32]. PrPSc formation in cell culture is initiated once PrPC has been translocated to the cell surface [33]. Substantial amounts of newly formed PrPSc are already detectable within minutes to hours post exposure [34, 35]. Importantly, transient PrPSc formation has been demonstrated in resistant cell lines, arguing that initial seeding of endogenous PrPSc does not necessarily lead to a successful prion infection ex vivo [34]. The continuous presence of PrPSc over multiple cell passages is indicative of a productive infection of the cell culture. Abnormal prion protein accumulation is routinely detected by western blot analysis of cell lysates treated with PK (50 μg/mL, 37°C, 1 hr) or by indirect immunofluorescence in fixed, prion infected cells following antigen retrieval by harsh denaturants such as guanidinium hydrochloride [36], formic acid [35] or partial proteolysis by proteinase K [36, 37]. In cultured cells, PrPSc is mainly confined to vesicles of the endocytic pathway, including early endosomes, recycling endosomes, and lysosomes [37–40]. Lipid rafts [41–43] and/or endocytic recycling compartments [40, 44] likely constitute sites of PrPSc formation. PrPSc produced in cell cultures has a half-life time of approximately 30 hrs [45]. Both lysosomes and autophagosomes have been implicated in PrPSc clearance [46–48]. Importantly, with one exception, prions in permanent cell lines do not induce visible morphological or pathological changes [49].
3. Sustained Propagation of Mammalian Prions in Culture
Vertical spreading from mother to daughter cells is a prominent feature of mammalian prions in tissue culture (Figure 1(a)) [50]. Under the right cell culture conditions, mammalian prions can be propagated ex vivo indefinitely. The mouse neuroblastoma cell line N2a infected with RML/Chandler strain in the late 1980's [51, 52] has been distributed throughout the world and still serves as the prototype cell line for studying cellular aspects of prion propagation. Cell division affects the aggregate load of the cell, diluting the number of infectious particles by half (Figure 2). The continuous prion propagation in cell culture implies that proper fragmentation and partitioning mechanisms are in place for seed multiplication. It is possible that large PrPSc aggregates are segregated by mechanical force, for example, during endocytosis, thereby producing smaller prion entities. Alternatively, unidentified cofactors catalyze the fragmentation of larger prion aggregates. Prion propagation in mammals is confined to the cell surface or endocytic vesicles, suggesting that cofactors necessary for aggregate fragmentation reside in the same cellular compartments. It is tempting to speculate that cellular quality control mechanisms are also exploited by mammalian prions to disassemble high molecular weight prion aggregates into smaller infectious entities. However, the disaggregase Hsp104 necessary for production of infectious prion entities in yeast does not exist in mammalian cells. Several chaperones have been identified that interact with or regulate PrP folding and misfolding in mammalian cells [53–55], but their contribution to prion propagation is unclear. Incomplete clearance of prion particles, for example, via lysosomes or autophagy, could potentially contribute to prion particle fragmentation. This hypothesis is supported by the observation that infection of autophagy-deficient mouse embryonic fibroblasts with mammalian prions was significantly enhanced by ectopic expression of autophagy-related protein ATG5 [56].
Figure 1.
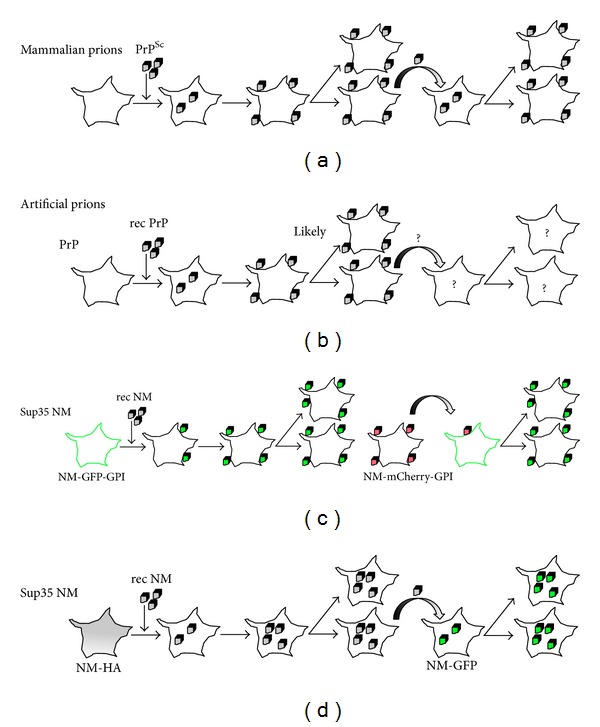
Induction of a PrP or Sup35 NM prion phenotype in mammalian cells. (a) Mammalian prions can infect selected cell cultures. Exposure of permissive cells to brain homogenate containing PrPSc leads to infection of cells. Prions persist mainly through stable inheritance of PrPSc aggregates by daughter cells. Prions also spread to adjacent cells by cell-to-cell contact or exosomes and induce productive infection in recipient cells. (b) Artificial prions produced in vitro from recombinant prion proteins. PrP aggregates derived from PrP and minimal components by PMCA are infectious to some cell lines. The increase in PrPSc over time suggests that prions propagate, likely by vertical transmission to daughter cells. Spreading to adjacent cells has not been studied so far. (c) Recombinant NM fibrils produce a heritable NM aggregation phenotype in N2a cells expressing a GPI-anchored NM-GFP fusion protein. When N2a cells that spontaneously form mCherry-tagged NM-GPI aggregates are cocultured with NM-GFP-GPI expressing cells, they induce NM aggregation in neighboring cells. (d) Cytosolically expressed NM-HA is soluble but can be induced to aggregate upon addition of recombinant NM fibrils. The NM aggregate phenotype is transmitted vertically and horizontally.
Figure 2.
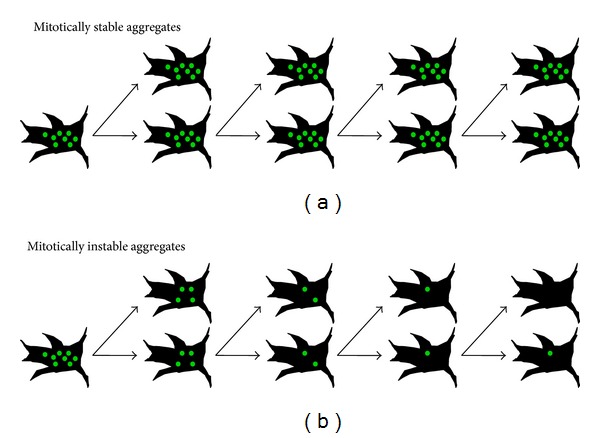
Mitotic stability of aggregate phenotypes is indicative of aggregate fragmentation. Dividing cells are a good model system to study propagation propensities of protein aggregates. (a) Proteins that can propagate as prions are mitotically stable in tissue culture, meaning that they are bidirectionally transmitted to daughter cells. Generation of infectious seeds that can self-propagate must be at least as fast as cell division. Prion aggregates must also be capable of escaping effective cellular clearance mechanisms. PrPSc, NM derived prions [57, 58], and some SOD1 mutants [59] fulfill these criteria. (b) Many protein aggregates are mitotically instable. Unidirectional segregation during cell division might represent an evolutionary conserved mechanism to protect a subset of the progeny cells from toxic effects of protein aggregates. Relatively poor mitotic stability has been reported for polyQ aggregates [60, 61].
4. Prion Infection Spreads to Adjacent Cells
Prions ex vivo are not only transmitted to progeny cells, but they also spread to neighboring cells (Figure 1(a)). Dissemination of mammalian prions in vitro involves at least two independent routes. Horizontal transmission of prions induces a prion phenotype in the recipient cells that spreads again both vertically and horizontally. In some cell culture models, prions were secreted into the cell culture supernatant [29, 49, 63]. Several studies demonstrated that prions are often associated with exosomes released by the donor cells. Exosomes containing PrPSc have been shown to efficiently initiate prion propagation in recipient cells [64–68]. How exosomes make contact with the recipient cells and how incorporated prions then induce infection is currently unknown. Direct proximity between donor and recipient cells drastically increased the infection in other cell culture models [69]. In some instances, prions travel through cytoplasmic bridges, so-called tunneling nanotubes that form transiently between cells [70]. These data suggest that prions can utilize several distinct routes for efficient cellular spreading. So far, it is unclear if the observed differences in spreading mechanisms are due to different cell culture models or strain-specific dissemination strategies. Of note, horizontal transmission of prions in cell culture is generally much less efficient than vertical transmission to daughter cells [50].
5. Not All PrP Aggregates Are Infectious
The conformational transition of cellular prion protein to a misfolded, aggregated isoform is believed to be the underlying principle of prion formation, but PrP expression levels, mutations, impairment of the cellular quality control mechanisms, and some chemicals also trigger formation of PrP aggregates in cell culture. PrPC expressed in cell culture is usually soluble, but overexpression increases the amount of insoluble protein (unpublished results). Prion proteins harboring familial mutations expressed in cell culture are often more abundant in detergent insoluble fractions and exhibit slightly enhanced PK resistance [71–76]. Some mutant PrP molecules linked to inherited prion diseases are retained in the cytosol, where they can aggregate into detergent-insoluble, partially PK-resistant assemblies following proteasome impairment [77]. Importantly, the moderate increase in PK resistance of PrP mutants expressed in tissue culture systems has so far not translated into infectious properties. It is possible that pathogenic mutations destabilize PrP and make it more prone to aggregate, and those misfolded proteins become refolded into an infectious PrPSc isoform in a secondary event during the very long incubation time in vivo [71, 74]. Of note, misfolding and aggregation are not confined to PrP with familial mutations, and replacements within the PrP coding sequence can alter PrP processing and increase PrP protease-resistance [78]. Truncated versions of PrP lacking the signal peptide and the GPI anchoring signal undergo spontaneous aggregate formation in the cytosol of mammalian cells [79]. Imbalances in cellular proteostasis can also alter the cellular localization of PrP and influence its solubility. Proteasome impairment increases the fraction of cytosolic PrP and triggers aggregation, but infectious properties of those aggregates have not been reported [80, 81]. Chemical compounds can alter the trafficking, cellular localization, and aggregation state of PrPC [82–84]. The most important implication from these studies is that PrP aggregates induced in cell culture by mutations or chemicals or triggered by changes in protein homeostasis have not acquired conformations that are self-propagating.
6. Recombinant Prions Induce Chronic Infections in Permissive Cell Cultures
Recently, it was shown that a synthetic prion strain, induced by the inoculation of β-sheet rich amyloid fibrils first into transgenic and subsequently into wild-type mice, was capable of replicating in mouse neuroblastoma cells with high fidelity [85]. New studies now demonstrate that synthetic prions derived from minimal components in vitro have the capacity to directly infect cell cultures (Figure 1(b)) [17, 62]. Synthetic prions produced by PMCA from recombinant mouse PrP mixed with synthetic anionic phospholipids and total liver RNA induced a sustained infection of murine SN56 cells (Figure 3(a)) [17]. Liver RNA could also be replaced by synthetic polyA RNA, leading to the formation of recombinant prions which are capable of chronically infecting a subclone of CAD cells [62]. The ex vivo propagation of recombinant synthetic prions produced by PMCA is surprising, given the restricted cell tropism of most prion strains. It is possible that the chosen cell lines generally represent highly susceptible substrates for prions [29, 86]. Future experiments may reveal if recombinant prions have an equally restricted host cell spectrum like natural prion strains.
Figure 3.
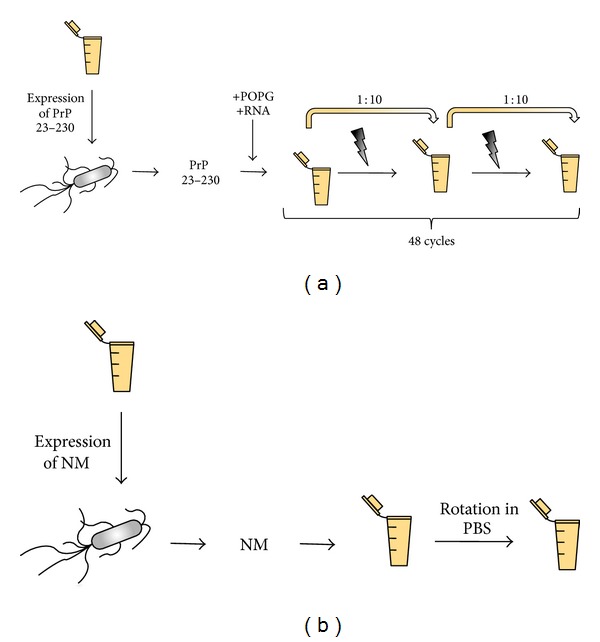
Generation of infectious protein polymers from minimal components in vitro. (a) For the generation of infectious PrP aggregates, purified recombinant murine PrP comprising amino acid residues 23 to 230 is mixed with lipid POPG and total liver RNA or synthetic polyA RNA. In a so-called protein misfolding cyclic amplification reaction (PMCA), intermittent pulses of sonication shear aggregates and increase the number of seeds. Preformed seeds are subsequently elongated and fragmented by serial dilution of samples into a new reaction buffer (supplemented with recombinant PrP and cofactors) and PMCA. Synthetic prions generated by this method have been shown to chronically infect SN56 and CAD cells [8, 62]. (b) The formation of Sup35 NM fibrils does not require additional cofactors. Purified recombinant NM is diluted to a concentration of 10 μM in phosphate buffered saline and left at room temperature overnight. Formation of NM fibrils is expedited by agitation. Addition of these fibrils to N2a cells stably expressing soluble NM induces a heritable NM prion phenotype [57, 58].
7. The Mammalian Cytosol Supports Propagation of Infectious Protein Aggregates
Prion diseases are the only known protein misfolding diseases that arise by aberrant folding of a GPI-anchored precursor protein. The strong association of PrPSc with membrane fractions and the attachment of PrPC to the outer leaflet of the cell membrane via a GPI moiety have led to the hypothesis that GPI-anchorage of amyloidogenic proteins might be key for the infectious properties of protein aggregates. The influence of the GPI anchor on prion-like properties of aggregation-prone proteins has recently been studied by tethering the yeast prion domain of the translation termination factor Sup35 to the N2a cell membrane via a GPI anchor [87]. The fusion protein of the prion domain NM with the GPI anchor either remained soluble or spontaneously aggregated, potentially dependent on its expression level or the fused fluorescent protein ([87] and unpublished results) (Figure 1(c)). Fluorescently labeled recombinant NM fibrils prepared in vitro were efficiently taken up by the cells and induced aggregation of the membrane-tethered soluble NM-GFP-GPI. Once switched into the aggregated state, this conformation was faithfully inherited by daughter cells. Coculture of cells expressing spontaneously aggregating NM-mCherry-GPI with cells expressing soluble GFP-tagged NM-GPI also induced a self-perpetuating prion phenotype in the latter.
Recent findings using the same Sup35 NM domain expressed in the cytosol of the same cell line, however, argue that cell membrane attachment via a GPI moiety is not a general requirement for infectious properties of protein aggregates in tissue culture (Figure 1(d)) [57]. In this experimental setting, the antibody epitope-tagged Sup35 NM domain was stably expressed in the cytosol as a soluble protein [79]. Spontaneous NM aggregation was not observed, not even under oxidative stress conditions [58]. Exposure of cells to in vitro produced fibrils from recombinant NM (Figure 3(b)) induced aggregation of the endogenous NM. Once induced, the aggregation state of NM was remarkably stable over multiple cell passages without any obvious loss of aggregate-bearing cells. NM aggregation could also be induced horizontally by direct transfer of NM aggregates from donor to acceptor through cell contact. The induced NM aggregation state in the acceptor cells was again heritable, strongly arguing that NM aggregates fulfill all criteria for prions in cell culture.
8. Prion-Like Properties of Proteins Associated with Neurodegenerative Diseases
Several neurodegenerative diseases are accompanied with intra- or extracellular deposition of amyloidogenic protein assemblies. While their primary sequences are diverse, aggregated proteins share a similar structure, consisting of an ordered arrangement of β-sheets [88–90]. Often, pathology begins locally, then progresses to other areas of the brain, reminiscent of the pathology spreading observed in prion diseases [91]. In contrast to prion diseases, neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), or Huntington's disease (HD) are not considered infectious diseases. Exciting research of the last years has demonstrated that aggregate pathology can spread to interconnected brain areas in several neurodegenerative disease models [91], suggesting that even nonprion protein aggregates somehow propagate and spread in tissue. These findings have led to the hypothesis that protein aggregates associated with neurodegenerative diseases share characteristic features of prions. Recent research has focused extensively on establishing cellular models for unraveling prion-like phenomena associated with neurodegenerative disease-related proteins. These studies convincingly demonstrated that aggregation-prone proteins can move between cells and seed protein aggregation in recipient cells. It is, however, a misconception that intercellular transmission and seeding of aggregates equal propagation. Propagation of prions—at least in vitro—is controlled by the rate of aggregate elongation, fragmentation, and degradation [15]. “Propagation” implies that transmitted seeds induce a self-templating aggregation state that will ultimately increase the number of protein aggregates per cell. Indeed, mammalian prions have found efficient ways to replicate, they produce enough seeds per cell that sustain the cellular clearance and then infect progeny and neighboring cells. This remarkable propagation efficiency is easily revealed in mitotically active cells. Here, constant cell division exponentially reduces the prion load, unless dilution and clearance effects can be compensated for by high rates of prion multiplication. The persistence of protein aggregates in dividing cells is, thus, indicative of how efficient a protein aggregate can propagate. In the following chapter we will discuss cell autonomous and noncell autonomous aggregation behaviors of the intracellular proteins Huntingtin (Htt), Tau, α-synuclein, and SOD1 in cell culture. We will particularly focus on the differences and similarities of PrP and other neurodegenerative disease-related proteins based on (i) their spontaneous aggregation propensities, (ii) their ability to aggregate upon addition of exogenous fibrillar seeds, (iii) their sustained propagation as induced aggregates, and (vi) their transmission in cell culture models. Of note, protein aggregates usually form in postmitotic neurons in vivo. However, mitotic stability ex vivo indicates how easily protein aggregates can propagate in a cellular environment.
9. Aggregation of Htt Polypeptides Encoded by Htt Exon 1
HD is a monogenic disease caused by expansion of CAG repeats in exon 1 of the Htt gene, resulting in an elongated polyglutamine (polyQ) region in the mutant protein [92]. Thus, the etiology of prion diseases crucially differs from that of HD. Mutant Htt undergoes proteolytic cleavage, giving rise to aminoterminal fragments (~the first 100–150 amino acid residues) that comprise the polyQ tract. Aggregated polyQ fragments are found in brains of HD patients and mouse models [93]. Therefore, HD pathogenesis is frequently modeled with proteins encoded by Htt exon 1.
(i) Aggregation Propensity When Expressed in Cell Culture. Normal nonmutant Htt protein (polyQ proteins with 6–35 glutamine residues) is usually soluble when expressed ex vivo. Expression of polyQ polypeptides causes spontaneous aggregate formation in a broad variety of cell types, including nonneuronal and neuronal cells lines as well as cultures of primary neurons [92]. Aggregation kinetics, subcellular localization, and shape of the aggregates are affected by repeat lengths, presence of Htt carboxy terminal regions, expression level, expression kinetics, and cell type [94–111].
(ii) Uptake and Seeding. Similar to PrPSc, recombinant polyQ peptides are internalized by cultured cells and can seed polymerization of a soluble Htt reporter protein. This has been shown by adding recombinant, fluorescently labeled polyQ fibrils to COS7, HEK293, CHO, HeLa, and N2A cells [60]. Interestingly, polyQ fibrils appeared to access the cytosol by direct penetration of the cell membrane. Internalized recombinant fibrils induced coaggregation of ectopically expressed soluble Htt fragments with nonmutant glutamine stretches.
(iii) Sustained Propagation. One hallmark of PrPSc is its stable inheritance in permanent cell cultures. For Htt, the mode of inheritance by daughter cells seems less stable. Asymmetric inheritance has been suggested based on the observation that big aggresome-like structures of polyQ aggregates are transmitted to only one daughter cell [61]. Interestingly, the number of cells with induced polyQ aggregates declined exponentially upon cell division, until reaching a low, but apparently persistent steady-state level of approximately 4% above background (Figure 2(b)) [60].
(iv) Cell-to-Cell Transmission. Coculture experiments of donor HEK cells expressing GFP-HttQ71 with cells expressing soluble HttQ25 revealed very inefficient aggregate induction in recipient cells, suggesting that Htt aggregates might not be transferred between cells at high rates [60]. Cell-to-cell transmission of polyQ proteins has also been demonstrated in cocultures of human H4 glioma and HEKT cells transfected with Htt exon-1 Q103 fused to halves of the Venus fluorescent reporter by bimolecular fluorescence complementation [99]. Importantly, in this experimental setup, no direct evidence for transfer of Htt aggregates has been provided, since the transferred protein species could also be monomeric or oligomeric. To date, it has not been shown if cell-to-cell transfer of polyQ aggregates induces ongoing replication of aggregates capable of propagating vertically and horizontally like prions.
10. Tau Aggregation in Cell Culture
Neurofibrillary tangles (NFTs) are a pathological hallmark of more than 20 so-called Tauopathies, including Alzheimer's disease (AD) and frontotemporal dementia. Accumulated hyperphosphorylated Tau protein aggregates into inclusions called paired helical filaments (PHF), which are the main component of NFTs. Tau is a microtubule-associated protein that stimulates and stabilizes microtubule assembly. Tau mutations cause familial neurodegenerative diseases. Tau is a naturally unfolded, highly soluble protein that exists as six isoforms [112, 113]. Alternative splicing generates Tau isoforms with three or four repeat domains (RD). The repeat domains are involved in microtubule binding and fibrillization. The mechanisms that trigger Tau conversion remain elusive. In AD brains, Tau is hyperphosphorylated, with a three- to four-fold increased phosphorylation level. Upon hyperphosphorylation, Tau dissociates from the microtubules and aggregates into the neurofibrillary tangles, resulting in microtubule destabilization and neurotoxicity [114, 115].
(i) Aggregation Propensity When Expressed in Cell Culture. Because of its high solubility, Tau—despite hyperphosphorylation—does not spontaneously aggregate upon overexpression in most cell lines [116, 117]. Spontaneous aggregation of overexpressed Tau constructs has been achieved using mutated full-length Tau and truncated Tau forms comprising the approximately 132 amino acid residue long aggregation-prone four-RD region [116, 118, 119].
(ii) Uptake and Seeding. Preformed recombinant aggregates derived from RD Tau or full-length Tau can be taken up by a variety of permanent cells and primary neurons [118, 120–122]. In primary neurons and HeLa cells, recombinant Tau was taken up by fluid-phase endocytosis [122]. Uptake of Tau appears to depend on its aggregation state, as monomeric or long fibrillar Tau is not internalized [122]. Similar to PrPSc, seeding properties of internalized Tau fibrils were shown. By applying preformed recombinant full-length or Tau RD fibrils to neuronal precursor cells, HEK cells, or primary neurons, formation of intracellular Tau aggregates by recruitment of soluble Tau was induced [118, 119, 123]. Importantly, recombinant Tau fibrils could also induce endogenous mouse Tau inclusions in wild-type hippocampal neurons, arguing that seeding does not require Tau overexpression [121].
(iii) Sustained Propagation. The propagation propensity of induced Tau aggregates in cell culture has not been elucidated so far. It is unclear if the transmitted aggregates also trigger a self-catalyzed propagation of Tau aggregates. If Tau aggregates faithfully replicate upon induction like prions, they should be maintained upon continuous culture.
(iv) Cell-to-Cell Transmission. Both spontaneously formed Tau RD aggregates and exogenously induced Tau full-length aggregates are transmitted from donor to acceptor cells in coculture experiments [118, 119]. Coculture of cells expressing differently tagged, spontaneously aggregating Tau RD variants revealed double stained inclusions, suggesting that Tau RD variants were exchanged between cells. This appears to depend on the release of fibrillar Tau RD species directly into the culture medium [119]. Transmission of HA-tagged Tau RD from the donor to a recipient cell line expressing both CFP- and YFP-tagged Tau RD variants increased fluorescent resonance energy transfer (FRET), suggesting that transmitted Tau RD increased endogenous Tau RD interaction and aggregate formation in the recipient. Of note, these experiments have been performed with nonphysiological, spontaneously aggregating Tau mutants, and the seeding of full-length Tau by cell-to-cell transmission has yet to be demonstrated.
11. Alpha-Synuclein Aggregation in Cell Culture
Parkinson's disease (PD) is characterized by deposition of the intracellular α-synuclein in dense Lewy bodies or Lewy neurites. α-synuclein is a 14 kDa protein that plays a role in synaptic neurotransmitter vesicle trafficking and releasing [124]. Increased α-synuclein expression and mutations in the coding region trigger misfolding and aggregation of α-synuclein into oligomers and amyloid fibrils in vivo [125, 126]. Posttranslational modifications such as phosphorylation, oxidation, nitration, and carboxy terminal truncation have been observed in vivo and could affect α-synuclein fibrillization [127].
(i) Aggregation Propensity When Expressed in Cell Culture. Recent studies in different mammalian cell lines suggest that endogenously or ectopically expressed human α-synuclein exists predominately as an unfolded monomer [128]. In some cell culture models, tetrameric or oligomeric species have been identified [129–132]. Overexpression of wild-type α-synuclein in diverse cell lines does not generally induce inclusion body formation [133], but mutant and/or truncated α-synuclein forms intracellular aggregates in some but not all cellular models [134]. Most often, α-synuclein aggregation is induced by treatment of the cells with oxidative stress and/or increased calcium levels [135–139]. α-synuclein aggregation in cell culture can also be promoted by coexpression of mutant leucine-rich repeat kinase, a protein linked to familial PD [140], or by treatment with mitochondrial inhibitor rotenone [141].
(ii) Uptake and Seeding. α-synuclein seeds prepared from recombinant protein ex vivo can be taken up by a variety of different cells [142–146]. Endocytosis has been proposed as a possible route for internalization of fibrillar recombinant human α-synuclein [142, 146]. In some instances, uptake of fibrils into the cells by physiological routes failed, and fibrils had to be introduced via cationic lipid transfection [145]. Variations in uptake efficiencies might be due to differences in fibril preparation, as has been shown for different α-synuclein oligomers [143, 144]. Exogenous α-synuclein oligomers or fibrils can seed the formation of intracellular inclusions in different permanent and primary cell models [143–146]. Overexpression of wild-type or mutant α-synuclein [146] is not generally required for noncell autonomous aggregate induction, but aggregation kinetics appear to be faster under α-synuclein overexpression conditions [144, 145].
(iii) Sustained Propagation. So far, sustained propagation of induced α-synuclein aggregates in cell culture has not been reported. Future experiments will be necessary to evaluate if α-synuclein aggregates, once triggered, gain self-propagating properties that allow them to continuously propagate over multiple passages. In analogy to mammalian prions, this would require that aggregate growth and fragmentation exceed the loss of aggregates by cellular clearance or cell division.
(iv) Cell-to-Cell Transmission. Coculturing experiments with cells producing α-synuclein aggregates with recipient cells revealed transfer of α-synuclein between cells with subsequent aggregate induction in the recipient cell [147, 148]. The exact mechanism of aggregate transfer has not been elucidated. Low amounts of soluble and aggregated forms of α-synuclein have been shown to be released by cells, potentially by unconventional endocytosis or packaged into exosomes [149–152].
12. Superoxide Dismutase 1 Aggregation in Cell Culture
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that usually occurs sporadically but can also be of genetic origin. During the course of the disease, motor neurons in spinal cord, brain stem, and brain degenerate [153]. More than 140 mutations in the copper/zinc superoxide dismutase (SOD1), an antioxidant enzyme, are linked to approx 2% of familial ALS cases (http://alsod.iop.kcl.ac.uk/). Misfolding of mutant SOD1 and cytoplasmic inclusion of body formation underlies disease pathology [154].
(i) Aggregation Propensity When Expressed in Cell Culture. Active SOD1 is located in the cytoplasm in the form of a homodimer. Overexpression of wild-type SOD1 usually does not lead to inclusion body formation in cell culture [155]. Still, misfolded states of the wild-type or mutant SOD1 overexpressed in cell culture could be revealed by antibody detection of normally buried antibody epitopes [156]. The aggregation propensities of various SOD1 mutants differ in cell culture [59, 155, 157].
(ii) Uptake and Seeding. Aggregated recombinant SOD1 can be taken up by neuroblastoma cells [59], and primary or permanent microglia cell cultures [158]. Uptake of aggregated SOD1 was decreased by either chemical inhibition of rafts and scavenger receptors, or by macropinocytosis inhibitors, indicating that uptake mechanisms might depend on the cell line or seed preparation [59, 158]. Recombinant mutant SOD1 aggregates added to the cell culture medium gained access to the cytosol of N2a cells, where they recruited the homotypic protein into small, dispersed aggregates [59].
(iii) Sustained Propagation. Unlike polyQ aggregates, seed-induced SOD1 aggregates exhibit a remarkable mitotic stability in cell culture [59]. The stable propagation of the SOD1 aggregation phenotype over multiple passages in cell culture is reminiscent of that of mammalian prions and suggests steady state levels of SOD1 aggregates.
(iv) Cell-to-Cell Transmission. Fluorescently tagged mutant SOD1 aggregates are efficiently taken up by N2a cells and subsequently transmitted to cocultured cells [59]. Direct cell contact between donor and acceptor cells was not required for transmission, suggesting that SOD1 was secreted into the cell culture medium. Secretion of endogenous SOD1 has been reported for a variety of different cell lines, including fibroblasts, neuroblastoma, motor neuron cell lines and primary spinal cord cultures [159–161]. It remains to be established if and how self-perpetuating SOD1 aggregates produced in cell culture spread horizontally and induce a SOD1 aggregation phenotype in the acceptor cells [59].
13. Concluding Remarks
Prions are proteinaceous infectious protein aggregates that have the capacity to enter the host cells and impose their abnormal conformational states onto their endogenous counterparts. Noncell autonomous aggregate induction through external seeds has recently also been demonstrated for a variety of different proteins associated with nonprion neurodegenerative diseases. The ability to invade cells and seed cytosolic aggregation appears to be a general feature of amyloidogenic proteins. Importantly, the aggregation state of a protein does not reflect its infectious properties and only a thorough investigation of its propagation ex vivo or in vivo can ultimately confirm infectious potentials. Prion replication proceeds through a process of seeded polymerization and secondary nucleation events by fibril fragmentation, and escape of those seeds from cellular clearance is crucial for prion maintenance. Mitotically active cells represent tractable models for studying aspects of prion fragmentation and clearance, as inefficient prion fragmentation or enhanced clearance result in rapid prion loss due to aggregate dilution by cell division. The fact that mammalian prions can successfully replicate mitotically active cells argues for a steady state between prion formation and prion reduction. The observed mitotic instability of other protein aggregates ex vivo could, thus, be due to inefficient fragmentation or enhanced clearance. In conclusion, the ability of a given protein aggregate to achieve a steady state of aggregate multiplication and reduction will ultimately affect its prion capacity.
Note. While in print persistent propagation of induced alpha-synuclein aggregates was reported in mouse neuroblastoma cell line N2a [162].
Acknowledgments
The authors thank Donato Di Monte and Daniele Bano for critical reading of the paper. They gratefully acknowledge the financial support by DFG Grant IV 1277/1-3 and Helmholtz Portfolio Wirkstoffforschung.
Abbreviations
- AD:
Alzheimer's disease
- ER:
Endoplasmic reticulum
- GdnHCl:
Guanidinium hydrochloride
- GPI:
Glycosylphosphatidylinositol
- HA:
Hemagglutinin antibody epitope
- HD:
Huntington's disease
- Htt:
Huntingtin
- NFT:
Neurofibrillary tangles
- NM:
Prion domain and middle domain of the yeast Sup35 translation termination factor
- PD:
Parkinson's disease
- PHF:
Paired helical filaments
- PK:
Proteinase K
- PMCA:
Protein misfolding cyclic amplification
- polyQ:
Polyglutamine
- PrP:
Prion protein
- PrPC:
Normal cellular isoform of the prion protein
- PrPSc:
Disease-associated isoform of the prion protein
- RD:
Repeat domain of Tau
- TSE:
Transmissible spongiform encephalopathy.
References
- 1.Aguzzi A. Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. Journal of Neurochemistry. 2006;97(6):1726–1739. doi: 10.1111/j.1471-4159.2006.03909.x. [DOI] [PubMed] [Google Scholar]
- 2.Dickinson AG, Meikle VM. A comparison of some biological characteristics of the mouse-passaged scrapie agents, 22A and ME7. Genetical Research. 1969;13(2):213–225. doi: 10.1017/s0016672300002895. [DOI] [PubMed] [Google Scholar]
- 3.Hecker R, Taraboulos A, Scott M, et al. Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes and Development. 1992;6(7):1213–1228. doi: 10.1101/gad.6.7.1213. [DOI] [PubMed] [Google Scholar]
- 4.Taraboulos A, Jendroska K, Serban D, Yang S-L, DeArmond SJ, Prusiner SB. Regional mapping of prion proteins in brain. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(16):7620–7624. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 6.Legname G, Baskakov IV, Nguyen H-OB, et al. Synthetic mammalian prions. Science. 2004;305(5684):673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 7.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(23):9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang F, Wang X, Yuan C-G, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327(5969):1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. Journal of Virology. 1994;68(12):7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Safar J, Wille H, Itri V, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nature Medicine. 1998;4(10):1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 11.Telling GC, Parchi P, DeArmond SJ, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274(5295):2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 12.Linden R, Martins VR, Prado MAM, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiological Reviews. 2008;88(2):673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 13.Pan K-M, Baldwin M, Nguyen J, et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(23):10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tzaban S, Friedlander G, Schonberger O, et al. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002;41(42):12868–12875. doi: 10.1021/bi025958g. [DOI] [PubMed] [Google Scholar]
- 15.Knowles TPJ, Waudby CA, Devlin GL, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326(5959):1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 16.Silveira JR, Raymond GJ, Hughson AG, et al. The most infectious prion protein particles. Nature. 2005;437(7056):257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 18.Wickner RB, Masison DC. Evidence for two prions in yeast: [URE3] and [PSI] Current Topics in Microbiology and Immunology. 1996;207:147–160. doi: 10.1007/978-3-642-60983-1_10. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442(7102):585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 20.Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. Mechanism of prion loss after Hsp104 inactivation in yeast. Molecular and Cellular Biology. 2001;21(14):4656–4669. doi: 10.1128/MCB.21.14.4656-4669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derdowski A, Sindi SS, Klaips CL, DiSalvo S, Serio TR. A size threshold limits prion transmission and establishes phenotypic diversity. Science. 2010;330(6004):680–683. doi: 10.1126/science.1197785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Béranger F, Mangé A, Solassol J, Lehmann S. Cell culture models of transmissible spongiform encephalopathies. Biochemical and Biophysical Research Communications. 2001;289(2):311–316. doi: 10.1006/bbrc.2001.5941. [DOI] [PubMed] [Google Scholar]
- 23.Piccardo P, Cervenakova L, Vasilyeva I, et al. Candidate cell substrates, vaccine production, and transmissible spongiform encephalopathies. Emerging Infectious Diseases. 2011;17(12):2262–2269. doi: 10.3201/eid1712.110607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubenstein R, Hui D, Race R, et al. Replication of scrapie strains in vitro and their influence on neuronal functions. Annals of the New York Academy of Sciences. 1994;724:331–337. doi: 10.1111/j.1749-6632.1994.tb38924.x. [DOI] [PubMed] [Google Scholar]
- 25.Nishida N, Harris DA, Vilette D, et al. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. Journal of Virology. 2000;74(1):320–325. doi: 10.1128/jvi.74.1.320-325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosque PJ, Prusiner SB. Cultured cell sublines highly susceptible to prion infection. Journal of Virology. 2000;74(9):4377–4386. doi: 10.1128/jvi.74.9.4377-4386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vorberg I, Raines A, Story B, Priola SA. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. Journal of Infectious Diseases. 2004;189(3):431–439. doi: 10.1086/381166. [DOI] [PubMed] [Google Scholar]
- 28.Lehmann S. Prion propagation in cell culture. Methods in Molecular Biology. 2005;299:227–234. doi: 10.1385/1-59259-874-9:227. [DOI] [PubMed] [Google Scholar]
- 29.Baron GS, Magalhães AC, Prado MAM, Caughey B. Mouse-adapted scrapie infection of SN56 cells: greater efficiency with microsome-associated versus purified PrP-res. Journal of Virology. 2006;80(5):2106–2117. doi: 10.1128/JVI.80.5.2106-2117.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grassmann A, Wolf H, Hofmann J, Graham J, Vorberg I. Cellular aspects of prion replication in vitro. Viruses. 2013;5(1):374–405. doi: 10.3390/v5010374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greil CS, Vorberg IM, Ward AE, Meade-White KD, Harris DA, Priola SA. Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology. 2008;379(2):284–293. doi: 10.1016/j.virol.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paquet S, Daude N, Courageot M-P, Chapuis J, Laude H, Vilette D. PrPc does not mediate internalization of PrPSc but is required at an early stage for de novo prion infection of Rov cells. Journal of Virology. 2007;81(19):10786–10791. doi: 10.1128/JVI.01137-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. The Journal of Biological Chemistry. 1991;266(27):18217–18223. [PubMed] [Google Scholar]
- 34.Vorberg I, Raines A, Priola SA. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. The Journal of Biological Chemistry. 2004;279(28):29218–29225. doi: 10.1074/jbc.M402576200. [DOI] [PubMed] [Google Scholar]
- 35.Goold R, Rabbanian S, Sutton L, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nature Communications. 2011;2(1, article 281) doi: 10.1038/ncomms1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taraboulos A, Serban D, Prusiner SB. Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. Journal of Cell Biology. 1990;110(6):2117–2132. doi: 10.1083/jcb.110.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veith NM, Plattner H, Stuermer CAO, Schulz-Schaeffer WJ, Bürkle A. Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. European Journal of Cell Biology. 2009;88(1):45–63. doi: 10.1016/j.ejcb.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. The Journal of Biological Chemistry. 1992;267(23):16188–16199. [PubMed] [Google Scholar]
- 39.McKinley MP, Taraboulos A, Kenaga L, et al. Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Laboratory Investigation. 1991;65(6):622–630. [PubMed] [Google Scholar]
- 40.Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathogens. 2009;5(5) doi: 10.1371/journal.ppat.1000426.e1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. The Journal of Biological Chemistry. 1997;272(10):6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- 42.Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. Journal of Cell Biology. 1990;110(3):743–752. doi: 10.1083/jcb.110.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vey M, Pilkuhn S, Wille H, et al. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(25):14945–14949. doi: 10.1073/pnas.93.25.14945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamasaki T, Suzuki A, Shimizu T, Watarai M, Hasebe R, Horiuchi M. Characterization of intracellular localization of PrPSc in prion-infected cells using a mAb that recognizes the region consisting of aa 119-127 of mouse PrP. Journal of General Virology. 2012;93(3):668–680. doi: 10.1099/vir.0.037101-0. [DOI] [PubMed] [Google Scholar]
- 45.Peretz D, Williamson RA, Kaneko K, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412(6848):739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 46.Aguib Y, Heiseke A, Gilch S, et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5(3):361–369. doi: 10.4161/auto.5.3.7662. [DOI] [PubMed] [Google Scholar]
- 47.Ertmer A, Gilch S, Yun S-W, et al. The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. The Journal of Biological Chemistry. 2004;279(40):41918–41927. doi: 10.1074/jbc.M405652200. [DOI] [PubMed] [Google Scholar]
- 48.Heiseke A, Aguib Y, Riemer C, Baier M, Schätzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. Journal of Neurochemistry. 2009;109(1):25–34. doi: 10.1111/j.1471-4159.2009.05906.x. [DOI] [PubMed] [Google Scholar]
- 49.Schätzl HM, Laszlo L, Holtzman DM, et al. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. Journal of Virology. 1997;71(11):8821–8831. doi: 10.1128/jvi.71.11.8821-8831.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghaemmaghami S, Phuan P-W, Perkins B, et al. Cell division modulates prion accumulation in cultured cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(46):17971–17976. doi: 10.1073/pnas.0708372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Butler DA, Scott MRD, Bockman JM, et al. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. Journal of Virology. 1988;62(5):1558–1564. doi: 10.1128/jvi.62.5.1558-1564.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Race RE, Caughey B, Graham K, Ernst D, Chesebro B. Analyses of frequency of infection, specific infectivity, and prion protein biosynthesis in scrapie-infected neuroblastoma cell clones. Journal of Virology. 1988;62(8):2845–2849. doi: 10.1128/jvi.62.8.2845-2849.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watts JC, Huo H, Bai Y, et al. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathogens. 2009;5(10) doi: 10.1371/journal.ppat.1000608.e1000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin T, Gu Y, Zanusso G, et al. The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. The Journal of Biological Chemistry. 2000;275(49):38699–38704. doi: 10.1074/jbc.M005543200. [DOI] [PubMed] [Google Scholar]
- 55.Xu F, Karnaukhova E, Vostal JG. Human cellular prion protein interacts directly with clusterin protein. Biochimica et Biophysica Acta. 2008;1782(11):615–620. doi: 10.1016/j.bbadis.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 56.Heiseke A, Aguib Y, Schatzl HM. Autophagy, prion infection and their mutual interactions. Current Issues in Molecular Biology. 2010;12(2):87–97. [PubMed] [Google Scholar]
- 57.Krammer C, Kryndushkin D, Suhre MH, et al. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(2):462–467. doi: 10.1073/pnas.0811571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofmann JP, Denner P, Nussbaum-Krammer C, et al. Cell-to-cell propagation of infectious cytosolic protein aggregates. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(15):5951–5956. doi: 10.1073/pnas.1217321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Münch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3548–3553. doi: 10.1073/pnas.1017275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren P-H, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nature Cell Biology. 2009;11(2):219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rujano MA, Bosveld F, Salomons FA, et al. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biology. 2006;4(12, article e417) doi: 10.1371/journal.pbio.0040417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang F, Zhang Z, Wang X, et al. Genetic informational RNA is not required for recombinant prion infectivity. Journal of Virology. 2012;86(3):1874–1876. doi: 10.1128/JVI.06216-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maas E, Geissen M, Groschup MH, et al. Scrapie infection of prion protein-deficient cell line upon ectopic expression of mutant prion proteins. The Journal of Biological Chemistry. 2007;282(26):18702–18710. doi: 10.1074/jbc.M701309200. [DOI] [PubMed] [Google Scholar]
- 64.Fevrier B, Vilette D, Archer F, et al. Cells release prions in association with exosomes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(26):9683–9688. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leblanc P, Alais S, Porto-Carreiro I, et al. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO Journal. 2006;25(12):2674–2685. doi: 10.1038/sj.emboj.7601162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alais S, Simoes S, Baas D, et al. Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biology of the Cell. 2008;100(10):603–615. doi: 10.1042/BC20080025. [DOI] [PubMed] [Google Scholar]
- 67.Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. Journal of Pathology. 2007;211(5):582–590. doi: 10.1002/path.2145. [DOI] [PubMed] [Google Scholar]
- 68.Paquet S, Langevin C, Chapuis J, Jackson GS, Laude H, Vilette D. Efficient dissemination of prions through preferential transmission to nearby cells. Journal of General Virology. 2007;88(2):706–713. doi: 10.1099/vir.0.82336-0. [DOI] [PubMed] [Google Scholar]
- 69.Kanu N, Imokawa Y, Drechsel DN, et al. Transfer of scrapie prion infectivity by cell contact in culture. Current Biology. 2002;12(7):523–530. doi: 10.1016/s0960-9822(02)00722-4. [DOI] [PubMed] [Google Scholar]
- 70.Gousset K, Schiff E, Langevin C, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nature Cell Biology. 2009;11(3):328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 71.Capellari S, Parchi P, Russo CM, et al. Effect of the E200K mutation on prion protein metabolism: comparative study of a cell model and human brain. American Journal of Pathology. 2000;157(2):613–622. doi: 10.1016/S0002-9440(10)64572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lehmann S, Harris DA. Two mutant prion proteins expressed in cultured cells acquire biochemical properties reminiscent of the scrapie isoform. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(11):5610–5614. doi: 10.1073/pnas.93.11.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petersen RB, Parchi P, Richardson SL, Urig CB, Gambetti P. Effect of the D178N mutation and the codon 129 polymorphism on the metabolism of the prion protein. The Journal of Biological Chemistry. 1996;271(21):12661–12668. doi: 10.1074/jbc.271.21.12661. [DOI] [PubMed] [Google Scholar]
- 74.Capellari S, Zaidi SIA, Long AC, Kwon EE, Petersen RB. The Thr183Ala mutation, not the loss of the first glycosylation site, alters the physical properties of the prion protein. Journal of Alzheimer’s Disease. 2000;2(1):27–35. doi: 10.3233/jad-2000-2104. [DOI] [PubMed] [Google Scholar]
- 75.Lorenz H, Windl O, Kretzschmar HA. Cellular phenotyping of secretory and nuclear prion proteins associated with inherited prion diseases. The Journal of Biological Chemistry. 2002;277(10):8508–8516. doi: 10.1074/jbc.M110197200. [DOI] [PubMed] [Google Scholar]
- 76.Zaidi SIA, Richardson SL, Capellari S, et al. Characterization of the F198S prion protein mutation: enhanced glycosylation and defective refolding. Journal of Alzheimer’s Disease. 2005;7(2):159–171. doi: 10.3233/jad-2005-7209. [DOI] [PubMed] [Google Scholar]
- 77.Zanusso G, Petersen RB, Jin T, et al. Proteasomal degradation and N-terminal protease resistance of the codon 145 mutant prion protein. The Journal of Biological Chemistry. 1999;274(33):23396–23404. doi: 10.1074/jbc.274.33.23396. [DOI] [PubMed] [Google Scholar]
- 78.Wegner C, Römer A, Schmalzbauer R, Lorenz H, Windl O, Kretzschmar HA. Mutant prion protein acquires resistance to protease in mouse neuroblastoma cells. Journal of General Virology. 2002;83(5):1237–1245. doi: 10.1099/0022-1317-83-5-1237. [DOI] [PubMed] [Google Scholar]
- 79.Krammer C, Suhre MH, Kremmer E, et al. Prion protein/protein interactions: fusion with yeast Sup35p-NM modulates cytosolic PrP aggregation in mammalian cells. FASEB Journal. 2008;22(3):762–773. doi: 10.1096/fj.07-8733com. [DOI] [PubMed] [Google Scholar]
- 80.Kristiansen M, Messenger MJ, Klöhn P-C, et al. Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. The Journal of Biological Chemistry. 2005;280(46):38851–38861. doi: 10.1074/jbc.M506600200. [DOI] [PubMed] [Google Scholar]
- 81.Orsi A, Fioriti L, Chiesa R, Sitia R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. The Journal of Biological Chemistry. 2006;281(41):30431–30438. doi: 10.1074/jbc.M605320200. [DOI] [PubMed] [Google Scholar]
- 82.Cohen E, Taraboulos A. Scrapie-like prion protein accumulates in aggresomes of cyclosporin A-treated cells. EMBO Journal. 2003;22(3):404–417. doi: 10.1093/emboj/cdg045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gilch S, Winklhofer KF, Groschup MH, et al. Intracellular re-routing of prion protein prevents propagation of PrPsc and delays onset of prion disease. EMBO Journal. 2001;20(15):3957–3966. doi: 10.1093/emboj/20.15.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nunziante M, Kehler C, Maas E, Kassack MU, Groschup M, Schätzl HM. Charged bipolar suramin derivatives induce aggregation of the prion protein at the cell surface and inhibit PrPSc replication. Journal of Cell Science. 2005;118(21):4959–4973. doi: 10.1242/jcs.02609. [DOI] [PubMed] [Google Scholar]
- 85.Ghaemmaghami S, Watts JC, Nguyen H-O, Hayashi S, Dearmond SJ, Prusiner SB. Conformational transformation and selection of synthetic prion strains. Journal of Molecular Biology. 2011;413(3):527–542. doi: 10.1016/j.jmb.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(52):20908–20913. doi: 10.1073/pnas.0710054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Speare JO, Offerdahl DK, Hasenkrug A, Carmody AB, Baron GS. GPI anchoring facilitates propagation and spread of misfolded Sup35 aggregates in mammalian cells. EMBO Journal. 2010;29(4):782–794. doi: 10.1038/emboj.2009.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perutz M. Polar zippers: their role in human disease. Protein Science. 1994;3(10):1629–1637. doi: 10.1002/pro.5560031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(8):5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. Journal of Molecular Biology. 1997;273(3):729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 91.Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends in Neurosciences. 2010;33(7):317–325. doi: 10.1016/j.tins.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 92.Rubinsztein DC. Lessons from animal models of Huntington’s disease. Trends in Genetics. 2002;18(4):202–209. doi: 10.1016/s0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- 93.Lunkes A, Lindenberg KS, Ben-Haem L, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Molecular Cell. 2002;10(2):259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 94.Sontag EM, Lotz GP, Agrawal N, et al. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington's disease models. The Journal of Neuroscience. 2012;32(32):11109–11119. doi: 10.1523/JNEUROSCI.0895-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schulte J, Littleton JT. The biological function of the Huntingtin protein and its relevance to Huntington's Disease pathology. Current Trends in Neurology. 2011;5:65–78. [PMC free article] [PubMed] [Google Scholar]
- 96.Roscic A, Baldo B, Crochemore C, Marcellin D, Paganetti P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. Journal of Neurochemistry. 2011;119(2):398–407. doi: 10.1111/j.1471-4159.2011.07435.x. [DOI] [PubMed] [Google Scholar]
- 97.Guzhova IV, Lazarev VF, Kaznacheeva AV, et al. Novel mechanism of Hsp70 chaperone-mediated prevention of polyglutamine aggregates in a cellular model of huntington disease. Human Molecular Genetics. 2011;20(20):3953–3963. doi: 10.1093/hmg/ddr314. [DOI] [PubMed] [Google Scholar]
- 98.Alba Sorolla M, Nierga C, José Rodríguez-Colman M, et al. Sir2 is induced by oxidative stress in a yeast model of Huntington disease and its activation reduces protein aggregation. Archives of Biochemistry and Biophysics. 2011;510(1):27–34. doi: 10.1016/j.abb.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 99.Herrera F, Tenreiro S, Miller-Fleming L, Outeiro TF. Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLOS Currents. 2011;3 doi: 10.1371/currents.RRN1210.RRN1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bocharova N, Chave-Cox R, Sokolov S, Knorre D, Severin F. Protein aggregation and neurodegeneration: clues from a yeast model of Huntington’s disease. Biochemistry. 2009;74(2):231–234. doi: 10.1134/s0006297909020163. [DOI] [PubMed] [Google Scholar]
- 101.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Human Molecular Genetics. 2002;11(9):1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 102.Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. Journal of Cell Science. 2008;121(10):1649–1660. doi: 10.1242/jcs.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gong B, Lim MCY, Wanderer J, Wyttenbach A, Morton AJ. Time-lapse analysis of aggregate formation in an inducible PC12 cell model of Huntington’s disease reveals time-dependent aggregate formation that transiently delays cell death. Brain Research Bulletin. 2008;75(1):146–157. doi: 10.1016/j.brainresbull.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 104.Sokolov S, Pozniakovsky A, Bocharova N, Knorre D, Severin F. Expression of an expanded polyglutamine domain in yeast causes death with apoptotic markers. Biochimica et Biophysica Acta. 2006;1757(5-6):660–666. doi: 10.1016/j.bbabio.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 105.Colby DW, Cassady JP, Lin GC, Ingram VM, Wittrup KD. Stochastic kinetics of intracellular huntingtin aggregate formation. Nature Chemical Biology. 2006;2(6):319–323. doi: 10.1038/nchembio792. [DOI] [PubMed] [Google Scholar]
- 106.Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, Monteiro MJ. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Human Molecular Genetics. 2006;15(6):1025–1041. doi: 10.1093/hmg/ddl017. [DOI] [PubMed] [Google Scholar]
- 107.Mukai H, Isagawa T, Goyama E, et al. Formation of morphologically similar globular aggregates from diverse aggregation-prone proteins in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(31):10887–10892. doi: 10.1073/pnas.0409283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC. Decreased cAMP response element-mediated transcription. An early event in exon 1 and full-length cell models of Huntington’s disease that contributes to polyglutamine pathogenesis. The Journal of Biological Chemistry. 2004;279(6):4988–4999. doi: 10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- 109.Sittler A, Lurz R, Lueder G, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Human Molecular Genetics. 2001;10(12):1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 110.Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(4):1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cooper JK, Schilling G, Peters MF, et al. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Human Molecular Genetics. 1998;7(5):783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- 112.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s Disease. Nature. 1963;197(4863):192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 113.Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-β structure. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):9034–9038. doi: 10.1073/pnas.1530287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Alonso ADC, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(1):298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gong C-X, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer’s disease. Journal of Neural Transmission. 2005;112(6):813–838. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- 116.Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, et al. Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Molecular Biology of the Cell. 2000;11(12):4093–4104. doi: 10.1091/mbc.11.12.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Khlistunova I, Biernat J, Wang Y, et al. Inducible expression of tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. The Journal of Biological Chemistry. 2006;281(2):1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- 118.Frost B, Jacks RL, Diamond MI. Propagation of Tau misfolding from the outside to the inside of a cell. The Journal of Biological Chemistry. 2009;284(19):12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. The Journal of Biological Chemistry. 2012;287(23):19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of α-synuclein and tau: cellular models of neurodegenerative diseases. The Journal of Biological Chemistry. 2010;285(45):34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guo JL, Lee VM. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Letters. 2013;587(6):717–723. doi: 10.1016/j.febslet.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu JW, Herman M, Liu L, et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. The Journal of Biological Chemistry. 2013;288(3):1856–1870. doi: 10.1074/jbc.M112.394528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guo JL, Lee VM-Y. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. The Journal of Biological Chemistry. 2011;286(17):15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nature Reviews Neuroscience. 2013;14(1):38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302(5646):p. 841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 126.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 127.Beyer K, Ariza A. Alpha-synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Molecular Neurobiology. 2013;47(2):509–524. doi: 10.1007/s12035-012-8330-5. [DOI] [PubMed] [Google Scholar]
- 128.Fauvet B, Mbefo MK, Fares MB, et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. The Journal of Biological Chemistry. 2012;287(19):15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu J, Kao S-Y, Lee FJS, Song W, Jin L-W, Yankner BA. Dopamine-dependent neurotoxicity of α-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nature Medicine. 2002;8(6):600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- 130.Mazzulli JR, Mishizen AJ, Giasson BI, et al. Cytosolic catechols inhibit α-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. The Journal of Neuroscience. 2006;26(39):10068–10078. doi: 10.1523/JNEUROSCI.0896-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yamakawa K, Izumi Y, Takeuchi H, et al. Dopamine facilitates α-synuclein oligomerization in human neuroblastoma SH-SY5Y cells. Biochemical and Biophysical Research Communications. 2010;391(1):129–134. doi: 10.1016/j.bbrc.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 132.Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kahle PJ, Neumann M, Ozmen L, Haass C. Physiology and pathophysiology of α-synuclein cell culture and transgenic animal models based on a Parkinson’s disease-associated protein. Annals of the New York Academy of Sciences. 2000;920:33–41. doi: 10.1111/j.1749-6632.2000.tb06902.x. [DOI] [PubMed] [Google Scholar]
- 134.Bodner RA, Outeiro TF, Altmann S, et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(11):4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. The Journal of Neuroscience. 2000;20(16):6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Follett J, Darlow B, Wong MB, Goodwin J, Pountney DL. Potassium depolarization and raised calcium induces alpha-synuclein aggregates. Neurotoxicity Research. 2013;23(4):378–392. doi: 10.1007/s12640-012-9366-z. [DOI] [PubMed] [Google Scholar]
- 137.Gerard M, Deleersnijder A, Daniëls V, et al. Inhibition of FK506 binding proteins reduces α-synuclein aggregation and Parkinson’s disease-like pathology. The Journal of Neuroscience. 2010;30(7):2454–2463. doi: 10.1523/JNEUROSCI.5983-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nath S, Goodwin J, Engelborghs Y, Pountney DL. Raised calcium promotes α-synuclein aggregate formation. Molecular and Cellular Neuroscience. 2011;46(2):516–526. doi: 10.1016/j.mcn.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 139.Goodwin J, Nath S, Engelborghs Y, Pountney DL. Raised calcium and oxidative stress cooperatively promote alpha-synuclein aggregate formation. Neurochemistry International. 2013;62(5):703–711. doi: 10.1016/j.neuint.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 140.Kondo K, Obitsu S, Teshima R. α-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biological and Pharmaceutical Bulletin. 2011;34(7):1078–1083. doi: 10.1248/bpb.34.1078. [DOI] [PubMed] [Google Scholar]
- 141.Lee H-J, Lee S-J. Characterization of cytoplasmic α-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. The Journal of Biological Chemistry. 2002;277(50):48976–48983. doi: 10.1074/jbc.M208192200. [DOI] [PubMed] [Google Scholar]
- 142.Bae EJ, Lee HJ, Rockenstein E, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. The Journal of Neuroscience. 2012;32(39):13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Danzer KM, Haasen D, Karow AR, et al. Different species of α-synuclein oligomers induce calcium influx and seeding. The Journal of Neuroscience. 2007;27(34):9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. Journal of Neurochemistry. 2009;111(1):192–203. doi: 10.1111/j.1471-4159.2009.06324.x. [DOI] [PubMed] [Google Scholar]
- 145.Luk KC, Song C, O’Brien P, et al. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(47):20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Desplats P, Lee H-J, Bae E-J, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(31):13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lee H-J, Suk J-E, Patrick C, et al. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. The Journal of Biological Chemistry. 2010;285(12):9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jang A, Lee H-J, Suk J-E, Jung J-W, Kim K-P, Lee S-J. Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. Journal of Neurochemistry. 2010;113(5):1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 150.Lee H-J, Patel S, Lee S-J. Intravesicular localization and exocytosis of α-synuclein and its aggregates. The Journal of Neuroscience. 2005;25(25):6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Emmanouilidou E, Melachroinou K, Roumeliotis T, et al. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. The Journal of Neuroscience. 2010;30(20):6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Alvarez-Erviti L, Seow Y, Schapira AH, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiology of Disease. 2011;42(3):360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nature Reviews Neuroscience. 2001;2(11):806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 154.Shibata N, Hirano A, Kobayashi M, et al. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. Journal of Neuropathology and Experimental Neurology. 1996;55(4):481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 155.Prudencio M, Durazo A, Whitelegge JP, Borchelt DR. Modulation of mutant superoxide dismutase 1 aggregation by co-expression of wild-type enzyme. Journal of Neurochemistry. 2009;108(4):1009–1018. doi: 10.1111/j.1471-4159.2008.05839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Grad LI, Guest WC, Yanai A, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(39):16398–16403. doi: 10.1073/pnas.1102645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Roberts BL, Patel K, Brown HH, Borchelt DR. Role of disulfide cross-linking of mutant SOD1 in the formation of inclusion-body-like structures. PLoS One. 2012;7(10) doi: 10.1371/journal.pone.0047838.e47838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Roberts K, Zeineddine R, Corcoran L, Li W, Campbell IL, Yerbury JJ. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia. 2013;61(3):409–419. doi: 10.1002/glia.22444. [DOI] [PubMed] [Google Scholar]
- 159.Mondola P, Annella T, Serù R, et al. Secretion and increase of intracellular CuZn superoxide dismutase content in human neuroblastoma SK-N-BE cells subjected to oxidative stress. Brain Research Bulletin. 1998;45(5):517–520. doi: 10.1016/s0361-9230(97)00438-3. [DOI] [PubMed] [Google Scholar]
- 160.Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien J-P. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nature Neuroscience. 2006;9(1):108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- 161.Gomes C, Keller S, Altevogt P, Costa J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neuroscience Letters. 2007;428(1):43–46. doi: 10.1016/j.neulet.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 162.Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nature Communications. 2013;4, article 2575 doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]