

Abstract
The protease of norovirus, an important human pathogen, is essential for the viral replication and, therefore, represents a potential drug target. A series of tripeptide-based inhibitors of the protease were designed, synthesized and tested, among which several potent inhibitors were identified with Ki values as low as 75 nM. The structure-activity relationships of these inhibitors are discussed.
Introduction
Norovirus is the leading cause of epidemic acute gastroenteritis in humans. Using PCR, more than 90% of outbreaks of nonbacterial gastroenteritis in the US are caused by noroviruses.1 Similar data are reported for the Netherlands and other countries in Europe.2–4 Norwalk virus (GI.1) in the genogroup I is the prototype of this group of viruses, while genogroup II viruses, such as Houston virus (GII.4), are now more prevalent.5 According to a recent CDC (Centers for Disease Control and Prevention) survey, noroviral infections account for 21 million cases of gastroenteritis annually in the US, among which ~2 million require outpatient treatment and ~70,000 hospitalization.6 While illness caused by norovirus is generally short-lived, mild, and self-healing in healthy people, it can be severe and sometimes fatal, especially among young children, the elderly and immunocompromised individuals.7 Each year noroviruses are responsible for the deaths of ~218,000 children less than 5 years old in developing countries.8 In developed countries, these viruses have been associated with deaths in the elderly in nursing homes, and are important pathogens causing chronic and sometimes severe illness in intestinal, solid organ and renal transplant patients.9–11 In addition, noroviruses are classified as class B biodefense agents in the US because of their high stability, extremely low infectious dose, and high rate of susceptibility in all age groups of humans, making them a potential threat to food and water supplies or the environment. To date, there have been no therapeutics or vaccines for the viral infections and there is therefore a pressing need to find effective countermeasures for noroviruses.
Theoretically, the best way to prevent a virus infection is by vaccination. While a noroviral virus-like particle vaccine is being developed, the broad-spectrum efficacy of this against the multiple types and emergence of new types of noroviruses remains to be determined,12 Even if vaccines are effective, antiviral drugs, would be useful to treat epidemics and chronic infections, such as acyclovir and oseltamivir used to treat herpes and influenza virus infections, respectively. These compounds have a broad antiviral activity because they target an essential viral protein, e.g., protease or neuraminidase, which often exhibits a high homology among the different serotypes of a viral family.
The genome of Norwalk virus (NV) consists of three open reading frames (ORFs). As shown in Figures 1a/b, ORF1 encodes a 200 kDa polyprotein, which is cleaved by a protease (NVPro) into six non-structural proteins, including NVPro itself.13 Because its function is essential for viral replication, NVPro represents a potential drug target for intervention.
Fig. 1.

(A) The polyprotein encoded by NV ORF1 is cleaved by NVPro into 6 functional proteins; (B) The peptide sequences of the five cleavage sites, with those highlighted in red being used for our inhibitor design; (C) A proposed mechanism of NVPro; (D) Structures and activities of NVPro inhibitors.
Our previous X-ray crystallographic study revealed NVPro is a chymotrypsin-like protease with the residues Cys139, His30 and Glu54 as the catalytic triad.14 A mechanism for the enzyme was proposed based on the structural information, as shown in Figure 1c. Cys139 functions as a nucleophile, His30 as a general base, and Glu54 is used to facilitate the correct orientation of the His30 imidazole ring. A series of peptidomimetic inhibitors of NVPro were recently reported, with the best compound in the series having an IC50 value of 870 nM (Figure 1d).15 We also determined the X-ray crystal structures of three tripeptide aldehyde inhibitors 1 - 3 (Figure 1d) in complex with NVPro.16 Here, we report the design, synthesis and activity of this series of tripeptide based inhibitors against two noroviral proteases from Norwalk and Houston viruses. Structure activity relationships (SAR) of these compounds are also discussed.
Results and Discussion
Inhibitor design and SAR
Based on the mechanism of NVPro, we first synthesized five tripeptide aldehyde compounds 2, 4 – 7, as shown in Table 1. This design exploited the P3-P1 sequences of the five cleavage sites for NVPro (Figure 1b) with the P1 amino acid being modified. The aldehyde functionality is to attract a possible nucleophilic attack by the –SH of Cys139 to form a covalent bond with the enzyme to increase the binding affinity. It is noted that since this covalent bond is reversible with rapid kinetics, these aldehyde compounds behave as the competitive inhibitors of NVPro, similar to those of rhinovirus 3C protease.17 The Gln side chain was chosen because the negatively charged Glu side chain could impair the cell membrane permeability of these compounds. In addition, two Me groups were used to mask the δ-CONH2 to avoid its cyclization with the aldehyde to form a six-membered hemiglutaminal species, as shown below in Scheme 1:
Table 1.
Structures and inhibitory activity (Ki) of NVPro inhibitors.
| cpd # | P3 | P2 | P1 | Ki(µM) | cpd # | P3 | P2 | P1 | Ki(µM) |
|---|---|---|---|---|---|---|---|---|---|
| 4 | 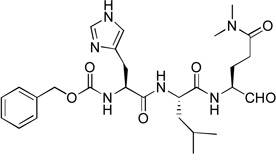 |
1.9 | 10 | 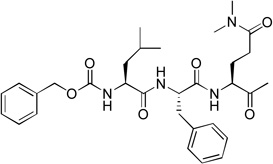 |
150 | ||||
| 5 | 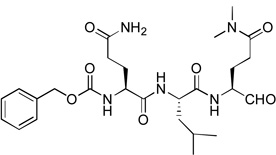 |
0.31 | 11 | 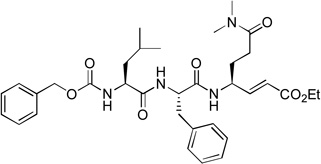 |
inactive | ||||
| 6 | 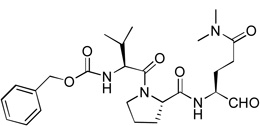 |
8.2 | 12 | 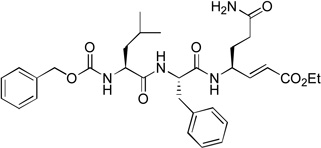 |
inactive | ||||
| 7 | 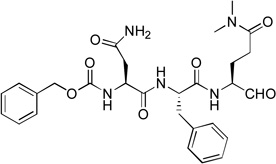 |
1.6 | 13 | 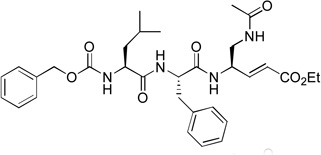 |
inactive | ||||
| 2 | 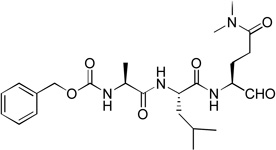 |
0.59 | 3 | 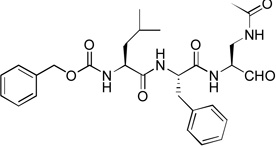 |
1.5 | ||||
| 1 | 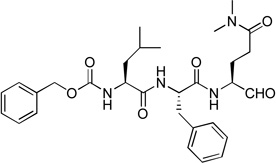 |
0.12 | 14 | 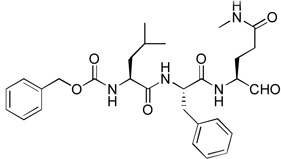 |
5.1 | ||||
| 8 | 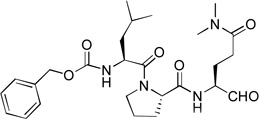 |
46 | 15 | 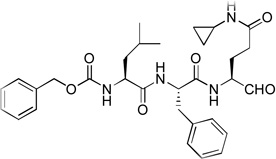 |
1.4 | ||||
| 9 | 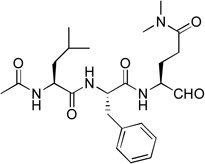 |
39 | 16 | 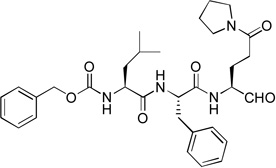 |
0.075 | ||||
Scheme 1.

Isomerization of glutaminal.
Similar inhibitor design has been applied to target the proteases of other RNA viruses such as rhinovirus and piconavirus.17–19 These five compounds were tested for their inhibitory activities against recombinant NVPro using our previously reported method14 and the results are shown in Table 1. All these five initial compounds were found to be NVPro inhibitors with their Ki values ranging from 0.31 – 8.2 µM. Compound 5 with a P3-P1 sequence of Gln-Leu-Gln(diMe) was found to be the strongest among these five, having a Ki value of 310 nM. Compound 2 with a sequence of Ala-Leu-Gln(diMe) is also a potent inhibitor with a Ki of 590 nM, while compounds 7 (Asn-Phe-Gln(diMe)) and 4 (His-Leu-Gln(diMe)) possess low µM inhibition (Ki: 1.6 and 1.9 µM, respectively). Compound 6 having a sequence of Val-Pro-Gln(diMe) exhibits the weakest activity with a Ki value of 8.2 µM among this series.
Since compounds 2, 4 and 5 contain a P2 Leu residue, we next synthesized compound 1 with a P2 Phe as well as 8 with a P2 Pro to compare their activities with those of compounds 7 and 5 with the same P2 residue, respectively. Compound 1 (Z-Leu-Phe-Gln(diMe)-CHO) was found to be a potent inhibitor of NVPro with a Ki value of 120 nM (Table 1), being ~3-5-fold more active than compounds 5 and 2 with the natural substrate sequences. On the other hand, compound 8 with a P3-P2 sequence of Leu-Pro is a very weak inhibitor (Ki = 46.1 µM).
The activity data from this small set of tripeptide aldehyde inhibitors suggest Leu or Phe is a more favorable P2 residue than Pro. For example, 1 is 380-fold more active than 8 with the same P3 and P1 residues, while the potencies of all inhibitors with P2=Leu or Phe (i.e., 1, 2, 4, 5 and 7) fall within a relatively narrow range of 0.1 – 1.9 µM. Our crystallographic studies show the P2 Leu or Phe side chain is deeply inserted into the hydrophobic S2 pocket of the protein well defined by I109, Q110, R112, and V114. However, the cyclic structure of Pro could restrict it from having deep contacts with the S2 pocket. Our SAR analysis further confirms that the van der Waals interactions between the inhibitor P2 and the protein S2 significantly affect the binding affinity. The second SAR is that the P3 residue seems to play a less important role, fine-tuning the inhibitory activity. For compounds 5, 2 and 4 with the same P2/P1, changing P3 residue from polar Gln to small/hydrophobic Ala as well as larger/bipolar His only results in ~6-fold activity decrease (0.31→0.59→1.9 µM). Similarly, ≤10× potency variation is found when altering the P3 Leu in compound 1 to the Asn in 7, or the P3 Val in 6 to the Leu in 8. This observation could be attributed to the less organized S3 pocket of NVPro, which is actually half exposed to the solvent.
Acetyl containing compound 9 was synthesized to find if the Z (benzyloxycarbonyl) protecting group is important for the inhibition. As compared to compound 1 with the same P3-P1 sequence, 9 (Ki = 39 µM) exhibits a large activity loss (>300-fold) for NVPro, showing Z contributes considerably to the tight binding of 1.
We next investigated the SARs with respect to the P1 residue. Since the aldehyde functionality is highly reactive and may be deactivated by cellular or plasma thiols such as glutathione,17 we synthesized compounds 10 and 11 with a methylketone and α, β-unsaturated ethyl ester as a nucleophile, respectively. These two functional groups have been successfully used in inhibitor discovery targeting other viral proteases. The P3-P1 sequence of 10 and 11 is the same as that of compound 1. However, ketone 10 only exhibits a very weak inhibitory activity with a Ki value of 150 µM, being 1,200-fold less active than 1 (Table 1). α, β-Unsaturated ester 11 was found to be essentially inactive. Because the α, β-unsaturated ester functionality is particularly attractive as it is chemically stable to non-specific nucleophilic attacks by cellular thiols,18,19 compounds 12 (with an unmasked Gln P1 residue) and 13 (with a β-acetyl diaminopropionic acid as the P1 residue) were also synthesized to test if a different P1 side chain affects the activity. Neither 12 nor 13 exhibits inhibitory activity against NVPro. These results show a highly reactive electrophile such as aldehyde is required to potently inhibit the protease.
Compounds 3, 14 – 16 were synthesized to optimize the P1 side chain. As compared with compound 1 with the same P3-P2 sequence of Leu-Phe, compound 3 with a reversed amide P1 side chain is ~12× less active (Ki = 1.5 µM). Compound 14 was designed to find if both N-Me of 1 is important for the activity. 14 was, however, found to possess a largely reduced inhibitory activity with a Ki value of 5.1 µM, being 42-fold less active than 1. Although the crystal structure of NVPro:1 indicates the N-Me trans to the carbonyl O atom in 1 has hydrophobic interactions with Ala160, it seems unlikely that the reduced van der Waals contacts for 14 (without the corresponding N-Me) is mainly responsible for such a large activity loss. More possibly, compound 14 exists as equilibrium between an aldehyde form and a cyclic hemiglutaminal (Scheme 1), with the latter species being inactive against NVPro. Indeed, the 1H NMR of compound 14 shows the aldehyde form accounts for <10%. Similarly, compound 15 (Ki = 1.4 µM) with an N-cyclopropyl-Gln side chain is also considerably less active than compound 1. As compared to 14, the ~3× improved inhibition for 15 might be attributed to a more favorable hydrophobic interaction between the cyclopropyl with the S1 pocket of NVPro. Compound 16, an analog of 1 containing a pyrrolidine-amide in the P1 position, was found to be the most potent inhibitor of NVPro with a Ki value of 75 nM. The slightly enhanced activity should be due to the additional van der Waals interactions of the 5-membered pyrrolidine ring with Lys162 and Ser163 in the S1 pocket.
Inhibition of Houston virus protease (HOVPro)
Houston virus (HOV) belongs to the genotype II noroviruses.5 As shown in Figure 2a, these two noroviral proteases exhibit a high sequence homology of 66% identity and 85% similarity. Because of its high prevalence in noroviral infections in recent years, we also tested 4 selected inhibitors, 1 – 3 and 5, against the protease of HOV (HOVPro) and the results are shown in Table 2. As compared with NVPro, these compounds exhibited reduced inhibitory activities against HOVPro with Ki values of 4.3 – 14 µM. Compound 1 with P2-P1 of Phe-Gln(diMe) shows ~60-fold activity reduction, while compounds 2 and 5 with a Leu at the P2 have ~10-fold decreases, being the best HOVPro inhibitors (Ki = 4.6 and 4.3 µM, respectively). Although having the same P3-P2 residues as 1, compound 3 with a β-acetyl diaminopropionic acid as the P1 residue also show ~10× activity loss.
Fig. 2.

(A) Alignment of NVPro and HOVPro protein sequence. The conserved residues that compose the S2 pockets of the two proteins are labeled with a bracket and a major difference is highlighted red letters; (B) Superposition of the crystal structure of NVPro (PDB: 2FYQ) shown in cyan and a homology model of HOVPro structure in gold. The S2 pocket loop (bII-cII) shows a different conformation in the HOV protease, which appears to be due to the Gly115 residue. In the NV protease the residue at the 115 position is His; (C) Tables depict the conserved residues that compose the S1, S2 and S4 pockets, in NV and HOV proteases.
Table 2.
Ki (µM) against NVPro and HOVPro.
| compound | NVPro | HOVPro |
|---|---|---|
| 1 | 0.12 | 7.5 |
| 2 | 0.59 | 4.6 |
| 3 | 1.5 | 14 |
| 5 | 0.31 | 4.3 |
To find out possible reasons for the activity changes, a homology modeling of HOVPro was performed using the program Phyre20 and the result is shown in Figure 2b, superimposed together with the crystal structure of NVPro. There is only one major conformational difference between the two structures. The S2 loop of HOVPro (104 – 115) moves more closely towards the protein active site, with the β-sheet kinked at Gly115. However, in NVPro, the side chain of the corresponding residue His115 forms a H-bond with Glu75, which stabilizes the NVPro S2 loop and prevents it from turning inwards (Fig. 2b). The consequence for the closer S2 loop of HOVPro is a smaller S2 pocket, which favours a smaller P2 residue of an inhibitor. This homology model seems to be reasonable, since compounds 2 and 5 with a smaller Leu as a P2 show better inhibitory activities than compound 1 with a bulkier Phe at this position (Table 2). The generally reduced activities of these inhibitors could also be due to the same reason. Gly115 renders the S2 loop of HOVPro more flexible (with a higher overall entropy), which could reduce the binding affinity of an inhibitor. In addition, as shown in Figure 2c, the S1 pocket of HOVPro contains more polar residues (i.e., Thr135 and 158 vs. Ile135 and A158 in NVPro). Our experimental results (~10× activity reduction for 3 vs. >60× for 1) suggest β-acetyl diaminopropionate at the P1 position could be more favourable for HOVPro.
Compound synthesis
The general methods for synthesis of compounds 1 – 16 are shown in Scheme 2 and detailed synthesis as well as compound characterization are described in Electronic Supplementary Information (ESI). EDC (N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride) and HOBT (1-hydroxybenzotriazole) were used as a general peptide/amide coupling method. Amino acid methyl ester 19, representing the P1 component of the final products, was synthesized from Z-Glu-OMe (α-Z-protected glutamic acid methyl ester) or α-Z-protected (S)-2,3-diaminopropionic acid methyl ester. Using an EDC/HOBT coupling reaction followed by hydrogenation to remove the Z protection group, tripeptide 21 was prepared from 19, via a dipeptide intermediate 20. The methyl ester of 21 was selectively reduced to a primary alcohol followed by Dess–Martin periodinane oxidation, to give compounds 1 – 8 and 14 – 16 having an aldehyde functionality. For synthesizing compounds 11 – 13, the corresponding aldehyde was treated with the sodium salt of triethyl phosphonoacetate in tetrahydrofuran to convert the aldehyde to α, β-unsaturated ethyl ester. Compound 10 was prepared by treating compound 1 with MeMgBr at −78 °C followed by Dess–Martin periodinane oxidation. For making acetyl containing compound 9, Ac-Leu-OH was used to react with compound 20 to obtain an acetyl protected tripeptide 21, which was reduced and oxidized to give compound 9.
Scheme 2.

General synthetic methods. Reagents and conditions: (i) X1X2NH, EDC, HOBT, triethylamine; (ii) H2, 5% Pd/C; (iii) Ac2O, AcOH; (iv) LiBH4; (v) Dess–Martin periodinane; (vi) triethyl phosphonoacetate, NaH; (vii) MeMgBr.
Conclusion
In conclusion, a series of 16 tripeptide based compounds were designed and synthesized targeting the protease of noroviruses, which is a potential drug target for treating noroviral infections. Compounds 1 and 16 with a general structure of Z-Leu-Phe-Gln(N,N-X1,X2)-CHO were found to be the most potent inhibitors of NVPro with Ki values of 120 and 75 nM, respectively. From the activities of this small set of compounds, SAR analysis shows that 1) a potent NVPro inhibitor requires to have an aldehyde group to be the P1 electrophile; 2) N,N-disubstituted Gln is the most favorable P1 side chain; 3) Phe or Leu is favorable for the P2, while Pro at this position is bad for the activity; and 4) the P3 residue seems to play a minor role in tight binding. Selected NVPro inhibitors were tested against the protease of the genotype II Houston virus. Despite a high homology of these two proteases, NVPro inhibitors exhibited a considerably reduced activity against HOVPro. A homology model of HOVPro was generated, which can rationalize the observed activity changes.
Supplementary Material
Acknowledgment
This work was supported by grants from the NIH (PO1 AI057788 to M.K.E., T.P. and B.V.V.P., and P30DK56338 to M.K.E.), the Robert Welch Foundation (Q1292 to B.V.V.P.), the Caroline Wiess Law Fund for Molecular Medicine and a seed grant from Texas Medical Center Digestive Disease Center (to Y.S.).
Footnotes
Electronic Supplementary Information (ESI) available: Detailed Experimental Section. See DOI: 10.1039/b000000x/
References
- 1.Patel MM, Hall AJ, Vinjé J, Parashar UD. J. Clin. Virol. 2009;44:1. doi: 10.1016/j.jcv.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Hale AD, Tanaka TN, Kitamoto N, Ciarlet M, Jiang X, Takeda N, Brown DW, Estes MK. J Clin. Microbiol. 2000;38:1656. doi: 10.1128/jcm.38.4.1656-1660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopman BA, Brown DW, Koopmans M. J. Clin. Virol. 2002;24:137. doi: 10.1016/s1386-6532(01)00243-8. [DOI] [PubMed] [Google Scholar]
- 4.Vinje J, Altena SA, Koopmans MP. J. Infect. Dis. 1997;176:1374. doi: 10.1086/517325. [DOI] [PubMed] [Google Scholar]
- 5.Green KY. In: Fields Virology. Knipe DM, editor. Philadelphia: Lippincott, Williams & Wilkins; 2007. pp. 949–980. [Google Scholar]
- 6.Hall AJ, Lopman BA, Payne DC, Patel MM, Gastañaduy PA, Vinjé J, Parashar UD. Emerg. Infect. Dis. 2013;19 doi: 10.3201/eid1908.130465. in press, DOI: 10.3201/eid1908.130465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bok K, Green KY. N Engl. J. Med. 2012;367:2126. doi: 10.1056/NEJMra1207742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel MM, Widdowson MA, Glass RI, Akazawa K, Vinje J, Parashar UD. Emerg. Infect. Dis. 2008;14:1224. doi: 10.3201/eid1408.071114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Florescu DF, Hill LA, McCartan MA, Grant W. Pediatr. Transplant. 2008;12:372. doi: 10.1111/j.1399-3046.2007.00875.x. [DOI] [PubMed] [Google Scholar]
- 10.Gallimore CI, Lewis D, Taylor C, Cant A, Gennery A, Gray JJ. J. Clin. Virol. 2004;30:196. doi: 10.1016/j.jcv.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Kaufman SS, Chatterjee NK, Fuschino ME, Magid MS, Gordon RE, Morse DL, Herold BC, LeLeiko NS, Tschernia A, Florman SS, Gondolesi GE, Fishbein TM. Am. J. Transplant. 2003;3:764. doi: 10.1034/j.1600-6143.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 12.Atmar RL, Estes MK. Expert Rev. Vaccines. 2012;11:1023. doi: 10.1586/erv.12.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asanaka M, Atmar RL, Ruvolo V, Crawford SE, Neill FH, Estes MK. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10327. doi: 10.1073/pnas.0408529102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeitler CE, Estes MK, Venkataram Prasad BV. J Virol. 2006;80:5050. doi: 10.1128/JVI.80.10.5050-5058.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiew KC, He G, Aravapalli S, Mandadapu SR, Gunnam MR, Alliston KR, Lushington GH, Kim Y, Chang KO, Groutas WC. Bioorg. Med. Chem. Lett. 2011;21:5315. doi: 10.1016/j.bmcl.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muhaxhiri Z, Deng L, Sreejesh SS, Sankaran B, Estes MK, Palzkill T, Song Y, Prasad BVV. J Virol. 2013;87:4281. doi: 10.1128/JVI.02869-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webber SE, Okano K, Little TL, Reich SH, Xin Y, Fuhrman SA, Matthews DA, Love RA, Hendrickson TF, Patick AK, Meador JW, 3rd, Ferre RA, Brown EL, Ford CE, Binford SL, Worland ST. J Med. Chem. 1998;41:2786. doi: 10.1021/jm980071x. [DOI] [PubMed] [Google Scholar]
- 18.Dragovich PS, Prins TJ, Zhou R, Webber SE, Marakovits JT, Fuhrman SA, Patick AK, Matthews DA, Lee CA, Ford CE, Burke BJ, Rejto PA, Hendrickson TF, Tuntland T, Brown EL, Meador JW, 3rd, Ferre RA, Harr JE, Kosa MB, Worland ST. J. Med. Chem. 1999;42:1213. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- 19.Dragovich PS, Webber SE, Babine RE, Fuhrman SA, Patick AK, Matthews DA, Reich SH, Marakovits JT, Prins TJ, Zhou R, Tikhe J, Littlefield ES, Bleckman TM, Wallace MB, Little TL, Ford CE, Meador JW, 3rd, Ferre RA, Brown EL, Binford SL, DeLisle DM, Worland ST. J. Med. Chem. 1998;41:2819. doi: 10.1021/jm9800696. [DOI] [PubMed] [Google Scholar]
- 20.Kelley LA, Sternberg MJE. Nat. Protoc. 2009;4:363. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.