

Abstract
Polycyclic aromatic hydrocarbons (PAHs), such as benzo[a]pyrene (BaP), are ubiquitous environmental contaminants that are implicated in causing lung cancer. BaP is a component of tobacco smoke that is transformed enzymatically to active forms that interact with DNA. We reported previously development of a sensitive stable isotope dilution LC/MS method for analysis of BaP metabolites. We now report efficient syntheses of 13C4-BaP and the complete set of its 13C4-labelled oxidized metabolites needed as internal standards They include the metabolites not involved in carcinogenesis (Group A) and the metabolites implicated in initiation of cancer (Group B). The synthetic approach is novel, entailing use of Pd-catalyzed Suzuki, Sonogashira, and Hartwig cross-coupling reactions combined with PtCl2-catalyzed cyclization of acetylenic compounds. This synthetic method requires fewer steps, employs milder conditions, and product isolation is simpler than conventional methods of PAH synthesis. The syntheses of 13C4-BaP and 13C4-BaP-8-ol each require only four steps, and the 13C-atoms are all introduced in a single step. 13C4-BaP-8-ol serves as the synthetic precursor of all the oxidized metabolites of 13C-BaP implicated in initiation of cancer. The isotopic purities of the synthetic 13C4-BaP metabolites were estimated to be ≥99.9%.
Keywords: Benzo[a]pyrene (BaP), Carcinogenic polycyclic aromatic, hydrocarbons (PAHs), Synthesis of 13C4-labelled BaP, 13C4-Labelled oxidized metabolites of BaP, Enzymatic activation of PAH carcinogens, Synthesis of PAHs via Pd-catalyzed cross-coupling reactions
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are widespread environmental pollutants that are implicated in initiation of lung cancer.1–3 PAHs are produced in the combustion of fossil fuels and other organic matter,1,3,4 and significant levels of PAHs are present in tobacco smoke,5 auto and diesel engine emissions,6 and in fried, smoked, and charbroiled meats.1,3
Metabolic activation of PAHs is required for expression of their carcinogenic activity.1,7,8 Benzo[a]pyrene (BaP)* has been most intensively investigated, and current evidence indicates that BaP is activated via three pathways, the diol epoxide path, the quinone path, and the radicalcation path (Fig. 1).1,3,7
Fig. 1.

Pathways of enzymatic activation of benzo[a]pyrene (BaP).
The diol epoxide path entails cytochrome P450 catalyzed oxidation of BaP to form metastable arene oxide metabolites that rearrange to phenols and/or undergo hydration to dihydrodiols.1 The (±)-trans-7,8-dihydrodiol of BaP (BaP 7,8-diol) (1) undergoes further enzyme-catalyzed oxidation to form highly mutagenic (±)-anti- and (±)-syn-diol epoxide metabolites (4 and 5) that react with DNA to form adducts.1,8 The quinone path entails aldo-keto reductase (AKR)-mediated oxidation of 1 to BaP 7,8-catechol (3). This enters into a redox cycle with O2 to form BaP 7,8-dione (2) and reactive oxygen species (ROS) that attack DNA to form 8′-hydroxy-2′-deoxyguanosine (8′-HO-2′-dGua) and cause DNA strand breaks.1,7c,9 The quinone 2 also combines with DNA to furnish stable and depurinating adducts. Collectively these events result in initiation of cancer. An analogous pathway involving quinone metabolites of steroids is involved in oestrogen-related carcinogenesis leading to breast cancer.10 The radicalcation path entails one-electron oxidation of BaP catalyzed by P450 monooxygenase or peroxidase to form a BaP radicalcation that attacks DNA to yield depurinating adducts.11a The signature metabolites formed via this pathway are BaP 1,6-dione (6) and BaP 3,6-dione (7). However, the involvement of the radicalcation pathway in carcinogenesis is disputed.1a,11b
Human bronchoalveolar H358 cells were examined recently as a model for study of the metabolism of BaP in normal human lung cells.12,13 The findings indicated that activation of 1 in these cells involves the AKR-mediated quinone pathway.14 More recently, we developed a stable isotope dilution atmospheric pressure chemical ionization tandem mass spectrometric method to assay quantitatively the metabolites formed by all three metabolic pathways.15 The 13C4-BaP metabolites whose syntheses are reported herein were employed as internal standards. In other studies, the syntheses of 13C2-BaP, 13C2-1, and 13C2-2 were also reported.16
2. Results
The aim of this investigation was to develop methods for efficient synthesis of the 13C4-labelled analogues of the complete set of oxidized metabolites of BaP. The BaP metabolites may be divided into two groups on the basis of their involvement in carcinogenesis. Group A includes the oxidized metabolites of BaP that current evidence indicates do not play a role in carcinogenesis [the 1-, 2-, 3-, 9-, and 12-phenol isomers of BaP, BaP-1,6-dione (6) and BaP-3,6-dione (7)] (Fig. 1). Group B includes the oxidized metabolites of BaP implicated in carcinogenesis [BaP 7,8-diol (1), BaP 7,8-dione (2), BaP 7,8-catechol diacetate (3), (±)-anti-BPDE (4), and (±)-syn-BPDE (5)] (Fig. 1) plus 8-HO-BaP and 9-HO-BaP. The 13C4-labelled BaP metabolites are needed as internal standards for LC/MS analysis of the BaP metabolites formed in human cells. This methodology is expected to provide a tool to assess the relative contributions of the three metabolic pathways to induction of cancer.
The methods of synthesis of the 13C4-labelled BaP metabolites involve the use of Pd-catalyzed cross-coupling reactions (Suzuki, Sonogashira, and/or Hartwig) in combination with PtCl2-catalyzed cyclization of acetylenic intermediates. This novel synthetic approach requires fewer steps and employs milder reaction conditions than the conventional methods for construction of PAH ring systems based on Friedel–Crafts chemistry. This synthetic method also has the advantage that the requisite 13C-labelled precursors are available from commercial sources.
2.1. Part I. BaP metabolites not implicated in carcinogenesis (Group A)
The initial synthetic targets were the 13C-labelled 1-, 2-, 3-, 9-, and 12-phenols of BaP. Exploratory studies to establish the feasibility of the planned synthetic approach were carried out with unlabelled precursors.
2.1.1. Synthesis of benzo[a]pyren-1-ol, -2-ol, and -3-ol
2.1.1.1. Benzo[a]pyren-3-ol (14f)
It was shown previously that 14f is the principal phenol metabolite of BaP formed in H358 human cells.13 Synthesis of 14f was carried out by the sequence in Scheme 1. Palladium-catalyzed Suzuki–Miyaura cross-coupling of 1-bromo-2-iodobenzene (8) with the 2-boronate ester of 7-methoxynaphthalene (9c) took place at the iodo position regiospecifically to furnish 2-(2-bromophenyl)-7-methoxynaphthalene (10c). Pd-catalyzed cross-coupling of 10c with BrZnCH2CO2R was carried out by a procedure based on Hartwig’s method.17 The choice of this route was dictated by the commercial availability of 13C2-BrCH2CO2Et. However, only tert-butyl esters were employed in the published examples of this reaction. Direct reaction of 10c with the zinc enolate of tert-butyl acetate afforded the expected tert-butyl ester adduct (11a) in moderate yield, but similar reaction of 10c with the zinc enolate of ethyl acetate failed to furnish the adduct of the ethyl ester (11b). However, cross-coupling of 10c with BrZnCH2CO2Et took place smoothly in the presence of Pd(dba)2 and Q-phos to yield ethyl 2-(7-methoxynaphthalenyl)phenylacetate (11b) in moderate yield (40%).
Scheme 1.

A brief study of this reaction was undertaken with the intent of improving the yield of 11b (Table 1). The yield was significantly improved by: (1) increasing the ratio of BrZnCH2CO2Et from 1.1 equiv to 2.0–3.0 equiv and (2) increasing the catalyst ratio from 1.0 mol % to 5.0 mol %. On the other hand, decreasing reaction time from 12 h to 1.5 h had minimal effect. The syntheses of the 13C-labelled compounds were carried out using the conditions in entry 5.
Table 1.
Effect of conditions on reaction of 10c with BrZnCH2CO2Eta
| Entry | Time (h) | Pd(dba)2/Q-phos (mol %) | BrZnCH2CO2 Et (equiv) | Yield 11b (%) |
|---|---|---|---|---|
| 1 | 12 | 1 | 1.1 | 40 |
| 2 | 12 | 5 | 1.1 | 52 |
| 3 | 12 | 1 | 3.0 | 62 |
| 4 | 1.5 | 1 | 3.0 | 92 |
| 5b | 1.5 | 5 | 2.0 | 85 |
| 6 | 1.5 | 10 | 3.0 | 90 |
| 7 | 1.5 | 20 | 3.0 | 95 |
Reactions were carried out by the method reported.17
Syntheses of 13C-labelled analogues were carried out under these conditions.
Treatment of 11b with NaOH in EtOH gave 2-(7-methoxynaphthalen-2-yl)phenylacetic acid (11c) (90%) (Scheme 1), and 11c underwent cyclization in the presence of MeSO3H at 50 °C to furnish 3-methoxychrysen-5-ol (12e) (77%). This was converted to the triflate ester (12f), by treatment with trifluoromethanesulfonic anhydride, and Sonogashira coupling18 of 12f with (trimethylsilyl) acetylene (TMSA) in the presence of Pd(PPh3)2Cl2, CuI, and TEA in DMF gave (3-methoxychrysen-5-ylethynyl)-trimethylsilane (13e) (89%). Desilylation of 13e with K2CO3 in MeOH/THF furnished 5-ethynyl-3-methoxychrysene (13f) (90%), and PtCl2-catalyzed cyclization19 of 13f afforded 3-methoxybenzo[a]pyrene (14e) (60%). Demethylation of 13f with BBr3 gave benzo[a]pyren-3-ol (14f).
2.1.1.2. Benzo[a]pyren-1-ol (14b) and benzo[a]pyren-2-ol (14d)
Syntheses of 14b and 14d were carried out by the method in Scheme 1. The 2-boronate ester of 5-methoxynaphthalene (9a) was prepared from 5-methoxy-2-naphthol,19,20 and Suzuki cross-coupling of 8 with 9a in the presence of Pd(OAc)2/PPh3 provided 2-(2-bromophenyl)-5-methoxynaphthalene (10a). Hartwig coupling17 of 10a with BrZnCH2CO2Et in the presence of Pd(dba)2 and Q-phos afforded ethyl 2-(5-methoxynaphthalenyl)phenylacetate (11b). Ethanolysis of 11b gave 11e, and acid-catalyzed cyclization of 11e furnished 1-methoxychrysen-5-ol (12a). Sonogashira coupling of the triflate ester (12b) with TMSA yielded (1-methoxychrysen-5-ylethynyl)trimethylsilane (13a). Removal of the TMS group followed by PtCl2-catalyzed cyclization18 afforded 1-methoxybenzo [a]pyrene (14a), and demethylation gave 14b. Synthesis of benzo [a]pyren-2-ol (14d) was carried out via an analogous sequence based on reaction of 8 with the 2-boronate ester of 6-methoxynaphthalene (9b) (Scheme 1).
2.1.2. Synthesis of benzo[a]pyren-9-ol (21b)
Synthesis of 21b was accomplished via an analogous sequence employing consecutive Suzuki, Hartwig, and Sonogashira cross-coupling reactions (Scheme 2). Pd-catalyzed Suzuki cross-coupling of 1-bromo-2-iodo-4-methoxybenzene (15)21 with naphthalene 2-boronic acid ester (16) furnished 2-(2-bromo-5-methoxyphenyl)naphthalene (17). Pd-catalyzed Hartwig coupling of 17 with BrZnCH2CO2Et provided ethyl 2-(napththalen-2-yl)-5-methoxyphenyl acetate (18a). Ethanolysis of 18a gave 18b, which underwent acid-catalyzed cyclization to 3-methoxychrysen-11-ol (19a) and esterification to the triflate ester 19b. Sonogashira coupling of 19b with TMSA furnished ((9-methoxychrysen-5-yl)ethynyl) trimethylsilane (20a), and removal of the trimethylsilyl group gave 5-ethynyl-9-methoxychrysene (20b). Finally, PtCl2-catalyzed cyclization of 20b furnished 9-methoxy-BaP (21a), and demethylation gave 21b.
Scheme 2.

In principle, benzo[a]pyren-8-ol and its 13C4-labelled analogue are accessible via an analogous sequence employing 1-bromo-2-iodo-5-methoxybenzene in place of 15. However, 8-HO-BaP was synthesized by the alternative method described in Part II.
2.1.3. Synthesis of benzo[a]pyren-12-ol (27b)
Synthesis of 27b was accomplished by consecutive application of the Suzuki, Hartwig, and Sonogashira cross-coupling methods (Scheme 3). 4-Methoxynaphthalene-2-boronate ester (22) was synthesized from 2-bromo-4-methoxynapthalene22 by modification of the method for preparation of 9c. Pd-catalyzed Suzuki cross-coupling of 22 with 8 gave 2-(2-bromophenyl)-4-methoxynaphthalene (23), and Pd-catalyzed cross-coupling of 23 with BrZnCH2CO2Et provided ethyl 2-(4-methoxynapththalen-2-yl)phenyl acetate (24a). Ethanolysis of 24a afforded the carboxylic acid (24b), which underwent acid-catalyzed cyclization to 12-methoxychrysen-5-ol (25a). This phenol was converted to the triflate ester (25b), and Sonogashira coupling of 25b with TMSA afforded 26a. Desilylation of 26a gave 26b, and PtCl2-catalyzed cyclization of the latter gave 12-methoxy-BaP (27a), which underwent demethylation to furnish 27b.
Scheme 3.

2.1.4. Synthesis of benzo[a]pyren-1,6-dione (6) and -3,6-dione (7)
The BaP-1,6- and 3,6-diones (6 and 7) were prepared by oxidation of BaP-1-ol (14b) and BaP-3-ol (14f) with bis(trifluoroacetoxy)iodobenzene (BTI) by the method reported.20,23
2.1.5. Synthesis of 13C4-labelled BaP and its Group A metabolites
BaP and 13C4-BaP were synthesized by two methods. Method A was modelled on the synthesis of the 1-, 2-, and 3-phenols of BaP (Scheme 1). Initial studies were conducted with unlabelled precursors (Scheme 4). Pd-catalyzed Suzuki coupling of 8 with naphthalene-2-boronate ester (28) gave 2-(2-bromophenyl) naphthalene (29), and cross-coupling of 29 with BrCH2CO2Et by the modified Hartwig method gave ethyl 2-(2-naphthalenyl)-phenylacetate (30a). Conversion of 30a to the carboxylic acid (30b) and acid-catalyzed cyclization of 30b gave chrysen-5-ol (31a). Sonogashira cross-coupling of the triflate ester (31b) with TMSA yielded (chrysen-5-ylethynyl)trimethyl silane (32a). Removal of the TMS group by treatment of 32a with K2CO3 in MeOH/THF afforded 32b, and PtCl2-catalyzed cyclization gave BaP.
Scheme 4.

*Sites of the 13C-atoms.
Synthesis of 13C4-BaP was accomplished in seven steps from 29 (Scheme 4). The 13C-atoms were incorporated in pairs, the first pair in the cross-coupling of the Reformatsky ester 13C2-BrZnCH2CO2R with 29, and the second pair in the Sonogashira cross-coupling24 of 13C2-TMSA with the triflate ester of 13C2-chrysen-5-ol (31b). The 13C-atoms in 13C4-BaPare located at the C-4,-5,-5a, and -6 aromatic ring positions.
The 13C4-BaP phenol isomers (Fig. 2) were synthesized by methods analogous to those used for synthesis of the unlabelled BaP phenols. The 13C-atoms were at the 4,5,5a, and 6-positions of BaP, the same as those of the 13C-atoms in 13C4-BaP. The methods for syntheses of 13C4-1-HO-BaP, 13C4-2-HO-BaP, and 13C4-3-HO-BaP were analogous to those used for preparation of 13C4-BaP (Scheme 4), using the appropriate methoxy-substituted derivatives (9a, 9b, and 9c) of the boronate ester in place of 28. Similarly, the syntheses of 13C4-HO-9-BaP and 13C4-12-HO-BaP were carried out by appropriate modification of the procedures for synthesis of unlabelled 9-HO-BaP (Scheme 2) and 12-HO-BaP (Scheme 3).
Fig. 2.
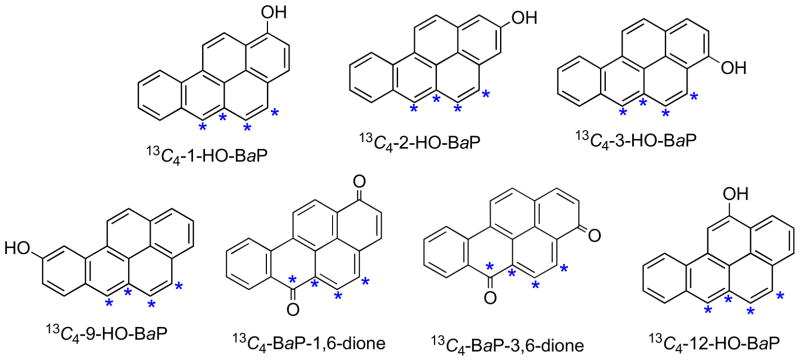
13C4-Labelled BaP phenols and quinones (*sites of 13C-atoms).
The 13C4-labelled 1,6- and 3,6-quinones of BaP (Fig. 2) were prepared by oxidation of 13C4-1-HO-BaP and 13C4-3-HO-BaP with bis-(trifluoroacetoxy)iodobenzene (TBI).20,23
2.2. Part II. BaP metabolites implicated in carcinogenesis (Group B)
The synthetic targets in this phase were the 13C4-labelled oxidized metabolites of BaP in Group B. They include the BaP metabolites implicated in initiation of cancer [Fig. 1: BaP 7,8-diol (1), BaP 7,8-dione (2), BaP 7,8-catechol diacetate (3), (±)-anti-BPDE (4), and (±)-syn-BPDE (5)] plus the 8- and 9-phenol isomers (37c and 37e).
2.2.1. Synthesis of BaP, benzo[a]pyren-8-ol (37c), and BaP-9-ol (37e) via Method B
The BaP metabolites 1–5 were shown previously to be accessible via a synthetic route based on benzo[a]pyren-8-ol (37c).20 Synthesis of BaP via an analogous route (designated Method B) was initially investigated. This method (Scheme 5) entailed Pd-catalyzed Suzuki–Miyaura cross-coupling of naphthylboronic acid (33a) with 1-bromo-2,6-dimethoxy benzene (34a). Reaction took place in the presence of Pd(OAc)2Cl2, biphenyl(di-tert-butylphosphine), and K3PO4 in THF at 40 °C to yield 2-(2,6-dimethoxyphenyl)naphthalene (35a). Demethylation of 35a with BBr3 yielded 2-(2,6-dihydroxyphenyl)naphthalene (35b), and treatment of the latter with triflic anhydride and pyridine afforded the triflate diester (35c). Sonogashira coupling24 of 35c with TMSA in the presence of Pd(Ph3)2Cl2, CuI, and TEA in DMF furnished 36a. Reaction of 36a with K2CO3 in MeOH/THF provided 2-(2,6-diethynylphenyl)naphthalene (36b), and PtCl2-catalyzed cyclization18 of 36b gave BaP (37a). This synthetic route to BaP is shorter than Method A (Scheme 4), and the availability of these two synthetic approaches provides the basis for the synthesis of two different pure 13C4-BaP isotopomers.
Scheme 5.

Benzo[a]pyren-8-ol (37c) was synthesized by an analogous sequence (Scheme 5). 1-Bromo-2,6-dibenzyoxybenzene (34c) was prepared by demethylation of 34a with BBr3 and base-catalyzed reaction of 1-bromo-2,6-dihydroxybenzene (34b) with benzyl bromide. Pd-catalyzed Suzuki cross-coupling of 34c with 6-methoxynaphthy lboronic acid (33b) furnished 2-(2,6-dibenzyloxyphenyl)-6-methoxynaphthalene (35d), and removal of the benzyl groups (by hydrogenation over a Pd/C catalyst) afforded 2-(2,6-dihydroxy phenyl)-6-methoxynaphthalene (35e). Treatment of 35e with triflic anhydride and pyridine provided the triflate diester (35f), and Pd-catalyzed Sonogashira coupling of 35e with TMSA furnished 36c. Reaction of 36c with K2CO3 in MeOH/THF afforded 2-(2,6-diethynylphenyl)naphthalene (36d), and PtCl2-catalyzed cyclization of 36d furnished 8-MeO-BaP (37b). Demethylation of 37b with BBr3 afforded 37c.
The synthetic approach in Scheme 5 was improved by use of 2,6-dibromo-1-iodobenzene (38)25 in place of 34a as the aryl halide reactant (Scheme 6). Compound 38 was prepared from 2,6-dibromoaniline by a modification of the literature method.25 Suzuki cross-coupling of 33a with 38 took place smoothly in the presence of Pd(PPh3)4 and KF in refluxing dioxane to provide 2-(2,6-dibromophenyl)naphthalene (39a). Double Sonogashira coupling of 39a with TMSA followed by removal of the TMS groups and PtCl2-catalyzed cyclization gave BaP (37a). Synthesis of BaP via this route requires only four steps.
Scheme 6.

Benzo[a]pyren-8-ol (37c) was synthesized by a similar sequence (Scheme 6). Pd-catalyzed Suzuki coupling of 33b with 38 provided 2-(2,6-dibromophenyl)-6-methoxynaphthalene (39b), and Pd-catalyzed double Sonogashira coupling of 39b with TMSA furnished 40c. This was transformed to 37c by removal of the TMS groups to give 40d, PtCl2-catalyzed cyclization to yield 37b, and demethylation to 37c.
Benzo[a]pyren-9-ol (37e) was synthesized by an analogous sequence (Scheme 6). Pd-catalyzed Suzuki–Miyaura cross-coupling of 33c with 38 furnished 39c, and this was converted to 9-HO-BaP (37e) via double Sonogashira coupling with TMSA, removal of the TMS groups, PtCl2-catalyzed cyclization, and demethylation. The boronate ester 2-(7-methoxynaphthalen-2-yl)-5,5-dimethyl-[1,3,2]dioxaborinane may be used in place of 33c.
2.2.2. Synthesis of BaP metabolites (1–5) implicated in carcinogenesis
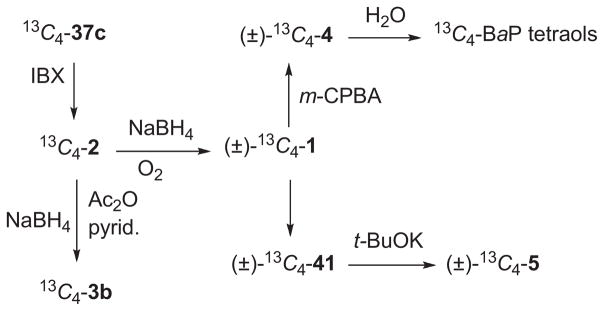
The BaP metabolites (1–5) implicated in initiation of cancer were shown previously to be synthetically accessible via a sequence based on 37c (Scheme 7).20,23 This approach was employed for the synthesis of the BaP metabolites 1–5 and their 13C4-labelled analogues. Oxidation of 37c with o-iodoxybenzoic acid (IBX) gave BaP 7,8-dione (2),19,20,23 and reduction of 2 with NaBH4/O2 furnished (±)-BaP 7,8-diol (1). Although BaP 7,8-catechol (3a) decomposes in air, it may be obtained pure as its diacetate derivative (3b) by reduction of 2 with NaBH4 in DMF and diacetylation with Ac2O/pyridine.20,23,26
Scheme 7.

anti-BPDE (4) is by definition the BaP diol epoxide isomer with the epoxide oxygen atom on the molecular face opposite the benzylic hydroxyl group, whereas syn-BPDE (5) bears these groups on the same face (Fig. 1).1b (±)-anti-BPDE was synthesized by epoxidation of 1 with m-chloroperbenzoic acid,1b,27,28 and (±)-syn-BPDE was prepared by conversion of 1 to the trans-bromohydrin (41) and base-catalyzed cyclization by established methods.27,28 The pure enantiomers of 1 are readily accessible by chromatographic separation of the diastereomeric (−)-menthoxyacetate or MTPA esters of 1.29 Small amounts of the (+) and (−)-enantiomers of 1 may be obtained by chromatography of the racemates on chiral HPLC columns.30
2.2.3. 13C4-Labelled metabolites of BaP
Syntheses of 13C4-BaP and its 1-, 2-, 3-, 9-, and 12-phenol isomers (with 13C at C-4, -5, -5a, and -6) ((Scheme 4 and Fig. 2) via Method B were described in Part I. Syntheses of 13C2-BaP and its key oxidized metabolites 13C2-BaP trans-7,8-diol (13C2-1) and 13C2-BaP-7,8-dione (13C2-2) with 13C at C-5,11) (Fig. 2) were reported previously.16
The structures of the 13C4-labelled BaP derivatives synthesized in Part II via Method B are shown in Fig. 3. They include 13C4-BaP, 13C4-8-HO-BaP (13C4-37c), and 13C4-9-HO-BaP (13C4-37e) (with 13C at C-4, -5, -11, and -12). Also included are the 13C4-labelled metabolites of BaP implicated in carcinogenesis [13C4-BaP trans-7,8-diol (13C4-1), 13C4-BaP 7,8-dione (13C4-2), BaP 7,8-catechol (13C4-3) and 13C4-(±)-anti-BPDE (13C4-4) with 13C at C-4, -5, -11, and -12] (Fig. 3). BaP 7,8-catechol (3) was previously shown to undergo decomposition in air.26 For this reason the 13C4-BaP 7,8-catechol was isolated as its stable diacetate (13C4-3 diacetate). And finally, the mixed 13C4-BaP tetraol isomers were prepared by hydrolysis of 13C4-(±)-anti-BPDE.
Fig. 3.
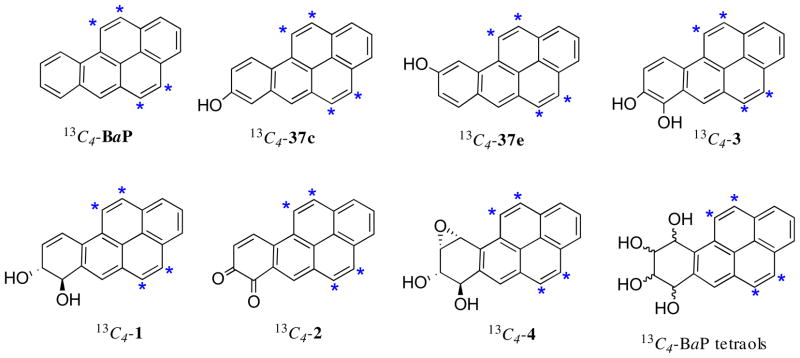
13C4-labelled BaP metabolites synthesized by Method B (*sites of 13C-atoms).
13C4-BaP (13C at C-4, -5, -11, and -12) was synthesized by a sequence similar to that for synthesis of unlabelled BaP (Scheme 8). Use of this method allowed incorporation of both pairs of 13C4-atoms to take place in a single step. Thus, Pd-catalyzed double Sonogashira coupling of 13C2-TMSA with 39a furnished 13C4-40a. Removal of the TMS groups by treatment of 13C4-40a with K2CO3 in MeOH/THF converted it to 13C4-40b, and PtCl2-catalyzed cyclization18 of the latter afforded 13C4-BaP (13C4-37a).
Scheme 8.

The 8- and 9-phenols of 13C4-BaP (13C4-37c and 13C4-37e with 13C at C-4, -5, -11, and -12) were synthesized from 39b and 39c via analogous sequences (Scheme 8). The 13C4-labelled carcinogenic metabolites [13C4-1, 13C4-2, 13C4-4, and 13C4-3 diacetate] were prepared from 13C4-37c (Scheme 9) by methods analogous to those for synthesis of the unlabelled BaP metabolites (Scheme 7). Since the 13C4-BaP metabolites derive from a common synthetic precursor (13C4-37c), their 13C-atoms are at the same sites (C-4, -5, -11, and -12).
Scheme 9.
The isotopic purity of the synthetic 13C4-labelled BaP metabolites was estimated by measurement of their product precursor ion transitions in the [12C] and [13C] channels (Supplemental Fig. 1). Based on a limit-of-detection (100), which is 10 fmol for the BaP-tetrol-1 and 6 fmol for all other BaP metabolites, and the injection of 10 pmol of each 13C4-labelled compound on column, it is estimated that BaP-tetrol-1 has an isotopic purity >99.9 % and for all other compounds the isotopic purity is >99.94 %.
3. Discussion
The principal aim of this investigation was to synthesize the complete set of 13C4-labelled oxidized metabolites of BaP needed as internal standards for a stable isotope dilution LC/MS method for their analysis.15 The BaP metabolites were divided into two groups (A and B) on the basis of their role in carcinogenesis. Group A includes the BaP metabolites that have no role in carcinogenesis [1-HO-, 2- HO-, 3- HO-, 9- HO-, and 12-HO-BaP, BaP-1,6-dione (6) and BaP-3,6-dione (7)] (Fig. 1), and the BaP metabolites in Group B are those implicated in initiation of cancer [BaP 7,8-diol (1), BaP 7,8-dione (2), BaP 7,8-catechol (diacetate) (3), (±)-anti-BPDE (4), and (±)-syn-BPDE (5)] (Fig. 1), plus 8- and 9-HO-BaP.
3.1. Synthesis of 13C4-labelled oxidized metabolites of BaP
This paper reports efficient syntheses of BaP and its oxidized metabolites in Groups A and B and their 13C4-labelled analogues. The synthetic design was influenced by: (1) the cost of the available 13C-labelled precursors; (2) the advantage of introducing the 13C-atoms late in the sequence; (3) the need to minimize the number of synthetic steps; and (4) the need for operational simplicity.
The Group A metabolites were synthesized via Method A (Suzuki, Sonogashira, and Hartwig cross-coupling reactions in combination with PtCl2-catalyzed cyclization of an acetylenic intermediate) (Scheme 1). The Group B metabolites were synthesized via Method B (Suzuki and Sonogashira cross-coupling reactions combined with PtCl2-catalyzed cyclization of a diacetylenic intermediate) (Scheme 8). The use of Suzuki cross-coupling for synthesis of biphenyls and other PAHs has been described,16,31 and the use of Sonogashira cross-coupling for synthesis of substituted phenanthrenes and terphenyls was reported.18,32
Synthesis of PAHs by transition metal-catalyzed cross-coupling chemistry has advantages over their synthesis via conventional Friedel–Crafts chemistry.1b,33,34 This approach requires fewer steps, employs milder reaction conditions (no Lewis acid catalysts), isomeric coproducts are not formed, and purification of products is relatively simple and straightforward.
The only compounds synthesized by both methods were BaP, 8-HO-BaP (37c), and their 13C4-labelled analogues (13C4-BaP and 13C4-37c). The synthesis of BaP by Method A requires eight steps (Scheme 1), whilst its synthesis by Method B requires only four steps (Scheme 6). Synthesis of the 13C4-labelled analogues of BaP and 37c by Method A affords 13C4-BaP and 13C4-37c (with 13C at C-4, -5, -5a, and -6) (Scheme 4), whilst their synthesis by Method B affords the isotopomers (with 13C at C-4, -5, -11, and -12) (Fig. 3). Method B has the major advantage that all four 13C-atoms are introduced simultaneously in a single step. The ease of synthesis of 13C4-37c via this route combined with the fact that 13C4-37c is a convenient synthetic precursor of all the 13C4-labelled active metabolites (13C4-1, 13C4-2, 13C4-3, 13C4-4, and 13C4-5) (Scheme 9) makes these compounds now all of them readily available for research in carcinogenesis.
3.2. Comparison with the syntheses of the 13C6-labelled BaP metabolites
Synthesis of 13C6-labelled analogues of several Group B BaP metabolites (1, 4, 5, and BaP tetraols) was reported by Diel et al.35 Their synthetic approach entailed multistep synthesis of 13C6-pyrene from 13C6-benzene followed by its use asstarting compound for synthesis of 13C6-9,10-dihydro-BaP (42) (Fig. 4) by Friedel–Crafts chemistry.1b,33,34
Fig. 4.
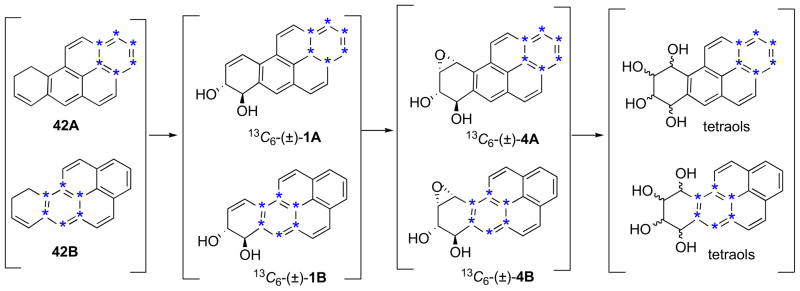
13C6-9,10-Dihydro-BaP was obtained as a mixture of isotopomers (42A and 42B) that were transformed into the (±)-dihydrodiols (13C6-1A and 13C6-1B), and they were converted into the anti- and syn-(±)-diol epoxides (only the anti-isomers, 13C6-4A and 13C6-4B, are shown). Sites of 13C-atoms are indicated by asterisks ‘*’.
As a consequence of the symmetry of 13C6-pyrene, 42 was obtained as a pair of isotopomers (42A and 42B) each possessing six 13C-atoms, but in different aromatic rings (Fig. 4). This mixture was converted into the mixed 13C6-BaP 7,8-diol isotopomers (13C6-1A and 13C6-1B), and this was further transformed into the mixed 13C6-(±)-anti-BPDEs (13C6-4A and 13C6-4B) by the established methods. The 13C6-(±)-syn-BPDEs (structures not shown) and the mixed 13C6-tetraols (from hydrolysis of the anti- and syn-BPDEs) were also prepared. The principal drawbacks to the use of these 13C6-labelled BaP analogues in biological studies are the large number of synthetic steps required, the 13C6-BaP metabolites are mixtures of isotopomers, and many of the likely 13C6-BaP metabolites (e.g., those in Group A) are not obtainable by this approach.
4. Conclusions
This paper reports efficient syntheses of the complete set of oxidized metabolites of the prototypical carcinogenic PAH BaP (Group A and Group B metabolites) and their 13C4-labelled analogues. The synthetic 13C4-BaP metabolites were required as standards for quantitation of the metabolic profiles of BaP in human bronchoalveolar (H358) cells by stable isotope dilution liquid chromatography.15 The syntheses of these polycyclic aromatic molecules were accomplished by a novel approach based on use of Pd-catalyzed Suzuki, Sonogashira, and Hartwig cross-coupling reactions in combination with PtCl2-catalyzed cyclization of acetylenic intermediates. This method requires fewer steps, employs milder conditions, and product isolation is simpler than the conventional methods of PAH synthesis based on Friedel–Crafts chemistry. It is also potentially applicable to the synthesis of a broad range of other PAH compounds and their 13C-labelled analogues.
5. Experimental section
5.1. Caution
Benzo[a]pyrene (BaP) has been designated a human carcinogen by the World Health Organization.2 It should be handled with caution following procedures recommended in the NIH Guidelines for the Laboratory Use of Chemical Carcinogens. Although the oxidized metabolites of BaP are not included in the official list of carcinogens, prudence suggests that they should also be handled with caution.
5.2. Synthesis of 1-, 2-, and 3-HO-BaP (14b, 14d, and 14f) and their 13C4 analogues
These phenols were synthesized by Pd-catalyzed Suzuki–Miyaura cross-coupling of 8 with the 2-boronate esters of 5-, 6-, and 7-methoxynaphthalene (9a, 9b, or 9c) (Scheme 1).
5.2.1. 2-(2-Bromophenyl)-7-methoxynaphthalene (10c)
To a solution of Pd(OAc)2 (101 mg, 0.45 mmol ), PPh3 (354 mg, 1.35 mmol), K2CO3 (2.76 g, 20.0 mmol) in DME (30 mL) and H2O (10 mL) at room temperature under argon was added 9c (3.0 g, 11 mol). The resulting solution was stirred for 10 min, then 8 (2.83 g, 10.0 mmol) was added, and the solution was heated at reflux for 23 h and monitored by TLC. The resulting solution was cooled to room temperature, EtOAc (100 mL) was added, and the solution was washed with a saturated brine solution and water, and dried over anhydrous Na2SO4. Following evaporation of the solvent under reduced pressure, the residue was purified by chromatography on a silica gel column eluted with hexane/EtOAc (150:1) to yield 10c (2.84 g, 91%): 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J=8.5 Hz, 1H), 7.82 (d, J=8.5 Hz, 1H), 7.81 (s, 1H), 7.76 (dd, J=8.0 and 1.0 Hz, 1H), 7.45–7.50 (m, 2H), 7.43 (dt, J=7.0 and 1.0 Hz, 1H), 7.22–7.30 (m, 3H), 3.97 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 157.9, 142.7, 139.1, 134.2, 133.1, 131.4, 129.2, 128.7, 128.1, 127.3, 127.1, 125.3, 122.7, 119.1, 106.0, 55.3; HRMS calcd for C17H14BrO [M+H]+ 313.0223, found 313.0225.
5.2.2. 2-(2-Bromophenyl)-5-methoxynaphthalene (10a)
Reaction of 9a with 8 gave 10a (70%): 1H NMR (500 MHz, CDCl3) δ 8.43 (d, J=8.5 Hz, 1H), 7.91 (d, J=1.5 Hz, 1H), 7.79 (dd, J=8.5 and 1.0 Hz, 1H), 7.66 (dd, J=8.5 and 1.5 Hz, 1H), 7.42–7.57 (m, 4H), 7.66 (td, J=7.5 and 1.5 Hz, 1H), 6.91 (d, J=7.5 Hz, 1H), 4.08 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 155.5, 142.6, 139.1, 134.1, 133.1, 131.5, 128.8, 127.8, 127.4, 126.9, 126.4, 124.7, 122.8, 121.7, 120.4, 104.2, 55.5; HRMS calcd for C17H14BrO [M+H]+ 313.0223, found 313.0253.
5.2.3. 2-(2-Bromophenyl)-6-methoxynaphthalene (10b)
Reaction of 9b with 8 gave 10b. Yield: 68%: 1H NMR (500 MHz, CDCl3) δ 7.80–7.90 (m, 3H), 7.75 (d, J=8.0 Hz, 1H), 7.59 (d, J=8.5 Hz, 1H), 7.39–7.50 (m, 2H), 7.20–7.30 (m, 3H), 3.98 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 158.0, 142.6, 136.4, 133.8, 133.1, 131.5, 129.6, 128.6, 128.5, 128.05, 128.02, 127.4, 126.2, 122.9, 119.1, 105.6, 55.3; HRMS calcd for C17H14BrO [M+H]+ 313.0223, found 313.0248.
5.2.4. Ethyl 2-(7-methoxynaphthalenyl)phenylacetate (11b)
To a solution of 10c (156 mg, 0.5 mmol), Pd(dba)2 (14.5 mg, 0.025 mmol), and Q-phos (18 mg, 0.025 mmol) in THF (0.5 mL) was added ZnBrCH2CO2Et (1M in THF, 1.5 mL) dropwise at room temperature under argon. The resulting mixture was stirred for 2 h, monitored by TLC, and diluted with EtOAc (20 mL). After evaporation of the solvent, the residue was purified by chromatography on a silica gel column. Elution with hexane/EtOAc (40:1 to 20:1) gave 11b (147 mg, 92%): 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J=8.0 Hz, 1H), 7.82 (d, J=9.0 Hz, 1H), 7.73 (s, 1H), 7.39–7.48 (m, 4H), 7.35 (dd, J=8.0 and 1.5 Hz, 1H), 7.21 (dd, J=8.5 and 2.5 Hz, 1H), 7.18 (d, J=2.5 Hz, 1H), 4.11 (q, J=7.0 Hz, 2H), 3.96 (s, 3H), 3.68 (s, 2H), 1.20 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 171.9, 157.9, 142.5, 138.1, 134.3, 132.0, 130.29, 130.25, 129.1, 127.8, 127.5, 127.4, 127.1, 126.9, 125.3, 118.8, 105.8, 60.6, 55.2, 39.0, 14.0; HRMS calcd for C21H21O3 [M+H]+ 321.1485, found 321.1483.
5.2.5. Ethyl 13C2-2-(7-methoxynaphthalenyl)phenylacetate (13C2-11b)
1H NMR (500 MHz, CDCl3) δ 7.84 (d, J=8.0 Hz, 1H), 7.82 (d, J=9.0 Hz, 1H), 7.73 (s, 1H), 7.48–7.39 (m, 4H), 7.35 (dd, J=8.0 and 1.5 Hz, 1H), 7.21 (dd, J=8.5 and 2.5 Hz, 1H), 7.18 (d, J=2.5 Hz, 1H), 4.11 (q, J=7.0 Hz, 2H), 3.96 (s, 3H), 3.68 (dd, J=129.0 and 8.0 Hz, 2H), 1.20 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 172.0 (d, J=228.0 Hz), 39.0 (d, J=228.0 Hz); HRMS calcd for 13C2-C21H20NaO3 [M+Na]+ 345.1372, found 345.1349.
5.2.6. Ethyl 2-(5-methoxynaphthalenyl)phenylacetate (11d)
Reaction of 10a with ZnBrCH2CO2Et gave 11d (93%): 1H NMR (500 MHz, CDCl3) δ 8.36 (d, J=8.5 Hz, 1H), 7.80 (d, J=1.5 Hz, 1H), 7.55–7.35 (m, 7H), 6.89 (dd, J=6.5 and 2.0 Hz, 1H), 4.12 (q, J=7.0 Hz, 2H), 4.07 (s, 3H), 3.68 (s, 2H), 1.22 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 171.9, 155.4, 142.4, 139.1, 134.2, 132.0, 130.31, 130.26, 127.6, 127.5, 127.1, 126.8, 126.3, 124.5, 121.9, 120.2, 103.9, 60.6, 55.5, 38.9, 14.0; HRMS calcd for C21H20NaO3 [M+Na]+ 343.1305, found 343.1330.
5.2.7. Ethyl 2-(6-methoxynaphthalenyl)phenylacetate (11f)
Reaction of 10b with ZnBrCH2CO2Et afforded 11f (90%): 1H NMR (500 MHz, CDCl3) δ 7.90–7.70 (m, 3H), 7.50–7.30 (m, 5H), 7.25–7.15 (m, 2H), 4.10 (q, J=7.0 Hz, 2H), 3.98 (s, 3H), 3.67 (s, 2H), 1.20 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 172.0, 157.9, 142.5, 136.4, 133.6, 132.2, 130.5, 130.4, 129.6, 128.7, 128.1, 127.9, 127.5, 127.2, 126.6, 119.2, 105.6, 60.8, 55.4, 39.1, 14.2; HRMS Calcd for C21H21O3 [M+H]+ 321.1485, found 321.1517.
5.2.8. 2-(7-Methoxynaphthalenyl)phenylacetic acid (11c)
To a solution of 11b (467 mg, 1.46 mmol) in EtOH (18 mL) and H2O (6 mL) was added NaOH (175 mg, 4.38 mmol). The resulting mixture was heated at reflux for 1 h, and reaction was monitored by TLC. This was evaporated to dryness, and the residue was diluted with water (50 mL), and acidified with 37% HCl. The solid was filtered off, and dried to provide 11c (385 mg, 90%): 1H NMR (500 MHz, acetone-d6) δ 8.02–7.83 (m, 2H), 7.76 (s, 1H), 7.50–7.44 (m, 1H), 7.44–7.29 (m, 5H), 7.19 (dd, J=9.0 and 2.5 Hz, 1H), 3.94 (s, 3H), 3.67 (s, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.3, 158.1, 142.4, 139.4, 134.6, 133.2, 131.3, 130.3, 129.6, 127.9, 127.4, 127.0, 125.4, 119.2, 106.7, 55.7; HRMS calcd for C19H16O3 [M]+ 292.1094, found 292.1061.
5.2.9. 2-(5-Methoxynaphthalenyl)phenylacetic acid (11e)
Hydrolysis of 11d gave 11e (92%): 1H NMR (500 MHz, DMSO-d6) δ 8.18 (d, J=9.0 Hz, 1H), 7.77 (s, 1H), 7.60–7.35 (m, 7H), 6.99 (dd, J=5.5 and 3.0 Hz, 1H), 3.99 (s, 3H), 3.60 (s, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.2, 155.3, 142.1, 139.3, 134.3, 133.1, 131.4, 130.3, 128.0, 127.7, 127.5, 127.2, 127.0, 124.2, 121.9, 120.6, 105.1, 56.1, 39.1; HRMS calcd for C19H17O3 [M+H]+ 293.1172, found 293.1143.
5.2.10. 2-(6-Methoxynaphthalenyl)phenylacetic acid (11g)
Hydrolysis of 11f gave 11g (90%): 1H NMR (500 MHz, DMSO-d6) δ 7.87 (d, J=8.0 Hz, 1H), 7.75 (s, 1H), 7.45–7.29 (m, 6H), 7.20 (d, J=8.0 Hz, 1H), 3.89 (s, 3H), 3.57 (s, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.3, 157.9, 142.3, 136.5, 133.7, 133.1, 131.3, 130.5, 129.9, 128.7, 128.2, 127.83, 127.75, 127.4, 127.0, 119.5, 106.2, 55.7, 39.1; HRMS calcd for C19H17O3 [M+H]+ 293.1172, found 293.1143.
5.2.11. 13C2-2-(7-Methoxynaphthalenyl)phenylacetic acid (13C2-11c)
1H NMR (500 MHz, DMSO-d6) δ 12.23 (br s, 1H), 8.00–7.83 (m, 2H), 7.87 (s, 1H), 7.50–7.29 (m, 6H), 7.19 (dd, J=9.0 and 2.5 Hz, 1H), 3.89 (s, 3H), 3.57 (dd, J=128.5 and 8.0 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.2 (d, J=218.0 Hz), 39.0 (d, J=218.0 Hz); HRMS calcd for 13C2-C19H17O3 [M+H]+ 295.1239, found 295.1211.
5.2.12. 3-Methoxychrysen-5-ol (12e)
A suspension of 11c (292 mg, 1 mmol) in MeSO3H was heated at 50 °C for 1 h and monitored by TLC, then cooled to room temperature, and poured onto crushed ice (50 g). The solid was filtered off, and dissolved in EtOAc (50 mL). The solution was washed with brine and water, and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the residue was chromatographed on a silica gel column. Elution with hexane/EtOAc (10:1 to 5:1) gave 12e (221 mg, 77%): 1H NMR (500 MHz, acetone-d6) δ 9.79 (s, 1H), 9.62 (d, J=3.0 Hz, 1H), 8.78 (d, J=11.0 Hz, 1H), 8.72 (d, J=11.0 Hz, 1H), 8.04 (d, J=11.5 Hz, 1H), 7.99 (d, J=11.0 Hz, 1H), 7.82 (d, J=11.0 Hz, 1H), 7.65–7.45 (m, 3H), 7.33 (dd, J=8.5 and 2.5 Hz, 1H), 4.00 (s, 3H); 13C NMR (125.8 MHz, acetone-d6) δ 158.1, 154.4, 133.3, 132.2, 131.5, 129.3, 128.1, 127.9, 126.8, 126.1, 126.0, 123.7, 123.4, 119.0, 116.6, 110.2, 108.6, 54.7; HRMS calcd for C19H15O2 [M+H]+ 275.1067, found 275.1062.
5.2.13. 13C2-3-Methoxychrysen-5-ol (13C2-12e)
1H NMR (500 MHz, DMSO-d6) δ 10.87 (s, 1H), 9.51 (d, J=2.5 Hz, 1H), 8.78 (d, J=8.5 Hz, 1H), 8.72 (d, J=9.0 Hz, 1H), 8.04 (d, J=9.0 Hz, 1H), 8.01 (d, J=9.0 Hz, 1H), 7.88–7.78 (m, 1H), 7.62–7.42 (m, 3H), 7.33 (dd, J=9.0 and 2.5 Hz, 1H), 3.96 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 154.9 (d, J=277.5 Hz), 108.7 (d, J=277.5 Hz); HRMS calcd for 13C2-C19H14O2 [M]+ 276.1055, found 276.1056.
5.2.14. 1-Methoxychrysen-5-ol (12a)
Similar acid-catalyzed cyclization of 11e gave 12a (80%): 1H NMR (500 MHz, acetone-d6) δ 9.66 (s, 1H), 9.65 (d, J=8.5 Hz, 1H), 8.86 (d, J=9.0 Hz, 1H), 8.81 (d, J=8.5 Hz, 1H), 8.40 (d, J=9.0 Hz, 1H), 7.82 (d, J=9.0 Hz, 1H), 7.65–7.49 (m, 4H), 7.17 (d, J=7.5 Hz, 1H), 4.09 (s, 3H); 13C NMR (125.8 MHz, acetone-d6) δ 155.2, 154.3, 133.2, 131.9, 131.3, 126.8, 126.2, 126.01, 125.96, 124.3, 123.8, 123.4, 121.5, 121.4, 120.5, 109.0, 108.9, 105.2, 55.2; HRMS calcd for C19H15O2 [M+H]+ 275.1067, found 275.1091.
5.2.15. 13C2-1-Methoxychrysen-5-ol (13C2-12a)
1H NMR (500 MHz, acetone-d6) δ 10.87 (s, 1H), 9.55 (d, J=9.0 Hz, 1H), 8.86 (d, J=9.0 Hz, 1H), 8.80 (d, J=8.5 Hz, 1H), 8.44 (d, J=9.5 Hz, 1H), 7.82–7.75 (m, 1H), 7.65–7.27 (m, 4H), 7.20 (d, J=7.5 Hz, 1H), 4.05 (s, 3H); 13C NMR (125.8 MHz, acetone-d6) δ 154.8 (d, J=277.5 Hz), 109.1 (d, J=277.5 Hz); HRMS calcd for 13C2-C19H15O2 [M+H]+ 277.1134, found 277.1093.
5.2.16. 2-Methoxychrysen-5-ol (12c)
Acid-catalyzed cyclization of 11g gave 12c (87%): 1H NMR (500 MHz, acetone-d6) δ 9.98 (d, J=9.5 Hz, 1H), 9.66 (s, 1H), 8.82 (d, J=9.0 Hz, 1H), 8.76 (d, J=8.0 Hz, 1H), 8.02 (d, J=9.0 Hz, 1H), 7.82 (d, J=7.5 Hz, 1H), 7.60–7.40 (m, 4H), 7.32 (dd, J=9.5 and 2.5 Hz, 1H), 3.99 (s, 3H); 13C NMR (125.8 MHz, acetone-d6) δ 157.7, 153.9, 134.9, 132.7, 130.8, 129.6, 127.9, 126.3, 126.2, 126.0, 125.4, 123.8, 123.0, 121.8, 121.4, 117.0, 109.0, 107.6, 54.7; HRMS calcd for C19H15O2 [M+H]+ 275.1067, found 275.1085.
5.2.17. 13C2-2-Methoxychrysen-5-ol (13C2-12c)
This unstable compound was used directly.
5.2.18. 3-Methoxychrysen-5-ol trifluoromethanesulfonate (12f)
To a solution of 12e (180 mg, 0.65 mmol) in CH2Cl2 (10 mL) was added pyridine (103 mg, 1.3 mmol), and the mixture was stirred for 10 min at room temperature. Then Tf2O (275 mg, 0.98 mmol) was added dropwise at −78 °C, and the mixture was warmed to room temperature, and stirred overnight. Then it was diluted with diethyl ether (50 mL), filtered, and the filtrate was washed with brine and water and dried over anhydrous Na2SO4. The solvent was evaporated, and the residue was purified by chromatography on a silica gel column eluted with hexane/EtOAc (40:1) to yield 12f (188 mg, 70%): 1H NMR (500 MHz, CDCl3) δ 8.67 (d, J=8.5 Hz, 1H), 8.59 (d, J=2.0 Hz, 1H), 8.55 (d, J=9.0 Hz, 1H), 7.96 (s, 1H), 7.94 (s, 1H), 7.92–7.85 (m, 2H), 7.78–7.65 (m, 2H), 7.34 (dd, J=8.5 and 2.5 Hz, 1H), 4.09 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 158.6, 145.8, 132.1, 130.7, 130.0, 129.8, 129.35, 129.28, 128.4, 128.0, 127.7, 127.6, 123.5, 120.7, 119.2, 118.72, 118.67 (q, J=1277.5 Hz), 118.2; HRMS calcd for C20H13F3NaO4S [M+Na]+ 429.0379, found 429.0365.
5.2.19. 13C2-3-Methoxychrysen-5-ol trifluoromethanesulfonate (13C2-12f)
This unstable compound was used directly in the next step.
5.2.20. 1-Methoxychrysen-5-ol trifluoromethanesulfonate (12b)
Esterification of 12a by a similar procedure gave 12b (75%): 1H NMR (500 MHz, CDCl3) δ 8.80–8.75 (m, 2H), 8.69 (d, J=9.0 Hz, 1H), 8.61 (d, J=9.5 Hz, 1H), 7.98 (d, J=7.5 Hz, 1H), 7.95 (s, 1H), 7.80–7.62 (m, 3H), 7.10 (d, J=9.0 Hz, 1H), 4.09 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 155.5, 145.6, 131.9, 130.8, 129.7, 129.2, 128.4, 127.9, 127.7, 127.3, 124.7, 123.6, 123.1, 121.3, 120.0, 119.8, 119.4, 118.7 (q, J=1277.0 Hz), 106.0, 55.8; HRMS calcd for C20H14F3O4S [M+H]+ 407.0559, found 407.0575.
5.2.21. 13C2-1-Methoxychrysen-5-ol trifluoromethanesulfonate (13C2-12b)
1H NMR (500 MHz, CDCl3) δ 8.80 (d, J=8.5 Hz, 1H), 8.77 (d, J=8.5 Hz, 1H), 8.73 (d, J=9.5 and 1.5 Hz, 1H), 8.63 (d, J=9.5 Hz, 1H), 8.18–7.78 (m, 3H), 7.78–7.62 (m, 2H), 7.10 (d, J=8.0 Hz, 1H), 4.11 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 145.6 (d, J=308.0 Hz), 120.1 (d, J=308.0 Hz); HRMS calcd for 13C2-labelled C20H13F3O4S (M+) 408.0553, found 408.0536.
5.2.22. 2-Methoxychrysen-5-ol trifluoromethanesulfonate (12d)
Esterification of 12c gave 12d (76%): 1H NMR (500 MHz, CDCl3) δ 9.10 (d, J=9.5 Hz, 1H), 8.68 (d, J=8.5 Hz, 1H), 8.62 (d, J=9.0 Hz, 1H), 8.00–7.80 (m, 3H), 7.74 (t, J=7.0 Hz, 1H), 7.67 (t, J=7.0 Hz, 1H), 7.38 (dd, J=9.5 and 3.0 Hz, 1H), 7.32 (d, J=2.5 Hz, 1H), 4.00 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 158.2, 145.3, 134.9, 130.3, 130.2, 129.8, 128.9, 128.7, 128.5, 127.9, 127.2, 123.2, 122.6, 121.7, 121.2, 120.1, 118.7 (q, J=1276.0 Hz), 118.0, 108.2, 55.4; HRMS calcd for C20H14F3O4S [M+H]+ 407.0559, found 407.0588.
5.2.23. 3-Methoxy-5-(trimethylsilylethynyl)chrysene (13e)
To a solution of 12f (406 mg, 1.0 mmol) in DMF (15 mL) were added Pd(Ph3)2Cl2 (35 mg, 0.05 mmol), CuI (9.5 mg, 0.05 mmol), TEA (1.3 mL), (trimethylsilyl)acetylene (120 mg, 1.2 mmol) under argon. The mixture was stirred for 2 h at room temperature and monitored by TLC. It was then diluted with EtOAc (100 mL), washed with brine and water, and dried over anhydrous Na2SO4. Following evaporation of the solvent under vacuum, the residue was chromatographed on a silica gel column. Elution with hexane/EtOAc (120:1) gave 13e (334 mg, 95%): 1H NMR (500 MHz, CDCl3) δ 9.96 (d, J=2.0 Hz, 1H), 8.80 (d, J=8.5 Hz, 1H), 8.55 (d, J=9.0 Hz, 1H), 8.34 (s, 1H), 8.00–7.92 (m, 2H), 7.90 (d, J=8.5 Hz, 1H), 7.76–7.62 (m, 2H), 7.34 (dd, J=8.5 and 2.0 Hz, 1H), 4.08 (s, 3H), 0.42 (s, 9H); 13C NMR (125.8 MHz, CDCl3) δ 157.8, 137.5, 132.2, 130.9, 130.5, 129.7, 129.4, 127.94, 127.88, 127.7, 127.6, 126.8, 126.1, 123.3, 118.8, 117.5, 117.4, 108.7, 108.1, 99.7, 55.9, 0.12; HRMS calcd for C24H22OSi (M+) 354.1440, found 354.1454.
5.2.24. 13C4-3-Methoxy-5-(trimethylsilylethynyl)chrysene (13C4-13e)
1H NMR (500 MHz, CDCl3) δ 9.95 (s, 1H), 8.73 (d, J=8.5 Hz, 1H), 8.60 (d, J=9.0 Hz, 1H), 8.33 (dd, J=99.5 and 7.5 Hz, 1H), 7.97 (d, J=8.0 Hz, 1H), 7.95–7.93 (m, 1H), 7.91 (d, J=9.0 Hz, 1H), 7.76–7.62 (m, 2H), 7.34 (dd, J=9.0 and 2.5 Hz, 1H), 4.07 (s, 3H), 0.38 (d, J=2.5 Hz, 9H); 13C NMR (125.8 MHz, CDCl3) δ 137.5 (dd, J=251.0 and 9.5 Hz), 117.5 (ddd, J=331.5, 251.0, and 36.0 Hz), 108.6 (dd, J=542.0 and 331.5 Hz), 99.7 (ddd, J=542.0, 36.0, and 9.5 Hz); HRMS calcd for 13C4-C24H22OSi (M+): 358.1575, found 358.1580.
5.2.25. 1-Methoxy-5-(trimethylsilylethynyl)chrysene (13a)
Synthesis from 12b by the foregoing procedure gave 13a (90%): 1H NMR (500 MHz, CDCl3) δ 10.11 (d, J=9.0 Hz, 1H), 8.80–8.60 (m, 2H), 8.55 (d, J=9.0 Hz, 1H), 8.36 (s, 1H), 7.93 (d, J=9.0 Hz, 1H), 7.71 (t, J=1.5 Hz, 1H), 7.66 (t, J=3.0 Hz, 1H), 7.59 (t, J=7.5 Hz, 1H), 7.07 (d, J=7.5 Hz, 1H), 4.09 (s, 3H), 0.45 (s, 9H); 13C NMR (125.8 MHz, CDCl3) δ 155.4, 136.8, 132.1, 130.9, 130.5, 129.6, 128.0, 127.7, 126.9, 126.8, 125.3, 124.5, 123.4, 121.8, 120.4, 119.4, 117.9, 108.6, 105.5, 99.6, 55.8, −0.11; HRMS calcd for C24H22OSi (M+) 354.1440, found 354.1460.
5.2.26. 13C4-1-Methoxy-5-(trimethylsilylethynyl)chrysene (13C4-13a)
1H NMR (500 MHz, CDCl3) δ 10.08 (d, J=9.0 Hz, 1H), 8.76 (d, J=8.5 Hz, 1H), 8.73 (d, J=8.5 Hz, 1H), 8.55 (d, J=9.5 Hz, 1H), 8.36 (dd, J=162.5 and 7.0 Hz, 1H), 7.95–7.90 (m, 1H), 7.71 (t, J=7.5 Hz, 1H), 7.65 (t, J=7.5 Hz, 1H), 7.57 (t, J=7.5 Hz, 1H), 7.07 (d, J=7.5 Hz, 1H), 4.10 (s, 3H), 0.40 (d, J=2.5 Hz, 9H); 13C NMR (125.8 MHz, CDCl3) δ 136.8 (dd, J=252.5 and 10.0 Hz), 117.8 (ddd, J=332.5, 252.5, and 36.0 Hz), 108.5 (dd, J=543.0 and 332.5 Hz), 99.6 (ddd, J=543.0, 36.0, and 10.0 Hz); HRMS calcd for 13C4-C24H22OSi (M+) 358.1575, found 358.1560.
5.2.27. 2-Methoxy-5-(trimethylsilylethynyl)chrysene (13c)
Synthesis from 12d by the foregoing procedure gave 13c (92%): 1H NMR (500 MHz, CDCl3) δ 10.46 (d, J=9.5 Hz, 1H), 8.64 (d, J=9.0 Hz, 1H), 8.62 (d, J=9.0 Hz, 1H), 8.35 (s, 1H), 7.91 (d, J=7.5 Hz, 1H), 7.88 (d, J=9.0 Hz, 1H), 7.68 (t, J=7.5 Hz, 1H), 7.61 (t, J=7.5 Hz, 1H), 7.38–7.20 (m, 2H), 4.01 (s, 3H), 0.48 (s, 9H); 13C NMR (125.8 MHz, CDCl3) δ 158.0, 136.8, 134.7, 130.7, 130.5, 128.7, 128.0, 127.8, 127.70, 127.68, 127.3, 126.4, 125.7, 123.0, 121.7, 117.3, 116.3, 108.6, 107.4, 99.5, 55.4, −0.07; HRMS calcd for C24H22OSi (M+) 354.1440, found 354.1426.
5.2.28. 3-Methoxy-5-ethynylchrysene (13f)
To a solution of 13e (124 mg, 0.35 mmol) in THF (3.6 mL) and MeOH (3.6 mL) was added K2CO3 (75 mg, 0.54 mmol). The resulting mixture was stirred for 1 h at room temperature and monitored by TLC. Evaporation of the solvent under reduced pressure and chromatography of the residue on a silica gel column eluted with hexane/EtOAc (40:1) gave 13f (94 mg, 92%): 1H NMR (500 MHz, CDCl3) δ 9.87 (d, J=2.5 Hz, 1H), 8.69 (d, J=8.5 Hz, 1H), 8.54 (d, J=9.0 Hz, 1H), 8.33 (s, 1H), 8.00–7.7.82 (m, 3H), 7.75–7.66 (m, 1H), 7.63 (t, J=7.0 Hz, 1H), 7.33 (dd, J=9.0 and 2.5 Hz, 1H), 4.04 (s, 3H), 3.72 (s, 1H); 13C NMR (125.8 MHz, CDCl3) δ 157.8, 137.3, 132.0, 130.7, 130.6, 129.7, 129.5, 128.0, 127.9, 127.74, 127.70, 126.8, 126.4, 123.3, 118.7, 117.9, 116.3, 107.2, 87.1, 82.1, 55.6; HRMS calcd for C21H14O (M+) 282.1045, found 282.1056.
5.2.29. 13C4-3-Methoxy-5-ethynylchrysene (13C4-13f)
1H NMR (500 MHz, CDCl3): δ 9.89 (d, J=2.5 Hz, H), 8.74 (d, J=8.5 Hz, 1H), 8.60 (d, J=9.0 Hz, 1H), 8.36 (dd, J=162.5 and 7.0 Hz, 1H), 8.00–7.85 (m, 3H), 7.75–7.55 (m, 2H), 7.33 (dd, J=8.5 and 2.5 Hz, 1H), 4.05 (s, 3H), 4.00–3.30 (m, 1H); 13C NMR (125.8 MHz, CDCl3) δ 137.3 (dd, J=253.0 and 11.5 Hz), 116.3 (ddd, J=347.0, 253.0, and 53.5 Hz), 87.2 (dd, J=704.5 and 347.0 Hz), 82.0 (ddd, J=704.5, 53.5, and 11.5 Hz); HRMS calcd for 13C4-C21H14O (M+): 286.1178, found 286.1190.
5.2.30. 1-Methoxy-5-ethynylchrysene (13b)
Similar reaction of 13a gave 13b (95%): 1H NMR (500 MHz, CDCl3) δ 10.02 (d, J=9.0 Hz, 1H), 8.73 (d, J=8.0 Hz, 1H), 8.70 (d, J=9.0 Hz, 1H), 8.55 (d, J=9.5 Hz, 1H), 8.37 (s, 1H), 7.92 (d, J=8.0 Hz, 1H), 7.71 (t, J=7.5 Hz, 1H), 7.70–7.55 (m, 2H), 7.06 (d, J=7.5 Hz, 1H), 4.08 (s, 3H), 3.70 (s, 1H); 13C NMR (125.8 MHz, CDCl3) δ 155.4, 137.6, 132.0, 130.9, 130.6, 129.6, 127.9, 127.8, 126.9, 126.8, 125.7, 124.5, 123.4, 121.9, 120.4, 119.1, 116.8, 105.5, 86.9, 82.5, 55.8; HRMS calcd for C21H14O (M+) 282.1045, found 282.1069.
5.2.31. 13C4-1-Methoxy-5-ethynylchrysene (13C4-13b)
1H NMR (500 MHz, CDCl3) δ 10.00 (d, J=9.0 Hz, 1H), 8.78 (d, J=8.5 Hz, 1H), 8.73 (d, J=9.0 Hz, 1H), 8.56(d, J=9.5 Hz, 1H), 8.37 (dd, J=162.5 and 7.0 Hz, 1H), 7.96–7.90 (m, 1H), 7.74 (t, J=7.5 Hz, 1H), 7.70–7.55 (m, 2H), 7.08 (d, J=8.0 Hz, 1H), 4.10 (s, 3H), 3.98–3.30 (m, 1H); 13C NMR (125.8 MHz, CDCl3) δ 137.5 (dd, J=253.0 and 11.5 Hz), 116.8 (ddd, J=346.5, 253.0, and 55.5 Hz), 86.8 (dd, J=706.0 and 346.5 Hz), 82.3 (ddd, J=706.0, 55.5, and 11.5 Hz); HRMS calcd for 13C4-C21H14O (M+) 286.1178, found 286.1167.
5.2.32. 2-Methoxy-5-ethynylchrysene (13d)
Similar reaction of 13c gave 13d (90%): 1H NMR (500 MHz, CDCl3) δ 10.34 (d, J=9.5 Hz, 1H), 8.67 (d, J=8.5 Hz, 1H), 8.64 (d, J=9.5 Hz, 1H), 8.35 (s, 1H), 7.95–7.88 (m, 2H), 7.70 (t, J=7.5 Hz, 1H), 7.61 (t, J=7.5 Hz, 1H), 7.38–7.29 (m, 2H), 4.00 (s, 3H), 3.68 (s, 1H); 13C NMR (125.8 MHz, CDCl3) δ 158.0, 137.6, 134.6, 130.8, 130.4, 128.3, 128.0, 127.83, 127.81, 127.78, 127.3, 126.5, 125.6, 123.0, 121.7, 116.6, 116.2, 107.7, 86.8, 82.4, 55.4; HRMS calcd for C21H15O [M+H]+ 283.1117, found 283.1116.
5.2.33. 3-Methoxybenzo[a]pyrene (14e)
To a solution of 13f (100 mg, 0.35 mmol) in toluene (5.2 mL) was added PtCl2 (9 mg, 0.035 mmol). The resulting mixture was heated overnight at 80 °C. After evaporation of the solvent under reduced pressure, the residue was purified by chromatography on a column of silica gel. Elution with hexane/EtOAc (40:1) gave 14e (70 mg, 70%). The 1H NMR spectrum of 7e matched that of an authentic sample.
5.2.34. 13C4-3-Methoxybenzo[a]pyrene (13C4-14e)
1H NMR (500 MHz, CDCl3) δ 9.01 (d, J=8.0 Hz, 1H), 8.88 (d, J=9.0 Hz, 1H), 8.65–7.72 (m, 8H), 7.62 (d, J=8.5 Hz, 1H), 4.19 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 130.3 (dd, J=247.0 and 219.0 Hz), 127.0 (ddd, J=255.0, 219.0, and 8.0 Hz), 123.5 (ddd, J=247.0, 26.0 and 8.0 Hz), 121.3 (ddd, J=255.0, 26.0, and 8.0 Hz); HRMS calcd for 13C4-labelled C21H24O (M+) 286.1178, found 286.1198.
5.2.35. 1-Methoxybenzo[a]pyrene (14b)
Analogous PtCl2-catalyzed reaction of 13b gave 14b (65%). The NMR spectral data matched that of an authentic sample.
5.2.36. 13C4-1-Methoxybenzo[a]pyrene (13C4-14b)
1H NMR (500 MHz, CDCl3) δ 9.05–8.95 (m, 2H), 8.69 (d, J=9.5 Hz, 1H), 8.59–8.22 (m, 2H), 8.04–7.92 (m, 2H), 7.82–7.75 (m, 2H), 7.68–7.62 (m, 1H), 7.42 (d, J=8.0 Hz, 1H), 4.19 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 130.3 (ddd, J=249.0, 212.5 and 3.5 Hz), 127.0 (ddd, J=256.0, 26.0, and 3.5 Hz), 125.5 (ddd, J=256.0, 212.5 and 8.0 Hz), 123.8 (ddd, J=249.0, 26.0, and 8.0 Hz); HRMS calcd for 13C4-C21H14O (M+) 286.1178, found 286.1167.
5.3. Synthesis of 9-HO-BaP (21b) (Scheme 2)
5.3.1. 2-(2-Bromo-4-methoxyphenyl)naphthalene (18)
Pd-catalyzed coupling of 15 with 16 by the method for preparation of 10c gave 2-(2-bromo-5-methoxyphenyl)naphthalene (17) (78%): 1H NMR (500 MHz, CDCl3) δ 7.98–7.89 (m, 4H), 7.69–7.60 (m, 2H), 7.57 (q, J=3.0 Hz, 2H), 7.55 (d, J=3.5 Hz, 1H), 7.03 (d, J=3.0 Hz, 1H), 6.86 (dd, J=8.5 and 3.0 Hz, 1H), 3.86 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 158.9, 143.4, 138.7, 133.8, 133.1, 132.7, 128.23, 128.21, 127.8, 127.6, 127.5, 126.3, 117.0, 114.9, 113.3, 55.6; HRMS calcd for C17H14BrO [M+H]+ 313.0223, found 313.0205.
5.3.2. Ethyl 2-(napththalen-2-yl)-5-methoxyphenyl acetate (18a)
Pd-catalyzed coupling of 17 with BrZnCH2CO2Et with 16 gave 18a (90%): 1H NMR (500 MHz, CDCl3) δ 7.98–7.89 (m, 4H), 7.60–7.50 (m, 3H), 7.41 (d, J=9.0 Hz, 1H), 7.10–7.00 (m, 2H), 4.15 (q, J=7.0 Hz, 2H), 3.89 (s, 3H), 3.65 (s, 2H), 1.23 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 172.3, 158.6, 143.6, 138.8, 133.3, 132.6, 131.6, 128.14, 128.05, 127.9, 127.8, 127.6, 126.4, 126.2, 124.5, 115.7, 113.5, 60.7, 55.4, 38.3, 14.2; HRMS calcd for C21H21O3 [M+H]+ 321.1485, found 321.1472.
5.3.3. Ethyl 13C2-2-(napththalen-2-yl)-5-methoxyphenyl acetate (13C2-18a)
1H NMR (500 MHz, CDCl3) δ 7.98–7.89 (m, 4H), 7.60–7.50 (m, 3H), 7.41 (d, J=9.0 Hz, 1H), 7.10–7.00 (m, 2H), 4.09 (qd, J=7.0 and 4.0 Hz, 2H), 3.85 (s, 3H), 3.56 (dd, J=127.0 and 8.0 Hz, 2H), 1.16 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 172.2 (d, J=229.0 Hz), 38.2 (d, J=229.0 Hz); HRMS calcd for 13C2-C21H21O3 [M]+ 322.1477, found 322.1514.
5.3.4. 2-(Napththalen-2-yl)-5-methoxyphenylacetic acid (18b)
Hydr olysis of 18a by the procedure for preparation of 11c gave 18b (91%): 1H NMR (500 MHz, acetone-d6) δ 9.30 (br s, 1H), 7.98–7.80 (m, 4H), 7.60–7.50 (m, 3H), 7.42 (d, J=8.0 Hz, 1H), 7.10–6.95 (m, 2H), 3.83 (s, 3H), 3.65 (s, 2H); 13C NMR (125.8 MHz, acetone-d6) δ 173.3, 158.7, 143.5, 139.0, 133.4, 132.6, 132.0, 128.1, 127.9, 127.8, 127.7, 127.5, 126.4, 126.2, 124.7, 115.4, 113.3, 54.5, 37.5; HRMS calcd for C19H16NaO3 [M+Na]+ 315.0992, found 315.0966.
5.3.5. 13C2-2-(Napththalen-2-yl)-5-methoxyphenylacetic acid (13C2-18b)
1H NMR (500 MHz, acetone-d6) δ 10.62 (br s, 1H), 8.10–7.90 (m, 3H), 7.88 (s, 1H), 7.60–7.50 (m, 3H), 7.38 (dd, J=8.0 and 4.0 Hz, 1H), 6.98 (dd, J=8.5 and 3.0 Hz, 1H), 6.82 (d, J=1.5 Hz, 1H), 3.83 (s, 3H), 3.57 (dd, J=128.0 and 8.0 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.5 (d, J=218.5 Hz), 38.2 (d, J=218.5 Hz); HRMS calcd for 13C2-C19H17O3 [M+H]+ 295.1239, found 295.1220.
5.3.6. 3-Methoxychrysen-11-ol (19a)
Acid-catalyzed cyclization of 18b by the method for preparation of 12e gave 19a (52%): 1H NMR (500 MHz, acetone-d6) δ 10.10 (d, J=8.5 Hz, 1H), 9.52 (s, 1H), 8.84 (d, J=9.0 Hz, 1H), 8.22 (s, 1H), 8.10–8.00 (m, 2H), 7.77 (d, J=9.0 Hz, 1H), 7.70–7.55 (m, 2H), 7.52 (s, 1H), 7.28 (dd, J=8.5 and 2.0 Hz, 1H), 4.04 (s, 3H); 13C NMR (125.8 MHz, acetone-d6) δ 156.8, 152.5, 133.1, 131.0, 130.2, 129.3, 128.1, 127.95, 127.92, 127.5, 127.1, 126.1, 126.0, 121.7, 121.4, 118.3, 109.2, 103.7, 54.9; HRMS calcd for C19H14O2 (M+) 274.0994, found 274.0976.
5.3.7. 13C2-3-Methoxychrysen-11-ol (13C2-19a)
1H NMR (500 MHz, CDCl3) δ 9.83 (d, J=8.5 Hz, 1H), 8.65 (d, J=9.0 Hz, 1H), 8.07 (s, 1H), 8.05–7.98 (m, 2H), 7.77 (d, J=9.0 Hz, 1H), 7.75–7.63 (m, 3H), 7.29 (d, J=2.0 Hz, 1H), 7.23 (dd, J=155.0 and 2.5 Hz, 1H), 5.61 (s, 1H), 4.06 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 150.9 (d, J=286.5 Hz), 109.7 (d, J=286.5 Hz); HRMS calcd for 13C2-C19H15O2 [M+H]+ 277.1134, found 277.1161.
5.3.8. 3-Methoxychrysen-11-ol triflate (19b)
Esterification of 19a with triflic anhydride and pyridine gave 19b (72%): 1H NMR (500 MHz, CDCl3) δ 9.18 (d, J=8.5 Hz, 1H), 8.41 (d, J=9.5 Hz, 1H), 8.00–7.60 (m, 7H), 7.30 (dd, J=8.5 and 1.5 Hz, 1H), 4.00 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 159.4, 143.7, 133.1, 131.2, 130.4, 129.8, 129.0, 128.7, 128.1, 127.2, 127.1, 127.0, 125.4, 121.8, 120.5, 119.9, 118.8, 118.7 (q, J=1276.5 Hz), 103.8, 55.4; HRMS calcd for C20H14F3O4S [M+H]+ 407.0559, found 407.0542.
5.3.9. 13C2-3-Methoxychrysen-11-ol triflate (13C2-3-19b)
1H NMR (500 MHz, CDCl3) δ 9.18 (d, J=8.5 Hz, 1H), 8.57 (d, J=9.5 Hz, 1H), 8.10–7.60 (m, 7H), 7.35 (dd, J=8.5and 1.5 Hz, 1H), 4.06(s, 3H);13C NMR (125.8 MHz, CDCl3) δ 143.8 (d, J=308.0 Hz), 120.0 (d, J=308.0 Hz); HRMS calcd for 13C2-labelled C20H13F3O4S (M+) 408.0553, found 408.0555.
5.3.10. ((9-Methoxychrysen-5-yl)ethynyl)trimethylsilane (20a)
Sonogashira coupling of 19b with (trimethylsilyl)acetylene afforded 20a (90%): 1H NMR (500 MHz, CDCl3) δ 10.70–10.55 (m, 1H), 8.54 (d, J=9.0 Hz, 1H), 8.29 (s, 1H), 8.00–7.85 (m, 3H), 7.82 (d, J=8.5 Hz, 1H), 7.73–7.64 (m, 2H), 7.28 (d, J=8.5 Hz, 1H), 4.03 (s, 3H), 0.47 (s, 9H); 13C NMR (125.8 MHz, CDCl3) δ 159.3, 136.6, 133.0, 132.0, 131.1, 129.6, 128.4, 128.1, 127.9, 127.6, 127.1, 126.6, 126.0, 125.3, 121.2, 118.0, 115.0, 108.8, 103.6, 98.8, 55.5, −0.03; HRMS calcd for C24H22OSi (M+) 354.1440, found 354.1446.
5.3.11. 13C4-((9-Methoxychrysen-5-yl)ethynyl)trimethylsilane (13C4-20a)
1H NMR (500 MHz, CDCl3) δ 10.70–10.55 (m, 1H), 8.62 (d, J=8.5 Hz, 1H), 8.30 (dd, J=162.0 and 6.5 Hz, 1H), 8.08–7.95 (m, 3H), 7.86 (dd, J=8.5 and 5.0 Hz, 1H), 7.71–7.62 (m, 2H), 7.30 (d, J=8.5 and 2.5 Hz, 1H), 4.07 (s, 3H), 0.41 (d, J=2.5 Hz, 9H); 13C NMR (125.8 MHz, CDCl3) δ 136.6 (dd, J=251.5 and 10.0 Hz), 115.0 (ddd, J=334.0, 251.5, and 37.0 Hz), 108.6 (dd, J=541.0 and 334.0 Hz), 98.8 (ddd, J=541.0, 37.0, and 10.0 Hz); HRMS calcd for 13C4-C24H22OSi (M+) 358.1575, found 358.1583.
5.3.12. 5-Ethynyl-9-methoxychrysene (20b)
Removal of the trimethylsilyl group of 20a gave 20b (92%): 1H NMR (500 MHz, CDCl3) δ 10.43 (d, J=8.5 Hz, 1H), 8.54 (d, J=9.0 Hz, 1H), 8.28 (s, 1H), 8.00–7.85 (m, 3H), 7.80 (d, J=8.5 Hz, 1H), 7.73–7.60 (m, 2H), 7.27 (dd, J=8.5 and 2.0 Hz, 1H), 4.03 (s, 3H), 3.67 (s, 1H); 13C NMR (125.8 MHz, CDCl3) δ 159.4, 137.3, 132.9, 132.1, 130.9, 129.6, 128.4, 128.2, 127.9, 127.6, 126.8, 126.6, 125.9, 125.7, 121.1, 118.1, 113.9, 103.6, 87.0, 81.9, 55.5; HRMS calcd for C21H15O [M+H]+ 283.1117, found 283.1105.
5.3.13. 13C4-5-Ethynyl-9-methoxychrysene (13C4-20b)
1H NMR (500 MHz, CDCl3) δ 10.42 (d, J=8.5 Hz, 1H), 8.62 (d, J=9.0 Hz, 1H), 8.32 (dd, J=162.5 and 7.0 Hz, 1H), 8.10–7.95 (m, 3H), 7.86 (dd, J=8.5 and 5.0 Hz, 1H), 7.75–7.64 (m, 2H), 7.31 (dd, J=8.5 and 2.0 Hz, 1H), 4.08 (s, 3H), 3.99–3.30 (m, 1H); 13C NMR (125.8 MHz, CDCl3) δ 137.3 (dd, J=252.5 and 11.5 Hz), 114.0 (ddd, J=350.0, 252.5, and 54.0 Hz), 86.9 (dd, J=706.0 and 350.0 Hz), 81.7 (ddd, J=706.0, 54.0, and 11.5 Hz); HRMS calcd for 13C4-C21H14O (M+): 286.1178, found 286.1194.
5.3.14. 9-Methoxybenzo[a]pyrene (21a)
Cyclization of 20b catalyzed by PtCl2 gave 21a (60%) whose 1H and 13C NMR spectra matched those of an authentic standard.
5.3.15. 13C4-9-Methoxybenzo[a]pyrene (13C4-21a)
1H NMR (500 MHz, CDCl3) δ 8.94 (d, J=9.0 Hz, 1H), 8.63–8.27 (m, 3H), 8.25–8.18 (m, 2H), 8.15–7.62 (m, 4H), 7.47 (dd, J=9.0 and 2.0 Hz, 1H), 4.15 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 128.4–124.4 (m, 4C); HRMS calcd for 13C4-C21H14O (M+): 286.1178, found 286.1190.
5.4. Synthesis of 12-HO-BaP (27b) (Scheme 3)
5.4.1. 2-(2-Bromophenyl)-4-methoxynaphthalene (23)
Pd-catalyzed Suzuki cross-coupling of 8 with 22 by the usual method furnished 23 (75%): 1H NMR (500 MHz, CDCl3) δ 8.47–8.38 (m, 1H), 7.95–7.88 (m, 1H), 7.80 (dd, J=8.0 and 0.5 Hz, 1H), 7.65–7.57 (m, 2H), 7.55–7.53 (m, 2H), 7.46 (td, J=8.0 and 1.0 Hz, 1H), 7.31 (td, J=8.0 and 1.0 Hz, 1H), 7.02 (d, J=0.5 Hz, 1H), 4.10 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 154.7, 142.8, 138.8, 134.0, 133.1, 131.4, 128.7, 127.7, 127.3, 126.7, 125.5, 124.8, 122.7, 121.9, 120.6, 106.0, 55.5; HRMS calcd for C17H14BrO [M+H]+ 313.0223, found 313.0250.
5.4.2. Ethyl 2-(4-methoxynapththalen-2-yl)phenyl acetate (24a)
Pd-catalyzed coupling of 23 with BrZnCH2CO2Et provided 24a (85%): 1H NMR (500 MHz, CDCl3) δ 8.32 (d, J=7.5 Hz, 1H), 7.83 (d, J=7.5 Hz, 1H), 7.60–7.50 (m, 2H), 7.50–7.35 (m, 5H), 6.89 (d, J=1.0 Hz, 1H), 4.13 (q, J=7.0 Hz, 2H), 4.04 (s, 3H), 3.68 (s, 2H), 1.21 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 171.9, 155.0, 142.7, 138.8, 134.1, 132.0, 130.3, 130.1, 127.53, 127.52, 127.0, 126.7, 125.2, 124.5, 121.8, 120.3, 105.9, 60.7, 55.5, 38.9, 14.0; HRMS calcd for C21H20NaO3 [M+Na]+ 343.1305, found 343.1307.
5.4.3. Ethyl 13C2-2-(4-methoxynapththalen-2-yl)phenyl acetate (13C2-24a)
1H NMR (500 MHz, DMSO-d6) δ 12.23 (br s, 1H), 8.00–7.83 (m, 2H), 7.87 (s, 1H), 7.50–7.29 (m, 6H), 7.19 (dd, J=9.0 and 2.5 Hz, 1H), 3.89 (s, 3H), 3.57 (dd, J=128.5 and 8.0 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.2 (d, J=218.0 Hz), 39.0 (d, J=218.0 Hz); HRMS calcd for 13C2-C19H17O3 [M+H]+ 295.1239, found 295.1211.
5.4.4. 2-(4-Methoxynapththalen-2-yl)phenylacetic acid (24b)
Ethan olysis of 24a provided 24b (91%): 1H NMR (500 MHz, acetone-d6) δ 9.30 (br s, 1H), 7.98–7.80 (m, 4H), 7.60–7.50 (m, 3H), 7.42 (d, J=8.0 Hz, 1H), 7.10–6.95 (m, 2H), 3.83 (s, 3H), 3.65 (s, 2H); 13C NMR (125.8 MHz, acetone-d6) δ 173.3, 158.7, 143.5, 139.0, 133.4, 132.6, 132.0, 128.1, 127.9, 127.8, 127.7, 127.5, 126.4, 126.2, 124.7, 115.4, 113.3, 54.5, 37.5; HRMS calcd for C19H16NaO3 [M+Na]+ 315.0992, found 315.0966.
5.4.5. 13C2-2-(4-Methoxynapththalen-2-yl)phenylacetic acid (13C2-24b)
1H NMR (500 MHz, DMSO-d6) δ 12.33(brs, 1H), 8.17 (d, J=8.5 Hz, 1H), 7.88 (d, J=8.0 Hz, 1H), 7.60–7.50 (m, 2H), 7.50–7.30 (m, 5H), 6.91 (s, 1H), 3.97 (s, 3H), 3.57 (dd, J=128.0 and 8.0 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.5 (d, J=218.5 Hz), 39.2 (d, J=218.5 Hz); HRMS calcd for 13C2-C19H17O3 [M+H]+ 295.1239, found 295.1219.
5.4.6. 12-Methoxychrysen-5-ol (25a)
Acid-catalyzed cyclization of 24b afforded 25a (82%): 1H NMR (500 MHz, acetone-d6) δ 10.06 (d, J=8.5 Hz, 1H), 9.65 (s, 1H), 8.79 (d, J=8.5 Hz, 1H), 8.47 (dd, J=8.5 and 1.0 Hz, 1H), 8.20 (s, 1H), 7.79 (d, J=7.5 Hz, 1H), 7.70–7.65 (m, 2H), 7.60–7.45 (m, 2H), 7.39 (s, 1H), 4.29 (s, 3H); 13C NMR (125.8 MHz, acetone-d6) δ 154.6, 154.2, 133.5, 132.1, 132.0, 129.0, 126.71, 126.68, 126.3, 126.0, 125.63, 125.60, 123.5, 123.3, 121.5, 116.2, 106.9, 98.3, 55.2; HRMS calcd for C19H15O2 [M+H]+ 275.1067, found 275.1094.
5.4.7. 13C2-12-Methoxychrysen-5-ol (13C2-25a)
1H NMR (500 MHz, CDCl3) δ 9.83 (d, J=8.5 Hz, 1H), 8.65 (d, J=9.0 Hz, 1H), 8.07 (s, 1H), 8.05–7.98 (m, 2H), 7.77 (d, J=9.0 Hz, 1H), 7.75–7.63 (m, 3H), 7.29 (d, J=2.0 Hz, 1H), 7.23 (dd, J=155.0 and 2.5 Hz, 1H), 5.61 (s, 1H), 4.06 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 150.9 (d, J=286.5 Hz), 109.7 (d, J=286.5 Hz); HRMS calcd for 13C2-C19H15O2 [M+H]+ 277.1134, found 277.1161.
5.4.8. 12-Methoxychrysen-5-ol triflate (25b)
Esterification of 25a with triflic anhydride and pyridine provided 25b (60%): 1H NMR (500 MHz, CDCl3) δ 9.15 (d, J=8.5 Hz, 1H), 8.65 (d, J=8.5 Hz, 1H), 8.51 (d, J=8.5 Hz, 1H), 7.98 (d, J=8.0 Hz, 1H), 7.92 (s, 1H), 7.82 (s, 1H), 7.80–7.65 (m, 4H), 4.24 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 155.6, 145.5, 132.7, 131.1, 129.2, 129.1, 128.6, 127.7, 127.6, 127.4, 127.0, 126.9, 126.7, 123.4, 122.4, 118.7 (q, J=1276.0 Hz), 117.5, 116.7, 97.4, 55.6; HRMS calcd for C20H13O4F3S (M+) 406.0485, found 406.0458.
5.4.9. 13C2-12-Methoxychrysen-5-ol triflate (13C2-25b)
1H NMR (500 MHz, DMSO-d6) δ 10.77 (s, 1H), 9.96 (d, J=8.5 Hz, 1H), 8.82 (d, J=8.0 Hz, 1H), 8.38 (d, J=8.0 Hz, 1H), 8.15 (s, 1H), 7.80–7.62(m, 3H), 7.56 (t, J=7.5 Hz, 1H), 7.48 (d, J=8.5 Hz, 1H), 7.33 (d, J=176.5 Hz, 1H), 4.23 (s, 3H); 13C NMR (125.8 MHz, DMSO-d6) δ 154.6 (d, J=277.5 Hz), 107.0 (d, J=277.5 Hz); HRMS calcd for 13C2-C19H15O2 [M+H]+ 277.1134, found 277.1155.
5.4.10. ((Chrysen-5-yl)ethynyl)trimethylsilane (26a)
Sonogashira coupling of 25b with (trimethylsilyl)acetylene afforded 26a (95%): 1H NMR (500 MHz, CDCl3) δ 10.48 (dd, J=6.5 and 3.5 Hz, 1H), 8.63 (d, J=8.5 Hz, 1H), 8.28 (dd, J=6.5 and 3.5 Hz, 1H), 8.22 (s, 1H), 7.93 (s, 1H), 7.91 (d, J=8.5 Hz, 1H), 7.75–7.60 (m, 4H), 4.23 (s, 3H), 0.43 (s, 9H); 13C NMR (125.8 MHz, CDCl3) δ 154.5, 134.5, 131.9, 131.1, 130.1, 130.0, 128.1, 127.2, 126.8, 126.7, 126.4, 126.3, 126.0, 123.1, 122.3, 121.8, 117.3, 108.5, 99.5, 97.8, 55.5, −0.12; HRMS calcd for C24H22OSi (M+) 354.1440, found 354.1417.
5.4.11. 13C4-((Chrysen-5-yl)ethynyl)trimethylsilane (13C4-26a)
1H NMR (500 MHz, CDCl3) δ 10.48 (dd, J=6.5 and 3.5 Hz, 1H), 8.61 (d, J=8.5 Hz, 1H), 8.48 (dd, J=6.5 and 3.5 Hz, 1H), 8.22 (dd, J=163.0 and 7.0 Hz, 1H), 7.93–7.87 (m, 2H), 7.72–7.58 (m, 4H), 4.21 (s, 3H), 0.44 (d, J=3.0 Hz, 9H); 13C NMR (125.8 MHz, CDCl3) δ 134.4 (dd, J=251.5 and 10.0 Hz), 117.3 (ddd, J=331.5, 251.5, and 36.0 Hz), 108.5 (ddd, J=541.5, 331.5, and 3.0 Hz), 99.4 (ddd, J=541.5, 36.0, and 10.0 Hz); HRMS calcd for 13C4-C24H22OSi (M+) 358.1575, found 358.1545.
5.4.12. 5-Ethynyl-12-methoxychrysene (26b)
Treatment of 26a with K2CO3 in MeOH/THF provided 26b (91%): 1H NMR (500 MHz, CDCl3) δ 10.37 (d, J=8.0 Hz, 1H), 8.65 (d, J=8.5 Hz, 1H), 8.48 (d, J=8.0 Hz, 1H), 8.25 (s, 1H), 7.95 (s, 1H), 7.92 (d, J=8.0 Hz, 1H), 7.78–7.60 (m, 4H), 4.24 (s, 3H), 3.67(s, 1H); 13C NMR (125.8 MHz, CDCl3) δ 154.6, 135.2, 131.8, 131.1, 130.3, 130.1, 128.1, 127.3, 126.8, 126.5, 126.4, 126.3, 123.2, 122.4, 121.9, 116.3, 97.8, 86.8, 82.3, 55.5; HRMS calcd for C21H14O (M+), 282.1045, found 282.1053.
5.4.13. 13C4-5-Ethynyl-12-methoxychrysene (13C4-26b)
1H NMR (500 MHz, CDCl3) δ 10.37 (d, J=8.5 Hz, 1H), 8.66 (d, J=8.5 Hz, 1H), 8.48 (dd, J=8.0 and 1.5 Hz, 1H), 8.26 (dd, J=163.0 and 7.0 Hz, 1H), 7.96 (s, 1H), 7.93 (t, J=6.5 Hz, 1H), 7.77–7.60 (m, 4H), 4.25 (s, 3H), 3.90–3.30 (m, 1H); 13C NMR (125.8 MHz, CDCl3) δ 135.3 (dd, J=252.5 and 11.5 Hz), 116.3 (ddd, J=345.0, 252.5, and 55.5 Hz), 86.9 (dd, J=704.5 and 345.0 Hz), 82.2 (ddd, J=704.5, 55.5, and 11.5 Hz); HRMS calcd for 13C4-C21H14O (M+) 286.1178, found 286.1167.
5.4.14. 12-Methoxybenzo[a]pyrene (27a)
PtCl2-catalyzed cyclization of 26b gave 27a (70%); the 1H and 13C NMR spectra matched those of an authentic sample.
5.4.15. 13C4-12-Methoxybenzo[a]pyrene (13C4-27a)
1H NMR (500 MHz, CDCl3) δ 8.95 (d, J=8.5 Hz, 1H), 8.63 (d, J=8.0 Hz, 1H), 8.37 (dd, J=159.0 and 5.0, 1H), 8.32–8.26 (m, 2H), 8.15–7.98 (m, 3H), 7.88–7.70 (m, 3H), 4.35 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ 132.0–127.1 (m, 3C), 122.4 (ddd, J=219.0, 25.0, and 16.0 Hz); HRMS calcd for 13C4-C21H14O (M+) 286.1178, found 286.1192.
5.5. Syntheses of BaP, 13C4-BaP, and the 13C4-BaP phenols
5.5.1. 2-(2-Bromophenyl)naphthalene (29)
Pd-catalyzed Suzuki cross-coupling of 8 with 28 by the method for preparation of 11c afforded 29 (87%): 1H NMR (500 MHz, CDCl3) δ 8.05–7.95 (m, 4H), 7.83 (dd, J=8.5 and 1.0 Hz, 1H), 7.70 (d, J=8.5 and 1.5 Hz, 1H), 7.65–7.58 (m, 2H), 7.53 (dd, J=7.5 and 1.5 Hz, 1H), 7.47 (td, J=7.5 and 1.5 Hz, 1H), 7.35 (td, J=7.5 and 1.5 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 142.5, 138.6, 133.1, 133.0, 132.6, 131.5, 128.8, 128.2, 128.1, 127.7, 127.6, 127.39, 127.36, 126.22, 126.20, 122.8; HRMS calcd for C16H11Br (M)+: 282.0044, found 282.0040.
5.5.2. Ethyl 2-(2-naphthalenyl)phenylacetate (30a)
Coupling of 29 with ZnBrCH2CO2Et by the method for preparation of 11b gave 30a (93 %): 1H NMR (500 MHz, CDCl3) δ 8.05–7.87 (m, 3H), 7.86 (s, 1H), 7.60–7.50 (m, 3H), 7.60–7.35 (m, 4H), 4.13 (q, J=7.0 Hz, 2H), 3.67 (s, 2H), 1.21 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 171.8, 142.3, 138.6, 133.1, 132.3, 132.1, 130.34, 130.30, 127.99, 127.96, 127.7, 127.62, 127.58, 127.5, 127.1, 126.2, 125.9, 60.7, 39.0, 14.0; HRMS calcd for C20H19O2 [M+H]+: 291.1380, found 291.1373.
5.5.3. Ethyl 13C2-2-(2-naphthalenyl)phenylacetate (13C2-30a)
1H NMR (500 MHz, CDCl3) δ 8.05–7.87 (m, 3H), 7.86 (s, 1H), 7.60–7.50 (m, 3H), 7.60–7.35 (m, 4H), 4.13 (qd, J=7.0 and 3.0 Hz, 2H), 3.67 (dd, J=129.0 and 8.0 Hz, 2H), 1.21 (t, J=7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3) δ 172.0 (d, J=229.0 Hz), 39.0 (d, J=229.0 Hz); HRMS calcd for 13C2-C20H18O2 [M]+ 292.1375, found 292.1374.
5.5.4. 2-(2-Naphthalenyl)phenylacetic acid (30b)
Hydrolysis of 30a by the usual method gave 30b (91%): 1H NMR (500 MHz, DMSO-d6) δ 12.25 (br s, 1H), 8.03–7.95 (m, 2H), 7.95–7.90 (m, 1H), 7.84 (s, 1H), 7.60–7.51 (m, 2H), 7.47 (dd, J=8.5 and 1.5 Hz, 1H), 7.45–7.30 (m, 4H), 3.59 (s, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.3, 142.2, 138.8, 133.3, 133.1, 132.4, 131.4, 130.4, 128.4, 128.1, 128.03, 128.02, 127.9, 127.8, 127.4, 126.9, 126.6, 39.1; HRMS Calcd for C18H15O2 [M+H]+ 263.1063, found 263.1012.
5.5.5. 13C2-2-(2-Naphthalenyl)phenylacetic acid (13C2-30b)
1H NMR (500 MHz, acetone-d6) δ 10.68 (br s, 1H), 8.03–7.90 (m, 3H), 7.87–7.82 (m, 1H), 7.60–7.44 (m, 4H), 7.43–7.34 (m, 3H), 3.67 (dd, J=128.5 and 8.0 Hz, 2H); 13C NMR (125.8 MHz, DMSO-d6) δ 173.2 (d, J=223.5 Hz), 39.1 (d, J=223.5 Hz); HRMS calcd for 13C2-C18H14NaO2 [M+Na]+ 287.0953, found 287.0933.
5.5.6. Chrysen-5-ol (31a)
Acid-catalyzed cyclization of 30b by the usual method gave 31a (87%): 1H NMR (500 MHz, acetone-d6) δ 10.12 (d, J=8.5 Hz, 1H), 9.80 (s, 1H), 8.83 (d, J=9.0 Hz, 1H), 8.76 (d, J=8.5 Hz, 1H), 8.10–8.00 (m, 2H), 7.85 (d, J=8.0 Hz, 1H), 7.75–7.63 (m, 2H), 7.61–7.55 (m, 2H), 7.55–7.50 (m, 1H); 13C NMR (125.8 MHz, acetone-d6) δ 154.4, 133.2, 133.1, 131.1, 130.9, 129.2, 128.5, 128.2, 126.8, 126.3, 126.09, 126.08, 126.0, 123.9, 123.3, 121.4, 121.1, 109.1; HRMS calcd for C18H13O [M+H]+ 245.0961, found 245.0970.
5.5.7. 13C2-Chrysen-5-ol (13C2-31a)
1H NMR (500 MHz, DMSO-d6) δ 10.87 (br s, 1H), 9.97 (d, J=8.5 Hz, 1H), 8.87 (d, J=9.0 Hz, 1H), 8.70 (d, J=8.5 Hz, 1H), 8.15–8.00 (m, 2H), 7.85–7.90 (m, 1H), 7.70–7.31 (m, 5H); 13C NMR (125.8 MHz, DMSO-d6) δ 154.8 (d, J=277.5 Hz), 109.2 (d, J=277.5 Hz); HRMS calcd for 13C2-C18H13O [M+H]+ 247.1032, found 247.1019.
5.5.8. Chrysen-5-ol triflate (31b)
Esterification of 31a gave 31b (80%): 1H NMR (500 MHz, CDCl3) δ 9.21 (d, J=8.5 Hz, 1H), 8.72 (d, J=8.5 Hz, 1H), 8.67 (d, J=9.0 Hz, 1H), 8.10–7.92 (m, 4H), 7.80–7.64 (m, 4H); 13C NMR (125.8 MHz, CDCl3) δ 145.5, 133.0, 131.6, 130.8, 129.73, 129.66, 128.7, 128.5, 128.1, 127.9, 127.7, 127.20, 127.16, 127.1, 123.4, 121.5, 120.6, 120.1, 118.7 (q, J=1276.5 Hz); HRMS calcd for C9H11F3O3S (M+) 376.0381, found 376.0385.
5.5.9. 13C2-Chrysen-5-ol triflate (13C2-31b)
1H NMR (500 MHz, CDCl3) δ 9.22 (d, J=9.0 Hz, 1H), 8.76 (d, J=8.0 Hz, 1H), 8.71 (d, J=9.0 Hz, 1H), 8.08 (d, J=9.0 Hz, 1H), 8.06–7.97 (m, 2H), 7.99 (dd, J=164.0 and 5.0 Hz, 1H), 7.80–7.64 (m, 4H); 13C NMR (125.8 MHz, CDCl3) δ 145.5 (d, J=358.0 Hz), 120.1 (d, J=358.0 Hz); HRMS calcd for 13C2-C19H11F3O3S (M+) 378.0429, found 378.0435.
5.5.10. (Chrysen-5-ylethynyl)trimethylsilane (32a)
Sonogashira coupling of 31b with trimethylsilylacetylene by the usual procedure gave 32a (91%): 1H NMR (500 MHz, CDCl3) δ 10.60–10.55 (m, 1H), 8.70–8.50 (m, 2H), 8.40 (s, 1H), 8.10–7.90 (m, 3H), 7.80–7.50 (m, 4H), 0.51 (s, 9H); 13C NMR (125.8 MHz, CDCl3) δ 136.7, 132.8, 130.9, 130.8, 130.4, 129.2, 128.3, 128.1, 127.9, 127.6, 127.0, 126.9, 126.7, 126.5, 125.3, 123.2, 121.1, 117.5, 108.4, 99.6, −0.15; HRMS calcd for C23H20Si (M+) 324.1334, found 324.1322.
5.5.11. 13C4-(Chrysen-5-ylethynyl)trimethylsilane (13C4-32a)
1H NMR (500 MHz, CDCl3) δ 10.52 (t, J=5.0 Hz, 1H), 8.75–8.70 (m, 2H), 8.36 (dd, J=163.0 and 7.0 Hz, 1H), 8.10–7.90 (m, 3H), 7.78–7.60 (m, 4H), 0.43 (d, J=3.0 Hz, 9H); 13C NMR (125.8 MHz, CDCl3) δ 136.8 (dd, J=251.5 and 10.5 Hz), 117.6 (ddd, J=332.0, 251.5, and 36.0 Hz), 108.6 (dd, J=542.5 and 332.0 Hz), 99.6 (ddd, J=542.5, 36.0, and 10.5 Hz); HRMS calcd for 13C4-C23H20Si (M+) 328.1468, found 328.1469.
5.5.12. 5-Ethynylchrysene (32b)
Removal of the TMS group of 32a gave 32b (85%): 1H NMR (500 MHz, CDCl3) δ 10.45 (d, J=8.5 Hz, 1H), 8.70–8.55 (m, 2H), 8.39 (s, 1H), 8.05–7.80 (m, 3H), 7.80–7.50 (m, 4H), 3.72 (s, 1H); 13C NMR (125.8 MHz, CDCl3) δ 137.6, 132.9, 130.9, 130.8, 130.6, 129.3, 128.5, 128.3, 128.0, 127.9, 127.1, 126.9, 126.7, 126.6, 125.8, 123.3, 121.1, 116.6, 86.8, 82.6; HRMS calcd for C20H13 [M+H]+ 253.1012, found 253.1006.
5.5.13. 13C4-5-Ethynylchrysene (13C4-32b)
1H NMR (500 MHz, CDCl3) δ 10.42 (d, J=8.5 Hz, 1H), 8.80–8.70 (m, 2H), 8.39 (dd, J=162.5 and 7.0 Hz, 1H), 8.05–7.80 (m, 3H), 7.80–7.50 (m, 4H), 4.10–3.30 (m, 1H); 13C NMR (125.8 MHz, CDCl3) δ 137.5 (dd, J=253.0 and 11.0 Hz), 116.5 (ddd, J=346.0, 253.0, and 56.0 Hz), 86.7 (ddd, J=706.0, 346.0, and 4.5 Hz), 82.4 (ddd, J=706.0, 56.0, and 11.0 Hz); HRMS calcd for 13C4-C20H12 (M+) 256.1073, found 256.1046.
5.5.14. Benzo[a]pyrene (BaP)
PtCl2-catalyzed cyclization of 32b furnished BaP (65%). The 1H and 13C NMR spectra were in good agreement with those of an authentic sample.
5.5.15. 13C4-Benzo[a]pyrene (13C4-BaP)
1H NMR (500 MHz, CDCl3) δ 9.12–9.02 (m, 2H), 8.72–8.22 (m, 4H), 8.20–7.94 (m, 3H), 7.90–7.75 (m, 3H); 13C NMR (125.8 MHz, CDCl3) δ 130.3–124.5 (m, 4C); HRMS calcd for 13C4-C20H14 (M+): 256.1073, found 256.1079.
5.6. BaP-1,6- and 3,6-dione (5 and 6) and their 13C4-labelled analogues (13C4-BaP-1,6-dione and 13C4-BaP-3,6-dione) (Fig. 1)
These quinones were synthesized by oxidation of BaP-1-ol (7b) and BaP-3-ol (7f) with BTI by the methods reported.20,24 Check refs!!
5.6.1. 13C4-BaP-1,6-dione and 13C4-BaP-3,6-dione
These quinones were synthesized by oxidation of 13C4-BaP-1-ol and 13C4-BaP-3-ol with BTI by methods analogous to those for oxidation of the unlabelled analogues.
13C4-BaP-1,6-dione
1H NMR (500 MHz, CDCl3) δ 8.60 (d, J=7.5 Hz, 1H), 8.51 (d, J=8.0 Hz, 1H), 8.44 (dd, J=3.5, 7 Hz), 8.31 (d, J=8 Hz), 8.05–8.00 (m, 1H), 7.99–7.71 (m, 3H), 7.66–7.61 (m, 1H), 6.76 (d, J=9.5 Hz); 13C NMR (125.8 MHz, CDCl3) δ 183.58, 183.55, 183.48, 183.42, 183.30, 183.17, 183.14, 183.10, 131.14, 131.10, 130.95, 130.82, 130.74, 130.70, 130.66, 130.35, 129.97, 129.92, 129.87, 129.80, 129.64, 129.49, 129.41, 129.35.
13C4-BaP-3,6-dione
1H NMR (500 MHz, CDCl3) δ 8.70–8.66 (m, 1H), 8.60–8.56 (m, 1H), 8.49–8.46 (m, 1H), 8.39 (d, J=7.5 Hz, 1H), 8.29 (d, J=8 Hz, 1H), 7.83–7.74 (m, 3H), 7.61 (t, J=7.5 Hz, 1H), 6.73 (d, J=10 Hz); 13C NMR (125.8 MHz, CDCl3) δ 183.60, 183.58, 183.56, 183.55, 183.17, 183.16, 183.14, 183.13, 132.53, 132.49, 132.10, 132.08, 132.07, 131.66, 131.62, 129.99, 129.95, 129.92, 129.55, 129.52, 129.48, 129.38, 129.36, 128.93, 128.92, 128.49, 128.48.
Supplementary Material
Acknowledgments
This investigation was supported by NIH grants R01-ES015857 and P30-ES013508 (awarded to T.M.P.) and R01-CA130038 (awarded to I.A.B.).
Abbreviations
- AKR1A1
aldo-keto reductase 1A1 enzyme
- BaP
benzo[a]pyrene
- BaP 7
8-diol, trans-7,8-dihydro-7,8-dihydroxy-BaP
- BaP 1
6-dione, benzo[a]pyren-1,6-dione
- BaP 3
6-dione, benzo[a]pyren-3,6-dione
- BaP 7
8-dione, benzo[a]pyren-7,8-dione
- BTI
bis-(trifluoroacetoxy)iodobenzene
- IBX
o-iodoxybenzoic acid
- anti-BPDE
trans-7,8-dihydroxy-7,8-dihydro-anti-9,10-epoxy-BaP
- syn-BPDE
trans-7,8-dihydroxy-7,8-dihydro-syn-9,10-epoxy-BaP
- 8′-HO-2′-dGua
8′-hydroxy-2′-deoxyguanosine
- 1-HO-BaP
benzo[a]pyren-1-ol
- 2-HO-BaP
benzo[a]pyren-2-ol
- 3-HOBaP
benzo[a]pyren-3-ol
- 9-HO-BaP
benzo[a]pyren-9-ol
- 12-HO-BaP
benzo[a]py-ren-12-ol
- 13C4-BaP
13C4-labelled-BaP (13C at C-4,-5,-5a, and -6 or at C-4,-5,-11, and -12, as specified)
- 13C4-1-HO-BaP
13C4-labelled-1-HO-BaP (13C at C-4,-5,-5a, and -6)
- 13C4-2-HO-BaP
13C4-labelled-2-HO-BaP (13C at C-4,-5,-5a, and -6)
- 13C4-3-HO-BaP
13C4-labelled-3-HO-BaP (13C at C-4,-5,-5a, and -6)
- 13C4-9-HO-BaP
13C4-labelled-9-HO-BaP (13C at C-4,-5,-5a, and -6)
- 13C4-12-HO-BaP
13C4-labelled-12-HO-BaP (13C at C-4,-5,-5a, and -6)
- PAH
polycyclic aromatic hydrocarbon
- ROS
reactive oxygen species
- TMSA
(trimethylsilyl)acetylene
Footnotes
Supplementary data associated with this article can be found in the online version, at doi:10.1016/j.tet.2012.05.130.
References and notes
- 1.(a) Harvey RG. In: Chemical Carcinogenesis. Penning TM, editor. Chapter 1. Humana; New York, NY: 2011. pp. 1–26. [Google Scholar]; (b) Harvey RG. Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity. Cambridge University Press; Cambridge, UK: 1991. [Google Scholar]; (c) Harvey RG, Geacintov N. Acc Chem Res. 1988;21:66–73. [Google Scholar]
- 2.Straif K, Boan R, Grosse Y, Secretan B, Ghissassi FE, Cogliano V. Nat Oncol. 2005;6:931–932. doi: 10.1016/s1470-2045(05)70458-7. [DOI] [PubMed] [Google Scholar]
- 3.International Agency for Research on Cancer. Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. IARC; Lyon, France: 1983. Polynuclear Aromatic Compounds, Part 1, Chemical, Environmental and Experimental Data; p. 32. [PubMed] [Google Scholar]
- 4.Grimmer G, editor. Environmental Carcinogens: Polycyclic Aromatic Hydrocarbons, Chemistry, Occurrence, Biochemistry, Carcinogenicity. CRC; Boca Raton, FL: 1983. The total quantity of BaP emitted into the atmosphere in the USA in 1979 was estimated to be ~1260 ton. [Google Scholar]
- 5.(a) International Agency for Research on Cancer. Tobacco Smoke and Involuntary Smoking. Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Vol. 83. IARC; Lyon, France: 2004. [PMC free article] [PubMed] [Google Scholar]; (b) World Health Organization. Tobacco or Health: A Global Status Report. WHO; Geneva: 1997. pp. 10–48. [Google Scholar]; (c) Pfiefer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht S, Hainaut P. Oncogene. 2002;21:7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]; (d) Armstrong B, Hutchinson E, Unwin J, Fletcher T. Environ Health Perspect. 2004;112:970–978. doi: 10.1289/ehp.6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marr LC, Kirschstetter TW, Hurley RA, Miguel AH, Haring SV, Hammond SK. Environ Sci Technol. 1999;3:3091–3099. [Google Scholar]
- 7.(a) Park JH, Mangal D, Frey AJ, Harvey RG, Blair IA, Penning TM. J Biol Chem. 2009;284:29725–29734. doi: 10.1074/jbc.M109.042143. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park JH, Mangal D, Tacka KA, Quinn AM, Harvey RG, Blair IA, Penning TM. Proc Natl Acad Sci USA. 2008;105:6845–6851. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Quinn AM, Harvey RG, Penning TM. Chem Res Toxicol. 2008;21:2207–2215. doi: 10.1021/tx8002005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ruan Q, Gelhaus S, Penning TM, Harvey RG, Blair IA. Chem Res Toxicol. 2007;20:424–431. doi: 10.1021/tx060180b. [DOI] [PubMed] [Google Scholar]
- 8.(a) Jennette KW, Jeffrey AM, Blobstein SH, Beland FA, Harvey RG, Weinstein IB. Biochemistry. 1977;16:932–938. doi: 10.1021/bi00624a019. [DOI] [PubMed] [Google Scholar]; (b) Weinstein IB, Jeffrey AM, Jennette K, Blobstein S, Harvey RG, Harris C, Autrup H, Kasai H, Nakanishi K. Science. 1976;193:592–595. doi: 10.1126/science.959820. [DOI] [PubMed] [Google Scholar]; (c) Jeffrey AM, Jennette KW, Blobstein SH, Weinstein IB, Beland FA, Harvey RG, Kasai H, Miura I, Nakanishi K. J Am Chem Soc. 1976;98:5714–5716. doi: 10.1021/ja00434a060. [DOI] [PubMed] [Google Scholar]
- 9.(a) Park JH, Mangal D, Frey AJ, Harvey RG, Blair IA, Penning TM. J Biol Chem. 2009;284:29725–29734. doi: 10.1074/jbc.M109.042143. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park JH, Mangal D, Tacka KA, Quinn AM, Harvey RG, Blair IA, Penning TM. Proc Natl Acad Sci USA. 2008;105:6845–6851. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ruan Q, Gelhaus S, Penning TM, Harvey RG, Blair IA. Chem Res Toxicol. 2007;20:424–431. doi: 10.1021/tx060180b. [DOI] [PubMed] [Google Scholar]; (d) Park JH, Gelhaus S, Vedantam S, Olivia A, Batra A, Blair IA, Field J. Chem Res Toxicol. 2008;21:1039–1049. doi: 10.1021/tx700404a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Flowers L, Ohnishi ST, Penning TM. Biochemistry. 1997;36:8640–8648. doi: 10.1021/bi970367p. [DOI] [PubMed] [Google Scholar]
- 10.Schultz CA, Quinn AM, Park JA, Harvey RG, Bolton JL, Maser E, Penning TM. Chem Res Toxicol. 2011;24:2153–2166. doi: 10.1021/tx200294c. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 11.(a) Cavalieri EL, Rogan EG. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]; (b) Melendez-Colon V, Luch A, Seidel A, Baird W. Carcinogenesis. 1999;20:1885–1891. doi: 10.1093/carcin/20.10.1885. [DOI] [PubMed] [Google Scholar]
- 12.Jiang H, Gelhaus SL, Mangal D, Harvey RG, Blair IA, Penning TM. Chem Res Toxicol. 2007;20:1331–1341. doi: 10.1021/tx700107z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruan Q, Gelhaus S, Penning TM, Harvey RG, Blair IA. Chem Res Toxicol. 2007;2:424–431. doi: 10.1021/tx060180b. [DOI] [PubMed] [Google Scholar]
- 14.Park JH, Mangal D, Tacka KA, Quinn A, Harvey RG, Blair IA, Penning TM. Proc Natl Acad Sci USA. 2008;105:6846–6851. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu D, Harvey RG, Blair IA, Penning TM. Chem Res Toxicol. 2011;24:1905–1914. doi: 10.1021/tx2002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Ran C, Xu D, Dai Q, Penning TM, Blair IA, Harvey RG. Tetrahedron Lett. 2008;49:4531–4533. doi: 10.1016/j.tetlet.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harvey RG, Dai Q, Ran C, Lim K, Blair IA, Penning TM. Polycyclic Aromat Compds. 2005;25:371–391. [Google Scholar]
- 17.Hama Y, Liu X, Culkin DA, Hartwig JF. J Am Chem Soc. 2003;125:11176–11177. doi: 10.1021/ja036792p. [DOI] [PubMed] [Google Scholar]
- 18.Furstner A, Musnam V. J Org Chem. 2002;67:6264–6267. doi: 10.1021/jo025962y. [DOI] [PubMed] [Google Scholar]
- 19.Xu D, Penning TM, Blair IA, Harvey RG. J Org Chem. 2009;74:597–604. doi: 10.1021/jo801864m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey RG, Dai Q, Ran C, Penning T. J Org Chem. 2004;69:2024–2032. doi: 10.1021/jo030348n. [DOI] [PubMed] [Google Scholar]
- 21.Orito K, Hatakey T, Takeo M, Suginome H. Synthesis. 1995:1273–1277. [Google Scholar]
- 22.Demirtas I, Erenler R, Cakmak O. J Chem Res, Synop. 2002:524–526. [Google Scholar]
- 23.Wu A, Duan Y, Xu D, Penning TM, Harvey RG. Tetrahedron. 2010;66:2111–2118. doi: 10.1016/j.tet.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chinchilla R, Najera C. Chem Rev. 2007;107:874–922. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 25.Kukyanek SM, Fang X, Jordan RF. Organometallics. 2009;28:300–305. [Google Scholar]
- 26.Cho H, Harvey RG. J Chem Soc, Perkin Trans 1. 1976:836–839. [PubMed] [Google Scholar]
- 27.Beland FA, Harvey RG. J Chem Soc, Chem Commun. 1976:84–85. doi: 10.1021/ja00432a042. [DOI] [PubMed] [Google Scholar]
- 28.Harvey RG, Fu PP. In: Polycyclic Hydrocarbons and Cancer, Environment, Chemistry, and Metabolism. Gelboin HV, Ts’o POP, editors. Vol. 1. Academic; New York, NY: 1978. pp. 133–165. [Google Scholar]
- 29.Harvey RG, Tang XQ. Tetrahedron Lett. 1995;36:2737–2740. [Google Scholar]; Harvey RG, Cho H. Anal Biochem. 1977;80:540–546. doi: 10.1016/0003-2697(77)90677-7. [DOI] [PubMed] [Google Scholar]; Yang SK, Gelboin HV, Weber ID, Sankaran V, Fischer DL, Engel JF. Anal Biochem. 1977;78:520–526. doi: 10.1016/0003-2697(77)90112-9. [DOI] [PubMed] [Google Scholar]; Yagi H, Akagi H, Thakker DR, Mah HD, Koreeda M, Jerina DM. J Am Chem Soc. 1976;99:2358–2359. doi: 10.1021/ja00449a066. [DOI] [PubMed] [Google Scholar]
- 30.Yang SK, Weems HB, Mushtaq M, Fu PP. J Chromatogr. 1994;316:569–584. doi: 10.1016/s0021-9673(00)96184-3. [DOI] [PubMed] [Google Scholar]
- 31.(a) Kumar S, Saravanan S. Polycyclic Aromat Compds. 2009;29:282–288. [Google Scholar]; (b) Ran C, Xu D, Dai Q, Penning TM, Blair IA, Harvey RG. Tetrahedron Lett. 2008;49:4531–4533. doi: 10.1016/j.tetlet.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sharma AK, Gowdahalli K, Gimbor M, Amin S. Chem Res Toxicol. 2008;21:1154–1162. doi: 10.1021/tx8000384. [DOI] [PubMed] [Google Scholar]; (d) Xu D, Duan Y, Blair IA, Penning T, Harvey RG. Org Lett. 2008;10:1059–1062. doi: 10.1021/ol7029323. [DOI] [PubMed] [Google Scholar]; (e) Feng X, Wu J, Ai M, Pisula W, Zhi L, Rabe JP, Mullen K. Angew Chem, Int Ed. 2007;46:3033–3036. doi: 10.1002/anie.200605224. [DOI] [PubMed] [Google Scholar]; (f) Wegner HA, Reisch H, Rauch K, Demeter A, Zachariasse KA, de Meijere A, Scott LT. J Org Chem. 2006;71:9080–9087. doi: 10.1021/jo0613939. [DOI] [PubMed] [Google Scholar]; (g) Desai D, Sharma AK, Lin J, Krzeminski J, Pementel M, El-Bayoumy K, Nesnow S, Amin S. Chem Res Toxicol. 2002;15:964–971. doi: 10.1021/tx0200111. [DOI] [PubMed] [Google Scholar]; (h) Zhang F, Cortez C, Harvey RG. J Org Chem. 2000;65:3952–3960. doi: 10.1021/jo9918044. [DOI] [PubMed] [Google Scholar]; (i) Kumar S. Tetrahedron Lett. 1996;37:6271–6274. [Google Scholar]
- 32.Chaumeil H, Le Drian C. Defois, A Synthesis. 2002:757–760. [Google Scholar]; Tovar JD, Swager TM. J Organomet Chem. 2002;653:215–222. [Google Scholar]
- 33.Harvey RG. Polycyclic Aromatic Hydrocarbons. Wiley-Interscience; New York, NY: 1997. [Google Scholar]
- 34.Harvey R. Curr Org Chem. 2004;8:303–323. [Google Scholar]
- 35.Diel BN, Han M, Kole PI, Boaz DB. J Labelled Compd Radiopharm. 2007;50:551–553. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.