

Abstract
Inhibition of the CDP-choline pathway during apoptosis restricts the availability of phosphatidylcholine (PtdCho) for assembly of membranes and synthesis of signaling factors. The N-terminal nuclear localization signal (NLS) in CTP:phosphocholine cytidylyltransferase (CCT)α is removed during apoptosis but the caspase(s) involved and the contribution to suppression of the CDP-choline pathway is unresolved. In this study we utilized siRNA silencing of caspases in HEK293 cells and caspase 3-deficient MCF7 cells to show that caspase 3 is required for CCTα proteolysis and release from the nucleus during apoptosis. CCTα-Δ28 (a caspase-cleaved mimic) expressed in CCTα-deficient Chinese hamster ovary cells was cytosolic and had increased in vitro activity. However, [3H]choline labeling experiments in camptothecin-treated MCF7 cells and MCF7 cells expressing caspase 3 (MCF7-C3) revealed a global suppression of the CDP-choline pathway that was consistent with inhibition of a step prior to CCTα. In camptothecin-treated MCF7 and MCF7-C3 cells, choline kinase activity was unaffected; however, choline transport into cells was reduced by 30 and 60%, respectively. We conclude that caspase 3-mediated removal of the CCTα NLS contributes minimally to the inhibition of PtdCho synthesis during DNA damage-induced apoptosis. Rather, the CDP-choline pathway is inhibited by caspase 3-independent and -dependent suppression of choline transport into cells.
Keywords: CTP:phosphocholine cytidylyltransferase, phosphatidylcholine, choline transporter, caspase 3, RNA interference
Phosphatidylcholine (PtdCho), a zwitterionic glycerophospholipid that comprises 40–60% of eukaryotic membrane mass, is an essential component of lung surfactant, bile, and lipoproteins, and a source for signaling molecules such as diacylglycerol (DAG) and phosphatidic acid (1). The de novo synthesis of PtdCho via the CDP-choline (Kennedy) pathway is initiated by choline transport into cells and subsequent phosphorylation by choline kinase (CK). Phosphocholine is converted to CDP-choline by the rate-limiting enzyme CTP:phosphocholine cytidylyltransferase (CCT), an amphitropic enzyme that is regulated by reversibly binding to membranes in response to changes in lipid composition (2). Finally, choline phosphotransferase (CPT) or choline/ethanolamine phosphotransferase (CEPT) in the Golgi and endoplasmic reticulum, respectively, synthesize PtdCho from CDP-choline and DAG (3). Because multiple enzyme isoforms catalyze each step in the pathway [reviewed in (4)], regulation and biosynthetic capacity is determined by unique developmental and tissue-specific patterns of enzyme expression. For example, knockout of the ubiquitous CCTα isoform in mice is embryonic lethal, indicating an essential developmental role (5). However, conditional CCTα deletion in tissues that express sufficient CCTβ or phosphatidylethanolamine N-methyltransferase is not lethal, resulting in phenotypes related to partial deduction in biosynthetic capacity (6–8).
The synthesis of PtdCho is increased during the G1 and S phases of the cell cycle to coincide with duplication of other cellular components in preparation for mitosis (9, 10). During the G0/G1 transition, PtdCho degradation supplies lipid activators of CCTα that promote increased activity throughout the S phase (10, 11). Depending on the cell type, the mode of CCTα activation involves increased enzyme dephosphorylation and activation (10) or export from the nucleus to the cytoplasm (12). Chinese hamster ovary (CHO) MT58 cells expressing a thermolabile CCTα mutant have reduced S phase content and undergo apoptosis at the nonpermissive temperature (13). This suggests that PtdCho imposes an essential checkpoint in the cell cycle that controls cell death and proliferation. Cancer cells have overcome this lipid-regulated brake by increasing overall lipogenic capacity by increased synthesis of fatty acids, sterols, and phospholipids. The activity and expression of fatty acid synthase and other lipogenic genes, such as ATP citrate lyase and acetyl CoA-carboxylase, are increased in a variety of invasive carcinomas, and are causal factors in cell transformation (14–17). The lipogenic phenotype also extends to synthesis of choline metabolites, primarily phosphocholine and PtdCho [reviewed in (18, 19)]. Choline transport (20), CCTα (20, 21), and/or CK activities (22, 23) are increased in ras-transformed cells and contribute to the oncogenic phenotype. CKα is frequently overexpressed in a variety of tumors, and inhibition by RNA interference (RNAi) or pharmacological methods reduces cancer cell proliferation. Increased PtdCho synthesis in transformed cells is frequently countered by phospholipase A2- and phospholipase C-mediated degradation, which could sustain the production of mitogenic signaling molecules such as DAG (24–26).
The importance of PtdCho to cell survival is further emphasized by the specific inhibition of CDP-choline pathway enzymes that occurs during apoptosis. A relatively early event in apoptosis is caspase cleavage of CCTα that removes the N-terminal nuclear localization signal (NLS) (27). Lipophilic activators, such as farnesol and oleyl alcohol, promote caspase cleavage of CCTα, export from the nucleus, and binding to cytoplasmic membranes (27, 28). CPT/CEPT is inhibited during apoptosis by cytotoxic drugs (29, 30), cellular acidification (30), and/or depletion of DAG (28, 31). Overexpression of CEPT prevents farnesol-induced inhibition of PtdCho synthesis but not apoptosis, indicating that inhibition of CEPT and PtdCho synthesis may not be directly linked to this mode of cell death (32). Apoptosis has not been reported to affect CK or choline transport activity.
It is evident that inhibition of the CDP-choline pathway is a common feature of apoptotic programs. However, a primary mechanism involving inhibition of CCTα and/or CPT/CEPT is at odds with metabolic labeling studies that consistently show a reduction in choline incorporation into the substrates and products of these enzymes (21, 28, 30). To reconcile this discrepancy, we compared CCTα, CK, and choline transport activities with the flux of radiolabeled choline through the CDP-choline pathway in apoptotic cells. This approach identified a caspase 3-dependent pathway for proteolysis and nuclear export of CCTα. However, caspase 3-dependent and -independent inhibition of choline transport was identified as the primary mechanism for suppression of the CDP-choline pathway.
MATERIALS AND METHODS
Materials
Rabbit polyclonal antibodies against polyADP-ribose polymerase (PARP), lamin A/C, and caspases 3, 6, and 7 were purchased from Cell Signaling Technologies (Boston, MA). Caspase 8 and V5 monoclonal antibodies were purchased from Cell Signaling Technologies and Invitrogen (Carlsbad, CA), respectively. A rabbit polyclonal CCTα anti-peptide antibody (PSPSFRWPFSGKTSP) was made by GenScript (Scotch Plains, NJ). Recombinant human caspases were purchased from Calbiochem (Gibbstown, NJ). Radioisotopes were purchased from Perkin-Elmer (Waltham, MA). TransIT-TKO was used for siRNA transfections (Mirus Corp., Nepean, ON, Canada). The caspase 8 siRNA (GGACAAAGUUUACCAAAUG) was from Dharmacon (Chicago, IL). Caspase 3 (GGAAUAUCCCUGGACAACA and GGUACUUUAAGACAUACUC), caspase 6 (GGGUUUGAUAUGGAGAAAC), and caspase 7 (GGAAUUGACUUACAUAGA) siRNAs were from Ambion/Life Technologies (Austin, TX).
A vector encoding CCTα with a deletion of amino acids 1–28 (CCTα-Δ28) was constructed by amplification of the rat cDNA with a forward primer containing a HindIII site and start codon prior to glycine 29 (CCAAGCTTCCACC ATG GGAATTCCTTCCAAAGT), and an internal reverse primer. The PCR product was digested with HindIII/EcoRV, cloned into HindIII/EcoRV-digested pcDNA-CCT-V5/His, and verified by restriction digestion and sequencing.
Cell culture
Human embryonic kidney (HEK)293 cells were cultured in DMEM containing 10% FBS (medium A). CHO-MT58 cells were cultured in DMEM with 5% FBS and proline (33 μg/ml). MCF7 cells stably expressing pBabe retroviral-encoded caspase 3 (MCF7-C3) or control vector (MCF7) were cultured in medium A containing puromycin (0.5 μg/ml) (kindly provided by Dr. Daum Tang, McMaster University, ON, Canada). HEK293 and MCF7 cells were incubated in a 5% CO2 atmosphere at 37°C. CHO-MT58 cells were maintained at 32°C but shifted to 42°C after cDNA transfection to reduce the expression of the endogenous temperature-sensitive CCTα.
HEK293 cells were transfected using TransIT-TKO reagent with 30 nM (caspase 6), 100 nM (caspase 8), or 100 nM (caspases 3 and 7) siRNAs. After 24 h, medium A was replaced and cells were cultured for another 24 h prior to the start of experiments. CHO-MT58 cells were transfected with pcDNA-CCT-V5/His or pcDNA-CCTΔ28-V5/His using Lipofectamine 2000 according to the manufacturer's instructions.
Fluorescence microscopy
CHO-MT58, MCF7, and MCF7-C3 cells (seeded on glass coverslips) were fixed in 4% paraformaldehyde in PBS for 10 min and quenched with 100 mM NH4Cl. Cells were permeabilized with 0.05% Triton X-100 (w/v) in PBS at 4°C for 10 min and blocked in PBS with 1% BSA (w/v). All primary and Alexa Fluor-conjugated secondary antibody incubations were in PBS/BSA for 1 h. Cells were incubated for 20 min with 10 μg/ml Hoechst 33258 (Invitrogen, Burlington, ON, Canada) in PBS/BSA, washed three times with PBS, and mounted on glass slides using Mowiol (Calbiochem). Images were captured using a Zeiss LSM510 Meta laser scanning confocal upright microscope with a plan-apochromat 100×/1.40 NA oil immersion objective.
Caspase cleavage of recombinant CCTα
CCTα and CCTα-D28E were in vitro transcribed/translated from pGEM4z-CCT and pGEM4z-CCTα-D28E using the TnT coupled reticulocyte lysate system (Promega Inc., Madison, WI) in the presence of [35S]methionine (27). [35S]-labeled CCT or CCTα-D28E (3 μl of translation reaction) was incubated with PBS or recombinant human caspase (5 units) in 20 μl of reaction buffer [50 mM HEPES (pH 7.4), 100 mM NaCl, 10% glycerol (v/v), 10 mM EDTA, 2 mM DTT, and 0.1% CHAPS (w/v)] (27). After 60 min at 37°C the reactions were terminated by addition of SDS-PAGE sample buffer [62.5 mM Tris-HCl (pH 6.8), 10% glycerol (v/v), 2% SDS (w/v), and 5% β-mercaptoethanol (v/v)], heated at 90°C for 5 min, resolved by SDS(8%)-PAGE and visualized by autoradiography.
Immunoblotting
HEK293, CHO-MT58, and MCF7 cells were rinsed once with PBS, lysed in SDS sample buffer, heated to 90°C for 5 min, and equivalent amounts of protein (10–20 μg) were resolved by SDS-PAGE and transferred to nitrocellulose. Membranes were incubated in blocking buffer with primary antibodies for 1 h at 20°C or overnight at 4°C. Proteins were visualized using secondary HRP-conjugated antibodies and chemiluminescence, or with IRDye800- or IRDye680-conjugated secondary antibodies and an Odyssey infrared imaging system. Depending on the method of protein visualization, expression was quantified by densitometry of film or fluorescence intensity.
[3H]choline metabolite analysis
Activity of the CDP-choline pathway in CHO-MT58 and MCF7 cells was analyzed by [3H]choline incorporation into PtdCho and water-soluble metabolites (33). At the end of the pulse or pulse-chase labeling period, cells were washed once with PBS, harvested in methanol:water (5:4, v/v), chloroform was added, and the phases were separated by centrifugation at 2,000 g for 3 min. The aqueous phase was removed for further analysis of water-soluble choline metabolites by thin-layer chromatography using a water:ethanol:ammonium hydroxide (95:48:6) solvent system. The organic solvent phase was washed twice with chloroform:0.58% NaCl:methanol (45:47:3), dried under nitrogen, and radioactivity was quantified by scintillation counting (PtdCho contains >97% of the radioactivity).
CCT, CK, and choline transporter assays
CCTα activity was assayed in the soluble and particulate (membrane) fractions prepared from CHO-MT58 cells expressing CCTα and CCTα-Δ28 (33, 34). Briefly, cells were homogenized in 20 mM Tris-HCl (pH 7.4) by 10–15 passages through a 25-gauge needle and sedimented at 100,000 g for 1 h. The pellet was resuspended in 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 250 mM sucrose. CCT activity was assayed in the soluble fraction in the presence of PtdCho/oleate vesicles over a range of CTP concentrations. The membrane fraction was assayed in a similar manner but in the absence of PtdCho/oleate vesicles.
MCF7 and MCF7-C3 cells were homogenized in 20 mM Tris-HCl (pH 7.4), 10 mM NaF, 1 mM EDTA, and 5 mM DTT by 10 passages through a 25-gauge needle. CK activity was assayed in the soluble fraction (prepared by centrifugation of the homogenate for 1 h at 100,000 g) based on the conversion of [3H]choline to phospho[3H]choline (33). High affinity choline transport activity in MCF and MCF7-C3 cells was measured by concentration-dependent uptake of [3H]choline for 10 min at 37°C as previously described (35, 36).
RESULTS
CCTα is a caspase 3 substrate
The NLS was removed from the N terminus of CCTα by caspase cleavage at TEED28 following induction of the intrinsic apoptotic pathway in cultured cells. Based on the consensus site and site-specific cleavage of in vitro-translated CCTα, caspase 6 or 8 was predicted to be involved (27). Given the variability of commercial caspase preparations, the limited scope of the initial screen, and the significant substrate redundancy of caspases under in vitro conditions (37), we tested whether [35S]methionine-labeled CCTα was a substrate for a wider range of recombinant caspases (Fig. 1). [35S]CCTα was incubated with recombinant caspases, resolved by SDS-PAGE, and visualized by autoradiography (Fig. 1). Incubation of [35S]CCTα with caspases 3, 6, 7, and 9 resulted in removal of the 28 N-terminal amino acids and appearance of a 37 kDa product (indicated by an arrowhead). Mutation of the caspase site in [35S]CCTα-D28E prevented proteolysis by all four caspases.
Fig. 1.
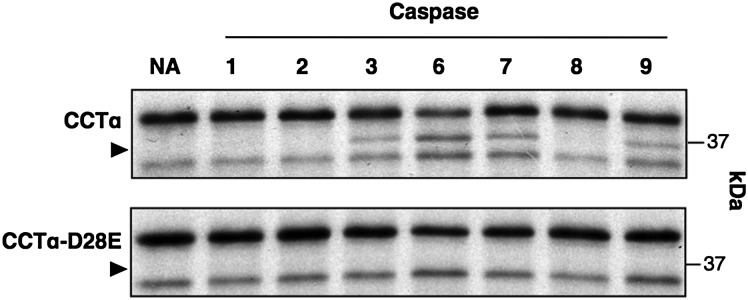
Proteolysis of in vitro translated CCTα by recombinant caspases. In vitro translated [35S]methionine-labeled CCTα was incubated with 5 units of recombinant caspase or no addition (NA) for 1 h at 37°C. Reactions were terminated by addition of SDS sample buffer, an aliquot was resolved by SDS(10%)-PAGE and the gel exposed to film at −80°C for 24 h. Processed CCTα is indicated by an arrowhead. Results are from a representative experiment that was repeated three other times with similar results.
The results of the in vitro assay served as the basis to identify the CCTα-specific caspase(s) in apoptotic HEK293 cells. Based on evidence from our lab and others (27, 38) and preferred caspase consensus sites (39), caspases 3, 6, 7, and 8 were selected for analysis. Our inability to identify the involvement of individual caspases using pharmacological approaches (results not shown) prompted the use of siRNAs to silence caspases 3, 6, 7, and 8 in HEK293 cells induced to undergo apoptosis with chelerythrine, a protein kinase C inhibitor that also profoundly inhibits the CDP-choline pathway (30). Caspase 6 displayed the highest activity toward CCTα in vitro and its major nuclear substrate is lamin A/C (40). Caspase 6 was silenced in HEK293 cells, apoptosis was induced with chelerythrine, and the cleavage of lamin A/C and CCTα was examined by immunoblotting (Fig. 2A). The caspase 6 siRNA reduced protein expression by 81.5 ± 6.7% (n = 4) after 48 h (Fig. 2A, Casp6 panel). In caspase 6 knockdown cells, lamin A/C was poorly processed to its 28 kDa cleavage product compared with nontargeting controls. However, the time course for processing of CCTα was similar in control and caspase 6-depleted cells, and there was no significant difference in the distribution of full-length and processed forms at 4 h (Fig. 2D). Caspase 8 is associated with the extrinsic death-receptor pathway but can also be activated through the intrinsic pathway (41). The involvement of caspase 8 was tested by silencing its expression in HEK293 cells, followed by induction of apoptosis with chelerythrine (Fig. 2B, C). Immunoblotting showed a 91.4 ± 5.6% (n = 3) reduction in caspase 8 expression in nonapoptotic HEK293 cells after RNAi treatment for 48 h (Fig. 2C). Cleavage of PARP was similar in apoptotic HEK293 cells transfected with nontargeting and caspase 8 siRNAs. Similarly, caspase 8 silencing did not inhibit CCTα processing during chelerythrine treatment (Fig. 2B, D). To address the possibility that individual knockdown of caspases 6 and 8 did not eliminate CCTα processing due to redundancy, both caspases were simultaneously suppressed by RNAi, and CCTα processing was determined after induction of apoptosis in HEK293 cells for 4 h (Fig. 2E). Silencing of caspases 6 and 8 caused a slight reduction in processed CCTα in treated cells, but this was not accompanied by an increase in full-length CCTα, suggesting that neither caspase 6 nor caspase 8 are primarily involved in CCTα proteolysis.
Fig. 2.

CCTα is not a substrate for caspase 6 or 8. A: HEK293 cells were transfected with a caspase 6 siRNA (siCasp6) (30 nM) or an equivalent amount of a nontargeting control (siNT) for 48 h. Cells then received medium A with 15 μM chelerythrine for the indicated times (0–8 h), lysates were prepared and resolved by SDS-PAGE and immunoblotted for caspase 6, lamin A/C, CCTα, and actin. B, C: HEK293 cells transfected with nontargeting or caspase 8 siRNAs (siCasp8) (100 nM) were treated as described above and the lysates immunoblotted for PARP, caspase 8, CCTα, and actin. D: Quantification of immunoblots in (A) and (B) showing the percent distribution of holo- and processed CCTα (pro-CCT) in lysates of HEK293 cells transfected with the indicated siRNAs and treated with chelerythrine for 4 h. Results are the mean and standard deviation of three experiments. E: Lysates from HEK293 transfected with siRNAs targeting caspases 6 and/or 8 (30 and 100 nM) were immunoblotted for CCTα, caspase 6, caspase 8, and actin. Symbols refer to the full-length (▶) and the caspase-cleaved (*) forms of CCTα, PARP, and lamin A/C.
We next considered executioner caspases 3 and 7, which are activated by intrinsic/extrinsic apoptotic signals but have consensus cleavage sites that would appear to exclude CCTα as a substrate. Nevertheless, both were tested by siRNA knockdown in HEK293 cells (Fig. 3). Two different caspase 3 siRNAs consistently reduced the expression of the 35 kDa caspase 3 by 80.6 ± 7.1% (n = 4) in untreated HEK293 cells (Fig. 3A). Compared with nontargeting controls, caspase 3 knockdown had only a minor effect on PARP processing after chelerythrine treatment for 4 h (Fig. 3B). This was not unexpected because PARP is a substrate for other executioner caspases, including caspase 7 (42–44). In contrast, caspase 3 depletion by both siRNAs inhibited CCTα processing at 2 and 4 h as indicated by reduction in the 37 kDa processed form and a reciprocal increase in the full-length enzyme (Fig. 3B). Quantification of CCTα processing at 4 h in response to siCasp3-1 is shown in Fig. 3D. To rule out the possibility of functional redundancy with caspase 7 (45), its expression was reduced with a siRNA, alone or in combination with siCasp3-1, and CCTα and PARP cleavage was assessed after chelerythrine treatment for 4 h (Fig. 3C and D). Reduction of caspase 7 expression by 82.7 ± 5.5% (n = 3) had no effect on PARP or CCTα processing relative to nontargeting controls, and combined knockdown of caspases 3 and 7 was no more effective than knockdown of caspase 3 alone. These siRNA experiments show that caspase 3 is primarily responsible for proteolysis of the N-terminal domain of CCTα.
Fig. 3.

Inhibition of CCTα cleavage by caspase 3 knockdown. HEK 293 cells were transfected with siRNAs (150 nM) targeting caspase 3 (A, B), caspase 7 (C), caspases 3 and 7 (C), or a nontargeting (siNT) control for 48 h. Apoptosis was induced with 15 μM chelerythrine for up to 4 h. At the indicated times, cell lysates were prepared, resolved by SDS(8%)-PAGE, and immunoblotted for PARP, caspase 3, caspase 7, CCTα, and actin as described in the Materials and Methods. Symbols indicate full-length (▶) and caspase-cleaved (*) forms of CCTα and PARP. D: Quantification of immunoblots in (B) and (C) showing the percent distribution of holo- and processed CCTα (pro-CCT) in lysates of HEK293 cells transfected with the indicated siRNAs and treated with chelerythrine for 4 h. Results are the mean and standard deviation of three experiments. *P < 0.05 using unpaired t-test compared with siNT.
MCF7 cells are partially resistant to apoptosis as a result of a deletion in the caspase 3 gene (46), but can be resensitized by ectopic expression of caspase 3 (47). This cell model was used to confirm that CCTα is a caspase 3 substrate and to dissect its involvement in inhibition of PtdCho synthesis. MCF cells stably expressing the empty retroviral vector (MCF7), or one encoding caspase 3 (MCF7-C3), were treated with camptothecin for up to 24 h and immunoblotted for caspase 3, PARP, and CCTα. Induction of the intrinsic DNA damage pathway by camptothecin inhibits PtdCho synthesis (30) and requires caspase 3 (46). Unlike control MCF7 cells, MCF7-C3 cells expressed holo-caspase 3 (p35), which was processed to the 17 kDa form during camptothecin treatment (Fig. 4A). There was extensive proteolysis of PARP in MCF7 cells treated with camptothecin for 24 h, but processing of CCTα was not evident (Fig. 4B, C). In contrast, 50% of CCTα was proteolyzed in MCF7-C3 cells by 24 h, with a reduction in total expression at 24 and 48 h (Fig. 4B, C). Activation of the extrinsic apoptotic pathways with tumor necrosis factor α (TNFα) also stimulated CCTα processing in MCF7-C3 cells but not in vector controls (Fig. 4D). Immunofluorescence was used to localize CCTα in the nucleus of untreated MCF and MCF7-C3 cells (Fig. 4E). The nuclei of MCF7 cells treated with camptothecin for 24 h were shrunken and irregularly shaped, but the nuclear envelope (NE) was uniformly stained with a nuclear pore complex (NPC) antibody and CCTα was retained in the nucleoplasm. Camptothecin-treated MCF7-C3 cells had shrunken nuclei, weak NPC staining of the NE, and CCTα was localized to the cytoplasm. CCTα had a similar localization in TNFα-treated MCF7 cells, but in MCF7-C3 cells there was more extensive localization of CCTα around or on the NE (Fig. 4E). This supports the conclusion that caspase 3 is responsible for CCTα processing and release from the nucleus.
Fig. 4.

Defective CCTα proteolysis in MCF7 cells is restored by stable expression of caspase 3. A: MCF7 (expressing empty vector) or MCF7-C3 (stably expressing caspase 3) cells were treated with 15 μM camptothecin (CMT). At the indicated times, total cell lysates were prepared and immunoblotted for caspase 3. The holo (p35) and processed form (p17) of caspase 3 are indicated. B: Cells were treated with camptothecin (15 μM) for the indicated times, harvested, and immunoblotted for CCTα, actin, or PARP. Caspase-processed forms of CCTα and PARP are indicated (▶). C: Quantification of immunoblots from (B) showing the percent distribution of holo- and processed CCTα (pro-CCT) after 24 h camptothecin treatment. Results are the mean and standard deviation of three experiments. *P < 0.05 using unpaired t-test compared with MCF7. D: Immunoblot analysis CCTα in MCF7 and MCF7-C3 treated with TNFα (10 ng/ml). E: Cells were treated with CMT (15 μM for 24 h), TNFα (10 ng/ml for 12 h), or no addition (NA) followed by immunostaining for CCTα and the NPC using a NUP62 monoclonal antibody. Images are 0.5 μm confocal sections. Results are representative of three separate experiments.
Enzyme activity of the caspase-cleaved mimic CCTα-Δ28
Endogenous caspase-cleaved CCTα is activated following induction of apoptosis with farnesol (27). However, farnesol is also a lipophilic activator of CCTα that increases membrane translocation and enzyme activity on liposomes and in cultured cells, making it difficult to directly determine the effect of N-terminal processing on enzyme activity. To assess whether caspase 3 processing affects CCTα activity independent of apoptotic induction, we assayed the activity of a constitutively cleaved mimic CCTα-Δ28. V5-tagged CCTα and CCTα-Δ28 had the expected molecular masses and were transiently expressed at similar levels in CHO-MT58 cells (Fig. 5A). Immunofluorescence analysis of transiently transfected CHO-MT58 cells showed that CCTα was expressed primarily in the nucleoplasm while CCTα-Δ28 was distributed in both the nucleoplasm and cytoplasm (Fig. 5B). To assess the activity of CCTα and CCTα-Δ28, each was transiently expressed in CHO-MT58 cells, and PtdCho and CDP-choline pathway metabolites were measured by [3H]choline labeling after shifting cells to 40°C to inactivate endogenous CCTα (Fig. 5C, D). [3H]choline incorporation into PtdCho was significantly increased in cells expressing CCTα-Δ28 compared with wild-type CCTα (Fig. 5C). Compared with cells expressing wild-type CCTα, CCTα-Δ28 expression caused a significant increase in [3H]choline incorporation into CDP-choline and a minor decrease in phosphocholine (Fig. 5D).
Fig. 5.

Transient expression of CCT-Δ28 in CHO MT58 cells increases PtdCho synthesis. A: Total lysates of CHO-MT58 cells transiently expressing CCTα or CCTα-Δ28 were immunoblotted with a V5 monoclonal antibody. B: Transiently transfected CHO-MT58 cells were immunostained using V5 monoclonal antibody and Alexa Fluor-488 antibodies. Hoechst 33258 was used to counterstain the nucleus. C, D: CHO-MT58 cells transiently expressing V5-tagged CCTα or CCTα-Δ28-V5 were cultured at 42°C. After 48 h, cells were labeled with [3H]choline (2 μCi/ml) for 4 h and PtdCho (C) and choline metabolites (D) were quantified. Choline incorporation was normalized to expression of CCTα and CCTα-Δ28 by densitometry of immunoblots of total cell lysates. Results are the mean and standard deviation for three experiments. *P < 0.05 using unpaired t-test compared with CCTα expressing cells.
To determine whether the kinetic properties of CCTα-Δ28 were altered compared with the wild-type enzyme, cDNAs were transiently overexpressed in CHO-MT58 cells and cytosolic and particulate (membrane) fractions were prepared after shifting cells to 40°C to inactivate endogenous CCTα. Kinetic constants were determined by assaying CCTα activity in soluble and membrane fractions with increasing concentrations of CTP in the presence or absence of PtdCho/oleate liposomes (Fig. 6). Km and Vmax constants for soluble CCTα and CCTα-Δ28 assayed in the presence of PtdCho/oleate liposomes were similar (Fig. 6A). Compared with the wild-type enzyme, membrane-associated CCTα-Δ28 had a Km for CTP that was reduced by 2-fold (Fig. 6B). Hence, increased activity of CCTα-Δ28 expressed in MT-58 cells (Fig. 5) could be the result of increased affinity for CTP by the membrane-associated form of the enzyme.
Fig. 6.

Kinetic analysis of CCTα-Δ28. Soluble (A) and membrane fractions (B) from CHO-MT58 cells (cultured at 42°C) transiently expressing CCTα or CCTα-Δ28 were assayed for CCTα activity at increasing concentrations of CTP (mM). Soluble CCTα activity was assayed in the presence of PtdCho/oleate vesicles. The activity of membrane fractions was assayed in the absence of PtdCho/oleate vesicles. Enzyme activity was normalized to expression of CCTα or CCTα-Δ28 in each fraction by immunoblotting and densitometry. Km and Vmax values (see inserts) are the mean and standard deviation for three experiments.
Caspase 3 cleavage of CCTα does not contribute to inhibition of PtdCho synthesis
CCTα is proteolyzed and exported into the cytoplasm of apoptotic MCF7-C3 cells. Because these two events do not occur in caspase 3-deficient MCF7 cells, we can directly ascertain the contribution of this caspase 3-dependent pathway to inhibition of PtdCho synthesis in apoptotic cells. A prior study indicated that camptothecin and other apoptotic agents inhibited PtdCho synthesis at the CEPT/CPT catalyzed step (30). However, the incorporation of radiolabeled-choline into CDP-choline and other CDP-choline pathway intermediates was reduced suggesting additional mechanisms. To identify which step(s) is inhibited, MCF7 and MCF7-C3 cells were treated with camptothecin for 24 h and pulse-labeled with [3H]choline. Incorporation of [3H]choline into PtdCho was inhibited by 50% and 30% in camptothecin-treated MCF7 and MCF7-C3 cells, respectively, although PtdCho synthesis was initially 40% lower in untreated MCF7-C3 cells (Fig. 7A). The reduced synthesis of PtdCho in untreated MCF-C3 cells was accompanied by increased phospho[3H]choline, which was reduced to a similar level in apoptotic MCF7 and MCF7-C3 cells (Fig. 7B). Isotope incorporation into CDP-choline, glycerophosphocholine, and choline were low relative to phosphocholine, and either unchanged or inhibited in apoptotic cells. [3H]choline incorporation into total CDP-choline pathway metabolites was reduced by 40 and 55% in MCF7 and MCF7-C3 cells, respectively, indicating both caspase 3-dependent and -independent inhibition of multiple steps in the pathway or a block in choline uptake.
Fig. 7.
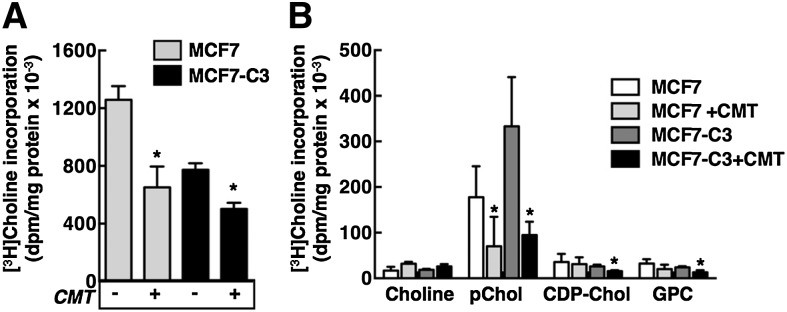
Suppression of the CDP-choline pathway by camptothecin-induced apoptosis in MCF7 and MCF7-C3 cells. MCF7 and MCF7-C3 cells were treated with camptothecin (15 μM) or solvent control for 24 h. During the last 3 h, cells were pulse-labeled with 2 μCi/ml [3H]choline in choline-free medium A. Isotope incorporation into PtdCho (A) and water-soluble choline metabolites (B) was determined as described in Materials and Methods. Results are the mean and standard deviation of five experiments. *P < 0.05 using an unpaired t-test compared with untreated cells.
[3H]choline pulse-chase experiments were also performed to assess the activity of CCTα in intact cells. One hour pulse-labeling of cells with [3H]choline resulted in accumulation of radiolabeled phosphocholine, which was then converted during a 3 h chase period to PtdCho as a function of the activity of the rate-limiting enzyme CCTα (Fig. 8). The rate of PtdCho synthesis was reduced by 50% in camptothecin-treated MCF7 cells compared with untreated controls (26,390 ± 7,320 dpm/h vs. 14,640 ± 4,330 dpm/h, respectively) (Fig. 8A, B). There was a similar 50% reduction in the rate of consumption of phosphocholine. In untreated MCF7-C3 cells, the initial rate of PtdCho synthesis (11,680 ± 5170 dpm/h) was reduced compared with untreated MCF7 cells, reflecting a larger pool of phospho[3H]choline in these cells (Fig. 8C). In camptothecin-treated MCF7-C3 cells (Fig. 8D), radiolabeled phosphocholine at the end of the pulse period (0 h) was reduced by 50% compared with untreated cells, and subsequent conversion to PtdCho was complete by 1 h. The estimated rate of PtdCho synthesis and phosphocholine conversion during this time was approximately 50% of that in untreated MCF7-C3 cells. Based on rates of phosphocholine conversion to PtdCho, CCTα activity was reduced by approximately 50% in camptothecin-treated MCF7 and MCF7-C3 cells. CDP-[3H]choline did not change during the chase period indicating that inhibition of CEPT/CPT is not a contributing factor.
Fig. 8.

[3H]choline pulse-chase analysis of CCT activity in apoptotic MCF7 and MCF7-C3 cells. MCF7 (A, B) and MCF7-C3 cells (C, D), cultured in the absence (A, C) or presence (B, D) of camptothecin (15 μM) for 24 h, were pulse-labeled with [3H]choline (2 μCi/ml) in choline-free medium for 1 h followed by replacement with medium containing 50 μM choline. Cells were harvested at the indicated times during the chase period and [3H]choline incorporation into PtdCho (•), phosphocholine (■), CDP-choline (▴), and glycerophosphocholine (GPC) (▾) was quantified as described in Materials and Methods. Results are the mean and standard deviation of three to four experiments.
Inhibition of choline uptake in camptothecin-treated cells
Metabolic labeling of phosphocholine is reduced in apoptotic MCF7 and MCF7-C3 cells despite an apparent reduction in CCTα activity (Fig. 8), which utilizes phosphocholine as a substrate. This suggests that the synthesis of phosphocholine is inhibited due to reduced choline transport and/or phosphorylation. CKα is the major isoform expressed in MCF7 cells and is overexpressed in many types of cancer (48). However, total CK activity in the cytosolic fractions from control and camptothecin-treated MCF7 and MCF7-C3 cells was similar (Fig. 9).
Fig. 9.
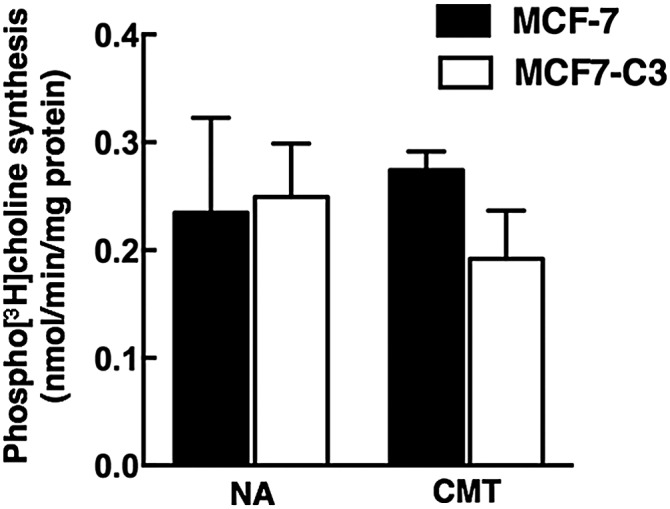
CK activity in apoptotic MCF7 and MCF7-C3 cells. CK activity was assayed in the cytosolic fractions from MCF7 and MCF7-C3 cells cultured in the absence [no addition (NA)] or presence of camptothecin (CMT, 15 μM) for 24 h. Results are the mean and standard deviation of three to four experiments.
Choline is taken up into mammalian cells by the high-affinity sodium-dependent choline transporter 1, intermediate affinity sodium-independent choline transporter-like proteins (CTLs), and low-affinity nonspecific sodium-independent organic cation transporters (OCTs) [reviewed in (49)]. To determine if choline transport was inhibited during apoptosis, MCF7 and MCF7-C3 cells were treated with camptothecin for 24 h and transport activity was measured based on uptake of 1–25 μM [3H]choline (Fig. 10A). The KD (22.5 ± 9.0 μM and 27.0 ± 9.1 μM) and maximal uptake (Bmax) values (321 ± 82 pmol/min/mg and 353 ± 78 pmol/min/mg) for saturable choline transport were similar in untreated MCF7 and MCF7-C3 cells, respectively. These kinetic parameters are also similar to those reported previously for MCF7 cells (50). Camptothecin treatment of MCF7 and MCF7-C3 cells did not affect the KD (17.7 ± 7.0 μM and 14.3 ± 6.3 μM) but caused a significant 30 and 60% reduction in the Bmax (219 ± 51 pmol/min/mg and 163 ± 39 pmol/min/mg, P < 0.05 compared with untreated controls). Based on mRNA expression, choline transport activity in MCF7 cells is mediated primarily by CTL1 and OCT2/1 (50). CTL1 is competitively inhibited by hemicholinium-3 (HC-3), whereas OCTs are not (49, 51). Treatment of MCF7 and MCF7-C3 with HC-3 inhibited choline uptake by 70–80% indicating CTL1 or a related family member is involved (Fig. 10B). HC-3 was then used to determine whether intermediate- and/or low-affinity choline transporter activities are inhibited by camptothecin-induced apoptosis (Fig. 10C, D). For these experiments, the relative inhibition by camptothecin of HC-3-sensitive and -insensitive choline transporters was measured in MCF7 and MCF7-C3 cells. In the case of MCF7 cells, camptothecin significantly inhibited choline transport activity in the absence but not the presence of HC-3, suggesting that a HC-3-sensitive CTL was affected (Fig. 10C). In contrast, camptothecin significantly inhibited approximately 70% of choline transport activity in MCF7-C3 cells in the absence and presence of HC-3. However, inhibition of the HC-3-sensitive transporter during apoptosis is more relevant because it accounts for >70% of activity in MCF7 cells. Thus, reduced choline incorporation into PtdCho in apoptotic MCF7 and MCF7-C3 cells (Figs. 7, 8) is primarily due to caspase 3-independent and -dependent inhibition of HC-3-sensitive CTL1 or related family members.
Fig. 10.

Choline transport activity is reduced in apoptotic MCF7 and MCF7-C3 cells. A: MCF7 and MCF7-C3 cells were treated with camptothecin (CMT, 15 μM) for 24 h. Cells were subsequently rinsed with Krebs-Ringer buffer and uptake of increasing concentrations of [3H]choline (1–25 μM) was measured at 37°C for 10 min as described in Materials and Methods. KD and Bmax values were determined by Scatchard analysis fit to a single binding site model. Results are the mean and standard deviation of triplicate measurements from five experiments. B: Uptake of 10 nM [3H]choline into MCF7 and MCF7-C3 cells was assayed for 10 min at 37°C in the presence of increasing concentrations HC-3. Results are the mean and standard deviation of three experiments. C, D: Uptake of 20 μM [3H]choline into MCF7 and MCF7-C3 cells (treated with control solvent or 15 μM CMT for 24 h) was assayed in the presence of 0, 100, and 200 μM HC-3 for 10 min at 37°C. Results are the mean and standard deviation of three experiments. *P < 0.05 using unpaired t-test compared with matched untreated MCF7 or MCF7-C3 cells.
DISCUSSION
Perturbation of membrane structure by alterations in the composition and topology of the phospholipid constituents is an important feature of apoptotic programs. In the case of PtdCho, synthesis by the CDP-choline pathway is inhibited (52), degradation by lipases is stimulated (53), and it is the source of the lysophosphatidylcholine “eat me” signal (54). Caspase processing of CCTα and inhibition of CEPT/CPT are implicated in the cessation of PtdCho synthesis but evidence is indirect and, based on radiolabeling experiments, the substrates for these enzymes (CDP-choline and phosphocholine) do not accumulate in apoptotic cells (28–30). In this study, we used two well-characterized apoptotic agents (camptothecin and chelerythrine) that strongly inhibit PtdCho synthesis to demonstrate that caspase 3 cleaves the NLS from CCTα. However, metabolic labeling and in vitro assays revealed that transport of choline into cells is inhibited by caspase 3-dependent and -independent mechanisms, leading to global suppression of choline incorporation into the CDP-choline pathway.
In vitro translated CCTα was a substrate for caspases 3, 6, 7, 8, and 9, but only caspase 3 was involved in processing the enzyme in cultured cells. The processing of in vitro translated CCTα by multiple caspases could be due to misfolding and loss of tertiary structure-specific context, lack of caspase substrate specificity in vitro (37), and/or loss of temporal and spatial regulatory elements in a cell-free system (43). The lack of involvement of caspase 6 in CCTα processing in cells was unexpected because it had the highest activity toward CCTα in vitro and is present in the nucleus where it cleaves lamin A (40), which also interacts with CCTα to regulate nucleoplasmic reticulum formation (55). Despite a poor consensus site match, siRNA silencing in apoptotic HEK293 cells showed that caspase 3 is primarily responsible for CCTα cleavage. This was confirmed by the restoration of CCTα processing in response to camptothecin and TNFα in MCF7 cells in which caspase 3 was reexpressed by viral transduction. Caspase 7 can trigger the intrinsic mitochondrial apoptotic cascade in the absence of caspase 3 (56–58); however, CCTα processing occurred in caspase 7-deficient HEK293 cells indicating that caspase 3 is primarily involved.
Caspase 3-replete and -deficient MCF7 cells were used to determine the role of this caspase in CCTα regulation and PtdCho synthesis. In apoptotic MCF7-C3 cells, CCTα was partially proteolyzed and excluded from the nucleus, effects that were completely absent in MCF7 cells. Nuclear export of CCTα does not require caspase cleavage but involves translocation to the NE in farnesol-treated cells (59). NE localization of CCTα was not evident in camptothecin-treated MCF7-C3 cells, which also had reduced staining of the NPC indicating that cytoplasmic CCTα could have resulted from loss of NE integrity. In this context it is difficult to determine whether caspase 3 cleavage occurred prior to or after release of CCTα from the nucleus. Nevertheless, it appears that NLS removal by caspase 3 is a mechanism to keep CCTα sequestered in the cytoplasm of apoptotic cells. The caspase-processed mimic CCTα-Δ28 was cytoplasmic, had increased PtdCho synthesis, and the membrane-associated enzyme had a reduced Km for CTP. Cytoplasmic CCTα-Δ28 had similar kinetic parameters as wild-type, suggesting that the increased activity of CCT-Δ28 results from activation on cytoplasmic membranes rather then a general stimulatory effect due to NLS deletion.
If caspase 3 processing of CCTα is involved in regulation of the CDP-choline pathway, there should have been notable differences in the distribution of [3H]choline-labeled metabolites in apoptotic MCF7 and MCF7-C3 cells. Instead, continuous [3H]choline pulse experiments (Fig. 7) indicated that isotope incorporation into PtdCho and other metabolites was reduced to a similar extent following camptothecin treatment of both cell lines. The exception was phosphocholine, which was elevated in untreated MCF7-C3 cells compared with controls but reduced to a similar level following camptothecin treatment. The reason for increased [3H]choline incorporation into phosphocholine in untreated MCF7-C3 cells is unclear because CCTα expression and localization were similar to MCF7 cells. Transient expression of caspase 3 in MCF7 cells did not cause accumulation of phosphocholine during a [3H]choline labeling experiment (results not shown), indicating the effect could be related to stable expression of caspase 3. The conclusion that CEPT inhibition is not a contributing factor in inhibition of PtdCho synthesis in apoptotic MCF7 cells is supported by both continuous-pulse and pulse-chase experiments. In both cases, there was no evidence of CDP-choline accumulation or reduced conversion of CDP-choline to PtdCho that would be indicative of CEPT/CPT inhibition. [3H]choline pulse-chase experiments did, however, indicate inhibition of CCTα activity based on a 50% reduction in phosphocholine conversion to PtdCho in apoptotic MCF7 and MCF7-C3 cells. This is in contrast to the caspase-processed mimic CCTα-Δ28, which had increased activity when transiently expressed in CHO cells. Loss of CCTα activity in apoptotic MCF7 cells could be due to a combination of factors including reduced substrate levels (phosphocholine and CTP), CCTα degradation, and/or the acidic environment in apoptotic cells.
The global suppression of [3H]choline incorporation into PtdCho and other metabolites in camptothecin-treated MCF7 and MCF-C3 cells indicated that an early step(s) in the CDP-choline pathway is inhibited. In vitro CK activity was unaffected, but maximal uptake by an intermediate affinity choline transporter was inhibited, suggesting that the number or activity of cell surface transporters is reduced in apoptotic cells. The inhibition of choline transport had both caspase 3-independent and -dependent components that each accounted for approximately 30%, which correlated well with the 30 and 55% inhibition of choline incorporation into total CDP-choline metabolites in apoptotic MCF7 and MCF7-C3 cells, respectively. Based on HC-3 inhibition (Fig. 10B–D), kinetic parameters (Fig. 10A), and mRNA expression profiles (50), we conclude that CTL1 is responsible for <70% of choline transport activity in MCF7 and MCF7-C3 cells and is inhibited by caspase 3-dependent and -independent processes. Based on metabolic labeling from other studies that show a general suppression of choline input in the CDP-choline pathway (21, 27, 30), we posit that inhibition of choline transport occurs in other apoptotic programs and, because of reduced input of choline into the pathway, secondary effects on CCTα and/or CEPT/CPT have a lesser contribution to inhibition of PtdCho synthesis.
Cancer cells have increased CKα expression and activity that is necessary for survival and proliferation (60–62). Phosphocholine is also elevated in cancer cells, where it has discrete oncogenic activities and contributes to PtdCho biogenesis and signaling (19). These hallmarks of proliferation are reversed in apoptotic cells, which display reduced choline transport and synthesis of phosphocholine. Inhibition of this initial step in the CDP-choline pathway efficiently blocks the synthesis of PtdCho and choline metabolites that are required for cell proliferation.
Acknowledgments
The authors thank Robert Zwicker for technical support of tissue culture.
Footnotes
Abbreviations:
- CCT
- CTP:phosphocholine cytidylyltransferase
- CEPT
- choline/ethanolamine phosphotransferase
- CHO
- Chinese hamster ovary
- CK
- choline kinase
- CPT
- choline phosphotransferase
- CTL
- choline transporter-like protein
- DAG
- diacylglycerol
- HC-3
- hemicholinium-3
- HEK
- human embryonic kidney
- MCF7-C3
- MCF7 cells expressing caspase 3
- NE
- nuclear envelope
- NLS
- nuclear localization signal
- NPC
- nuclear pore complex
- OCT
- organic cation transporter
- PARP
- polyADP-ribose polymerase
- PtdCho
- phosphatidylcholine
- RNAi
- RNA interference
This work was supported by a grant from the Canadian Institutes of Health Research. C.C.M. was the recipient of a Studentship from the Nova Scotia Health Research Foundation.
REFERENCES
- 1.Li Z., Vance D. E. 2008. Phosphatidylcholine and choline homeostasis. J. Lipid Res. 49: 1187–1194 [DOI] [PubMed] [Google Scholar]
- 2.Cornell R. B., Taneva S. G. 2006. Amphipathic helices as mediators of the membrane interaction of amphitropic proteins, and as modulators of bilayer physical properties. Curr. Protein Pept. Sci. 7: 539–552 [DOI] [PubMed] [Google Scholar]
- 3.Henneberry A. L., Wright M. M., McMaster C. R. 2002. The major sites of cellular phospholipid synthesis and molecular determinants of fatty acid and lipid head group specificity. Mol. Biol. Cell. 13: 3148–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vance D. E., Vance J. E. 2009. Physiological consequences of disruption of mammalian phospholipid biosynthetic genes. J. Lipid Res. 50: S132–S137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L., Magdaleno S., Tabas I., Jackowski S. 2005. Early embryonic lethality in mice with targeted deletion of the CTP:phosphocholine cytidylyltransferase alpha gene (Pcyt1a). Mol. Cell. Biol. 25: 3357–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D., Tang W., Yao P. M., Yang C., Xie B., Jackowski S., Tabas I. 2000. Macrophages deficient in CTP:phosphocholine cytidylyltransferase-alpha are viable under normal culture conditions but are highly susceptible to free cholesterol-induced death. Molecular genetic evidence that the induction of phosphatidylcholine biosynthesis in free cholesterol- loaded macrophages is an adaptive response. J. Biol. Chem. 275: 35368–35376 [DOI] [PubMed] [Google Scholar]
- 7.Tian Y., Zhou R., Rehg J. E., Jackowski S. 2007. Role of phosphocholine cytidylyltransferase alpha in lung development. Mol. Cell. Biol. 27: 975–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs R. L., Devlin C., Tabas I., Vance D. E. 2004. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J. Biol. Chem. 279: 47402–47410 [DOI] [PubMed] [Google Scholar]
- 9.Jackowski S. 1996. Cell cycle regulation of membrane phospholipid metabolism. J. Biol. Chem. 271: 20219–20222 [DOI] [PubMed] [Google Scholar]
- 10.Jackowski S. 1994. Coordination of membrane phospholipid synthesis with the cell cycle. J. Biol. Chem. 269: 3858–3867 [PubMed] [Google Scholar]
- 11.Ng M. N., Kitos T. E., Cornell R. B. 2004. Contribution of lipid second messengers to the regulation of phosphatidylcholine synthesis during cell cycle re-entry. Biochim. Biophys. Acta. 1686: 85–99 [DOI] [PubMed] [Google Scholar]
- 12.Northwood I. C., Tong A. H., Crawford B., Drobnies A. E., Cornell R. B. 1999. Shuttling of CTP:phosphocholine cytidylyltransferase between the nucleus and endoplasmic reticulum accompanies the wave of phosphatidylcholine synthesis during the G(0) → G(1) transition. J. Biol. Chem. 274: 26240–26248 [DOI] [PubMed] [Google Scholar]
- 13.Cui Z., Houweling M., Chen M. H., Record M., Chap H., Vance D. E., Terce F. 1996. A genetic defect in phosphatidylcholine biosynthesis triggers apoptosis in Chinese hamster ovary cells. J. Biol. Chem. 271: 14668–14671 [DOI] [PubMed] [Google Scholar]
- 14.Swinnen J. V., Brusselmans K., Verhoeven G. 2006. Increased lipogenesis in cancer cells: new players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care. 9: 358–365 [DOI] [PubMed] [Google Scholar]
- 15.Menendez J. A., Vellon L., Mehmi I., Oza B. P., Ropero S., Colomer R., Lupu R. 2004. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA. 101: 10715–10720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porstmann T., Griffiths B., Chung Y. L., Delpuech O., Griffiths J. R., Downward J., Schulze A. 2005. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 24: 6465–6481 [DOI] [PubMed] [Google Scholar]
- 17.Swinnen J. V., Van Veldhoven P. P., Timmermans L., De Schrijver E., Brusselmans K., Vanderhoydonc F., Van de Sande T., Heemers H., Heyns W., Verhoeven G. 2003. Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem. Biophys. Res. Commun. 302: 898–903 [DOI] [PubMed] [Google Scholar]
- 18.Glunde K., Ackerstaff E., Mori N., Jacobs M. A., Bhujwalla Z. M. 2006. Choline phospholipid metabolism in cancer: consequences for molecular pharmaceutical interventions. Mol. Pharm. 3: 496–506 [DOI] [PubMed] [Google Scholar]
- 19.Ridgway N. D. 2013. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 48: 20–38 [DOI] [PubMed] [Google Scholar]
- 20.Geilen C. C., Wieder T., Boremski S., Wieprecht M., Orfanos C. E. 1996. c-Ha-ras oncogene expression increases choline uptake, CTP: phosphocholine cytidylyltransferase activity and phosphatidylcholine biosynthesis in the immortalized human keratinocyte cell line HaCaT. Biochim. Biophys. Acta. 1299: 299–305 [DOI] [PubMed] [Google Scholar]
- 21.Arsenault D. J., Yoo B. H., Rosen K. V., Ridgway N. D. 2013. ras-Induced up-regulation of CTP:phosphocholine cytidylyltransferase alpha contributes to malignant transformation of intestinal epithelial cells. J. Biol. Chem. 288: 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teegarden D., Taparowsky E. J., Kent C. 1990. Altered phosphatidylcholine metabolism in C3H10T1/2 cells transfected with the Harvey-ras oncogene. J. Biol. Chem. 265: 6042–6047 [PubMed] [Google Scholar]
- 23.Momchilova A., Markovska T., Pankov R. 1999. Ha-ras-transformation alters the metabolism of phosphatidylethanolamine and phosphatidylcholine in NIH 3T3 fibroblasts. Cell Biol. Int. 23: 603–610 [DOI] [PubMed] [Google Scholar]
- 24.Larrodera P., Cornet M. E., Diaz-Meco M. T., Lopez-Barahona M., Diaz-Laviada I., Guddal P. H., Johansen T., Moscat J. 1990. Phospholipase C-mediated hydrolysis of phosphatidylcholine is an important step in PDGF-stimulated DNA synthesis. Cell. 61: 1113–1120 [DOI] [PubMed] [Google Scholar]
- 25.Bjørkøy G., Overvatn A., Diaz-Meco M. T., Moscat J., Johansen T. 1995. Evidence for a bifurcation of the mitogenic signaling pathway activated by Ras and phosphatidylcholine-hydrolyzing phospholipase C. J. Biol. Chem. 270: 21299–21306 [DOI] [PubMed] [Google Scholar]
- 26.Martin A., Duffy P. A., Liossis C., Gomez-Munoz A., O'Brien L., Stone J. C., Brindley D. N. 1997. Increased concentrations of phosphatidate, diacylglycerol and ceramide in ras- and tyrosine kinase (fps)-transformed fibroblasts. Oncogene. 14: 1571–1580 [DOI] [PubMed] [Google Scholar]
- 27.Lagace T. A., Miller J. R., Ridgway N. D. 2002. Caspase processing and nuclear export of CTP:phosphocholine cytidylyltransferase alpha during farnesol-induced apoptosis. Mol. Cell. Biol. 22: 4851–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagace T. A., Ridgway N. D. 2005. Induction of apoptosis by lipophilic activators of CTP:phosphocholine cytidylyltransferase alpha (CCTalpha). Biochem. J. 392: 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miquel K., Pradines A., Terce F., Selmi S., Favre G. 1998. Competitive inhibition of choline phosphotransferase by geranylgeraniol and farnesol inhibits phosphatidylcholine synthesis and induces apoptosis in human lung adenocarcinoma A549 cells. J. Biol. Chem. 273: 26179–26186 [DOI] [PubMed] [Google Scholar]
- 30.Anthony M. L., Zhao M., Brindle K. M. 1999. Inhibition of phosphatidylcholine biosynthesis following induction of apoptosis in HL-60 cells. J. Biol. Chem. 274: 19686–19692 [DOI] [PubMed] [Google Scholar]
- 31.Voziyan P. A., Goldner C. M., Melnykovych G. 1993. Farnesol inhibits phosphatidylcholine biosynthesis in cultured cells by decreasing cholinephosphotransferase activity. Biochem. J. 295: 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright M. M., Henneberry A. L., Lagace T. A., Ridgway N. D., McMaster C. R. 2001. Uncoupling farnesol-induced apoptosis from its inhibition of phosphatidylcholine synthesis. J. Biol. Chem. 276: 25254–25261 [DOI] [PubMed] [Google Scholar]
- 33.Storey M. K., Byers D. M., Cook H. W., Ridgway N. D. 1997. Decreased phosphatidylcholine biosynthesis and abnormal distribution of CTP:phosphocholine cytidylyltransferase in cholesterol auxotrophic Chinese hamster ovary cells. J. Lipid Res. 38: 711–722 [PubMed] [Google Scholar]
- 34.Cornell R., Vance D. E. 1987. Binding of CTP: phosphocholine cytidylyltransferase to large unilamellar vesicles. Biochim. Biophys. Acta. 919: 37–48 [DOI] [PubMed] [Google Scholar]
- 35.Okuda T., Okamura M., Kaitsuka C., Haga T., Gurwitz D. 2002. Single nucleotide polymorphism of the human high affinity choline transporter alters transport rate. J. Biol. Chem. 277: 45315–45322 [DOI] [PubMed] [Google Scholar]
- 36.Yuan Z., Wagner L., Poloumienko A., Bakovic M. 2004. Identification and expression of a mouse muscle-specific CTL1 gene. Gene. 341: 305–312 [DOI] [PubMed] [Google Scholar]
- 37.McStay G. P., Salvesen G. S., Green D. R. 2008. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 15: 322–331 [DOI] [PubMed] [Google Scholar]
- 38.Henderson F. C., Miakotina O. L., Mallampalli R. K. 2006. Proapoptotic effects of P. aeruginosa involve inhibition of surfactant phosphatidylcholine synthesis. J. Lipid Res. 47: 2314–2324 [DOI] [PubMed] [Google Scholar]
- 39.Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., et al. 1997. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272: 17907–17911 [DOI] [PubMed] [Google Scholar]
- 40.Orth K., Chinnaiyan A. M., Garg M., Froelich C. J., Dixit V. M. 1996. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J. Biol. Chem. 271: 16443–16446 [PubMed] [Google Scholar]
- 41.Tang D., Lahti J. M., Kidd V. J. 2000. Caspase-8 activation and bid cleavage contribute to MCF7 cellular execution in a caspase-3-dependent manner during staurosporine-mediated apoptosis. J. Biol. Chem. 275: 9303–9307 [DOI] [PubMed] [Google Scholar]
- 42.Del Bello B., Valentini M. A., Comporti M., Maellaro E. 2003. Cisplatin-induced apoptosis in melanoma cells: role of caspase-3 and caspase-7 in Apaf-1 proteolytic cleavage and in execution of the degradative phases. Ann. N. Y. Acad. Sci. 1010: 200–204 [DOI] [PubMed] [Google Scholar]
- 43.Germain M., Affar E. B., D'Amours D., Dixit V. M., Salvesen G. S., Poirier G. G. 1999. Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J. Biol. Chem. 274: 28379–28384 [DOI] [PubMed] [Google Scholar]
- 44.Slee E. A., Adrain C., Martin S. J. 2001. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276: 7320–7326 [DOI] [PubMed] [Google Scholar]
- 45.Walsh J. G., Cullen S. P., Sheridan C., Luthi A. U., Gerner C., Martin S. J. 2008. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. USA. 105: 12815–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jänicke R. U., Sprengart M. L., Wati M. R., Porter A. G. 1998. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273: 9357–9360 [DOI] [PubMed] [Google Scholar]
- 47.Yang X. H., Sladek T. L., Liu X., Butler B. R., Froelich C. J., Thor A. D. 2001. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 61: 348–354 [PubMed] [Google Scholar]
- 48.Gallego-Ortega D., Ramirez de Molina A., Ramos M. A., Valdes-Mora F., Barderas M. G., Sarmentero-Estrada J., Lacal J. C. 2009. Differential role of human choline kinase alpha and beta enzymes in lipid metabolism: implications in cancer onset and treatment. PLoS ONE. 4: e7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel V., Yuan Z., Ramsubir S., Bakovic M. 2006. Choline transport for phospholipid synthesis. Exp. Biol. Med. (Maywood). 231: 490–504 [DOI] [PubMed] [Google Scholar]
- 50.Eliyahu G., Kreizman T., Degani H. 2007. Phosphocholine as a biomarker of breast cancer: molecular and biochemical studies. Int. J. Cancer. 120: 1721–1730 [DOI] [PubMed] [Google Scholar]
- 51.O'Regan S., Traiffort E., Ruat M., Cha N., Compaore D., Meunier F. M. 2000. An electric lobe suppressor for a yeast choline transport mutation belongs to a new family of transporter-like proteins. Proc. Natl. Acad. Sci. USA. 97: 1835–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui Z., Houweling M. 2002. Phosphatidylcholine and cell death. Biochim. Biophys. Acta. 1585: 87–96 [DOI] [PubMed] [Google Scholar]
- 53.Atsumi G., Murakami M., Kojima K., Hadano A., Tajima M., Kudo I. 2000. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. Proteolytic fragment of type IVA cytosolic phospholipase A2alpha inhibits stimulus-induced arachidonate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J. Biol. Chem. 275: 18248–18258 [DOI] [PubMed] [Google Scholar]
- 54.Mueller R. B., Sheriff A., Gaipl U. S., Wesselborg S., Lauber K. 2007. Attraction of phagocytes by apoptotic cells is mediated by lysophosphatidylcholine. Autoimmunity. 40: 342–344 [DOI] [PubMed] [Google Scholar]
- 55.Gehrig K., Cornell R. B., Ridgway N. D. 2008. Expansion of the nucleoplasmic reticulum requires the coordinated activity of lamins and CTP:phosphocholine cytidylyltransferase alpha. Mol. Biol. Cell. 19: 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guerrero A. D., Chen M., Wang J. 2008. Delineation of the caspase-9 signaling cascade. Apoptosis. 13: 177–186 [DOI] [PubMed] [Google Scholar]
- 57.Lakhani S. A., Masud A., Kuida K., Porter G. A., Jr, Booth C. J., Mehal W. Z., Inayat I., Flavell R. A. 2006. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 311: 847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wright K. M., Vaughn A. E., Deshmukh M. 2007. Apoptosome dependent caspase-3 activation pathway is non-redundant and necessary for apoptosis in sympathetic neurons. Cell Death Differ. 14: 625–633 [DOI] [PubMed] [Google Scholar]
- 59.Gehrig K., Morton C. C., Ridgway N. D. 2009. Nuclear export of the rate-limiting enzyme in phosphatidylcholine synthesis is mediated by its membrane binding domain. J. Lipid Res. 50: 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yalcin A., Clem B., Makoni S., Clem A., Nelson K., Thornburg J., Siow D., Lane A. N., Brock S. E., Goswami U., et al. 2010. Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene. 29: 139–149 [DOI] [PubMed] [Google Scholar]
- 61.Rodríguez-González A., Ramírez de Molina A., Fernández F., Ramos M. A., del Carmen Núñez M., Campos J., Lacal J. C. 2003. Inhibition of choline kinase as a specific cytotoxic strategy in oncogene-transformed cells. Oncogene. 22: 8803–8812 [DOI] [PubMed] [Google Scholar]
- 62.Glunde K., Raman V., Mori N., Bhujwalla Z. M. 2005. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res. 65: 11034–11043 [DOI] [PubMed] [Google Scholar]