

Abstract
Background. Although data from rodent systems are extremely useful in providing insights into possible mechanisms of age-related bone loss, concepts evolving from animal models need to ultimately be tested in humans.
Methods. This review provides an update on mechanisms of age-related bone loss in humans based on the author’s knowledge of the field and focused literature reviews.
Results. Novel imaging, experimental models, biomarkers, and analytic techniques applied directly to human studies are providing new insights into the patterns of bone mass acquisition and loss as well as the role of sex steroids, in particular estrogen, on bone metabolism and bone loss with aging in women and men. These studies have identified the onset of trabecular bone loss at multiple sites that begins in young adulthood and remains unexplained, at least based on current paradigms of the mechanisms of bone loss. In addition, estrogen appears to be a major regulator of bone metabolism not only in women but also in men. Studies assessing mechanisms of estrogen action on bone in humans have identified effects of estrogen on RANKL expression by several different cell types in the bone microenvironment, a role for TNF-α and IL-1β in mediating effects of estrogen deficiency on bone, and possible regulation of the Wnt inhibitor, sclerostin, by estrogen.
Conclusions. There have been considerable advances in our understanding of age-related bone loss in humans. However, there are also significant gaps in knowledge, particularly in defining cell autonomous changes in bone in human studies to test or validate concepts emerging from studies in rodents.
Decision Editor: Luigi Ferrucci, MD, PhD
Key Words: Osteoporosis, Bone, Estrogen, Aging.
There has been considerable progress in recent years in our understanding of both the pattern and underlying pathogenesis of age-related bone loss in humans. Because fracture risk in elderly individuals depends, in part, on the amount of bone acquired during growth, there is also increasing attention focused on the determinants of peak bone mass acquisition. This review provides an update, based on human studies, on our understanding of both the gain and the loss of bone mass with aging and the potential underlying mechanisms mediating age-related bone loss.
Acquisition of Peak Bone Mass
It is clear that peak bone mass attained during childhood and adolescent growth is a major determinant of bone mass later in life. For example, Cooper and coworkers (1) used the Finnish national databases to link birth and childhood growth data to later hospital discharge records for hip fracture in ~7,000 women and men born at the Helsinki University Central Hospital during 1924–1933. Body size at birth, along with 10 measurements of height and weight throughout childhood, was recorded for each subject. Following adjustment for age and gender, a low rate of childhood growth (height, p = .006; weight, p = .01) was a significant determinant of hip fracture risk in late life. Additional analyses demonstrated that in boys, there was a constant deficit in height and weight between ages 7 and 15 years among those who sustained hip fractures later; in girls, there was a progressively increasing deficit in weight but a delayed height gain among those who later sustained fractures.
Skeletal mass increases steadily during childhood through linear growth and changes in bone density and dimensions. The pubertal growth spurt, however, is associated with a marked acceleration in bone mass acquisition, with 25%–50% of the peak bone mass of adulthood accumulated during this period. Interestingly, the pubertal growth spurt is also associated with a marked increase in the incidence of fractures during childhood, most notably fractures of the distal forearm (2– 4). In order to better understand the structural basis for this rise in distal forearm fractures during adolescence, we studied healthy 6–21-year-old girls and boys using high-resolution peripheral quantitative computed tomography (HRpQCT) (5). At the distal radius, trabecular parameters (bone volume fraction, trabecular number and thickness) did not change through puberty in girls, but increased in boys from late puberty onward. Cortical thickness and density decreased from pre- to mid-puberty in girls, but were unchanged in boys, before rising to higher levels at the end of puberty in both sexes. Total bone strength, assessed using microfinite element models, increased linearly across bone age groups in both sexes, with boys showing greater bone strength than girls after mid-puberty. Interestingly, the proportion of load borne by cortical bone, and the ratio of cortical to trabecular bone volume, decreased transiently during mid- to late-puberty, with apparent cortical porosity peaking during this time. These findings thus indicated that regional deficits in cortical bone during growth may underlie the adolescent peak in forearm fractures.
In further studies, we also examined the relationship of these changes in bone microstructure during growth to serum sclerostin levels (6). Sclerostin, produced by osteocytes, is a potent inhibitor of Wnt signaling and bone formation. Serum sclerostin levels were higher in boys as compared to girls and declined in both sexes following the onset of puberty. There was no consistent relationship between sclerostin levels and trabecular bone parameters in either sex. However, serum sclerostin levels were inversely associated with cortical volumetric bone mineral density and cortical thickness in girls and positively associated with the cortical porosity index in both girls and boys. Thus, sclerostin production during growth may play an important role in defining cortical structure.
To summarize, much of adult bone mass is achieved during childhood and particularly during adolescent growth. During this period, there is also a reproducible (across multiple populations) increase in fractures, predominantly of the distal radius, which appears to be due to transient decreases in cortical thickness and increase in cortical porosity (5), at least at this site. In addition, sclerostin, produced by osteocytes, may be an important regulator of cortical microstructure during growth.
Patterns of Bone Loss in Women and Men
Although dual-energy x-ray absorptiometry (DXA) is a very useful clinical tool, it does have significant limitations in the research setting. Specifically, it cannot separate the more metabolically active trabecular bone from the more structurally important cortical bone. Thus, recent studies have used quantitative computed tomography (QCT), along with newer image analysis tools, to assess age- and sex-specific changes in volumetric bone mineral density (vBMD, a true three-dimensional density), bone size, geometry, and structure at various skeletal sites. In one such study, we used central (at the lumbar spine and femoral neck) and peripheral (at the distal radius and tibia) QCT in an age- and sex-stratified population sample of 373 women and 323 men (age, 20–97 years) and found that in young adulthood, men had 35%–42% larger bone areas than women, consistent with their larger body size (7). Bone area increased in both sexes over life by ~15%, consistent with ongoing apposition of periosteal bone during adult life. While cortical vBMD at multiple sites remained stable until mid-life and then decreased in both sexes (Figure 1A), decreases in trabecular vBMD began in young adulthood and continued throughout life (Figure 1B). Cortical vBMD decreased over life more in women (~25%) than in men (~18%), consistent with menopausal-induced increases in bone turnover and bone porosity. Average decreases in trabecular vBMD between the ages of 20 and 90 years were greater in women (−55%) than in men (−46%) at the central sites, but were similar (−24% and −26%) at peripheral sites.
Figure 1.
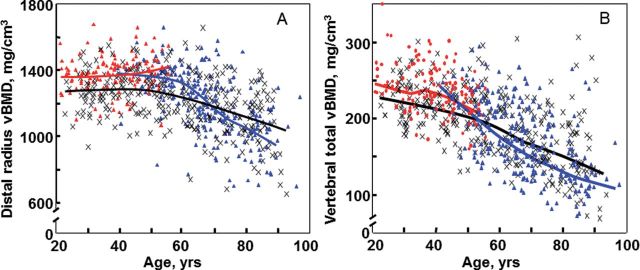
(A) Values for vBMD (mg/cm3) of cortical bone at the distal radius in a population sample of Rochester, Minnesota women and men between the ages of 20 and 97 years. Individual values and smoother lines are given for premenopausal women in red, for postmenopausal women in blue, and for men in black. (B) Values for vBMD of the total vertebral body in the same cohort. Color code is as in Panel (A). All changes with age were significant (p < .05). Reproduced from Riggs and coworkers (7), with permission.
These cross-sectional data were subsequently confirmed by a longitudinal analysis of the same cohort (8). For this, vBMD of trabecular and cortical bone was measured annually for 3 years at the distal radius and distal tibia, and trabecular vBMD was measured at baseline and 3 years at the lumbar spine. Consistent with the cross-sectional findings, substantial cortical bone loss began in middle life in women but began mainly after age 70–75 years in men. By contrast, substantial trabecular bone loss began in young adult women and men at all three skeletal sites and continued throughout life with acceleration during perimenopause in women. Women experienced 37% and men experienced 42% of their total lifetime trabecular bone loss before age 50 years compared with 6% and 15%, respectively, for cortical bone. Taken together, these cross-sectional and longitudinal data indicated that the late onset of cortical bone loss is temporally associated with sex steroid deficiency, but the early-onset, substantial trabecular bone loss in both sexes during sex steroid sufficiency is unexplained.
The recent application of HRpQCT at the distal radius and tibia has also provided important new information on changes in trabecular and cortical microstructure with aging. Using HRpQCT at the wrist in a population-based cross-sectional study involving 324 women and 278 men of age 21–97 years (9), we found that relative to young women (age 20–29 years), young men had greater trabecular bone volume/tissue volume (by 26%) and trabecular thickness (by 28%) but similar values for trabecular number and trabecular separation. Between ages 20 and 90 years, cross-sectional decreases in trabecular bone volume/tissue volume were similar in women (−27%) and in men (−26%), but whereas women had significant decreases in trabecular number (−13%) and increases in trabecular separation (+24%), these parameters had little net change over life in men. However, trabecular thickness decreased to a greater extent in men (−24%) than in women (−18%). These population-based structural data thus demonstrated that although decreases in trabecular bone volume/tissue volume with age are similar in men and women, the structural basis for this decrease is quite different between the sexes. Over life, women undergo loss of trabeculae with an increase in trabecular separation, whereas men begin young adult life with thicker trabeculae and primarily sustain trabecular thinning, with no net change in trabecular number or trabecular separation. This has important biomechanical consequences, because decreases in trabecular number have been shown to have a much greater impact on bone strength as compared with decreases in trabecular thickness (10). These findings may help explain the lower life-long risk of fractures in men and specifically their virtual immunity to age-related increases in distal forearm fractures.
Additional studies utilized the power of HRpQCT to define the possible BMD-independent effects of age on bone fragility (11). Thus, although previous studies using DXA have demonstrated that age is a major predictor of bone fragility and fracture risk independent of BMD (12), this BMD-independent effect of age on bone “quality” remains poorly defined. To address this issue, 44 women < 50 years old (mean age, 41.0 years) were matched to 44 women ≥ 50 years old (mean age, 62.7 years) by ultradistal radius BMD, and 57 men < 50 years (mean age, 41.3 years) were matched to 57 men ≥ 50 years (mean age, 68.1 years). In these subjects matched for BMD by DXA, there were no sex-specific differences in trabecular microstructural parameters. However, older women and men had significant increases in cortical porosity, total cortical pore volume, and mean cortical pore diameter, as demonstrated by representative images from young and elderly women and men matched for DXA BMD (Figure 2). These findings thus indicated that younger and older women and men matched for DXA BMD have similar trabecular microstructure but clearly different cortical microstructure, at least at an appendicular site represented by the radius.
Figure 2.

Representative cross-sectional images from HRpQCT scans from younger and older women (top panels) and younger and older men (bottom panels) with similar UD radius vBMD values (within 3% of each other) but marked differences in cortical porosity. Reproduced from Nicks and coworkers (11), with permission.
To summarize, recent data using QCT have demonstrated that trabecular bone mass seems to “peak” in early adult life, with decreases in trabecular bone evident in both sexes as early as the third decade, although these decreases clearly accelerate around the time of the menopause in women. By contrast, cortical bone remains stable in both sexes until the menopause in women and somewhat later in life in men, with subsequent decreases in cortical bone present in both sexes. At a microstructural level, women lose bone primarily via decreases in trabecular numbers (ie, complete loss of trabeculae), whereas men principally undergo trabecular thinning; the former is structurally much more destabilizing, and may account, at least, in part, for the higher lifetime risk of fractures in women as compared to men. In addition, age-related increases in cortical porosity, which are not evident using DXA, may contribute significantly to the effect of age on fracture risk independent of BMD.
Role of Menopause in Women and Sex Steroid Deficiency in Men
Role of Menopause and Estrogen Deficiency in Women
Fuller Albright (13) demonstrated almost 70 years ago that osteoporosis in women was related to estrogen deficiency and that estrogen treatment improved calcium balance in postmenopausal women. Using photon absorptiometry at the metacarpals, Lindsay and coworkers subsequently validated the original Albright hypothesis by demonstrating that the accelerated bone loss induced by ovariectomy in women could be prevented by estrogen therapy (14). Subsequent work has now clearly demonstrated the importance of the menopause and estrogen deficiency as perhaps the major contributor to age-related bone loss in women. The observed decrease in bone mass at multiple sites is associated with marked increases in biochemical markers of bone resorption, whereas markers of bone formation increase to a lesser extent, consistent with increased bone resorption as well as a relative deficit in bone formation in the setting of estrogen deficiency that leads to bone loss.
Because the final effector system regulating the differentiation and lifespan of osteoclasts is the RANKL/OPG/RANK system (reviewed in detail in [15]), there has been considerable interest in defining possible direct or indirect effects of estrogen on the components of this system. To assess this in humans, our group (16) isolated bone marrow mononuclear cells expressing RANKL on their surfaces by two-color flow cytometry using FITC-conjugated OPG-Rc as a probe. The cells were characterized as preosteoblastic marrow stromal cells (MSCs), B lymphocytes, or T lymphocytes by using antibodies against alkaline phosphatase, CD20, and CD3, respectively, in 12 premenopausal women (Group A), 12 early postmenopausal women (Group B), and 12 age-matched, estrogen-treated postmenopausal women (Group C). We found that the fluorescence intensity of OPG-Fc-FITC, an index of the surface concentration of RANKL per cell, was increased in Group B over Groups A and C by twofold to threefold for MSCs, B cells, T cells, and total RANKL-expressing cells. Moreover, in the merged groups, RANKL expression per cell correlated directly with the bone resorption markers, serum C-telopeptide of type I collagen and urine N-telopeptide of type I collagen, in all three cell types and inversely with serum estradiol levels for total RANKL-expressing cells. These data thus demonstrated that upregulation of RANKL on bone marrow cells is an important determinant of increased bone resorption induced by estrogen deficiency. In a subsequent study, Taxel and colleagues (17) used a specific antibody to RANKL and flow cytometry to demonstrate that, compared with premenopausal women, postmenopausal women had more than threefold greater percentage of bone marrow cells expressing RANKL. Moreover, treatment of the postmenopausal women with estrogen reduced the percentage of RANKL-expressing cells to those present in premenopausal women. Collectively, these studies provide evidence that estrogen deficiency is associated with increased RANKL production and an expansion in RANKL-expressing cells in the bone microenvironment. However, given the very recent evidence from rodent studies that osteocytes are the major source of RANKL in bone (18,19), further studies in humans are needed to evaluate the effects of estrogen on osteocytic RANKL production.
Estrogen also appears to have effects on RANK signaling in osteoclast precursors (20), although it does not seem to affect the concentration of RANK on the cell surface. Thus, in studies in postmenopausal women, although estrogen treatment decreased the proportion of bone marrow mononuclear cells expressing the late osteoclast phenotype marker, the calcitonin receptor, there were no significant effects of estrogen on cell surface expression of RANK, TNF receptor 1, or receptors for the osteoclast costimulatory molecules, TREM2 or OSCAR (21).
Despite the evidence noted earlier for effects of estrogen on the RANKL/OPG/RANK system, it remains unclear what proportion of these effects are direct versus indirect effects of estrogen. For example, RANKL production is upregulated in osteoblastic cells by TNF-α, IL-1β, IL-11, and PGE2 (22,23) and is downregulated by TGF-β (24). Thus, estrogen regulation of RANKL and, in turn, bone resorption could be indirectly mediated via suppression of one or more of these cytokines. In addition, a number of these cytokines act upstream of RANKL in expanding the pool of preosteoclastic cells (25). Perhaps the most definitive evidence supporting a role for TNF-α and IL-1β in mediating estrogen deficiency bone loss in vivo in humans comes from a study in which transdermal estradiol was administered to 42 early postmenopausal women for 60 days to suppress bone resorption, following which estrogen treatment was then discontinued, and the subjects were randomly assigned to intervention groups receiving 3 weeks of injections with saline, the IL-1 receptor 1 blocker, anakinra, or the soluble p75 TNF receptor (which binds and thereby inhibits TNF action), etanercept (26). As shown in Figure 3, either IL-1 or TNF-α blockade reduced the estrogen deficiency-induced increase in bone resorption by ~50%, although TNF-α blockade appeared to be more effective (due to potential toxicity, both blockers could not be administered simultaneously). These data in humans are thus consistent with considerable work in rodents demonstrating important roles for these cytokines in mediating bone loss following estrogen deficiency (27).
Figure 3.
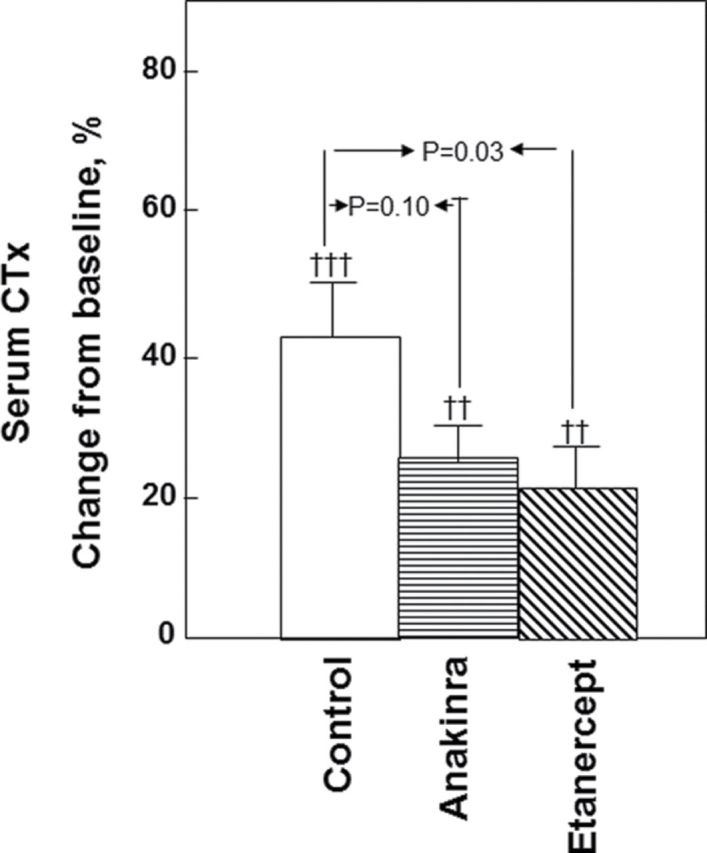
Proportional change (%) in serum C-telopeptide of type I collagen in postmenopausal women treated for 60 days with transdermal estradiol, made acutely estrogen deficient, and treated with saline (control), an IL-1 blocker (anakinra), or a TNF blocker (etanercept). From Charatcharoenwitthaya and coworkers (26), with permission.
In addition to the effects of estrogen in regulating bone resorption, there is considerable interest in defining possible effects of estrogen in humans on maintaining bone formation. The strongest evidence for estrogen effects on bone formation come from studies of acute estrogen deprivation, where bone formation markers decrease significantly (26). Conversely, Hannon and colleagues (28) have shown that although chronic estrogen treatment is associated with reductions in bone resorption and formation markers, early after estrogen treatment, bone resorption markers fall, whereas bone formation markers actually increase. Collectively, these data in women, which are further supported by studies in men (see later), provide convincing evidence that estrogen not only suppresses bone resorption but is also critical for the maintenance of bone formation. Following estrogen deficiency, bone resorption increases and, due to the coupling of bone resorption and bone formation (29), bone formation also increases over time; however, due to the absence of estrogen, there is a persistent gap between bone resorption and bone formation, leading to the observed bone loss.
In order to assess the possible direct effects of estrogen on osteoblastic cells in vivo in humans, we recently assess the effects of 4 months of transdermal estradiol treatment (0.05mg/day) of postmenopausal women as compared to no treatment on the expression of genes in prespecified pathways in freshly isolated bone marrow osteoprogenitor cells (hematopoietic lineage [lin]-/Stro-1+) (30). We also evaluated whether estrogen treatment modulated peripheral blood or bone marrow plasma levels of the Wnt antagonist, sclerostin (31). Estrogen treatment led to a significant decrease in the expression of several proliferation markers (cyclin B1, cyclin E1, E2F1) and increase in adhesion molecules (N-cadherin) in bone marrow lin-/Stro1+ cells as compared to cells obtained from untreated women. Interestingly, sclerostin levels were significantly lower in the estrogen-treated women as compared to the control women both in peripheral serum (by 32%) and in bone marrow plasma (by 34%). Thus, consistent with previous studies in mice (32), estrogen suppresses the proliferation of human bone marrow lin-/Stro1+ cells, which likely represent early osteoprogenitor cells. Further animal and human studies are needed to define the role of the changes we observed in mRNAs for adhesion molecules in these cells and in local sclerostin production in bone in mediating the effects of estrogen on bone metabolism in humans.
Although menopausal bone loss is clearly related to estrogen deficiency, there are a number of additional hormonal changes during the menopause that may contribute to bone loss. Thus, the observation that early menopausal bone loss begins even when serum estradiol levels are normal led to the hypothesis a number of years ago by Prior and colleagues (33) that luteal phase defects and reductions in progesterone levels during perimenopause contribute to bone loss during this period. In addition to progesterone, androgen levels also decrease during the menopausal transition (34) and could contribute to bone loss. Recently, Perrien and colleagues (35) demonstrated marked reductions in serum inhibins (A and B) during the menopause in women and found that the decreases in inhibin levels were associated with increases in bone turnover markers. In addition, Ebeling and colleagues (36) have found that increases in bone resorption markers in perimenopausal women were best correlated not with serum estradiol levels but rather with follicle stimulating hormone (FSH) levels. Subsequent data from the Study of Women’s Health Across the Nation showed that spine and hip vBMD losses during the menopause transition were most strongly related to the interaction between initial FSH levels and longitudinal FSH changes and not to estradiol or androgen levels (37). These findings raised the possibility that FSH may have direct effects on bone; however, as noted by the authors of that study (37), FSH could also be a better predictor of BMD changes during the perimenopause than estradiol because it may serve as a more robust proxy measure of ovarian dynamics involving estradiol than single estradiol measurements. In rodent studies, Sun and coworkers examined the skeletal phenotype of FSH receptor null (FORKO) mice and found that despite being hypogonadal, these mice had normal bone mass (38). These investigators also found that osteoclasts and their precursors possessed FSH receptors and that FSH (but not luteinizing hormone) increased osteoclast formation and function in vitro and they concluded that high circulating FSH levels caused hypogonadal bone loss (38). By contrast, Gao and colleagues (39) subsequently found that the FORKO mice did have reduced bone mass; moreover, bilateral ovariectomy reduced the elevated circulating testosterone levels in the FORKO mice and decreased bone mass to levels indistinguishable from those in ovariectomized controls. These investigators came to the opposite conclusions from that of Sun and coworkers (38), namely, that sex steroids regulated bone turnover in the FORKO mice independently of any bone resorptive action of FSH. Consistent with this, we found that suppression of FSH levels in postmenopausal women into the premenopausal range for 4 months failed to reduce bone resorption markers, indicating that FSH is likely not an important regulator of bone resorption in humans (40). Although FSH is unlikely to contribute to postmenopausal bone loss, the precise roles of changes in progesterone, androgen, and inhibin levels in enhancing the effects of estrogen deficiency on bone loss during the perimenopausal period remain to be fully defined.
Role of Sex Steroid Deficiency in Men
Although men do not have the equivalent of the menopause, total testosterone levels do decline with aging (41,42). More importantly, a number of studies have now demonstrated that the biologically available fraction of testosterone and estrogen (ie, the fraction not bound to sex hormone binding globulin) declines markedly with aging in men, due in large part to a near doubling in sex hormone binding globulin levels over life, combined with an inadequate compensatory response by the aging hypothalamic–pituitary–testicular axis to appropriately compensate for the declining bioavailable sex steroid levels (41,42). Thus, in a population-based sample of 350 men between the ages of 20 and 90 years, we found that bioavailable testosterone decreased over life by 64%, bioavailable estrogen by 47%, and sex hormone binding globulin rose by 124% (41).
Although both serum-free or bioavailable testosterone and estradiol levels decline with age in men, it had generally been believed that because testosterone is the major sex steroid in men, it was the decrease in bioavailable testosterone levels that would be associated most closely with bone loss in men. However, Slemenda and colleagues (43) found that BMD (assessed by DXA) at various sites in 93 healthy men more than age 55 years correlated with serum estradiol levels (correlation coefficients, depending on the site, of +0.21 to +0.35, p = .01 to .05) and, in fact, inversely with serum testosterone levels (correlation coefficients of −0.20 to −0.28, p = .03 to .10). Subsequent to this report, other similar cross-sectional studies have demonstrated significant positive associations between BMD by DXA and estrogen levels in men (41,44–49), particularly circulating bioavailable estradiol levels. These cross-sectional findings have subsequently been validated by longitudinal data. Thus, we (50) studied, in a longitudinal manner, elderly (60–90 years) men in whom rates of change in BMD using DXA at various sites over 4 years were related to sex steroid levels. Forearm sites (distal radius and ulna) provided the clearest data, perhaps because of the greater precision of peripheral site measurements as compared with central sites, such as the spine or hip. BMD at the forearm sites declined by 0.49% to 0.66% per year in these men, and these decreases were associated with serum bioavailable estradiol levels more closely than with bioavailable testosterone levels. Moreover, further analysis of the data suggested that there may be a threshold bioavailable estradiol level of approximately 40 pmol/L (11 pg/mL), below which the rate of bone loss in these men clearly was associated with bioavailable estradiol levels. Above this level, there did not appear to be any relationship between the rate of bone loss and bioavailable estradiol levels. In these older men, the bioavailable estradiol level of 40 pmol/L (11 pg/mL) represented the median bioavailable estradiol level in these men and corresponded to a total estradiol level of approximately 114 pmol/L (31 pg/mL), which is close to the middle of the reported normal range for estradiol levels in men (10–50 pg/mL). In further studies using QCT at various sites, we (51) found that in elderly men, bioavailable estradiol was the most consistent predictor of vBMD and some of the geometric variables related to bone size and that the possible “threshold” for skeletal estrogen deficiency was most evident at cortical sites.
Although these studies helped to establish that estrogen levels are associated with skeletal maintenance in men, they could not definitively establish causal relationships. In order to address this issue, we (52) performed a direct interventional study to distinguish between the relative contributions of estrogen versus testosterone in regulating bone resorption and formation in normal elderly men. Endogenous estrogen and testosterone production were suppressed in 59 elderly men using a combination of a long-acting GnRH agonist and an aromatase inhibitor. Physiologic estrogen and testosterone levels were maintained by simultaneously placing the men on estrogen and testosterone patches delivering doses of sex steroids that mimicked circulating estradiol and testosterone levels in this age group. After baseline measurements of bone resorption [urinary deoxypyridinoline and N-telopeptide of type I collagen] and bone formation [serum osteocalcin and amino-terminal propeptide of type I collagen] markers, the subjects were randomized to 1 of 4 groups: Group A (−T, −E) discontinued both the testosterone and estrogen patches; Group B (−T, +E) discontinued the testosterone patch but continued the estrogen patch; Group C (+T, −E) discontinued the estrogen patch but continued the estrogen patch; and Group D (+T, +E) continued both patches. Because gonadal and aromatase blockade was continued throughout the 3-week period, separate effects of estrogen versus testosterone [in the absence of aromatization to estrogen] on bone metabolism could be delineated. We found that bone resorption markers increased significantly in the absence of both hormones and were unchanged in men receiving both hormones. By two-factor ANOVA, estrogen played the major role in preventing the increase in the bone resorption markers, whereas testosterone had no significant effect. By contrast, serum osteocalcin, a bone formation marker, decreased in the absence of both hormones, and both estrogen and testosterone maintained osteocalcin levels. These findings thus demonstrated that in aging men, estrogen is the dominant sex steroid regulating bone resorption, whereas both estrogen and testosterone are important in maintaining bone formation. Using a somewhat different design, Leder and coworkers (53) confirmed an independent effect of testosterone on bone resorption, although the data in the aggregate clearly favor a more prominent effect of estrogen on the control of bone resorption in men.
Cell Autonomous Changes in Bone With Aging and Potential Secondary Causes
In addition to the key effects of sex steroid deficiency in women and in men described earlier, it is clear that there are intrinsic changes in skeletal metabolism that contribute to age-related bone loss (reviewed in [54]). For example, Zhou and coworkers (55) have demonstrated that there are intrinsic effects of age in human marrow stromal cells (hMSCs), which have the potential to differentiate into osteoblasts. These investigators evaluated cultured hMSCs from subjects aged 17–90 years and found that fourfold more human bone hMSCs were positive for senescence-associated β-galactosidase in samples from older as compared to younger subjects. Doubling time of hMSCs was 1.7-fold longer in cells from the older than the younger subjects and correlated with age. In the cells from aged individuals, more cells were apoptotic, and there were age-related increases in expression of p53 and its pathway genes, p21 and BAX. In addition, there was a significant age-related decrease in the generation of osteoblasts both in the Stro-1+ cells and in adherent hMSCs. Collectively, these data indicated that there are intrinsic alterations in hMSCs with aging that include a decrease in proliferation and differentiation, an increase in senescence associated β-galactosidase-positive cells and apoptosis in hMSCs, and an upregulation of the p53 pathway. In addition to these changes, other investigators have described age-related decreases in telomere length, increases in oxidative stress and DNA damage, and creation of an inflammatory microenvironment (perhaps by senescent osteoblastic cells), all contributing to impaired osteoblast function and an age-related impairment in bone formation (Figure 4) (reviewed in [54]).
Figure 4.
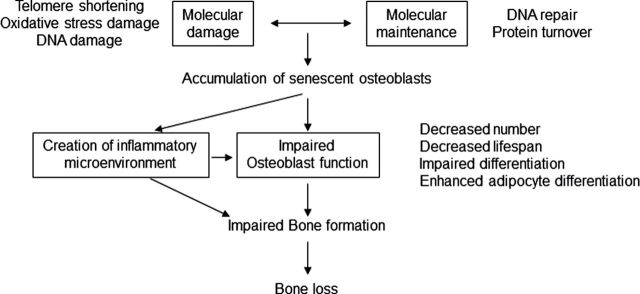
Intrinsic mechanisms that are involved in osteoblast senescence and resulting in decreased bone formation and bone loss associated with aging. Reproduced from Kassem and coworkers (54), with permission.
Given the critical role of Wnt signaling in regulating bone formation and bone mass (31), one of the more intriguing recent findings has been potential changes in sclerostin production with aging. Thus, serum sclerostin levels, which correlate well with bone marrow plasma sclerostin levels (30,56) increase markedly with age in women (by 2.4-fold) and in men (by 4.6-fold) (Figure 5) (57). In addition, for a given total body bone mineral content, elderly subjects (age ≥ 60 years) have higher serum sclerostin levels compared with young subjects (age 20–39 years). These data suggest the hypothesis that with aging, there is increased sclerostin production by individual osteocytes. However, whether increased skeletal sclerostin production with aging explains, at least in part, the known age-related impairment in bone formation (58) warrants further investigation.
Figure 5.

Serum sclerostin levels versus age in (A) women and (B) men. Correlation coefficients are as noted. Reproduced from Mödder and coworkers (57), with permission.
A number of secondary causes of bone loss are often superimposed on the effects of sex steroid deficiency and aging and contribute to fracture risk in specific individuals. As an estimate, these secondary causes may contribute to fracture risk in about 40% of aging men and 20% of women (59). These include various genetic disorders, endocrine diseases, use of certain drugs such as corticosteroids, diseases such as malabsorption, anorexia nervosa, renal hypercalciuria, vitamin D deficiency, and behavioral factors such as smoking, alcohol abuse, and physical inactivity. A detailed description of these is beyond the scope of the present discussion, but this has been previously reviewed (see Lowe and colleagues [60]).
Summary and Conclusions
There is a growing understanding, based directly on studies in humans, of the pathogenesis of age-related bone loss. Clearly, optimizing peak bone mass during growth is critical for minimizing fracture risk late in life. Studies using QCT and HRpQCT have provided novel insights into the differing patterns of bone loss with aging in trabecular versus cortical bone. Sex steroids, particularly estrogen, play a key role in regulating bone metabolism and age-related bone loss in both women and men. Despite these advances, however, there are considerable gaps in knowledge, particularly in defining possible cell autonomous changes in bone in human studies to test or validate evolving concepts from studies in rodents.
Funding
Supported by National Institutes of Health (Grants AG004875 and AR027065).
Acknowledgment
The authors would like to thank James Peterson for help with the figures.
References
- 1. Cooper C, Eriksson JG, Forsen T, Osmond C, Tuomilehto J, Barker DJP. Maternal height, childhood growth and risk of hip fracture in later life: a longitudinal study Osteoporos Int 2001;12:623–629 [DOI] [PubMed] [Google Scholar]
- 2. Landin LA. Fracture patterns in children. Analysis of 8,682 fractures with special reference to incidence, etiology and secular changes in a Swedish urban population 1950–1979 Acta Orthop Scand Suppl 1983;202:1–109 [PubMed] [Google Scholar]
- 3. Kramhoft M, Bodtker S. Epidemiology of distal forearm fractures in Danish children Acta Orthop Scand. 1988;59:557–559 [DOI] [PubMed] [Google Scholar]
- 4. Bailey DA, Wedge JH, McCulloch RG, Martin AD, Bernhardson SC. Epidemiology of fractures of the distal end of the radius in children as associated with growth J Bone Joint Surg Am. 1989;71:1225–1231 [PubMed] [Google Scholar]
- 5. Kirmani S, Christen D, van Lenthe GH, et al. Bone structure at the distal radius during adolescent growth J Bone Miner Res 2009;24:1033–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kirmani S, Amin S, McCready LK, et al. Sclerostin levels during growth in children Osteoporos Int. 2012;23:1123–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riggs BL, Melton LJ, III, Robb RA, et al. Population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites J Bone Miner Res. 2004;19:1945–1954 [DOI] [PubMed] [Google Scholar]
- 8. Riggs BL, Melton LJI, Robb RA, et al. A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men J Bone Miner Res. 2008;23:205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khosla S, Riggs BL, Atkinson EJ, et al. Effects of sex and age on bone microstructure at the ultradistal radius: a population-based noninvasive in vivo assessment J Bone Miner Res. 2006;21:124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Silva MJ, Gibson LJ. Modeling the mechanical behavior of vertebral trabecular bone: effects of age-related changes in microstructure Bone. 1997;21:191–199 [DOI] [PubMed] [Google Scholar]
- 11. Nicks KM, Amin S, Atkinson EJ, Riggs BL, Melton LJI, Khosla S. Relationship of age to bone microstructure independent of areal bone mineral density J Bone Miner Res. 2012;27:637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hui SL, Slemenda W, Johnston CC., Jr. Age and bone mass as predictors of fracture in a prospective study J Clin Invest. 1988;81:1804–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albright F, Smith PH, Richardson AM. Postmenopausal osteoporosis JAMA. 1941;116:2465–2474 [Google Scholar]
- 14. Lindsay R, Aitken JM, Anderson JB, Hart DM, MacDonald EB, Clarke AC. Long-term prevention of postmenopausal osteoporosis by oestrogen: evidence for an increased bone mass after delayed onset of oestrogen treatment Lancet. 1976;1:1038–1041 [DOI] [PubMed] [Google Scholar]
- 15. Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappab ligand and osteoprotegerin regulation of bone remodeling in health and disease Endocr Rev. 2008;29:155–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women J Clin Invest. 2003;111:1221–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taxel P, Kaneko H, Lee S-K, Aguila HL, Raisz LG, Lorenzo JA. Estradiol rapidly inhibits osteoclastogenesis and RANKL expression in bone marrow cultures in postmenopausal women: a pilot study Osteoporos Int. 2008;19:193–199 [DOI] [PubMed] [Google Scholar]
- 18. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’ Brien CA. Matrix-embedded cells control osteoclast formation Nat Med. 2011;17:1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression Nat Med. 2011;17:1231–1234 [DOI] [PubMed] [Google Scholar]
- 20. Shevde NK, Bendixen AC, Dienger KM, Pike JW. Estrogens suppress RANK ligand-induced osteoclast differentiation via a stromal cell independent mechanism involving c-Jun repression Proc Natl Acad Sci U S A. 2000;97:7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clowes JA, Eghbali-Fatourechi GZ, McCready L, Oursler MJ, Khosla S, Riggs BL. Estrogen action on bone marrow osteoclast lineage cells of postmenopausal women in vivo Osteoporos Int. 2009;20:761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yasuda H, Shima N, Nakagawa N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro Endocrinology. 1998;39:1329–1337 [DOI] [PubMed] [Google Scholar]
- 23. Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells Bone. 1999;25(3):255–259 [DOI] [PubMed] [Google Scholar]
- 24. Takai H, Kanematsu M, Yano K, et al. Transforming growth factor-b stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells J Biol Chem. 1998;273:27091–27096 [DOI] [PubMed] [Google Scholar]
- 25. Pacifici R. Estrogen, cytokines, and pathogenesis of postmenopausal osteoporosis J Bone Miner Res. 1996;11:1043–1051 [DOI] [PubMed] [Google Scholar]
- 26. Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL. Effect of blockade of TNF-alpha and interleukin-1 action on bone resorption in early postmenopausal women J Bone Miner Res. 2007;22:724–729 [DOI] [PubMed] [Google Scholar]
- 27. Pfeilschifter J, Koditz R, Pfohl M, Schatz H. Changes in proinflammatory cytokine activity after menopause Endocr Rev. 2002;23:90–119 [DOI] [PubMed] [Google Scholar]
- 28. Hannon R, Blumsohn A, Naylor K, Eastell R. Response of biochemical markers of bone turnover to hormone replacement therapy: impact of biological variability J Bone Miner Res. 1998;13:1124–1133 [DOI] [PubMed] [Google Scholar]
- 29. Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption Trends Mol Med. 2005;11:76–81 [DOI] [PubMed] [Google Scholar]
- 30. Modder UI, Roforth MM, Hoey K, et al. Effects of estrogen on osteoprogenitor cells and cytokines/bone regulatory factors in postmenopausal women Bone. 2011;49:202–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khosla S, Westendorf JJ, Oursler MJ. Building bone to reverse osteoporosis and repair fractures J Clin Invest. 2008;118:421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gregorio GB, Yamamoto M, Ali AA, et al. Attenuation of the self-renewal of transit-amplifying osteoblast progenitors in the murine bone marrow by 17 beta-estradiol J Clin Invest. 2001;107:803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prior JC. Progesterone as a bone-trophic hormone Endocr Rev. 1990;11:386–398 [DOI] [PubMed] [Google Scholar]
- 34. Wierman ME, Basson R, Davis SR, et al. Androgen therapy in women: an endocrine society clinical practice guideline J Clin Endocrinol Metab 2006;91:3697–3710 [DOI] [PubMed] [Google Scholar]
- 35. Perrien DS, Achenbach SJ, Bledsoe SE, Walser B, Suva LJ, Khosla DG. Bone turnover across the menopause transition: correlations with inhibins and follicle-stimulating hormone J Clin Endocrinol Metab. 2006;91:1848–1854 [DOI] [PubMed] [Google Scholar]
- 36. Ebeling PR, Atley LM, Guthrie JR, et al. Bone turnover markers and bone density across the menopausal transition J Clin Endocrinol Metab. 1996;81:3366–3371 [DOI] [PubMed] [Google Scholar]
- 37. Sowers MFR, Jannausch M, McConnell D, et al. Hormone predictors of bone mineral density changes during the menopausal transition J Clin Endocrinol Metab. 2006;91:1261–1267 [DOI] [PubMed] [Google Scholar]
- 38. Sun L, Peng Y, Sharrow AC, et al. / FSH directly regulates bone mass Cell. 2006;125:247–260 [DOI] [PubMed] [Google Scholar]
- 39. Gao J, Tiwari-Pandey R, Samadfam R, et al. Altered ovarian function affects skeletal homeostasis independent of the action of follicle-stimulating hormone (FSH) Endocrinology. 2007;148:2613–2621 [DOI] [PubMed] [Google Scholar]
- 40. Drake MT, McCready L, Hoey KA, Atkinson EJ, Khosla S. Effects of suppression of follicle stimulating hormone secretion on bone resorption markers in postmenopausal women J Clin Endocrinol Metab. 2010;95:5063–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khosla S, Melton LJ, III, Atkinson EJ, O’Fallon WM, Klee GG, Riggs BL. Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen J Clin Endocrinol Metab. 1998;83:2266–2274 [DOI] [PubMed] [Google Scholar]
- 42. Orwoll E, Lambert LC, Marshall LM, et al. Testosterone and estradiol among older men J Clin Endocrinol Metab 2006;91:1336–1344 [DOI] [PubMed] [Google Scholar]
- 43. Slemenda CW, Longcope C, Zhou L, Hui SL, Peacock M, Johnston C. Sex steroids and bone mass in older men: positive associations with serum estrogens and negative associations with androgens J Clin Invest. 1997;100:1755–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greendale GA, Edelstein S, Barrett-Connor E. Endogenous sex steroids and bone mineral density in older women and men: the Rancho Bernardo study J Bone Miner Res. 1997;12:1833–1843 [DOI] [PubMed] [Google Scholar]
- 45. Center JR, Nguyen TV, Sambrook PN, Eisman JA. Hormonal and biochemica parameters in the determination of osteoporosis in elderly men J Clin Endocrinol Metab. 1999;84:3626–3635 [DOI] [PubMed] [Google Scholar]
- 46. Ongphiphadhanakul B, Rajatanavin R, Chanprasertyothin S, Piaseau N, Chailurkit L. Serum oestradiol and oestrogen-receptor gene polymorphism are associated with bone mineral density independently of serum testosterone in normal males Clin Endocrinol. 1998;49:803–809 [DOI] [PubMed] [Google Scholar]
- 47. van den Beld AW, de Jong FH, Grobbee DE, Pols HAP, Lamberts SWJ. Measures of bioavailable serum testosterone and estradiol and their relationships with muscle strength, bone density, and body composition in elderly men J Clin Endocrinol Metab. 2000;85:3276–3282 [DOI] [PubMed] [Google Scholar]
- 48. Amin S, Zhang Y, Sawin CT, et al. Association of hypogonadism and estradiol levels with bone mineral density in elderly men from the Framingham study Ann Intern Med. 2000;133:951–963 [DOI] [PubMed] [Google Scholar]
- 49. Szulc P, Munoz F, Claustrat B, Garnero P, Marchand F. Bioavailable estradiol may be an important determinant of osteoporosis in men: the Minos study J Clin Endocrinol Metab. 2001;86:192–199 [DOI] [PubMed] [Google Scholar]
- 50. Khosla S, Melton LJ, Atkinson EJ, O’Fallon WM. Relationship of serum sex steroid levels to longitudinal changes in bone density in young versus elderly men J Clin Endocrinol Metab. 2001;86:3555–3561 [DOI] [PubMed] [Google Scholar]
- 51. Khosla S, Melton LJ, III, Robb RA, et al. Relationship of volumetric BMD and structural parameters at different skeletal sites to sex steroid levels in men J Bone Miner Res. 2005;20:730–740 [DOI] [PubMed] [Google Scholar]
- 52. Falahati-Nini A, Riggs BL, Atkinson EJ, O’Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men J Clin Invest. 2000;106:1553–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leder BZ, LeBlanc KM, Schoenfeld DA, Eastell R, Finkelstein JS. Differential effects of androgens and estrogens on bone turnover in normal men J Clin Endocrinol Metab. 2003;88:204–210 [DOI] [PubMed] [Google Scholar]
- 54. Kassem M, Marie PJ. Senescence-associated intrinsic mechanisms of osteoblast dysfunctions Aging Cell. 2011;10:191–197 [DOI] [PubMed] [Google Scholar]
- 55. Zhou S, Greenberger JS, Epperly MW, et al. Age-related intrinsic changes in human bone marrow-derived mesenchymal stem cells and their differentiation to osteoblasts Aging Cell 2008;7:335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Drake MT, Srinivasan B, Modder UI, et al. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women J Clin Endocrinol Metab. 2010;95:5056–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Modder UI, Hoey KA, Amin S, et al. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men J Bone Miner Res. 2011;26:373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lips P, Courpron P, Meunier PJ. Mean wall thickness of trabecular bone packets in the human iliac crest: changes with age Calcif Tissue Res. 1978;26:13–17 [DOI] [PubMed] [Google Scholar]
- 59. Riggs BL, Melton LJ. Medical progress series: involutional osteoporosis N Engl J Med. 1986;314:1676–1686 [DOI] [PubMed] [Google Scholar]
- 60. Lowe H, Shane E. Osteoporosis associated with illnesses and medications.In: Marcus R, Feldman D, Nelson DA, Rosen CJ. eds. Osteoporosis San Diego, CA: Elsevier Press; 2008:. II: 1283–1314 [Google Scholar]