

Abstract
Sphenostylisins A–C (1–3), three complex dimeric compounds representing two novel carbon skeletons, along with an additional eight new compounds, sphenostylisins D–K (4–11), were isolated from the active chloroform-soluble extract of the root bark of S. marginata ssp. erecta using a bioactivity-guided isolation approach. The structures were elucidated by means of detailed spectroscopic analysis, including NMR and HRESIMS analysis, with tandem MS fragmentation utilized to further support the structures of 1–3. The absolute configuration of sphenostylisin C (3) was established by electronic circular dichroism analysis. Plausible biogenetic relationships between the modified isoflavonoids 1–11 are proposed, with a cyclization reaction of 9 conducted to support one of the biogenetic proposals made. All these pure isolates were evaluated against a panel of in vitro bioassays, and, among the results obtained, sphenostylisin A (1) was found to be a very potent NF-κB inhibitor (IC50 6 nM).
INTRODUCTION
Sphenostylis marginata E. Mey. ssp. erecta (Baker f.) Verdc. (syn.: Dolichos erectus Baker f., Sphenostylis erecta (Baker f.) Hutch. ex Baker f.; Fabaceae; African yellow pea), is a medicinal plant used as an antiseptic and for the treatment of abdominal pain, diarrhea, edema, and fever.1 In addition, the edible tubers, flowers, and starchy fruits of S. marginata are utilized as a food source in some African countries.2 Although the carbohydrate, amino acid, and protein composition profiles of Sphenostylis species have been studied previously,2 there has been only one reported study of secondary metabolites - four antifungal pterocarpans isolated from the root bark of S. marginata ssp. erecta.3
Scavenging reactive oxygen species by antioxidants and enhancing carcinogen detoxification via induction of phase II enzymes such as quinone reductase (QR) are two important cancer chemopreventive strategies, while inhibition of nuclear factor kappa B (NF-κB) is a promising approach for both cancer chemotherapy and chemoprevention.4,5 In our search for naturally occurring compounds that combat cancer, we found that the chloroform-soluble extract of the root bark of S. marginata ssp. erecta collected in Zimbabwe showed both hydroxyl radical-scavenging and QR-inducing activities. Assays that measured these two activities were used in tandem to guide compound isolation. Herein, we report the isolation and structure elucidation of sphenostylisins A–C (1–3), representative of two novel carbon skeletons, and an additional eight new compounds, sphenostylisins D–K (4–11), as well as the biological evaluation of all isolates obtained using the hydroxyl radical-scavenging, QR-inducing, and NF-κB inhibition assays. Compounds 1–11 were also evaluated for their cytotoxicity against HT-29 human colon cancer cell line.
RESULTS AND DISCUSSION
The methanol extract of the root bark of S. marginata ssp. erecta was suspended in H2O, and then partitioned sequentially with hexanes, CHCl3, EtOAc, and n-BuOH. All the partitions obtained were evaluated in the in vitro hydroxyl radical-scavenging and QR-inducing assays, and the activities were presented as ED50 (test partition concentration scavenging hydroxyl radicals by 50%) and CD (test partition concentration that doubles quinone reductase activity) values, respectively. Among these partitions, the CHCl3 partition exhibited the most potent activity in both the hydroxyl radical-scavenging (ED50 3.3 μg/mL) and QR-inducing (CD 9.6 μg/mL) assays. Therefore, it was selected for further purification and afforded 11 major fractions. Fraction F05 was active in the hydroxyl radical-scavenging and QR-inducing assays, with ED50 and CD values of 1.7 and 5.2 μg/mL, respectively. Accordingly, fraction F05 was subsequently fractionated, leading to the isolation of eleven new modified isoflavonoids, sphenostylisins A–K (1–11), comprising three complex dimeric compounds representative of two novel carbon skeletons, four 3-phenylcoumarins, three deoxybenzoins, and one isoflavone.
The molecular formula of sphenostylisin A (1) was determined as C40H34O10 based on the sodiated molecular ion peak at m/z 697.2035 (calcd 697.2050) in the HRESIMS. The analysis of the 1H, 13C, DEPT, 1H-1H COSY, 1H-13C HSQC and HMBC NMR spectra (Table S1 and Figure S1, Supporting Information) suggested that the molecule of 1 has two moieties (fragments A and B), each including a 15-carbon skeleton with an α,α-dimethylallyl side chain. In the 1H NMR spectrum, fragment A showed five aromatic singlets at δH 7.73 (1H, H-4), 7.52 (1H, H-6′), 7.46 (1H, H-5), 6.77 (1H, H-8), and 6.35 (1H, H-3′), of which H-4 at δH 7.73 was observed as a characteristic proton signal of a 3-phenylcoumarin skeleton. The 1H NMR spectrum of fragment A also showed three hydroxy group singlets at δH 10.53 (1H, OH-7), 10.04 (1H, OH-4′), and 9.85 (1H, OH-2′), while resonances at δH 6.26 (1H, dd, J = 17.5, 10.7 Hz, H-10), 4.97 (1H, br d, J = 10.7 Hz, H-11a), 4.95 (1H, br d, J = 17.5 Hz, H-11b), and 1.48 (6H, s, CH3 × 2, H-12/13) were attributed to an α,α-dimethylallyl side chain. The 13C NMR spectrum of fragment A showed 20 carbon signals, which were classified from the DEPT and HSQC data as two methyl carbons, six quaternary carbons, six tertiary sp2 carbons, one secondary sp2 carbon, four oxygen-bearing tertiary sp2 carbons, and a conjugated lactone carbonyl resonance at δC 159.9. The characteristic NMR data of fragment A were comparable to those of known 3-phenylcoumarins, e.g., licoarylcoumarin, licopyranocoumarin, licofuranocoumarin, and glycycoumarin isolated from Glycyrrhiza (licorice) species.6 The carbon signal at δC 159.9 (C-2), attributed to the conjugated lactone carbonyl based on its correlation with H-4 (δH 7.73) in the HMBC spectrum (Figure 2), combined with the HMBC correlations of H-6′ to C-3 (δC 120.4) and H-4 to C-1′ (δC 114.6), confirmed the presumed 3-phenylcoumarin skeleton. Fragment A showed a non-oxygenated signal at C-5 (δC 126.6), a position at which most known 3-phenylcoumarins isolated from licorice species have a hydroxy or methoxy group. Thus, a long-range correlation in the HMBC spectrum showed that the proton at δH 7.46, attributed to H-5, correlated with C-4 (δC 142.2), C-7 (δC 159.3) and C-8a (δC 153.1), while the proton at δH 6.77 (H-8) correlated with C-4a (δC 111.1) and C-6 (δC 131.6). The α,α-dimethylallyl group was placed at C-6 (δC 131.6), based on the HMBC correlations of H-10, H-12, and H-13 to C-6, and was confirmed by the HMBC cross-peak between H-5 and C-9 (δC 39.9). Another key difference is that the 3-phenyl ring of fragment A only exhibited two singlets at δH 7.52 (H-6′) and 6.35 (H-3′), which, on comparison to the ABX spin system reported for H-6′, H-5′, and H-3′ in the 3-phenyl ring of known 3-phenylcoumarins isolated from licorice species, indicated that C-5′ is the carbon of attachment to fragment B. The assignments of H-6′ and H-3′ were confirmed from the HMBC correlations of H-6′ to C-2′ (δC 157.8) and C-4′ (δC 155.9), and of H-3′ to C-1′ (δC 114.6) and C-5′ (δC 108.4) (Figure 2). When fragment B is considered, observed in the 1H NMR spectrum were signals for three isolated spin systems: a 1,2,4-trisubstituted benzene ring [δH 7.33 (1H, d, J = 8.4 Hz, H-4″), 6.98 (1H, d, J = 1.6 Hz, H-7″), and 6.78 (1H, dd, J = 8.4, 1.6 Hz, H-5″)], a 1,2,4,5-tetrasubstituted benzene ring [δH 7.32 (1H, s, H-14″) and 6.34 (1H, s, H-11″)], and an α,α-dimethylallyl group [δH 5.84 (1H, dd, J = 17.5, 10.7 Hz, H-16″), 4.74 (1H, br d, J = 10.7 Hz, H-17″a), 4.67 (1H, br d, J = 17.5 Hz, H-17″b), and 1.09 (6H, s, CH3 × 2, H-18″/19″)], in addition to three hydroxy group singlets at δH 12.66 (1H, OH-10″), 10.54 (1H, OH-12″), and 9.67 (1H, OH-6″). The 13C NMR spectrum of fragment B showed 20 carbon signals, including two methyls, five quaternary carbons, six tertiary sp2 carbons, one secondary sp2 carbon, five oxygen-bearing tertiary sp2 carbons, and a carbonyl carbon, as classified from the DEPT and HSQC spectra. The NMR data of fragment B were comparable to those of morachalcone B, a chalcone derivative fused with a furan ring isolated from Morus alba.7 Comparison of the 1D- and 2D-NMR spectra with those of morachalcone B revealed a major change in the side chain, with a prenyl group in morachalcone B being replaced by an α,α-dimethylallyl group in fragment B of 1. The location of the α,α-dimethylallyl group was determined to be at C-13″ (δC 125.8), according to the HMBC correlations of H-16″, H-18″, and H-19″ to C-13″. The singlet at δH 7.32 correlated with C-8″ (δC 194.7), C-10″ (δC 162.5), C-12″ (δC 163.2), and C-15″ (δC 39.2), while the other singlet at δH 6.34 correlated with C-9″ (δC 112.9) and C-13″ (δC 125.8) in the HMBC spectrum, and thus were assigned to H-14″ and H-11″, respectively. The ABX spin system at δH 7.33, 6.98, and 6.78 was attributed to H-4″, H-7″, and H-5″ in the benzofuran ring, respectively, based on their splitting pattern and coupling constants, and were confirmed by the HMBC correlations of H-4″ to C-3″ (δC 114.0), C-6″ (δC 155.8), and C-7″a (δC 154.2), of H-7″ to C-3″a (δC 120.1) and C-5″ (δC 112.7), and of H-5″ to C-3″a (δC 120.1) and C-7″ (δC 97.4) (Figure 2). All six hydroxy group proton signals of 1 were also assignable based on their HMBC correlations (Table S1, Supporting Information), which facilitated the assignments of the remaining protons and carbons, and further supported the structure of 1 as shown. Among these hydroxy group protons, the signal of OH-10″ appeared downfield at δH 12.66 due to hydrogen-bonding with the carbonyl group (C-8″, δC 194.7). The connectivity of fragments A and B, determined to be through a carbon-carbon linkage between C-5′ (δC 108.4) and C-2″ (δC 152.2), was confirmed by direct evidence of a key HMBC correlation between H-6′ and C-2″ (Figure 2; Figure S1f, Supporting Information). The structure of 1 elucidated via NMR spectroscopy was supported by the tandem mass spectrum. The highly conjugated structure o.f 1 did not fragment appreciably through carbon-carbon bond cleavage when collision-induced dissociation (CID) was used, so that only one predominant fragment peak from the parent ion (m/z 697.22 [M + Na]+) was observed at m/z 679.22 [M + Na – H2O]+ (Figure S13l, Supporting Information). This may be generated through a proton rearrangement similar to a McLafferty rearrangement, and with cyclization occurring to form a stable six-membered ring and the loss of one water molecule8 (Figure 3). Therefore, the structure of 1 as shown in Figure 1 was elucidated unambiguously. This compound bears a novel carbon skeleton formed through a carbon-carbon bond linkage of a 3-phenylcoumarin skeleton and a 3-arylbenzofuran unit, and was accorded the trivial name sphenostylisin A.
Figure 2.
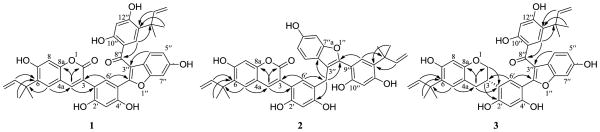
Key HMBC correlations of compounds 1–3 with novel carbon skeletons.
Figure 3.

Tandem MS fragmentation pathway of compounds 1 and 2 with novel carbon skeletons.
Figure 1.

Structures of the new compounds isolated from S. marginata ssp. erecta.
Sphenostylisin B (2) was determined to have a molecular formula of C40H36O9 based on the HRESIMS [M + Na]+ ion peak at m/z 683.2269 (calcd 683.2257), representing one degree of unsaturation less than compound 1. The 1H and 13C NMR spectra of 2 were very similar to those of compound 1. On comparison of the 1H NMR data of these two compounds, an additional methylene resonance appeared at δH 3.71 (2H, s, H-8″) in compound 2. Correspondingly, an additional carbon signal, classified as a methylene carbon from the DEPT NMR spectrum, resonated at δC 22.6 (C-8″) in the 13C NMR spectrum of 2 and showed a cross-peak with H-8″ in the HSQC spectrum, while the carbonyl carbon (δC 194.7) of compound 1 was absent in compound 2. Thus, it was inferred that the carbonyl group of compound 1 is replaced by a methylene group in compound 2. In addition, the HMBC correlation of the proton at δH 7.01 (1H, s, H-14″) to C-2″ (δC 149.3) allowed the establishment of the connectivity of C-9″ (δC 108.1) to C-2″, and supported the presence of a 2-arylbenzofuran skeleton. In turn, the HMBC correlations of H-6′ (δH 6.67) to C-3 (δC 121.4) and H-4 (δH 7.56) to C-1′ (δC 113.7) confirmed the presence of a 3-phenylcoumarin skeleton. These two units were linked through C-5′ and C-3″ by the methylene group (δH 3.71, δC 22.6), which was established on the basis of the key HMBC correlations of H-8″ (δH 3.71) to C-4′ (δC 155.5), C-6′ (δC 131.4), C-2″ (δC 149.3) and C-3″a (δC 121.9) (Figure 2). H-6′ and H-14″ in compound 2 showed upfield shifts of 0.85 and 0.31 ppm, respectively, when compared to compound 1 in which H-6′ and H-14″ are each at a β-position to a double bond conjugated with an electron-withdrawing group, the carbonyl group (C-8″). The absence of this carbonyl group at C-8″ in compound 2 resulted in the lack of inductive deshielding effects on the β-protons. In addition, the spatial arrangement of H-6′ relative to the carbonyl group (C-8″) in compound 1 may also contribute to the anisotropic deshielding effect on H-6′, while there is no such effect in compound 2 since this carbonyl group was absent at C-8″. All other protons and carbons of compound 2 were assigned based on the comparison of the NMR data of compounds 1 and 2, and were confirmed by the detailed analysis of their HMBC correlations (Table S1 and Figure S2, Supporting Information). In the ESIMS/MS of 2, the molecular ion peak at m/z 683.24 [M + Na]+ was dissociated between the methylene group and the 2-arylbenzofuran skeleton through a quinone methide fragmentation9,10 to give two fragments at m/z 373.14 [M + Na – C19H18O4]+ and m/z 333.14 [M + Na – C21H18O5]+, accounting for the most abundant daughter ions (Figure 3; Figure S13m, Supporting Information). This fragmentation pathway is similar to that previously reported for some hydroxyphenylflavanones, in which the fragmentation occurred between the methylene group and the flavanone skeleton.11 Hence, the structure of 2 was established unambiguously as shown in Figure 1. This compound (sphenostylisin B) has a novel carbon skeleton different from that of compound 1, with a 3-phenylcoumarin moiety coupled with a 2-arylbenzofuran unit through a methylene group.
The molecular formula of sphenostylisin C (3) was assigned as C40H38O9 on the basis of the [M + Na]+ ion peak at m/z 685.2404 (calcd 685.2414), indicating two degrees of unsaturation less than that in compound 1. Analysis of the 1D- and 2D-NMR spectra of 3 (Table S1 and Figure S3, Supporting Information), which were closely comparable to those of compound 1, revealed that 3 is also comprised of two fragments (A and B), with each including a 15-carbon skeleton with an α,α-dimethylallyl side chain. The 1H NMR spectrum of fragment A clearly indicated the presence of an ABMXY spin system in the heterocyclic region [δH 4.09 (1H, br d, J = 9.7 Hz, H-2β), 3.77 (1H, t, J = 9.7 Hz, H-2α), 3.25 (1H, m, H-3), 2.87 (1H, dd, J = 15.1, 11.7 Hz, H-4α), and 2.64 (1H, dd, J = 15.1, 3.5 Hz, H-4β)], characteristic of the spin pattern of an isoflavan heterocycle.12,13 This assignment was supported by the 1H-1H COSY correlations between H-2α/H-2β, H-2α/H-3, H-2β/H-3, H-3/H-4α, H-3/H-4β, and H-4α/H-4β, along with the HMBC correlations of H-2α and H-2β to C-3 (δC 31.4), C-4 (δC 29.7), C-8a (δC 152.6), and C-1′ (δC 118.6), of H-3 to C-1′ (δC 118.6), C-2′ (δC 157.8), and C-6′ (δC 128.1), and of H-4α and H-4β to C-2 (δC 69.2), C-5 (δC 127.7), C-8a (δC 152.6), and C-1′ (δC 118.6) (Figure 2). Compared to compound 1, the replacement of the 3-phenylcoumarin skeleton by a similar isoflavan skeleton in compound 3 resulted in the lack of an α,β-unsaturated ketone functional group, and thus accounted for the two less degrees of unsaturation and led to the upfield shifts of H-5 (δH 6.81), H-8 (δH 6.24), and H-6′ (δH 7.23). These proton signals were assigned from the HMBC correlations of H-5 to C-4 (δC 29.7), C-7 (δC 154.5), and C-8a (δC 152.6), of H-8 to C-4a (δC 111.6) and C-6 (δC 126.1), and of H-6′ to C-3 (δC 31.4), C-2′ (δC 157.8), and C-4′ (δC 154.7). The structure of fragment B of compound 3 was elucidated as being the same as that in compound 1 based on the comparison of their 1H and 13C NMR data, and was confirmed from the DEPT, 1H-1H COSY, HSQC, and HMBC data (Table S1 and Figure S3, Supporting Information). According to the key HMBC correlation between H-6′ and C-2″ (Figure 2; Figure S3g, Supporting Information), the connectivity between fragments A and B of compound 3 was established as being the same as that of compound 1. In the tandem mass spectrum of 3, both the protonated and sodiated molecular ion peaks were observed before fragmentation at m/z 663.22 [M + H]+ and 685.21 [M + Na]+, respectively. The protonated molecular ion peak at m/z 663.22 [M + H]+ was fragmented in a more facile manner under the dissociation conditions used, and thus was isolated for MS/MS fragmentation. A fragment peak at m/z 645.21 [M + H – H2O]+ was observed (Figure S13n, Supporting Information), which was produced through loss of one water molecule from the parent ion via the same fragmentation mechanism as that of compound 1. Besides this fragment, two predominant daughter ions occurred at m/z 429.05 [M + H – H2O – C14H16O2]+ and m/z 485.13 [M + H – H2O – C11H12O]+, respectively, and resulted from heterocyclic ring fissions of the isoflavan skeleton.9,14,15 In addition, another fragment arising through retro-Diels-Alder fragmentation was observed at m/z 191.08 [M + H – H2O – C28H22O6]+ (Figure S14, Supporting Information). Thus, the full planar structure of 3 was assigned as shown.
The absolute configuration at C-3 of compound 3 was determined by electronic circular dichroism (ECD) analysis. In addition to the absolute configuration at C-3, the position of the conformational equilibrium of the dihydropyran ring also markedly influences the optical activity of isoflavans,16 and thus analysis of the conformation that the dihydropyran ring may adopt was performed before ECD measurement. The favored conformation of the dihydropyran ring of the isoflavan skeleton is proposed to be the half-chair based on the minimization of torsional strain as established previously,12,16,17 and thus results in four possible conformers, the (3S)-eq-conformer, the (3S)-ax-conformer, the (3R)-eq-conformer, and the (3R)-ax-conformer. The coupling pattern of the dihydropyran ring observed in the 1H NMR spectrum of 3 showed that H-3 exhibited much larger coupling constants with H-2ax (J = 9.7 Hz) and H-4ax (J = 11.7 Hz), when compared to those with H-2eq (J too small to be observed) and H-4eq (J = 3.5 Hz) (Figure S3b, Supporting Information). This indicated that H-3 is in an axial orientation while the larger 3-phenyl substitution is equatorial (Figure 4). This deduction was consistent with the coupling patterns and constants previously reported for isoflavans with the 3-phenyl group orientated at the equatorial position,12,17 which was also supported by the NOESY correlations between H-2eq/H-3, H-2ax/H-4ax, and H-3/H-4eq (Figure 4; Figure S3i, Supporting Information). The (S)-absolute configuration at C-3 was proposed from the ECD spectrum, which demonstrated a positive Cotton Effect in the 1La transition region (230–250 nm), and a negative Cotton Effect in the 1Lb region (276–300 nm), as shown in Figure S15 (Supporting Information), comparable with the reported ECD data of a structurally similar 3S isoflavan-isoflavone dimer12 and other (3S)-eq-isoflavan conformers with oxygenation at both the A and B rings.16,18 Therefore, the full structure of sphenostylisin C (3) was established. This compound shares the same novel carbon skeleton as that of compound 1.
Figure 4.

(3S)-eq-half chair conformer of the dihydropyran ring of compound 3, showing the observed proton coupling constants and key NOESY correlations.
The HRESIMS of sphenostylisin D (4) gave a sodiated molecular ion peak at m/z 361.1046 (calcd 361.1052), consistent with a molecular formula of C20H18O5. The UV spectrum of 4 was typical of a 3-phenylcoumarin skeleton.6 The 1H NMR spectrum of 4 showed a singlet at δH 7.80 (1H, H-4), indicating it to be a characteristic proton located at C-4 in a 3-phenylcoumarin skeleton. The remaining signals observed in the 1H NMR spectrum showed the presence of a 1,2,4-trisubstituted benzene ring [δH 7.03 (1H, d, J = 8.3 Hz, H-6′), 6.35 (1H, d, J = 2.2 Hz, H-3′), and 6.26 (1H, dd, J = 8.3, 2.2 Hz, H-5′)], a 1,3,4,6-tetrasubstituted benzene ring [δH 7.45 (1H, s, H-5) and 6.74 (1H, s, H-8)], and an α,α-dimethylallyl group [δH 6.23 (1H, dd, J = 17.5, 10.7 Hz, H-10), 4.95 (1H, dd, J = 10.7, 1.2 Hz, H-11a), 4.93 (1H, dd, J = 17.5, 1.2 Hz, H-11b), and 1.45 (6H, s, CH3 × 2, H-12/13)]. The 13C NMR spectrum of 4 showed 20 carbon signals, which were classified as two methyls, five quaternary carbons, seven tertiary sp2 carbons, one secondary sp2 carbon, four oxygen-bearing tertiary sp2 carbons, and a lactone carbonyl group, based on the DEPT and HSQC data. The characteristic NMR data of 4 were found to be very similar to those of fragment A of compound 1, while the absence of any fragment B resulted in an additional proton signal (H-5′) as well as an upfield shift of 0.49 ppm of H-6′ in compound 4. The H-3′, H-5′, and H-6′ resonances of the trisubstituted aromatic ring were assigned on the basis of the coupling constants and from the 1H-1H COSY spectrum, and corroborated by the HMBC correlations of H-3′ to C-1′ (δC 113.8), C-2′ (δC 156.1) and C-5′ (δC 106.2), of H-5′ to C-1′ (δC 113.8), C-3′ (δC 102.6), and C-6′ (δC 131.5), and of H-6′ to C-2′ (δC 156.0) and C-4′ (δC 158.3). In addition, the HMBC cross-peak between H-6′ and C-3 (δC 121.0) confirmed that this trisubstituted benzene ring is linked at C-3. The assignments of the two singlets at δH 7.45 (H-5) and 6.74 (H-8) were based on the HMBC correlations of H-5 to C-4 (δC 142.2), C-7 (δC 159.1) and C-8a (δC 153.0), and of H-8 to C-4a (δC 111.2) and C-6 (δC 131.6). The linkage of the α,α-dimethylallyl group at C-6 (δC 131.6) was established from the observed HMBC correlations of H-10, H-12, and H-13 to C-6, and was confirmed by the HMBC correlation of H-5 to C-9 (δC 39.9). Thus, sphenostylisin D (4) was assigned as 7-hydroxy-6-(2-methylbut-3-en-2-yl)-2′,4′-dihydroxy-3-phenylcoumarin.
Sphenostylisins E–G (5–7) were found to share the same molecular formula, C20H18O6, based on analysis of their HRESIMS data, which showed [M + Na]+ ion peaks at m/z 377.1007, 377.0991, and 377.0993, respectively (calcd 377.1001). When compared to compound 4, this molecular formula indicated that compounds 5–7 have the same degree of unsaturation with one more oxygen present, implying that a carbon in 4 has been oxygenated to give compounds 5–7. The NMR spectroscopic data of 5–7 (Table 2; Figures S5–S7, Supporting Information) were closely comparable to those of 4, with the only differences evident in signals for the side chain at C-6. Thus, an α,α-dimethylallyl group at C-6 in 4 was replaced by a 3-methylbut-1,2-diol moiety in 5. The signals of the latter unit occurred at δH 4.45 (1H, dd, J = 6.3, 5.3 Hz, H-10), 3.86 (1H, br d, J = 5.3 Hz, H-11a), 3.85 (1H, br d, J = 6.3 Hz, H-11b), 1.44 (3H, s, H-12), and 1.25 (3H, s, H-13) in the 1H NMR spectrum as well as at δC 43.7 (C, C-9), 95.6 (CH, C-10), 61.9 (CH2, C-11), 28.2 (CH3, C-12), and 23.2 (CH3, C-13) in the 13C NMR spectrum. In addition, key HMBC correlations of H-10 to C-9, C-11, C-12 and C-13, as well as H-11, H-12, and H-13 to C-9 and C-10, supported the structure assigned for the side-chain moiety in 5. Moreover, this side chain formed a dihydrofuran ring with C-6 (δC 137.2) and C-7 (δC 162.8), as suggested by the HMBC correlations of H-10 to C-6 and C-7, and of H-12 and H-13 to C-6. Thus, the structure of sphenostylisin E (5) was established as 6-(2,7-epoxy-3-methylbut-1-ol-3-yl)-2′,4′-dihydroxy-3-phenylcoumarin.
Table 2.
1H and 13C NMR Spectroscopic Data of Compounds 4–8a
| no | 4 | 5 | 6 | 7 | 8 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| δHb | δCc | δHb | δCc | δHb | δCc | δHb | δCc | δHb | δCc | |
| 2 | 160.3 | 163.7 | 164.0 | 160.3 | 8.10, s | 156.3 | ||||
| 3 | 121.0 | 122.9 | 122.9 | 120.9 | 124.1 | |||||
| 4 | 7.80, s | 142.1 | 7.87, s | 144.2 | 7.79, s | 144.1 | 7.80, s | 142.2 | 179.2 | |
| 4a | 111.2 | 115.3 | 113.7 | 111.3 | 117.0 | |||||
| 5 | 7.45, s | 126.4 | 7.38, s | 122.6 | 7.33, s | 132.7 | 7.45, s | 128.3 | 8.13, s | 127.4 |
| 6 | 131.6 | 137.2 | 124.4 | 129.1 | 133.3 | |||||
| 7 | 159.1 | 162.8 | 161.4 | 159.2 | 164.1 | |||||
| 8 | 6.74, s | 102.3 | 6.76, s | 98.4 | 6.75, s | 103.4 | 6.72, s | 102.3 | 6.86, s | 104.0 |
| 8a | 153.0 | 156.0 | 155.2 | 153.0 | 158.5 | |||||
| 9 | 39.9 | 43.7 | 75.3 | 46.1 | 48.0 | |||||
| 10 | 6.23, dd (17.5, 10.7) | 147.1 | 4.45, dd (6.3, 5.3) | 95.6 | 6.01, dd (17.3, 10.8) | 146.0 | 6.26, dd (17.6, 10.8) | 144.1 | 6.33, dd (17.4, 10.6) | 144.6 |
| 11a | 4.95, dd (10.7, 1.2) | 110.5 | 3.86, br d (5.3) | 61.9 | 5.20, dd (17.3, 1.2) | 112.2 | 5.02, dd (10.8, 1.1) | 112.5 | 5.14, br d (10.6) | 113.8 |
| 11b | 4.93, dd (17.5, 1.2) | 3.85, br d (6.3) | 5.00, dd (10.8, 1.2) | 4.92, dd (17.6, 1.1) | 5.04, br d (17.4) | |||||
| 12a | 1.45, s | 26.9 | 1.44, s | 28.2 | 2.92, d (13.9) | 43.6 | 3.79, d (10.3) | 67.1 | 4.03, d (10.7) | 69.3 |
| 12b | 2.87, d (13.9) | 3.72, d (10.3) | 3.89, d (10.7) | |||||||
| 13 | 1.45, s | 26.9 | 1.25, s | 23.2 | 1.26, s | 27.0 | 1.41, s | 21.8 | 1.53, s | 22.4 |
| 1′ | 113.8 | 115.4 | 115.6 | 113.7 | 112.3 | |||||
| 2′ | 156.0 | 157.4 | 157.4 | 156.0 | 158.0 | |||||
| 3′ | 6.35, d (2.2) | 102.6 | 6.38, d (2.3) | 104.0 | 6.38, d (2.3) | 104.0 | 6.35, d (2.2) | 102.6 | 6.40, d (2.2) | 104.7 |
| 4′ | 158.3 | 160.0 | 160.0 | 158.3 | 160.2 | |||||
| 5′ | 6.26, dd (8.3, 2.2) | 106.2 | 6.35, dd (8.2, 2.3) | 107.9 | 6.35, dd (8.2, 2.3) | 107.9 | 6.25, dd (8.3, 2.2) | 106.2 | 6.38, dd (8.2, 2.2) | 108.3 |
| 6′ | 7.03, d (8.3) | 131.5 | 7.12, d (8.2) | 132.7 | 7.11, d (8.2) | 132.7 | 7.02, d (8.3) | 131.5 | 7.05, d (8.2) | 132.9 |
NMR data obtained in DMSO-d6 for 4 and 7, and in CD3OD for 5, 6 and 8. Assignments are based on 1H–1H COSY, HSQC, and HMBC spectroscopic data.
Measured at 400 MHz for 1H NMR; δ in ppm, mult. (J in Hz).
Measured at 100 MHz for 13C NMR; δ in ppm.
Compound 6 showed side-chain signals at δH 6.01 (1H, dd, J = 17.3, 10.8 Hz, H-10), 5.20 (1H, dd, J = 15.3, 1.2 Hz, H-11a), 5.00 (1H, dd, J = 10.8, 1,2 Hz, H-11b), 2.92 (1H, d, J = 13.9 Hz, H-12a), 2.87 (1H, d, J = 13.9 Hz, H-12b), and 1.26 (3H, s, H-13) in the 1H NMR spectrum, along with corresponding 13C NMR data at δC 75.3 (C, C-9), 146.0 (CH, C-10), 112.2 (CH2, C-11), 43.6 (CH2, C-12), and 27.0 (CH3, C-13). The downfield shift of C-9 to δC 75.3 suggested that C-9 is oxygenated. These signals together indicated a 2-methylbut-3-en-2-ol side chain in 6 when compared to those of 4. This side chain was connected to C-6 (δC 124.4), as determined by the long-range correlations of H-12 to C-5 (δC 132.7) and C-7 (δC 161.4), and of H-5 (δH 7.33) to C-12 (δC 43.6) in the HMBC spectrum. Therefore, the structure of sphenostylisin F (6) was determined as 7-hydroxy-6-(2-hydroxy-2-methylbut-3-en-1-yl)-2′,4′-dihydroxy-3-phenylcoumarin.
On data comparison, the NMR spectra showed that one of the geminal dimethyl groups of the side chain in 4 was replaced by a hydroxymethyl group (δH 3.79, 3.72; δC 67.1) in 7, which was the only difference apparent between these two compounds. The 1H NMR signals at δH 6.26 (1H, dd, J = 17.6, 10.8 Hz, H-10), 5.02 (1H, dd, J = 10.8, 1.1 Hz, H-11a), 4.92 (1H, dd, J = 17.6, 1.1 Hz, H-11b), 3.79 (1H, d, J = 10.3 Hz, H-12a), 3.72 (1H, d, J = 10.3 Hz, H-12b), and 1.41 (3H, s, H-13), and the 13C NMR signals at δC 46.1 (C, C-9), 144.1 (CH, C-10), 112.5 (CH2, C-11), 67.1 (CH2, C-12), and 21.8 (CH3, C-13) were assigned to the side chain in 7, a 1-hydroxymethyl-1-methylallyl group. The linkage of this side chain at C-6 (δC 129.1) was confirmed by the HMBC correlations of H-10, H-12, and H-13 to C-6, and of H-5 (δH 7.45) to C-9. Hence, sphenostylisin G (7) was established structurally as 7-hydroxy-6-(1-hydroxy-2-methylbut-3-en-2-yl)-2′,4′-dihydroxy-3-phenylcoumarin.
Compounds 5–7 are isomers with different arrangements only in their respective side chain at C-6. These side chains are considered to be derived from an α,α-dimethylallyl group through oxidation, rearrangement, and cyclization (Scheme S1, Supporting Information). To the best of our knowledge, this is the first report of the presence of these side chains among the naturally occurring 3-phenylcoumarins.
The molecular formula of sphenostylisin H (8) was determined as C20H18O6 from the molecular ion peak [M + Na]+ at m/z 377.0995 (calcd 377.1001) in the HRESIMS, the same as those of compounds 5–7. However, the diagnostic H-2 vinylic singlet at δH 8.10 (H-2) in the 1H NMR spectrum of 8 indicated the presence of an isoflavone skeleton instead of a coumarin skeleton. The B-ring protons of the 1H NMR spectrum displayed an ABX spin system at δH 7.05 (1H, d, J = 8.2 Hz, H-6′), 6.40 (1H, d, J = 2.2 Hz, H-3′), and 6.38 (1H, dd, J = 8.2, 2.2 Hz, H-5′). These chemical shifts suggested that the B-ring is a 2′,4′-hydroxybenzene unit rather than a 3′,4′-hydroxybenzene moiety, according to the literature,19 which was confirmed by the HMBC spectrum in which only H-6′ showed an HMBC correlation to C-3 (δC 124.1). The aromatic region of the 1H NMR spectrum also exhibited two one-proton singlets at δH 8.13 and 6.86, which were assigned to H-5 and H-8 of the isoflavone A-ring, respectively, based on the HMBC correlations of H-5 to C-4 (δC 179.2) and C-8a (δC 158.5), and of H-8 to C-4a (δC 117.0). In addition, the 1H NMR signals at δH 6.33 (1H, dd, J = 17.4, 10.6 Hz, H-10), 5.14 (1H, br d, J = 10.6 Hz, H-11a), 5.04 (1H, br d, J = 17.4 Hz, H-11b), 4.03 (1H, d, J = 10.7 Hz, H-12a), 3.89 (1H, d, J = 10.7 Hz, H-12b), and 1.53 (3H, s, H-13), along with the 13C NMR signals at δC 48.0 (C, C-9), 144.6 (CH, C-10), 113.8 (CH2, C-11), 69.3 (CH2, C-12), and 22.4 (CH3, C-13), were characteristic of a 1-hydroxymethyl-1-methylallyl side chain, the same as that in 7. Moreover, H-10, H-12, and H-13 correlated to C-6 (δC 133.3), and H-5 (δH 8.13) correlated to C-9 in the HMBC spectrum, which established the connectivity of this side chain to C-6. Accordingly, sphenostylisin H (8) was established structurally as 7-hydroxy-6-(1-hydroxy-2-methylbut-3-en-2-yl)-2′,4′-dihydroxyisoflavone.
The molecular formula of sphenostylisin I (9), C19H20O6, was determined by HRESIMS, which gave a [M + Na]+ ion peak at m/z 367.1151 (calcd 367.1158). The characteristic UV spectrum and the typical 1H NMR signals at δH 4.11 (1H, d, J = 16.3 Hz, H-8a) and 4.03 (1H, d, J = 16.3 Hz, H-8b), combined with the corresponding 13C NMR signals at δC 202.9 (C-7) and 38.7 (C-8), revealed that 9 has a deoxybenzoin skeleton.20,21 Additional 1H NMR signals observed for this skeleton included an ABX aromatic spin system [δH 7.94 (1H, d, J = 8.8 Hz, H-6), 6.38 (1H, dd, J = 8.8, 2.3 Hz, H-5), and 6.24 (1H, d, J = 2.3 Hz, H-3)] and two aromatic protons at para positions [δH 6.83 (1H, s, H-6′) and 6.23 (1H, s, H-3′)], while the corresponding 13C NMR signals observed were three quaternary carbons [δC 112.3 (C-1), 113.4 (C-1′), and 127.6 (C-5′)], five tertiary sp2 carbons [δC 102.4 (C-3), 108.0 (C-5), 133.1 (C-6), 96.8 (C-3′), and 124.2 (C-6′)], and four oxygen-bearing tertiary sp2 carbons [δC 164.4 (C-2), 164.7 (C-4), 154.8 (C-2′), and 157.5 (C-4′)]. These NMR data were very similar to those of maackiaphenone, a known deoxybenzoin compound isolated from Maackia tenuifolia,21 with the only change being evident in the side chain. The side-chain resonances of 9 occurred at δH 4.16 (1H, dd, J = 6.7, 5.2 Hz, H-8′), 3.65 (2H, m, H-9′), 1.27 (3H, s, H-10′), and 1.02 (3H, s, H-11′), as well as at δC 42.1 (C, C-7′), 92.8 (CH, C-8′), 60.0 (CH2, C-9′), 27.2 (CH3, C-10′), and 23.2 (CH3, C-11′), consistent with a 3-methylbut-1,2-diol moiety fused with C-4′ and C-5′ to form a dihydrofuran ring, the same as that in 5, and was confirmed by HMBC correlations (Table S3 and Figure S9, Supporting Information). Thus, the structure of sphenostylisin I (9) was determined as 2,4-dihydroxy-2′-dihydroxy-5′-(2,4′-epoxy-3-methylbut-1-ol-3-yl)deoxybenzoin.
Sphenostylisin J (10) and sphenostylisin K (11) gave the same molecular formula, C19H20O6, as that of 9 based on the analysis of their HRESIMS data. Comparison of their 1D- and 2D-NMR spectra with those of 9 revealed the presence of the same skeleton in both 10 and 11, with the only difference being due to rearrangements of the isoprenyl side chain at C-5′. Compound 10 has a 2-methylbut-3-en-2-ol side chain, the same as that in 6, while compound 11 exhibited a 1-hydroxymethyl-1-methylallyl substituent, also present in 7 and 8. Hence, the structures of 10 and 11 were established as 2,4-dihydroxy-2′,4′-dihydroxy-5′-(2-hydroxy-2-methylbut-3-en-1-yl)deoxybenzoin and 2,4-dihydroxy-2′,4′-dihydroxy-5′-(1-hydroxy-2-methylbut-3-en-2-yl)deoxybenzoin, respectively, by comparison of their NMR data with those of 9, and were confirmed by detailed 2D-NMR spectroscopic data analysis (Table S3 and Figures S9 and S10, Supporting Information). To the best of our knowledge, the side chains of 9–11 have not been reported to be present in other naturally occurring deoxybenzoin derivatives to date.
A plausible biogenetic pathway involved in the generation of sphenostylisins A–K (1–11) is proposed in Scheme S1 (Supporting Information). From a biogenetic perspective, 1–11 may be all considered as isoflavonoid derivatives in a broad sense, despite their different structures. It has been validated in previous investigations that isoflavone is involved in the first step of isoflavonoid biosynthesis, with the basic skeleton being constructed for different subclasses of isoflavonoid before any isoprenoid substituents are added.22 Accordingly, 2′-hydroxydaidzein, an isoflavone previously isolated from some Fabaceae species,23 was proposed as a precursor. Simple prenylation and hydroxylation of this precursor could lead to the generation of compound 8. As relatively well characterized by previous investigations, the isoflavan (fragment A of 3) and 3-phenylcoumarin (4, fragment A of 1 and 2) were biosynthesized from isoflavone,22 followed by prenylation and then diverse modifications of the prenyl side chains24 in the formation of 5–7. The class of arylbenzofuran (fragment B of 1–3) was proposed to be produced by 4-pyrone ring cleavage, recyclization, and then reduction.25–27 The assembly mechanism of fragments A and B via carbon-carbon linkage in compounds 1–3 is believed to be comparable to that of existing bioflavonoids, although this is still a matter of conjecture. The deoxybenzoins (9–11) appear to be derived by 4-pyrone ring opening, oxidation of aldehyde to carboxylic acid, and then loss of one carbon atom through decarboxylation.25,28 It is interesting that this class of compounds has been reported to usually co-occur with various structurally related isoflavonoids,20–22 and their stability during isolation was considered due to the hydrogen-bonding of the ortho OH-2 to the carbonyl. It has been demonstrated that the lack of an ortho OH-2 group resulted in spontaneous cyclization to benzofuran derivatives.25 Based on these observations, it was assumed in previous investigations that deoxybenzoins could be the precursors of arylbenzofurans.20,21 Thus, in the present study, a cyclization reaction of 9 was performed on acid treatment with Amberlyst® 15 resin, and the expected 2-arylbenzofuran skeleton was generated (Scheme 1), which supported the assumption mentioned above to suggest a possible link among different classes of isoflavonoid compounds, and was also supportive of the structure elucidation of the deoxybenzoin skeleton characterized for compounds 9–11 in the present study.
Scheme 1.
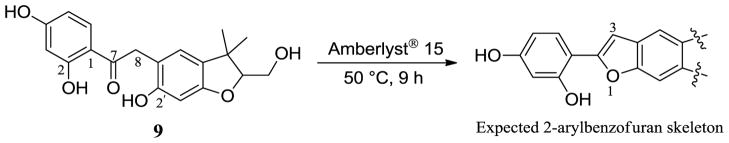
Cyclization Reaction of 9 to Form A 2-Arylbenzufuran Skeleton
All eleven new compounds isolated in the present investigation (1–11) were evaluated biologically. As shown in Table 4, eight compounds (1–4 and 8–11) exhibited potent hydroxyl radical-scavenging activity (ED50 values range from 0.60 to 2.6 μM) comparable to the positive control, quercetin (ED50 1.2 μM), with sphenostylisin J (10, ED50 0.60 μM) being the most active. Six compounds (1–4, 8, and 9) exhibited quinone reductase-inducing activity, with sphenostylisin C (3, CD 2.1 μM) exhibiting the most potent effect. Eight compounds (1–8) showed NF-κB p65 inhibitory activity. In particular, sphenostylisin A (1), representative of a novel carbon skeleton, was found to be a very potent NF-κB p65 inhibitor that exhibited an IC50 value of 6 nM and was >10 times more potent than the positive control, rocaglamide. A comparison of the NF-κB p65 inhibitory activities of compounds 1–11 showed an interesting preliminary structure-activity relationship (SAR) among those compounds. The 3-phenylcoumarin derivatives (4–7) with different side chains exhibited similarly potent NF-κB inhibitory activity, which implied that the 3-phenylcoumarin skeleton is important in mediating this type of activity. In contrast, the deoxybenzoins (9–11) that lack a lactone ring lost activity, indicating that a lactone ring is also important for activity. This deduction was supported by the observation that compound 1, incorporating a 3-phenylcoumarin moiety, exhibited 75 times more potency than compound 3, which contains an isoflavan unit instead of a 3-phenylcoumarin unit. This striking difference in potency between 1 and 3 in the NF-κB inhibition assay can be rationalized through the following analysis. First, the carbonyl group (C-2) of the lactone, present in 1 but absent in 3, may form hydrogen-bonding interactions with key residues in the active sites of the proteins associated with NF-κB p65 inhibition. Secondly, energy-minimized conformational models of 1 and 3 generated using the LigPrep/ConfGen software suite suggested that 1 bears a more extended conformation than that of 3 (Figure 5). The half-chair conformation of the dihydropyran ring in 3 sets the 3-phenyl ring almost perpendicular to the dihydropyran ring while the 3-phenyl ring in 1 is comparatively closer to being planar with the lactone ring due to the presence of the double bond between C-3 and C-4. The dihedral angles between C-3, C-4, C-1′ and C-6′ are 138.2° and 82.9° in 1 and 3, respectively. The distance between C-6 and C-6″ in 1 is about 1 Å longer than that in 3. It is conjectured that bioactive small molecules favor extended conformations that intuitively expose more hydrophobic surfaces into contact with the receptor, as compared to folded conformations.29,30 Thus, the more extended conformation of 1 may imply more extensive interaction with protein residues. Another interesting result observed in the NF-κB p65 inhibitory activity is that compound 4, the 3-phenylcoumarin moiety of 1, retains one-tenth of the activity of 1, which indicated that the 3-phenylbenzofuran moiety of 1 either facilitates the binding affinity or improves the penetration of the compound into the cell. In addition to the potent NF-κB p65 inhibitory activity, sphenostylisin A (1) also showed cytotoxicity (IC50 1.6 μM) against the HT-29 cell line.
Table 4.
Hydroxyl radical-scavenging, quinone reductase-inducing, NF-κB inhibitory, and cytotoxic activities of compounds 1–11.
| compound | hydroxy radical- scavenging | quinone reductase induction | NF-κB inhibition | cytotoxicity | ||
|---|---|---|---|---|---|---|
|
| ||||||
| ED50a (μM) | CDb (μM) | IC50c (μM) | CId | IC50e (μM) | IC50f(μM) | |
| 1 | 0.71 | 7.4 | 9.2 | 1.3 | 0.006 | 1.6 |
| 2 | 0.73 | 3.0 | 10.8 | 3.6 | 1.5 | 4.5 |
| 3 | 0.94 | 2.1 | 15.6 | 7.4 | 0.44 | 4.2 |
| 4 | 2.6 | 13.3 | 41.3 | 3.1 | 0.06 | >10 |
| 5 | 18.5 | >20 | >100 | NAg | 0.70 | >10 |
| 6 | >20 | >20 | >100 | NAg | 0.34 | >10 |
| 7 | >20 | >20 | >100 | NAg | 0.23 | >10 |
| 8 | 2.2 | 13.0 | >100 | >7.7 | 0.45 | >10 |
| 9 | 1.0 | 19.6 | 74.3 | 3.8 | >20 | >10 |
| 10 | 0.60 | >20 | >100 | NAg | >20 | >10 |
| 11 | 0.63 | >20 | >100 | NAg | >20 | >10 |
| quercetinh | 1.2 | |||||
| L-sulforaphanei | 0.71 | 15.2 | 21.4 | |||
| rocaglamidej | 0.08 | |||||
| paclitaxelk | 0.0006 | |||||
ED50, concentration scavenging hydroxyl radical by 50%. Compounds with ED50 values of <20 μ M are considered active.
CD, concentration required to double quinone reductase activity. Compounds with CD values of <20 μM are considered active.
IC50, concentration inhibiting hepa1c1c7 cell growth by 50%.
CI, Chemoprevention Index (= IC50/CD).
IC50, concentration inhibiting NF-κB p65 by 50%. Compounds with IC50 values of <20 μ M are considered active.
IC50, concentration inhibiting HT-29 cell growth by 50%. Compounds with IC50 values of <10 μ M are considered active.
NA, not applicable.
Positive control for hydroxyl radical-scavenging assay.
Positive control for quinone reductase induction assay.
Positive control for NF-κB p65 inhibition assay.
Positive control for cytotoxicity assay against HT-29 cell line.
Figure 5.
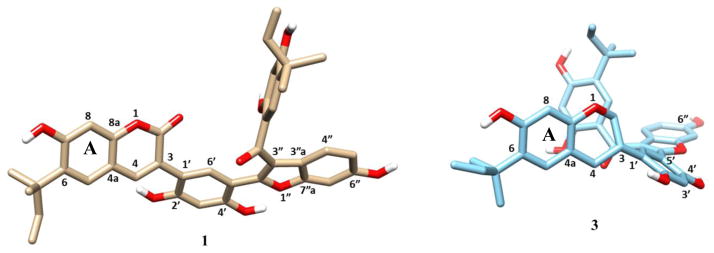
Conformational models of 1 and 3 produced using the LigPrep/ConfGen software suite, with A ring placed in the same plane.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured on a polarimeter. UV spectra were run on a spectrophotometer. Electronic circular dichroism (ECD) spectra were recorded on a spectropolarimeter. IR spectra were obtained on an IR spectrometer. NMR spectroscopic data were recorded at room temperature on 400, 600 and 800 MHz spectrometers. High-resolution electrospray ionization mass spectra (HRESIMS) were obtained on a Q-Tof mass spectrometer operated in the positive-ion mode, with sodium iodide being used for mass calibration. The tandem mass spectrometric analysis was performed on an ion-trap mass spectrometer operated in the positive-ion mode. Column chromatography was performed with LH-20, silica gel, and 40–63 μm C18-RP silica gel. Analytical thin-layer chromatography (TLC) was conducted on precoated 250 μm thickness Si gel F254 glass plates. A semi-preparative C18 column (5 μm, 150 mm × 10 mm i.d.) with a guard column (5 μm, 10 mm × 10 mm i.d.), and a preparative C18 column (5 μm, 150 mm × 19 mm i.d.) with a guard column (5 μm, 10 mm × 19 mm i.d.) were used for HPLC, along with a diode array detector.
Plant Material
The root bark of S. marginata ssp. erecta was collected in 1991 and recollected in 1996 at the Mazoe Hills in Zimbabwe by T. E. C., who also identified this plant. A voucher specimen (T.E. Chagwedera 187) was deposited in the National Herbarium Botanic Garden, Harare, Zimbabwe.
Extraction and Isolation
The air-dried and milled root bark (2 kg) of S. marginata ssp. erecta was extracted with methanol (3 × 8 L) at room temperature for two days each to afford an extract (739 g), which was suspended in H2O (2 L), and then partitioned in turn with hexanes (3 × 2 L), CHCl3 (3 × 2 L), EtOAc (3 × 2 L), and n-BuOH (3 × 2 L) to furnish dried hexanes (21 g), CHCl3 (60 g), EtOAc (30 g), n-BuOH (350 g), and H2O-soluble (278 g) extracts. The CHCl3-soluble extract, the most potent among these extracts in the in vitro hydroxyl radical-scavenging and QR-inducing assays, was subjected to chromatography over coarse silica gel, and eluted with a CH2Cl2–acetone gradient (40:1, 15:1, 10:1, 8:1, 6:1, 4:1, 3:1, 2:1, 1:1, 1:2, and pure acetone) to afford 11 fractions (F01–F11). Fraction F05 (7.2 g), active in the hydroxyl radical-scavenging and QR-inducing assays, was chromatographed over a silica gel column with a CHCl3–MeOH solvent system (30:1, 25:1, 20:1, 15:1, 12:1, 10:1, 8:1, 6:1, 5:1, 3:1, 2:1, and 1:1) to give 15 subfractions (F0501–F0515). F0505 (eluted with CHCl3–MeOH, 12:1; 168 mg) was further purified by HPLC, using a preparative C18 column (5 μm, 150 mm × 19 mm i.d.) with MeOH–H2O (65:35) at a flow rate of 8.0 mL/min, to yield compound 4 (tR = 25.5 min; 22.0 mg). F0507 (eluted with CHCl3–MeOH, 10:1; 980 mg) was purified by sephadex LH-20 column chromatography, with elution by MeOH–H2O (20:80, 40:60, 60:40, 80:20, and 100:0), to afford a further subfraction, F050703. This subfraction (357 mg) was then purified using the same HPLC column with MeOH–H2O (48:52, flow rate 8.0 mL/min), to yield compounds 5 (tR = 22.2 min; 5.0 mg), 6 (tR = 35.8 min; 23.4 mg), 8 (tR = 30.6 min; 3.0 mg), 9 (tR = 43.4 min; 5.6 mg), and 10 (tR = 49.1 min; 6.4 mg). F0508 (eluted with CHCl3–MeOH, 8:1; 485 mg) was chromatographed initially over a LH-20 column, with elution by MeOH–H2O gradient mixtures (0:100, 25:75, 50:50, 75:25, and 100:0), to afford five subfractions (F050801–F050805). F050803 (42 mg) was then purified by HPLC on a semi-preparative C18 column (5 μm, 150 mm × 10 mm i.d.), with isocratic elution (21% CH3CN–79% H2O, flow rate 4.0 mL/min), to yield compounds 7 (tR = 35.0 min; 5.3 mg) and 11 (tR = 46.8 min; 1.7 mg). F0512 (eluted with CHCl3–MeOH, 5:1; 1.2 g) was subjected to passage over a LH-20 column, using MeOH H2O–mixtures (0:100, 20:80, 40:60, 60:40, 80:20, and 100:0) as eluting solvents, to afford eight subfractions (F051201–F051208). F051204 (176 mg) was then purified using the same HPLC semi-preparative C18 column with isocratic elution (43% CH3CN–57% H2O, containing 0.05% TFA in H2O), at a flow rate of 4.0 mL/min, to yield compounds 1 (tR = 37.6 min; 3.4 mg) and 3 (tR = 46.9 min; 4.7 mg), and a further subfraction, F05120403. This subfraction (69 mg) was purified again using the same HPLC C18 column, eluted with MeOH–H2O (62:38, flow rate 4.0 mL/min), to yield compound 2 (tR = 45.0 min; 6.8 mg).
Sphenostylisin A (1): yellow amorphous solid; UV (MeOH) λmax (log ε) 216 (4.60), 253 (4.32), 308 (4.38), 343 (4.39) nm; IR (film) νmax 3252, 2964, 2923, 1693, 1615, 1571, 1492, 1387, 1275, 1191 1118, 1064, 908, 841 cm−1; 1H and 13C NMR data shown in Table 1; HRESIMS obsd m/z 697.2035 [M+Na]+ (calcd for C40H34O10Na, 697.2050).
Table 1.
1H and 13C NMR Spectroscopic Data of Compounds 1–3a
| no. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
|
| ||||||
| δHb | δCc | δHb | δCc | δHb | δCc | |
| 2α | 159.9 | 160.1 | 3.77, t (9.7) | 69.2 | ||
| 2β | 4.09, br d, (9.7) | |||||
| 3 | 120.4 | 121.4 | 3.25, m | 31.4 | ||
| 4α | 7.73, s | 142.2 | 7.56, s | 141.8 | 2.87, dd (15.1, 11.7) | 29.7 |
| 4β | 2.64, dd (15.1, 3.5) | |||||
| 4a | 111.1 | 111.0 | 111.6 | |||
| 5 | 7.46, s | 126.6 | 7.37, s | 126.4 | 6.81, s | 127.7 |
| 6 | 131.6 | 131.5 | 126.1 | |||
| 7 | 159.3 | 159.3 | 154.5 | |||
| OH-7 | 10.53, s | 9.07, s | ||||
| 8 | 6.77, s | 102.4 | 6.68, s | 102.2 | 6.24, s | 103.4 |
| 8a | 153.1 | 153.0 | 152.6 | |||
| 9 | 39.9 | 39.8 | 39.5 | |||
| 10 | 6.26, dd (17.5, 10.7) | 147.1 | 6.21, dd (17.5, 10.7) | 147.1 | 6.24, dd (17.5, 10.7) | 148.0 |
| 11a | 4.97, br d (10.7) | 110.6 | 4.93, br d (10.7) | 110.5 | 4.92, br d (17.5) | 109.6 |
| 11b | 4.95, br d (17.5) | 4.91, br d (17.5) | 4.90, br d (10.7) | |||
| 12 | 1.48, s | 26.9 | 1.43, s | 26.9 | 1.40, s | 26.9 |
| 13 | 1.48, s | 26.9 | 1.43, s | 26.9 | 1.40, s | 26.9 |
| 1′ | 114.6 | 113.7 | 118.6 | |||
| 2′ | 157.8 | 153.9 | 157.8 | |||
| OH-2′ | 9.85, s | 9.89, s | ||||
| 3′ | 6.35, s | 102.8 | 6.41, s | 103.9 | 6.32, s | 102.6 |
| 4′ | 155.9 | 155.5 | 154.7 | |||
| OH-4′ | 10.04, s | 9.75, s | ||||
| 5′ | 108.4 | 116.8 | 108.3 | |||
| 6′ | 7.52, s | 131.7 | 6.67, s | 131.4 | 7.23, s | 128.1 |
| 2″ | 152.2 | 149.3 | 153.0 | |||
| 3″ | 114.0 | 114.7 | 113.7 | |||
| 3″a | 120.1 | 121.9 | 120.1 | |||
| 4″ | 7.33, d (8.4) | 120.6 | 7.19, d (8.4) | 119.8 | 7.38, d (8.4) | 120.4 |
| 5″ | 6.78, dd (8.4, 1.6) | 112.7 | 6.60, dd (8.4, 1.8) | 111.0 | 6.77, dd (8.4, 1.6) | 112.6 |
| 6″ | 155.8 | 154.8 | 155.7 | |||
| OH-6″ | 9.67, s | 9.65, s | ||||
| 7″ | 6.98, d (1.6) | 97.4 | 6.80, d (1.8) | 97.3 | 6.97, d (1.6) | 97.4 |
| 7″a | 154.2 | 154.7 | 154.2 | |||
| 8″ | 194.7 | 3.71, s | 22.6 | 194.6 | ||
| 9″ | 112.9 | 108.1 | 112.8 | |||
| 10″ | 162.5 | 154.6 | 162.4 | |||
| OH-10″ | 12.66, s | 12.64, s | ||||
| 11″ | 6.34, s | 103.2 | 6.44, s | 102.4 | 6.29, s | 103.1 |
| 12″ | 163.2 | 157.2 | 163.0 | |||
| OH-12″ | 10.54, s | 10.47, s | ||||
| 13″ | 125.8 | 124.7 | 125.6 | |||
| 14″ | 7.32, s | 131.4 | 7.01, s | 129.5 | 7.21, s | 131.1 |
| 15″ | 39.2 | 39.4 | 39.0 | |||
| 16″ | 5.84, dd (17.5, 10.7) | 147.2 | 6.10, dd (17.5, 10.7) | 147.9 | 5.78, dd (17.5, 10.7) | 147.2 |
| 17″a | 4.74, br d (10.7) | 110.2 | 4.78, br d (17.5) | 109.6 | 4.73, br d (10.7) | 110.0 |
| 17″b | 4.67, br d (17.5) | 4.75, br d (10.7) | 4.61, br d (17.5) | |||
| 18″ | 1.09, s | 26.6 | 1.27, s | 26.7 | 1.04, s | 26.7 |
| 19″ | 1.09, s | 26.6 | 1.27, s | 26.7 | 1.04, s | 26.7 |
NMR data obtained in DMSO-d6 for 1–3. Assignments are based on 1H–1H COSY, HSQC, and HMBC spectroscopic data.
Measured at 800 MHz for 1H NMR; δ in ppm, mult. (J in Hz).
Measured at 150 MHz for 13C NMR; δ in ppm.
Sphenostylisin B (2): yellow amorphous solid; UV (MeOH) λmax (log ε) 218 (4.58), 258 (4.15), 318 (4.25), 340 (4.19) nm; IR (film) νmax 3286, 2966, 2926, 1686, 1617, 1577, 1491, 1442, 1364, 1293, 1191, 1149, 1121, 1022, 842 cm−1; 1H and 13C NMR data shown in Table 1; HRESIMS obsd m/z 683.2269 [M+Na]+ (calcd for C40H36O9Na, 683.2257).
Sphenostylisin C (3): yellow amorphous solid; [α]20D +23 (c 0.18, MeOH); UV (MeOH) λmax (log ε) 210 (4.87), 271 (4.37), 294 (4.42), 309 (4.39), 342 (sh, 4.12) nm; ECD (MeOH) λmax ([θ]) 212 (+41693), 240 (+4312), 268 (+6305), 289 (−4626), 315 (+5501) nm; IR (film) νmax 3419, 2967, 2930, 1622, 1587, 1494, 1392, 1273, 1143, 1118, 1082, 900, 838 cm−1; 1H and 13C NMR data shown in Table 1; HRESIMS obsd m/z 685.2404 [M+Na]+ (calcd for C40H38O9Na, 685.2414).
Sphenostylisin D (4): yellow amorphous solid; UV (MeOH) λmax (log ε) 206 (4.69), 248 (3.92), 350 (4.20) nm; IR (film) νmax 3308, 2968, 1683, 1615, 1575, 1508, 1423, 1366, 1285, 1188, 1166, 1102, 980, 846, 550 cm−1; 1H and 13C NMR data shown in Table 2; HRESIMS obsd m/z 361.1046 [M+Na]+ (calcd for C20H18O5Na, 361.1052).
Sphenostylisin E (5): yellow amorphous solid; [α]20D +13 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 206 (4.64), 248 (3.86), 350 (4.14) nm; IR (film) νmax 3319, 2961, 2926, 1699, 1621, 1576, 1482, 1390, 1285, 1204, 1146, 1099, 1026, 979, 843 cm−1; 1H and 13C NMR data shown in Table 2; HRESIMS obsd m/z 377.1007 [M+Na]+ (calcd for C20H18O6Na, 377.1001).
Sphenostylisin F (6): yellow amorphous solid; [α]20D −6 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 206 (4.70), 247 (4.02), 349 (4.26) nm; IR (film) νmax 3289, 2976, 2927, 1691, 1621, 1581, 1497, 1456, 1368, 1292, 1241, 1152, 1021, 981, 932, 848, 543 cm−1; 1H and 13C NMR data shown in Table 2; HRESIMS obsd m/z 377.0991 [M+Na]+ (calcd for C20H18O6Na, 377.1001).
Sphenostylisin G (7): yellow amorphous solid; [α]20D +9 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 206 (4.70), 248 (3.99), 350 (4.27) nm; IR (film) νmax 3273, 2974, 1689, 1616, 1578, 1509, 1461, 1366, 1291, 1232, 1165, 1102, 1022, 980, 846, 551 cm−1; 1H and 13C NMR data shown in Table 2; HRESIMS obsd m/z 377.0993 [M+Na]+ (calcd for C20H18O6Na, 377.1001).
Sphenostylisin H (8): yellow amorphous solid; [α]20D −5 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 221 (4.34), 250 (4.24), 288 (4.10) nm; IR (film) νmax 3354, 2934, 1615, 1575, 1464, 1374, 1268, 1116, 1024, 847 cm−1; 1H and 13C NMR data shown in Table 2; HRESIMS obsd m/z 377.0995 [M+Na]+ (calcd for C20H18O6Na, 377.1001).
Sphenostylisin I (9): colorless resin; [α]20D −3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 210 (4.24), 224sh (4.01), 280 (3.90), 316 (3.73) nm; IR (film) νmax 3310, 2962, 2933, 1625, 1497, 1445, 1363, 1293, 1232, 1143, 1024, 845, 802 cm−1; 1H and 13C NMR data shown in Table 3; HRESIMS obsd m/z 367.1151 [M+Na]+ (calcd for C19H20O6Na, 367.1158).
Table 3.
1H and 13C NMR Spectroscopic Data of Compounds 9–11a
| no | 9 | 10 | 11 | |||
|---|---|---|---|---|---|---|
|
| ||||||
| δHb | δCc | δHb | δCc | δHb | δCc | |
| 1 | 112.3 | 112.2 | 112.2 | |||
| 2 | 164.4 | 164.5 | 164.5 | |||
| 3 | 6.24, d (2.3) | 102.4 | 6.23, d (2.3) | 102.4 | 6.19, d (2.3) | 102.4 |
| 4 | 164.7 | 164.6 | 164.8 | |||
| 5 | 6.38, dd (8.8, 2.3) | 108.0 | 6.34, dd (8.9, 2.3) | 108.0 | 6.32, dd (8.9, 2.3) | 108.1 |
| 6 | 7.94, d (8.8) | 133.1 | 7.89, d (8.9) | 133.2 | 7.91, d (8.9) | 133.2 |
| 7 | 202.9 | 203.2 | 203.1 | |||
| 8a | 4.11, d (16.3) | 38.7 | 4.01, d (15.7) | 38.4 | 3.99, br s | 38.5 |
| 8b | 4.03, d (16.3) | 3.96, d (15.7) | ||||
| 1′ | 113.4 | 111.9 | 111.3 | |||
| 2′ | 154.8 | 153.9 | 153.9 | |||
| 3′ | 6.23, s | 96.8 | 6.30, s | 102.7 | 6.29, s | 103.5 |
| 4′ | 157.5 | 155.2 | 155.2 | |||
| 5′ | 127.6 | 114.9 | 121.6 | |||
| 6′ | 6.83, s | 124.2 | 6.73, s | 134.2 | 6.83, s | 130.5 |
| 7′ | 42.1 | 2.57, s | 42.1 | 45.6 | ||
| 8′ | 4.16, dd (6.7, 5.2) | 92.8 | 73.5 | 6.20, dd (17.6, 10.6) | 145.0 | |
| 9′a | 3.65, m | 60.0 | 5.89, dd (17.3, 10.7) | 146.0 | 4.96, br d (10.6) | 111.7 |
| 9′b | 4.89, br d (17.6) | |||||
| 10′a | 1.27, s | 27.2 | 5.08, dd (17.3, 1.9) | 110.7 | 3.67, d (10.5) | 67.6 |
| 10′b | 4.87, dd (10.7, 1.9) | 3.65, d (10.5) | ||||
| 11′ | 1.02, s | 23.2 | 1.07, s | 26.4 | 1.29, s | 21.4 |
NMR data obtained in DMSO-d6 for 9–11. Assignments are based on 1H–1H COSY, HSQC, and HMBC spectroscopic data.
Measured at 400 MHz for 1H NMR; δ in ppm, mult. (J in Hz).
Measured at 100 MHz for 13C NMR; δ in ppm.
Sphenostylisin J (10): colorless resin; [α]20D −4 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 212 (4.30), 224sh (4.09), 279 (4.04), 316 (3.85) nm; IR (film) νmax 3288, 2971, 2939, 1625, 1506, 1446, 1359, 1290, 1232, 1179, 1140, 1105, 1021, 927, 848, 801 cm−1; 1H and 13C NMR data shown in Table 3; HRESIMS obsd m/z 367.1153 [M+Na]+ (calcd for C19H20O6Na, 367.1158).
Sphenostylisin K (11): colorless resin; [α]20D −4 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 211 (4.28), 224sh (4.05), 279 (3.96), 316 (3.81) nm; IR (film) νmax 3197, 2968, 2924, 1624, 1507, 1456, 1237, 1177, 1135, 1023, 999, 849 cm−1; 1H and 13C NMR data shown in Table 3; HRESIMS obsd m/z 367.1149 [M+Na]+ (calcd for C19H20O6Na, 367.1158).
Cyclization Reaction of 9 to Form a 2-Arylbenzofuran Skeleton
To a 5 mL round-bottomed flask under nitrogen containing 9 (1.0 mg), were added freshly dried and degassed THF (1 mL), Amberlyst® 15 resin (3.9 mg), and freshly dried, powdered 4 Å molecular sieve (3.0 mg). After being stirred at 50 °C for 9 h under a N2 atmosphere, TLC (7:1 CHCl3–MeOH) indicated the reaction to be completed. The crude reaction solution was evaporated in vacuo, and dissolved in CD3OD for 1H NMR spectroscopic measurement, which showed the presence of extra singlets at δH 7.02 and 7.20 (H-3) in the products and the disappearance of the H-8 signal (δH 4.07) of the starting material (9). This indicated that the cyclization of 9 between the OH-2′ and the carbonyl carbon (C-7) was successful, and the expected 2-arylbenzofuran skeleton was generated (Figure S12a, Supporting Information). Then, the product mixture was purified by HPLC on a semi-preparative C18 column (5 μm, 150 mm × 10 mm i.d.), using a MeOH/H2O gradient (50–70% MeOH from 0 to 40 min, and 70–100% MeOH from 40 to 50 min, flow rate 4.0 mL/min), to be separated as three major peaks (peak 1, tR = 8.1 min; peak 2, tR = 22.8 min; peak 3, tR = 31.0 min). The UV absorbances of peaks 1–3 were recorded by a PDA detector coupled with the HPLC, and the characteristic UV spectra (Figure S12b, Supporting Information) of peaks 2 (λmax 218, 295sh, 320, 341 nm) and 3 (λmax 218, 295sh, 323, 343 nm) were comparable to those of the 2-arylbenzofuran derivatives previously reported,31,32 which further supported the generation of the expected 2-arylbenzofuran skeleton in the reaction. Due to the very small amount of 9 available for the reaction, the amount of the products purified from peaks 2 and 3 was not enough for full structural characterization via NMR spectroscopy.
Evaluation of Hydroxyl Radical-scavenging Activity
Hydroxyl radical-scavenging activity was performed according to a method described previously33,34 (Protocol S1, Supporting Information).
Evaluation of Quinone Reductase-inducing Activity
The potential QR-inducing activity of the extracts, fractions, and pure isolates was assayed as described previously34,35 (Protocol S2, Supporting Information).
Evaluation of NF-κB Inhibition Activity
The NF-κB assay was carried out according to an established protocol36 (Protocol S3, Supporting Information).
Evaluation of Cytotoxicity
The cytotoxic activity of the pure compounds isolated was assayed based on a method described previously37,38 (Protocol S4, Supporting Information).
Conformational Analysis of 1 and 3 by LigPrep/ConfGen software suite
The 2D SDF file format was converted to a 3D format employing LigPrep program (version 2.5, Schrödinger LLC, New York, NY). LigPrep accounts for the different tautomeric and stereoisomeric states of the compounds as well as a low energy ring conformation sampling. Possible ionization states of the compounds at pH 7±2 were generated using the Epic module. Energy minimization was performed using the OPLS2005 force field to obtain geometry-optimized 3D coordinates. The output of LigPrep was passed onto further conformational sampling utilizing a ConfGen advanced module (version 2.3, Schrödinger LLC). Energy minimization calculations also used the OPLS2005 force field. A GB/SA solvation model of water was used with a setting of constant dielectric for the electrostatic treatment and 1.0 for the dielectric constant. A maximum of 100 steps of truncated Newton conjugate gradient (TNCG) minimizations proceeded until the rms force dropped below a speci ed threshold (0.05 kcal/mol Å). Finally a rapid search mode was implemented to produce conformers of compounds 1 and 3.
Supplementary Material
Acknowledgments
We wish to acknowledge financial support for this work from projects U01/U19 CA52956 and P01 CA125066, funded by NCI/NIH. We would like to thank Mr. John W. Fowble (College of Pharmacy, the Ohio State University) for facilitating the acquisition of NMR data. Mr. Mark Apsega from the Mass Spectrometry & Proteomics Facility at the Ohio State University is acknowledged for MS training.
Footnotes
The authors declare no competing financial interest.
Supporting Information. 1H, 13C, 13C DEPT 135, 1H–1H COSY, HSQC, and HMBC NMR spectra as well as tables of NMR data for compounds 1–11; UV, ECD, and NOESY spectra for compound 3; 1H NMR spectra and HPLC separation chromatogram of the crude reaction solution in Scheme 1; HRESIMS for compounds 1–11; ESIMS/MS for compounds 1–3; tandem MS fragmentation scheme for compound 3; plausible biogenetic relationships between compounds 1–11; protocols of hydroxyl radical-scavenging, quinone reductase-inducing, NF-κB inhibition, and cytotoxicity assays. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Sohni YR, Mutangadura-Mhlanga T, Kale PG. Mutat Res. 1994;322:133–140. doi: 10.1016/0165-1218(94)00021-2. [DOI] [PubMed] [Google Scholar]
- 2.Malaisse F, Parent G. Ecol Food Nutr. 1985;18:43–82. [Google Scholar]
- 3.Gunzinger J, Msonthi JD, Hostettmann K. Helv Chim Acta. 1988;71:72–76. [Google Scholar]
- 4.Cuendet M, Oteham CP, Moon RC, Pezzuto JM. J Nat Prod. 2006;69:460–463. doi: 10.1021/np050362q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarkar FH, Li Y. Front Biosci. 2008;13:2950–2959. doi: 10.2741/2900. [DOI] [PubMed] [Google Scholar]
- 6.Hatano T, Aga Y, Shintani Y, Ito H, Okuda T, Yoshida T. Phytochemistry. 2000;55:959–963. doi: 10.1016/s0031-9422(00)00244-2. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Zhang T, Xiao L, Yang L, Chen R. Fitoterapia. 2010;81:614–616. doi: 10.1016/j.fitote.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Cuyckens F, Claeys M. J Mass Spectrom. 2004;39:1–15. doi: 10.1002/jms.585. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal R, Jayasinghe L, Kuhnert N. J Mass Spectrom. 2012;47:502–515. doi: 10.1002/jms.2954. [DOI] [PubMed] [Google Scholar]
- 10.Li HJ, Deinzer ML. Anal Chem. 2007;79:1739–1748. doi: 10.1021/ac061823v. [DOI] [PubMed] [Google Scholar]
- 11.Pan L, Matthew S, Lantvit DD, Zhang X, Ninh TN, Chai H, Carcache de Blanco EJ, Soejarto DD, Swanson SM, Kinghorn AD. J Nat Prod. 2011;74:2193–2199. doi: 10.1021/np200557e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bezuidenhoudt BCB, Brandt EV, Steenkamp JA, Roux DG, Ferreira D. J Chem Soc, Perkin Trans 1. 1988:1227–1235. [Google Scholar]
- 13.Rohwer MB, van Heerden PS, Brandt EV, Bezuidenhoudt BCB, Ferreira D. J Chem Soc, Perkin Trans 1. 1999:3367–3374. [Google Scholar]
- 14.Rufer CE, Glatt H, Kulling SE. Drug Metab Dispos. 2006;34:51–60. doi: 10.1124/dmd.105.004929. [DOI] [PubMed] [Google Scholar]
- 15.Pinheiro PF, Justino GC. Structural Analysis of Flavonoids and Related Compounds - A Review of Spectroscopic Applications. In: Rao V, editor. Phytochemicals - A Global Perspective of Their Role in Nutrition and Health. InTech; Rijeka: 2012. pp. 42–44. [Google Scholar]
- 16.Slade D, Ferreira D, Marais JP. Phytochemistry. 2005;66:2177–2215. doi: 10.1016/j.phytochem.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Kurosawa K, Ollis WD, Redman BT, Sutherland IO. Chem Commun. 1968:1265–1267. [Google Scholar]
- 18.Kim M, Kim S, Kim Y, Han J. Bull Kor Chem Soc. 2009;30:415–418. [Google Scholar]
- 19.Yoo HS, Lee JS, Kim CY, Kim J. Arch Pharm Res. 2004;27:544–546. doi: 10.1007/BF02980129. [DOI] [PubMed] [Google Scholar]
- 20.Kiuchi F, Chen X, Tsuda Y. Heterocycles. 1990;31:629–636. [Google Scholar]
- 21.Zeng JF, Zhu DY. Acta Bot Sin. 1999;41:997–1001. [Google Scholar]
- 22.Dewick PM. In: The Flavonoids: Advances in Research since 1986. Harborne JB, editor. Chapman and Hall; London: 1994. pp. 195–206. [Google Scholar]
- 23.Ko HH, Weng JR, Tsao LT, Yen MH, Wang JP, Lin CN. Bioorg Med Chem Lett. 2004;14:1011–1014. doi: 10.1016/j.bmcl.2003.11.074. [DOI] [PubMed] [Google Scholar]
- 24.Boonnak N, Khamthip A, Karalai C, Chantrapromma S, Ponglimanont C, Kanjana-Opas A, Tewtrakul S, Chantrapromma K, Fun HK, Kato S. Aust J Chem. 2010;63:1550–1556. [Google Scholar]
- 25.Whalley WB, Lloyd G. J Chem Soc. 1956:3213–3224. [Google Scholar]
- 26.Li L, Cheng XF, Leshkevich J, Umezawa T, Harding SA, Chiang VL. Plant Cell. 2001;13:1567–1586. doi: 10.1105/TPC.010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alphey MS, Yu W, Byres E, Li D, Hunter WN. J Biol Chem. 2005;280:3068–3077. doi: 10.1074/jbc.M411069200. [DOI] [PubMed] [Google Scholar]
- 28.Wei Y, Lin M, Oliver DJ, Schnable PS. BMC Biochem. 2009;10:7. doi: 10.1186/1471-2091-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diller DJ, Merz KM., Jr J Comput Aided Mol Des. 2002;16:105–112. doi: 10.1023/a:1016320106741. [DOI] [PubMed] [Google Scholar]
- 30.Perola E, Charifson PS. J Med Chem. 2004;47:2499–2510. doi: 10.1021/jm030563w. [DOI] [PubMed] [Google Scholar]
- 31.Halabalaki M, Aligiannis N, Papoutsi Z, Mitakou S, Moutsatsou P, Sekeris C, Skaltsounis AL. J Nat Prod. 2000;63:1672–1674. doi: 10.1021/np000071b. [DOI] [PubMed] [Google Scholar]
- 32.Yenesew A, Midiwo JO, Guchu SM, Heydenreich M, Peter MG. Phytochemistry. 2002;59:337–341. doi: 10.1016/s0031-9422(01)00459-9. [DOI] [PubMed] [Google Scholar]
- 33.LeBel CP, Ischiropoulos H, Bondy SC. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Deng Y, Yuan C, Pan L, Chai H, Keller WJ, Kinghorn AD. J Agric Food Chem. 2012;60:11551–11559. doi: 10.1021/jf303712e. [DOI] [PubMed] [Google Scholar]
- 35.Su BN, Jung PE, Vigo JS, Graham JG, Cabieses F, Fong HH, Pezzuto JM, Kinghorn AD. Phytochemistry. 2003;63:335–341. doi: 10.1016/s0031-9422(03)00112-2. [DOI] [PubMed] [Google Scholar]
- 36.Deng Y, Balunas MJ, Kim JA, Lantvit DD, Chin YW, Chai H, Sugiarso S, Kardono LB, Fong HH, Pezzuto JM, Swanson SM, Carcache de Blanco EJ, Kinghorn AD. J Nat Prod. 2009;72:1165–1169. doi: 10.1021/np9001724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan L, Kardono LB, Riswan S, Chai H, Carcache de Blanco EJ, Pannell CM, Soejarto DD, McCloud TG, Newman DJ, Kinghorn AD. J Nat Prod. 2010;73:1873–1878. doi: 10.1021/np100503q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan L, Acuna UM, Li J, Jena N, Ninh TN, Pannell CM, Chai H, Fuchs JR, Carcache de Blanco EJ, Soejarto DD, Kinghorn AD. J Nat Prod. 2013;76:394–404. doi: 10.1021/np3007588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.