

Abstract
Fibroblast growth factor 2 (FGF2) induces endothelial cell migration and angiogenesis through two classes of receptors: receptor tyrosine kinases, such as FGF receptor 1 (FGFR1), and heparan sulfate proteoglycans, such as syndecan 4 (S4). We examined the distinct contributions of FGFR1 and S4 in shaping the endothelial response to FGF2. S4 determined the kinetics and magnitude of FGF2-induced mitogen-activated protein kinase (MAPK) signaling by promoting the macropinocytosis of the FGFR1-S4-FGF2 signaling complex. Internalization of the S4 receptor complex was independent of clathrin and dynamin, proceeded from lipid raft–enriched membranes, and required activation of the guanosine triphosphatases RhoG and Rab5. Genetic knockout of S4, disruption of S4 function, or inhibition of Rab5 led to increased endocytosis and MAPK signaling. These data define the mechanism by which FGFR1 and S4 coordinate downstream signaling upon FGF2 stimulation: FGFR1 initiates MAPK signaling, whereas S4-dependent FGFR1 macropinocytosis modulates the kinetics of MAPK activation. Our studies identify S4 as a regulator of MAPK signaling and address the question of how distinct classes of FGFRs individually contribute to signal transduction in endothelial cells.
INTRODUCTION
Syndecan 4 (S4) is a transmembrane proteoglycan involved in the regulation of various cellular processes, including cell adhesion and migration (1, 2). This broad spectrum of activity is derived from the ability of S4 to regulate the signaling of fibroblast growth factor (FGF) receptors (FGFRs) and integrins and to signal independently as a growth factor receptor.
As with other syndecans, S4 bears heparan sulfate chains on its extracellular domain that can bind various heparan-binding growth factors and other families of transmembrane growth factor receptors, such as integrins (2, 3). S4 signals largely through its short intracellular domain, which includes a C-terminal PDZ (postsynaptic density, Discs large, zona occludens 1) binding region in one of its two conserved domains (4). This PDZ-binding domain binds various intracellular partners, including synectin (5). A variable region that is unique to S4 enables S4 to bind and activate protein kinase C α (PKCα) (6). The PDZ-binding domain has been implicated in orchestrating endothelial migration through the Rho family guanosine triphosphatases (GTPases) RhoG and Rac1 (7), whereas the interaction between S4 and PKCα promotes mTOR (mammalian target of rapamycin) complex 2 assembly and Akt activity (8).
In vivo disruption of signaling initiated by S4 or its binding partners affects various physiological processes such as arterial development (9, 10), post-infarct myocardial dysfunction (11), recovery from endotoxic shock (12), wound healing (13), and neural crest development (14). The mechanism by which S4 regulates FGFR signaling has not been established. Typically, syndecans and other heparan sulfate–carrying proteins are thought to bind FGFs through their heparan sulfate chains, thereby facilitating FGF-FGFR binding and stabilizing the formation of the receptor-ligand complex (15). However, recent studies of the cytoplasmic signaling capabilities of S4 have suggested that there may be additional mechanisms of proteoglycan-mediated regulation (1, 16, 17).
One mechanism investigated in the present study is receptor trafficking. Although cell surface receptors may initiate signaling cascades from the membrane, numerous signaling events require cytoplasmic localization, and the process of endocytosis can exert fine spatiotemporal control over signaling (18). Analogous to specialized cell membrane microenvironments that facilitate the formation of signaling complexes and receptor activation [such as cholesterol and sphingolipid-enriched lipid rafts (19)], cytoplasmic signaling is likewise thought to occur at specialized signaling compartments (20, 21). In the case of FGFR1 signaling, receptor activation occurs at the cell membrane upon ligand binding (15), although intracellular activation of the mitogen-activated protein kinase (MAPK) pathway has also been reported (22).
Other proteins affecting receptor trafficking are the Rab family of GTPases, which play a key role in regulating vesicle maturation and in determining whether vesicles are recycled or undergo degradation (23, 24). Rab5 in particular has been implicated in the preliminary stages of vesicular development into early signaling endosomes and thus links receptor endocytosis and signaling (25, 26).
Given that S4 forms a ternary complex with its co-receptor (FGFR1) and their shared ligand (FGF2), we examined the role of S4 in the regulation of FGFR1 endocytosis and signaling. We report that FGFR1 uptake in response to FGF2 proceeds through a macropinocytic pathway that is directly controlled by S4-dependent activation of RhoG. Furthermore, whereas canonical MAPK signaling is initiated by FGFR1, its kinetics and magnitude are regulated by S4-directed endocytosis. Thus, the control of FGFR1 trafficking by S4 represents a previously unknown mechanism of MAPK signaling regulation.
RESULTS
Here, we tested the hypothesis that FGF2-mediated FGFR1 signaling is regulated by receptor-initiated endocytosis and that S4 controls this process. We first examined how FGFR1 becomes internalized upon ligand binding. Because specific and functionally inactive antibodies directed against extracellular FGFR epitopes are lacking, we created an FGFR1 construct containing an extracellular hemagglutinin (HA) tag (FGFR1-HA) and expressed it in rat fat pad endothelial cells (RFPECs), which have low amounts of native FGFR.
FGFR1-HA tyrosine autophosphorylation and subsequent ERK1/2 (extracellular signal–regulated kinases 1 and 2) activation were detected within 30 s, and ERK1/2 activation occurred within 2.5 min of FGF2 treatment (fig. S1A). Because divalent anti-HA antibodies were used to determine subcellular FGFR1-HA localization in this system, we examined whether these antibodies alone were sufficient to induce FGFR1-HA oligomerization and receptor activation. Incubation with anti-HA antibodies or the same antibodies followed by secondary antibodies did not appear to induce appreciable receptor phosphorylation and ERK1/2 activation, as compared with a positive control of FGF2 stimulation (fig. S1B). Thus, divalent anti-HA antibodies did not mimic ligand-induced receptor cross-linking and activation, rendering them suitable for receptor localization studies.
To exclude the possibility that the addition of an HA tag interferes with signal sequence function or receptor localization to the cell membrane, we stained RFPECs expressing the FGFR1-HA construct with anti-HA antibodies without first subjecting them to membrane permeabilization (fig. S1C). This revealed extensive membrane localization of the tagged construct. We also verified the ability to wash bound anti-HA antibodies from the cell surface by washing the antibodies with phosphate-buffered saline (PBS) at pH 2.5 (fig. S1D). Finally, because the modification of extracellular receptor epitopes frequently diminishes effective ligand binding, we sought to determine whether FGF2 binding was also preserved in the FGFR1-HA construct. After 10 min of fluorophore-labeled FGF2 incubation, ligand was endocytosed and extensively colocalized with FGFR1-HA (fig. S1E). A previously characterized S4–Fc receptor (S4-FcR) chimeric construct, which contains the cytoplasmic and transmembrane domains of S4 fused to an extracellular human FcR (27), was also used in these cells to confirm that the FGFR1 co-receptor S4 associated with FGFR1 upon FGF2 treatment. These cells showed extensive colocalization of FGFR1-HA and S4-FcR after FGF2 treatment (fig. S1F). Together, these data indicate that RFPECs with stable FGFR1-HA expression exhibit appropriate receptor activation, signaling co-receptor association, and endocytosis in response to ligand binding.
We next concentrated on defining the mechanisms of endothelial FGF2–induced FGFR1 internalization. Signaling complexes are often organized in lipid rafts, discrete regions of the plasma membrane enriched in cholesterol and sphingolipids (28). Because S4 is localized to lipid rafts and taken up through the macropinocytic pathway (29), we evaluated the role of lipid rafts and macropinocytosis in the uptake of FGFR1. FGFR1 colocalized with two markers of lipid raft–enriched membrane regions, cholera toxin B and a glycosylphosphatidylinositol (GPI)–anchored green fluorescent protein (GFP) construct (30), thus demonstrating that FGFR1 was localized to these membrane microdomains (Fig. 1, A and B). Furthermore, cholesterol depletion by methyl β-cyclodextrin revealed that FGFR1 endocytosis was inhibited in the absence of membrane-associated cholesterol (Fig. 1C). We conclude that FGFR1 is present in lipid rafts, and dissolution of these membrane subdomains by cholesterol depletion precludes receptor uptake.
Fig. 1.

FGFR1 endocytosis originates at lipid rafts and occurs via macropinocytosis. (A) Lipid raft–enriched regions of the plasmamembrane were labeled in RFPECs with Alexa Fluor 594–conjugated cholera toxin B. n = 8 cells in three independent experiments. (B) RFPECs expressing FGFR1-HA were transfected with GFPGPI and treated with FGF2 to initiate endocytosis. n = 12 cells per condition in three independent experiments. (C) Cells were treated with vehicle or methyl β-cyclodextrin to deplete membrane cholesterol and then labeled with anti-HA antibodies and treated with FGF2 to initiate FGFR1-HA endocytosis. Staining is shown after 15 min of endocytosis. Quantification is for eight cells per condition. P < 0.01 by χ2 test. DIC, differential interference contrast. (D) Primary murine pulmonary endothelial cells were incubated with neutral Texas Red–70-kD dextran and Alexa Fluor 647–FGF2. (E) Quantification of FGF2 and dextran colocalization from (D). (F) Cells were prepared for FGFR1 endocytosis assay and preincubated with either vehicle (left panels) or amiloride (right panels) and then treated with FGF2, Texas Red–conjugated 70-kD dextran, and Alexa Fluor 647–conjugated transferrin. (G) Quantification of (F) with eight cells per condition. *P < 0.01, **P < 0.01 by χ2 test. Scale bar, 10 μm. Insets represent magnifications of bordered regions.
To evaluate the contribution of macropinocytosis to FGFR1 uptake, we used labeled neutral 70-kD dextran, a marker for macropinocytosis (31, 32). FGF2 was localized in macropinocytic vesicles (Fig. 1D), and treatment with amiloride, an inhibitor of macropinocytosis (33), resulted in decreased uptake of both 70-kD dextran and FGFR1 (Fig. 1, E and F). Transferrin, a marker of clathrin-mediated endocytosis, was used as a control in these studies to demonstrate that amiloride selectively inhibits macropinocytosis, leaving transferrin uptake unaffected.
To further confirm the key role of macropinocytosis in FGFR1 trafficking, we examined the potential contributions of other endocytic pathways to FGFR1 uptake, beginning with clathrin-mediated endocytosis. Cells transfected with a GFP-tagged clathrin light chain construct exhibited minimal colocalization of this construct with FGFR1-HA after 5 and 15 min of FGF2 incubation (Fig. 2A). This lack of colocalization was also observed between FGF2 and clathrin (Fig. 2B) and between FGF2 and transferrin (a marker of clathrin-mediated endocytosis) in primary endothelial cells (Fig. 2C). The adaptor protein AP180 is also an integral component of clathrin-mediated endocytosis (34). Transfection with a dominant-negative form of AP180 (AP180C) had no effect on FGFR1-HA uptake (Fig. 2D), further demonstrating that clathrin-mediated endocytosis is dispensable in FGFR1 uptake. Because the use of anti-HA antibodies might influence the endocytic pathway taken by the FGFR1-HA construct, we incubated cells with FGF2 alone and assessed the localization of both FGFR1-HA and GFP-tagged clathrin light chain after cell fixation and permeabilization (Fig. 2E). As in Fig. 2A, we did not observe substantial colocalization of clathrin and FGFR1-HA. Finally, knockdown of clathrin heavy chain with small interfering RNA (siRNA) in primary endothelial cells did not affect the rate or degree of FGFR1 internalization (Fig. 2F). Thus, FGF2-induced FGFR1 endocytosis in endothelial cells is clathrin-independent.
Fig. 2.
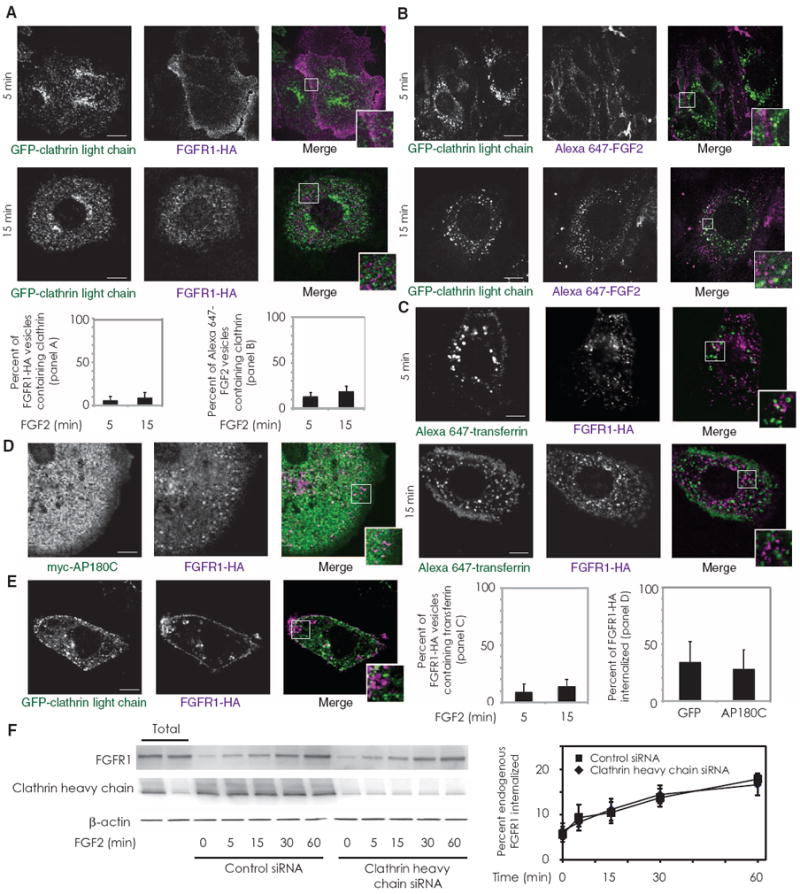
FGFR1 endocytosis is clathrin-independent. (A) RFPECs stably expressing FGFR1-HA were transfected with GFP-tagged clathrin light chain, serum-starved, and treated with FGF2 for the indicated times. Internalized FGFR1-HA was stained with anti-HA antibodies. Twenty-two cells were imaged in three experiments. (B) Primary murine pulmonary endothelial cells were transfected with GFP-tagged clathrin light chain, serum-starved, and treated with Alexa Fluor 647–labeled FGF2 for the indicated times. Thirteen cells were imaged in three experiments. (C) RFPECs with stable FGFR1-HA expression were treated with FGF2 and Alexa Fluor 647–transferrin for the indicated time points. Twenty-one cells were imaged in three experiments. (D) RFPECs stably expressing FGFR1-HA were transfected with myc-tagged AP180C and treated with FGF2, as above. Eleven cells were imaged in two experiments. (E) RFPECs expressing FGFR1 and GFP-tagged clathrin light chain were treated with FGF2 and labeled with anti-HA antibodies. Minimal colocalization of the two constructs was observed except in perinuclear vesicles (white). Sixteen cells were imaged in three experiments. Scale bar, 10 μm in all images. Insets represent magnifications of the indicated regions. (F) Primary murine pulmonary endothelial cells were transfected with either clathrin or control siRNA, serum-starved, exposed to surface biotinylation, and treated with FGF2 for the indicated times. Western blotting was performed to measure biotinylated internalized FGFR1. Quantification represents data from three Western blots.
We next examined whether dynamin 2 is involved in FGFR1 endocytosis. Expression of a dominant-negative (K44A) form of GFP–dynamin 2 did not impede the process of FGFR1 internalization (Fig. 3, A and B), whereas transferrin uptake was inhibited (Fig. 3C). Similarly, siRNAs directed against dynamin 2 in primary murine endothelial cells did not affect FGFR1 uptake (Fig. 3D).
Fig. 3.
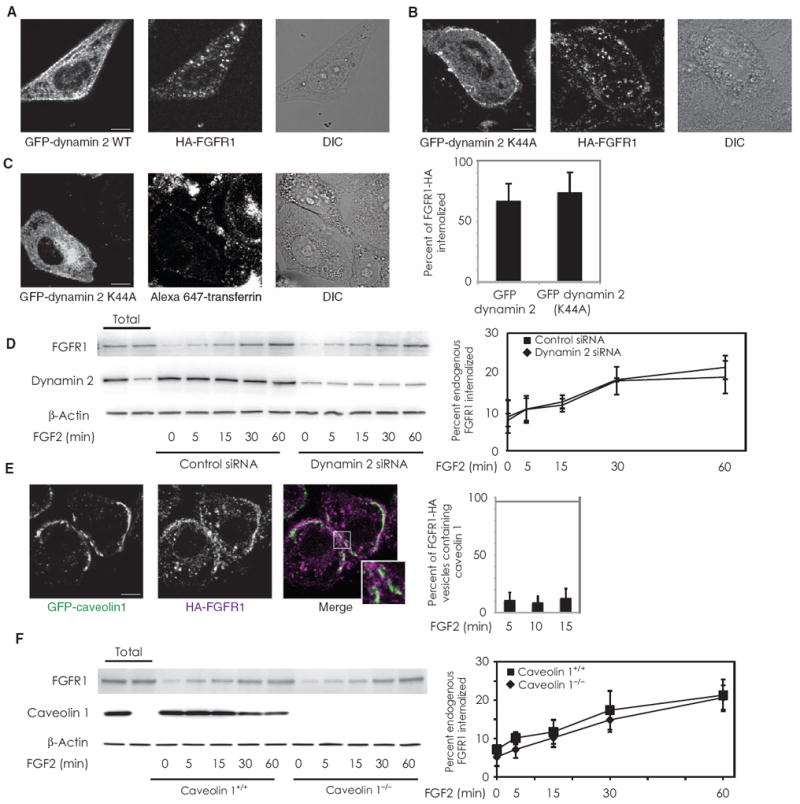
FGFR1 endocytosis is dynamin- and caveolin-independent and involves Rab5 activation. (A and B) Internalized FGFR1-HA was imaged in RFPECs stably expressing FGFR1-HA that were transfected with either GFP-tagged wild-type (WT) dynamin 2 or GFP-tagged dynamin (K44A), serum-starved, and treated with FGF2. Quantification is for 13 cells in each condition over three experiments. (C) Cells transfected with GFP-tagged dynamin 2 (K44A) were prepared as above and incubated with Alexa Fluor 647–conjugated transferrin as a positive control. n = 11 cells over two experiments. (D) Primary murine pulmonary endothelial cells were transfected with either dynamin 2 or control siRNA, serum-starved, exposed to surface biotinylation, and treated with FGF2 for the indicated times. Western blot analysis was performed to measure biotinylated internalized FGFR1. Quantification represents data from threeWestern blots. (E) RFPECs expressing FGFR1-HA were transfected with GFP-tagged caveolin 1 and serum-starved before FGF2-induced receptor endocytosis. Images shown were after 10 min of FGF2 treatment. Quantification represents 12 cells per condition over three experiments. (F) Primary pulmonary endothelial cells from WT or caveolin 1 knockout mice were serum-starved, exposed to surface biotinylation, and treated with FGF2 for the indicated times. Western blot analysis was performed to measure biotinylated internalized FGFR1. Quantification represents data from three Western blots.
Another mode of endocytic trafficking originates from flask-shaped membrane structures denoted caveolae and is associated with the caveolin protein family (35). To exclude the possibility that FGFR1 endocytosis is caveolin-dependent, we examined whether FGFR1 localizes with caveolin 1. We did not observe substantial colocalization between FGFR1-HA and GFP–caveolin 1, particularly at the cell membrane (Fig. 3E). Furthermore, endocytosis of FGFR1 was not affected in pulmonary endothelial cells from caveolin 1 knockout mice (Fig. 3F).
We next addressed the role of macropinocytosis of FGFR in FGF2-mediated signaling by assessing activation of ERK1/2. The GTPase Rab5, which is required for vesicular maturation, is critical for macropinocytosis (26) and FGFR1-mediated activation of ERK1/2 (36). We therefore explored the possibility that Rab5 is specifically required to link FGF2-initiated internalization with downstream signaling. In agreement with previous observations in HeLa cells (36), dominant-negative Rab5 inhibited FGF2-mediated activation of ERK1/2 in endothelial cells (fig. S2A). In endothelial cells, FGFR1-HA colocalized extensively with GFP-Rab5 (Fig. 4A, second panel from the right). Overexpression of cytosolic fluorescent proteins can result in nonspecific targeting toward multiple cytoplasmic compartments; to exclude the possibility that Rab5-GFP and FGFR1-HA colocalization represented an overexpression artifact, we concurrently introduced a red fluorescent protein (RFP) construct as a control for nonspecific fluorescent protein localization. Calculation of the GFP-Rab5/RFP ratio at each pixel demonstrates that GFP-Rab5 was specifically enriched at FGFR1-HA–positive endosomes (Fig. 4B, right-most panel).
Fig. 4.
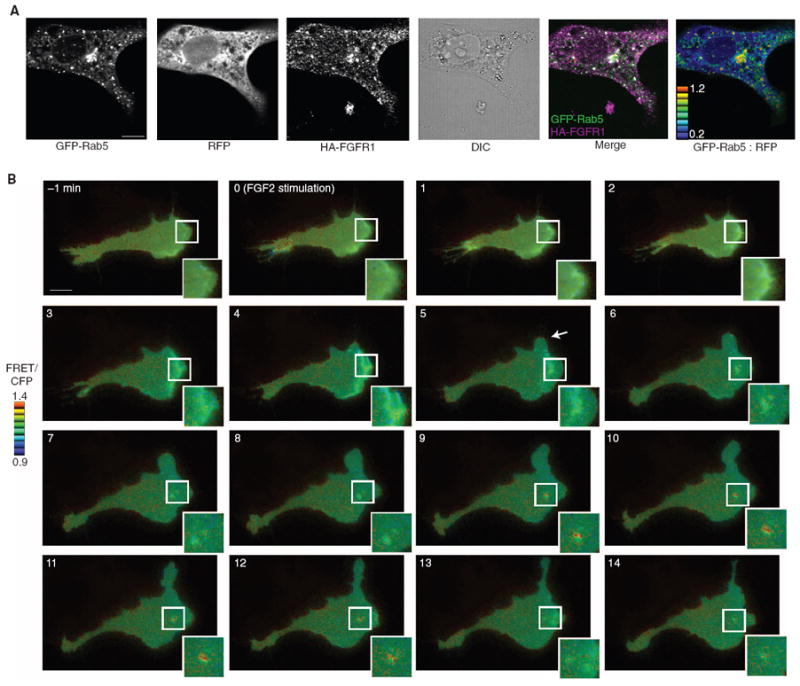
FGFR1 endocytosis involves Rab5 activation. (A) RFPECs stably expressing FGFR1-HA were cotransfected with RFP and GFP-Rab5. Cells were serum-starved, labeled with mouse anti-HA antibody, stimulated with FGF2, and stained with Alexa Fluor 647–conjugated anti-mouse antibodies. Five cells were imaged over two experiments. (B) Live-cell wide-field imaging was performed on RFPECs expressing FGFR1-HA that were transfected with Raichu-Rab5, and then cells were serum-starved and treated with FGF2. FGF2-initiated cellular protrusions are indicated by an arrowhead. Seven cells were imaged over three independent experiments. Scale bar, 10 μm.
Rab5 must be activated after it localizes to macropinosomes to facilitate vesicular maturation to early or signaling endosomes (25). We used fluorescence resonance energy transfer (FRET)–based imaging to visualize the activity of Rab5 (25) in living cells after FGF2 treatment. The formation of cellular protrusions (Fig. 4B, arrowhead) and membrane ruffling (inset) became apparent after 5 min of FGF2 treatment, and the internalization of macropinosomes became prominent by 8 min (Fig. 4B, inset). Rab5 activity became a predominant feature at macropinosomes after 9 min and exhibited an oscillating pattern of activation and deactivation consistent with previous reports of live-cell Rab5 imaging during phagocytosis (25). Vesicular Rab5 activity ceased after 13 min because the vesicle is transported toward recycling or degradative endosomes. These results indicate that Rab5 localizes to FGFR1-HA–positive vesicles, becomes activated at these vesicles, and is necessary for downstream ERK1/2 activation after FGF2 stimulation. Rab5-dependent maturation of nascent vesicles into signaling endosomes thus represents the indispensable link between FGF2-mediated endocytosis and canonical MAPK signaling.
Given that S4 promotes RhoG activation (7) and the role of RhoG in macropinocytosis (37), we next concentrated on the role of S4 in regulating the endocytosis and signaling of FGFR1. The amount of receptor uptake at 15 min was increased by expression of a constitutively active RhoG construct (V12) and decreased by the RhoG-blocking peptide RhoGIP122 (38) (Fig. 5A). To further assess whether RhoG is involved in FGFR1-HA uptake, we introduced wild-type or mutant forms of SGEF, a RhoG-specific guanine nucleotide exchange factor (GEF) (37). After 15 min of FGF2 stimulation, the presence of dominant-negative SGEF inhibited uptake of FGFR1-HA (Fig. 5B).
Fig. 5.
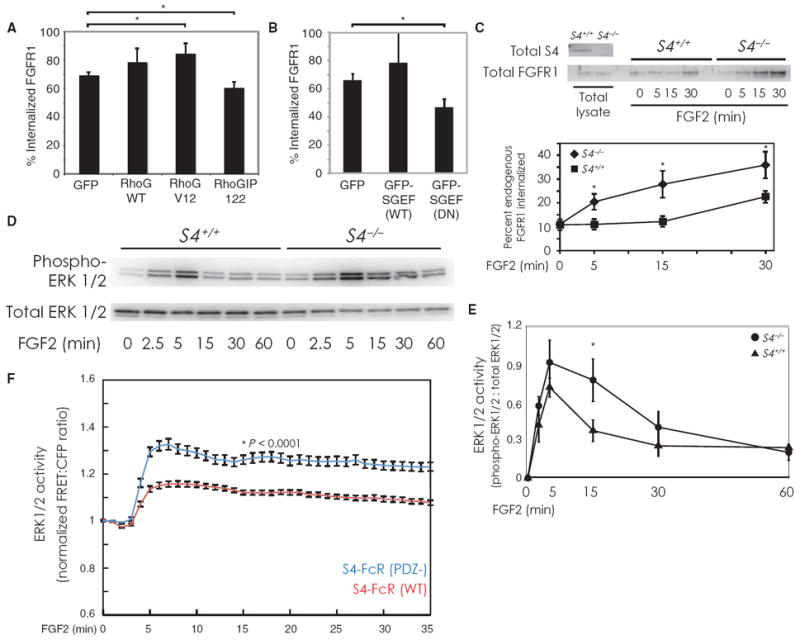
S4 regulates macropinocytosis and ERK activation. (A and B) Endocytosis was quantitatively measured in RFPECs stably expressing FGFR1-HA with flow cytometry. Cells in (A) were transfected with GFP, along with empty vector, RhoG WT, RhoG V12 (a constitutively active form), or the RhoG blocking peptide RhoGIP122. In (B), cells were transfected with either GFP, GFP SGEF (WT) (a RhoG-specific GEF), or GFP-SGEF (DN) (a dominant-negative form). Bars represent percentage of total fluorescently labeled FGFR1-HA internalized after 15 min. Panels represent four samples per condition with 10,000 GFP-positive cells in each sample. Error bars are SEM. Asterisks represent two-tailed P values of 0.05 and 0.067 [(A) lower and upper bracket, respectively] and 0.046 (B) by χ2 test. (C) Primary lung endothelial cells from wild-type or S4 knockout mice were surface-biotinylated and treated with FGF2 for the indicated times. Western blotting was performed to measure internalized biotinylated endogenous FGFR1. Quantification (lower panel) shows the results from three independent experiments. S4 knockout cells exhibit faster FGFR1 endocytosis kinetics than wild-type cells. Paired, two-tailed P values from left to right as follows: 0.005, 0.007, and 0.01. (D) Cells were prepared as in (C). FGF2 was added for the indicated times, after which cells were lysed and subjected to Western blot analysis to measure ERK1/2 phosphorylation. (E) Quantification of Western blotting performed as in (D) (n = 5 experiments). Error bars are SEM. Paired, two-tailed P value at t = 15 min is 0.069. (F) HeLa cells expressing FGFR1-HA were transfected with a FRET-based biosensor and a chimera of the human FcR extracellular domain, the S4 transmembrane domain, and either the wild-type cytoplasmic domain of S4 (S4-FcR; red line; n = 31 cells) or the cytoplasmic lacking the terminal amino acid (PDZ−; blue line; n = 39 cells). Cells were imaged for FRET fluorescence and stimulated with FGF2. The average FRET/CFP ratio over the entire imaged region of a cell was measured and plotted over time. Error bars are SEM. Paired, two-tailed P value at t = 15 min is <0.0001. Traces for individual cells are shown in fig. S2D.
Because RhoG activity is increased in the absence of S4 (7), we hypothesized that macropinocytosis would be constitutively increased in S4−/− endothelial cells. Indeed, uptake of endogenous FGFR1 was accelerated in S4−/− compared to wild-type primary lung endothelial cells (Fig. 5C). Total endogenous FGFR1 abundance did not appear to differ between wild-type and S4 knockout endothelial cells, thus suggesting that the differences in uptake of FGFR1 were not due to variation in receptor abundance (fig. S2B).
Given that S4 forms a signaling complex with its co-receptor FGFR1, we examined the role of S4 in the regulation of FGFR1 endocytosis. Using the S4-FcR chimeric construct (FcR-S4) (27), we quantified FGFR1 uptake upon antibody-mediated chimera oligomerization of the FcR-S4 construct (fig. S2C). S4 assembles a complex that inhibits RhoG activity through S4’s intracellular PDZ domain by binding to synectin at this site. Deleting the terminal cytoplasmic amino acid abrogates the binding ability of the PDZ domain and results in constitutively high RhoG activity (7). We measured the effect of disrupting the PDZ domain in S4 (FcR-PDZ−) and of synectin knockdown, both of which result in increased RhoG activity (7). FcR-S4 uptake was increased in both cases, indicating that synectin and the PDZ domain of S4 (both of which are required for the suppression of RhoG activity) likewise regulate FGFR1 endocytosis. Collectively, these findings demonstrate that S4 controls the rate of FGFR1 and S4 co-receptor macropinocytosis through RhoG.
Consistent with the notion that the internalization of activated receptors into signaling endosomes is required for effective ERK1/2 activation (21, 39), we found that ERK1/2 activation was enhanced and its deactivation was delayed in S4 knockout cells (Fig. 5, D and E). Because previous studies have focused on FGFR1 activation and Rho GTPase activity in epithelial cells, we conducted additional experiments to determine whether the effect of S4 on FGF2-mediated ERK1/2 activation is an endothelial cell–specific phenomenon. Using a well-characterized epithelial cell line (HeLa), we used a FRET-based biosensor to measure ERK activity (40) after FGF2 stimulation. Compared to Western blotting, this approach affords greater temporal resolution and the ability to monitor activity changes in living cells. Cells expressing the form of S4-FcR lacking the PDZ-binding motif (PDZ−) exhibited a prolonged time course of ERK1/2 activation compared with cells expressing the form of S4-FcR with the full-length cytoplasmic tail. These data confirm our Western blot analyses of ERK1/2 activation and demonstrate that S4’s influence over ERK activity is not unique to endothelial cells.
Our findings demonstrate that S4 directs signaling and endocytosis of FGFR1 through RhoG- and Rab5-dependent macropinocytosis of the ligand and co-receptor complex. Abrogation of S4 signaling (either by genetic knockout or by mutation of the PDZ domain) leads to heightened RhoG activity and a higher basal rate of receptor uptake, resulting in enhanced ERK1/2 activation and slower ERK1/2 deactivation kinetics. Thus, S4 controls the duration of ERK1/2 activation. These results establish a unified model by which S4 and FGFR1 influence the cell’s response to FGF2 (Fig. 6).
Fig. 6.
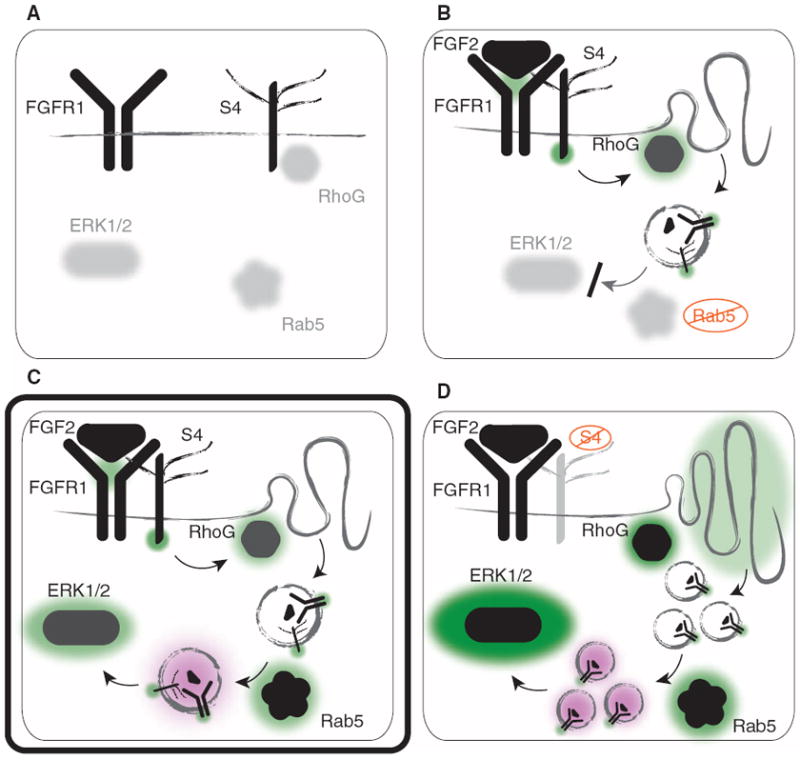
S4 controls FGF2-mediated signaling through RhoG-dependent macropinocytosis of the FGFR1-S4-FGF2 signaling complex. (A) Before FGFR1 and S4 are activated by FGF2, RhoG is kept inactive by S4 (in complex with synectin and RhoGDI1), and Rab5 activity and phosphorylation of ERK1/2 are low at baseline. (B) Upon ligand binding, FGFR1 and S4 form a signaling complex, and independent signaling pathways are initiated by each co-receptor: Tyrosine kinase phosphorylation of FGFR1 activates several parallel signaling networks, and S4 releases inactive RhoG to become activated by GEFs such as SGEF. This induces membrane ruffling and subsequent macropinocytosis of the entire receptor complex. In the absence of functional Rab5, nascent vesicles cannot mature and become fully functional signaling endosomes, which leads to inadequate activation of the MAPK pathway (as indicated by diminished phosphorylation of ERK1/2). (C) Physiological signaling (designated by a double-border) is restored as functional Rab5 localizes to nascent macropinosomes containing FGFR1-S4-FGF2 and becomes activated, facilitating vesicle maturation. This permits effective activation of the MAPK pathway (as indicated by ERK1/2 phosphorylation). (D) In the absence of S4 or S4’s cytoplasmic PDZ domain, RhoG activity is high at baseline, causing increased membrane ruffling and macropinocytosis. Enhanced macropinocytosis leads to increased co-receptor internalization. Rab5 promotes vesicular maturation, leading to potentiated ERK1/2 activity with slower deactivation kinetics upon FGF2 stimulation. Green and black denote activation, and purple denotes the process of vesicular maturation into early/signaling endosomes.
DISCUSSION
Receptor endocytosis, trafficking, and signaling are inextricably linked processes that influence the cellular response to growth factor stimulation. In the endothelium, FGF signaling promotes cell survival and vascular stability (41). Although the magnitude and kinetics of the involved signaling networks are stringently regulated, the mechanisms by which these parameters are modulated have thus far remained poorly understood. Here, we show that two FGFRs—a tyrosine kinase receptor FGFR1, the predominant FGFR in endothelial cells (42), and a non–tyrosine kinase receptor S4—cooperate to modulate MAPK activation. The cooperation is achieved by S4-dependent modulation of FGFR1 endocytosis through RhoG- and Rab5-dependent macropinocytosis.
Previous studies of FGFR trafficking have relied on indirect evidence from FGFR-GFP fusion constructs (43) or immunohistochemistry with antibodies against the cytoplasmic domains of FGFR (44) to determine subcellular localization. However, neither method can distinguish between FGFRs originating at the cell membrane and those in nascent cytoplasmic pools of recycling endosomes. Likewise, techniques of labeled ligand tracking do not effectively isolate particular FGFR isoforms because these ligands bind to, and are internalized with, all four high-affinity tyrosine kinase receptors and heparan sulfate proteoglycans (45).
We focused on S4-dependent activation of RhoG because RhoG promotes membrane ruffling and macropinocytosis (37, 46). The effects of endocytic regulation upon ERK1/2 activation highlight the importance of S4 and RhoG in FGF signaling. In agreement with enhanced activation of RhoG and, consequently, of macropinocytosis in S4−/− cells, we observed enhanced ERK activation and delayed deactivation with both a FRET-based approach and conventional Western blotting. These findings contrast with a recent study using S4 siRNA in human umbilical vein endothelial cells (HUVECs), which showed by Western blot analysis that activation of ERK is diminished in the absence of S4 (11). These discrepancies could result from a nonspecific siRNA effect on relevant signaling molecules. They might also represent disparities in the signaling between endothelial cells derived from different tissues or species. In our study, we used two endothelial models (primary endothelial cells derived from S4+/+ and S4−/− mice and immortalized rat endothelial cells with S4 chimeric constructs to isolate the effect of its critical PDZ-binding domain) and both Western blotting analysis and a more sensitive FRET-based quantitative approach to independently characterize the role of S4 in shaping the dynamics of ERK1/2 signaling.
Another controversy involves the endocytic fate of activated FGFR1. Using endothelial cells, we observed FGFR1 uptake exclusively through a macropinocytic pathway. However, other studies have shown that FGFR1 is also able to undergo clathrin-mediated endocytosis. For example, a synaptotagmin-related protein E-Syt2 has been reported to be involved in FGFR1 uptake through clathrin-mediated endocytosis in Xenopus embryos and human embryonic kidney (HEK) 293T cells (47). Another study with U2OS cells likewise suggested that uptake of FGF1 is diminished by clathrin knockdown, although receptor uptake was not directly measured (48). Finally, a modestly increased surface–to–cytoplasmic localization ratio of enhanced GFP (EGFP)–fused FGFR1 in PC12 cells was observed after inhibition of clathrin-mediated endocytosis with chlorpromazine (49). However, other studies support the present finding of clathrin-independent uptake of FGFR, including the observation of FGFR4 in caveolin-containing compartments upon FGF1 stimulation (50) and the lack of effect of a dominant-negative Eps15 construct on the nuclear translocation of FGFR1 (44).
Three interpretations potentially reconcile these findings. First, depending on cell type, both clathrin-dependent endocytosis and macropinocytosis might represent predominant modes of receptor uptake. Second, both pathways may exist as endocytic alternatives within the same cell, depending on ligand type and concentration, specific co-receptor association, or other protein factors that influence intracellular fate. Other transmembrane receptors, such as the transforming growth factor–β receptors (TGFβRs), can be internalized by both pathways (51). Similarly, the epidermal growth factor receptor (EGFR) is internalized by different pathways, including clathrin-dependent endocytosis [reviewed in (52)] and a pathway that depends on membrane ruffling (53), potentially mirroring the endocytic possibilities available to FGFR1. Finally, clathrin-mediated endocytosis might represent an early stage of receptor endocytosis, whereas macropinocytosis might encompass a delayed, more inclusive mechanism of receptor internalization.
With these possibilities in mind, our studies in primary and immortalized endothelial cells comprehensively address the endocytic regulation of FGFR1 signaling and suggest that clathrin- and caveolin-mediated endocytosis are dispensable, whereas macropinocytosis represents the predominant mode of both endocytosis and signal modulation of FGFR1 by S4. Similarly, S4 can initiate caveolin-dependent endocytosis of integrins, another class of S4 co-receptors (54). The study further highlights S4’s role as a regulator of co-receptor internalization, in this case by a separate endocytic pathway. Thus, it appears that S4 can direct various endocytic fates for receptors that are determined by specific co-receptor binding.
Lipid rafts have been reported to be essential components of macropinocytosis (55, 56). Previous studies have yielded indirect evidence to support the notion that cholesterol-enriched membrane microdomains are required for effective FGFR1 signaling (57), supporting our findings that FGFR1 localizes to lipid raft regions of the cell membrane and that these microdomains are required for receptor uptake.
Finally, in our studies, Rab5 emerged as a salient feature of FGF2-mediated signaling and endocytosis. Rab5 is a requisite component of macropinocytosis (26) and is required for the maturation of nascent vesicles (25), leading to appropriate sequential trafficking. Rab5 is an essential component of FGF signaling (36) and nuclear translocation of FGFR1 (44). Using live-cell imaging, we demonstrated the ability of Rab5 to specifically localize to vesicles containing FGFR1, S4, and FGF2 and to become activated at these nascent macropinosomes, and confirmed the requirement for FGF2-mediated signal transduction through the MAPK pathway. Thus, Rab5 represents an indispensable link between receptor endocytosis and signaling. Its role in vesicular maturation suggests that, as has been speculated for various systems involving ERK activation, FGFR1-mediated signal transduction is not optimally active at the membrane, but instead from within a mature signaling endosome (21, 58).
Thus, FGFR1 and S4 are independent receptors with co-dependent signaling roles. S4-mediated modulation of FGF2-induced FGFR1 endocytosis and MAPK signaling represents a previously unappreciated mechanism of crosstalk between the two receptors binding the same ligand.
METHODS
Cell culture and transfection
RFPECs and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Cambrex) containing 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml) (Mediatech). RFPECs with stable expression of FGFR1-HA were selected by culturing cells in G418 (1 mg/ml) with subsequent fluorescence-activated cell sorting. All experiments were performed with these pooled cell populations. RFPECs with stable expression of the S4-FcR and S4-FcR (PDZ−) chimeras (27) were used for S4 clustering studies. The S4-FcR (PDZ−) mutant includes a single amino acid truncation at the cytoplasmic terminus (59). Primary pulmonary or cardiac murine endothelial cells from wild-type and S4 knockout mice were isolated by harvesting murine lungs or hearts and subjecting them to fine mincing and digestion in 25 ml of 0.2% (w/v) collagenase at 37°C for 45 min. The crude cell preparation was pelleted and resuspended in Dulbecco’s PBS (DPBS). The cell suspension was incubated with PECAM-1 (platelet endothelial cell adhesion molecule 1)–coated beads [immunoglobulin G (IgG) Dynal beads, Dynal Corporation] at room temperature for 10 min with end-over-end rotation. The bead-bound cells were recovered with a magnetic separator, washed with DMEM containing 20% FBS, suspended in 12 ml of complete culture medium [DMEM containing 20% fetal calf serum supplemented with heparin (100 μg/ml), endothelial cell growth factor growth supplement (100 μg/ml, Biomedical Technologies), nonessential amino acids, sodium pyruvate, l-glutamine, and antibiotics at standard concentrations], and then plated in fibronectin-coated 75-cm2 tissue culture flasks. Transfection of RFPECs was performed with FuGENE 6 (Roche) or Amaxa (Amaxa Inc.), and transfection of HeLa cells was performed with 293Fectin (Invitrogen) according to the manufacturers’ protocols.
Complementary DNA constructs, antibodies, and reagents
FGFR1-HA was created with complementary DNA for human FGFR1 IIIc. The HA sequence (5′-TACCCATACGAtGTtCCAGATTACGCT-3′) was cloned distal to the first extracellular immunoglobulin loop of the receptor by Hind III and Not I restriction digestion and subsequently confirmed by nucleotide sequencing.
Mouse monoclonal anti-HA IgG was obtained from Covance. Rabbit monoclonal phospho-ERK1/2 and mouse monoclonal total ERK1/2 antibodies were from Cell Signaling. Rabbit polyclonal β-actin antibodies were from Abcam. Rabbit polyclonal anti-FGFR1 was purchased from Santa Cruz. GFP-tagged clathrin light chain was a gift from J. Keen (Thomas Jefferson University). Myc-tagged AP180C was a gift from H. McMahon (Scripps Institute). Wild-type dynamin 2 and K44A constructs were a gift from M. McNiven (Mayo Clinic). GFP-tagged caveolin 1 was a gift from R. Stan (Dartmouth Medical School). Alexa Fluor 647–labeled transferrin and Texas Red–labeled 70-kD dextran were from Invitrogen. All Rab5 constructs and the EKAR (ERK activity reporter) FRET biosensor were described previously (25, 40). RhoG wild type, V12, and A37 mutants were a gift from H. Katoh (Kyoto University). RhoGIP122 was a gift from A. Blangy (Montpelier University). GFP-SGEF and GFP-SGEF (DN) were a gift from M. Schwartz (Yale University). GFP-GPI was a gift from S. Mayor (National Centre for Biological Sciences, India). Methyl β-cyclodextrin and amiloride were purchased from Sigma-Aldrich. Lipid rafts were labeled with cholera toxin B from Invitrogen according to the manufacturer’s protocols.
FGF2 was labeled with Alexa Fluor 488 (Invitrogen) as follows. To protect the S4 binding sites, we adsorbed FGF2 (1 mg) to 0.5-ml bed volume heparin-Sepharose beads (Amersham Biosciences). The beads were washed with ice-cold 0.1 M NaHCO3 and resuspended in 1 ml of ice-cold 0.1 M NaHCO3 (pH 8.2). Alexa Fluor 647 succinimidyl ester was added (0.5 mg in 0.2 ml of 0.1 M NaHCO3) and the mixture was shaken for 1 hour at 4°C. The supernatant was drained and the column was washed with 4 ml of tris-buffered saline (TBS) and 2 ml of 10 mM tris/0.3 M NaCl (pH 7.5). Labeled FGF2 was eluted with 2 ml of 10 mM tris/2 M NaCl (pH 7.5) and desalted with two Hi-Trap desalting cartridges connected in series and elution with tris-buffered saline. The FGF2-containing fraction was concentrated in an Amicon 10-kD centrifugal filter (Millipore) and protein concentration was determined by spectrophotometry. The dye/FGF2 labeling ratio was 0.5:1, which was intended to be low so as not to interfere with the biological function of FGF2.
Western blotting and biotinylation studies
For Western blotting, cells were placed on ice and lysed with ice-cold NP-40 buffer containing Complete Mini Protease Inhibitor Cocktail (Roche). The lysate was added to sample buffer (Pierce), boiled for 5 min, and analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE). For all Western blots, 12% polyacrylamide gels (Bio-Rad) were used.
Biotinylation studies were performed as follows. Primary murine heart endothelial cells were plated on fibronectin-coated dishes, grown to confluence, and starved 24 hours before assay. The cells were placed on ice and were treated with biotin for 30 min before being washed with ice-cold medium three times. Cells were then treated with FGF2 (50 ng/ml) and returned to 37°C for the indicated times. Biotin remaining on the cell surface was then stripped by reducing with glutathione, and glutathione was then quenched by rinsing with PBS containing iodoacetamide (5 mg/ml). Cells were then lysed with NP-40 buffer containing protease inhibitors, as above. The lysate was incubated with 75 μl of NeutrAvidin agarose overnight at 4°C. The agarose beads were then washed three times with PBS, boiled in sample buffer, and analyzed by SDS-PAGE.
Endocytosis assays
For microscopic imaging of endocytosis, cells were plated on glass-bottomed dishes, grown to 75% confluence, transfected 48 hours before assay with the indicated constructs, and serum-starved in medium containing 0.5% FBS for 24 hours before assay. Cells were treated with FGF2 (50 ng/ml) and anti-HA mouse IgG (1 μg/ml) to initiate endocytosis for the indicated time points. They were then placed on ice and washed once with ice-cold acidic PBS (pH 2.5) and three times with ice-cold PBS. Fixed sections were prepared by transfer of cells growing on fibronectin-coated glass-bottomed dishes to ice, washing once with ice-cold PBS, and fixation with 4% parafomaldehyde/PBS at room temperature for 10 min. Samples were then washed three times with PBS and incubated with 0.1% Triton X-700 for 10 min for permeabilization where indicated. They were blocked with 1% bovine serum albumin (BSA)/PBS for 30 min at room temperature before antibody incubation. Primary antibody incubation with anti-mouse IgG (1 μg/ml) and other indicated antibodies was 1 hour at room temperature followed by three washes with PBS, a secondary antibody incubation (as indicated) for 1 hour at room temperature, and three successive washes with PBS before imaging.
Quantitative analyses of microscopy images were performed by imaging cells under identical image acquisition settings for each experiment. Regions of interest were selected, and mean luminance was measured as an estimate of endocytosed fluorophore. Standard statistical analyses were then performed as described in each condition. Results were normalized to positive control cells with maximal endocytosis measured at 45 min.
For quantitative analyses of endocytosis by flow cytometry, cells were plated on fibronectin-coated dishes, grown to 75% confluence, transfected 48 hours before assay with the indicated constructs, and serum-starved in medium containing 0.5% FBS for 24 hours before assay. Cells were placed on ice and incubated with anti-HA mouse IgG (1 μg/ml) for 10 min to label cell surface FGFR1-HA. They were then washed three times with ice-cold PBS and incubated with FGF2 (50 ng/ml) and anti-mouse IgG (1 μg/ml) at 37°C for the indicated times. The cells were then returned to ice and washed once with ice-cold acidic PBS (pH 2.5) and three times with ice-cold PBS. They were then trypsinized, incubated with PBS containing 5% BSA, and analyzed by flow cytometry.
Microscopy
Live-cell imaging of FRET probes was performed by excitation of cyan fluorescent protein (CFP) and measurement of both CFP and yellow fluorescent protein (YFP) emission in cells transfected with the indicated probes (60). These studies were performed with an Olympus IX-81 environment-controlled (set to 37°C) wide-field microscope with an Olympus oil immersion 60× objective [numerical aperture (NA) = 1.4]. MetaMorph software was used for acquisition. The filters used for the dual-emission imaging were obtained from Omega Optical: an XF1071 (440AF21) excitation filter, an XF2034 (455DRLP) dichroic mirror, and two emission filters [XF3075 (480AF30) for CFP and XF3079 (535AF26) for FRET]. The imaging medium was phenol red–free DMEM/F12 (1:1 ratio) supplemented with 1% BSA and covered by mineral oil (Sigma-Aldrich) to prevent evaporation. The camera used for these studies was a CoolSNAP HQ model (Roper Scientific).
Confocal imaging of fixed cells was performed at room temperature with PBS as imaging medium by means of an Olympus FV1000 system equipped with an Olympus oil immersion 60× objective (NA = 1.35) with Olympus FluoView software for acquisition. All figures were assembled with Adobe Photoshop and Illustrator software.
Quantitative analyses
Western blots were scanned with either the G:Box (Syngene) or the Odyssey (Li-Cor) and quantified with GelEval software (FrogDance). FRET ratio and total fluorescence ratio analyses were performed with MetaMorph software.
Supplementary Material
Fig. S1. HA-FGFR1 construct validation.
Fig. S2. Endocytosis and ERK1/2 activation in RFPECs expressing S4-FcR.
Acknowledgments
We thank K. Aoki, Y. Kamioka, and N. Komatsu (Kyoto University); R. Stan (Dartmouth Medical School); and W. Sessa, J. Rhodes, M. Murakami, and F. Corti (Yale Cardiovascular Research Center) for their expertise, invaluable assistance, and thoughtful contributions. We also thank all members of the Simons and Matsuda laboratories for their guidance. Funding: This work was supported by the Global Center of Excellence Postdoctoral Fellowship and a Grant-in-Aid for Scientific Research on Innovative Areas “Fluorescence Live Imaging” (number 22113002) from the Ministry of Education, Sports, Science and Technology, Japan (A.E., M.M.). A.Y. and T.X.Z. were supported by the Dartmouth College Presidential Scholarship. M.S. is supported by the National Heart, Lung, and Blood Institute (HL62289 and HL53793).
Footnotes
Author contributions: A.E., M.S., and M.M. developed the project and designed the experiments. A.E. cloned the FGFR1-HA construct and performed all microscopy. A.E., A.L., T.X.Z., A.Y., and E.T. performed all other experiments. A.E., M.S., and M.M. wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Elfenbein A, Simons M. Auxiliary and autonomous proteoglycan signaling networks. Methods Enzymol. 2010;480:3–31. doi: 10.1016/S0076-6879(10)80001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan MR, Humphries MJ, Bass MD. Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol. 2007;8:957–969. doi: 10.1038/nrm2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tkachenko E, Rhodes JM, Simons M. Syndecans: New kids on the signaling block. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 4.Bass MD, Humphries MJ. Cytoplasmic interactions of syndecan-4 orchestrate adhesion receptor and growth factor receptor signalling. Biochem J. 2002;368:1–15. doi: 10.1042/BJ20021228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao Y, Li M, Chen W, Simons M. Synectin, syndecan-4 cytoplasmic domain binding PDZ protein, inhibits cell migration. J Cell Physiol. 2000;184:373–379. doi: 10.1002/1097-4652(200009)184:3<373::AID-JCP12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 6.Murakami M, Horowitz A, Tang S, Ware JA, Simons M. Protein kinase C (PKC) δ regulates PKCα activity in a Syndecan-4-dependent manner. J Biol Chem. 2002;277:20367–20371. doi: 10.1074/jbc.M202501200. [DOI] [PubMed] [Google Scholar]
- 7.Elfenbein A, Rhodes JM, Meller J, Schwartz MA, Matsuda M, Simons M. Suppression of RhoG activity is mediated by a syndecan 4–synectin–RhoGDI1 complex and is reversed by PKCα in a Rac1 activation pathway. J Cell Biol. 2009;186:75–83. doi: 10.1083/jcb.200810179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Partovian C, Ju R, Zhuang ZW, Martin KA, Simons M. Syndecan-4 regulates subcellular localization of mTOR complex2 and Akt activation in a PKCα-dependent manner in endothelial cells. Mol Cell. 2008;32:140–149. doi: 10.1016/j.molcel.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chittenden TW, Claes F, Lanahan AA, Autiero M, Palac RT, Tkachenko EV, Elfenbein A, Ruiz de Almodovar C, Dedkov E, Tomanek R, Li W, Westmore M, Singh JP, Horowitz A, Mulligan-Kehoe MJ, Moodie KL, Zhuang ZW, Carmeliet P, Simons M. Selective regulation of arterial branching morphogenesis by synectin. Dev Cell. 2006;10:783–795. doi: 10.1016/j.devcel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Dedkov EI, Thomas MT, Sonka M, Yang F, Chittenden TW, Rhodes JM, Simons M, Ritman EL, Tomanek RJ. Synectin/syndecan-4 regulate coronary arteriolar growth during development. Dev Dyn. 2007;236:2004–2010. doi: 10.1002/dvdy.21201. [DOI] [PubMed] [Google Scholar]
- 11.Matsui Y, Ikesue M, Danzaki K, Morimoto J, Sato M, Tanaka S, Kojima T, Tsutsui H, Uede T. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ Res. 2011;108:1328–1339. doi: 10.1161/CIRCRESAHA.110.235689. [DOI] [PubMed] [Google Scholar]
- 12.Ishiguro K, Kadomatsu K, Kojima T, Muramatsu H, Iwase M, Yoshikai Y, Yanada M, Yamamoto K, Matsushita T, Nishimura M, Kusugami K, Saito H, Muramatsu T. Syndecan-4 deficiency leads to high mortality of lipopolysaccharide-injected mice. J Biol Chem. 2001;276:47483–47488. doi: 10.1074/jbc.M106268200. [DOI] [PubMed] [Google Scholar]
- 13.Alexopoulou AN, Multhaupt HAB, Couchman JR. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol. 2007;39:505–528. doi: 10.1016/j.biocel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Matthews HK, Marchant L, Carmona-Fontaine C, Kuriyama S, Larraín J, Holt MR, Parsons M, Mayor R. Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development. 2008;135:1771–1780. doi: 10.1242/dev.017350. [DOI] [PubMed] [Google Scholar]
- 15.Nugent MA, Iozzo RV. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:115–120. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- 16.Bass MD, Morgan MR, Humphries MJ. Syndecans shed their reputation as inert molecules. Sci Signal. 2009;2:pe18. doi: 10.1126/scisignal.264pe18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer L, Schaefer RM. Proteoglycans: From structural compounds to signaling molecules. Cell Tissue Res. 2010;339:237–246. doi: 10.1007/s00441-009-0821-y. [DOI] [PubMed] [Google Scholar]
- 18.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: An emerging feature of cancer. Nat Rev Cancer. 2008;8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 19.Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: Domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–385. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 21.Teis D, Wunderlich W, Huber LA. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev Cell. 2002;3:803–814. doi: 10.1016/s1534-5807(02)00364-7. [DOI] [PubMed] [Google Scholar]
- 22.Sandilands E, Akbarzadeh S, Vecchione A, McEwan DG, Frame MC, Heath JK. Src kinase modulates the activation, transport and signalling dynamics of fibroblast growth factor receptors. EMBO Rep. 2007;8:1162–1169. doi: 10.1038/sj.embor.7401097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: Achieving specificity in membrane traffic. Proc Natl Acad Sci U S A. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 25.Kitano M, Nakaya M, Nakamura T, Nagata S, Matsuda M. Imaging of Rab5 activity identifies essential regulators for phagosome maturation. Nature. 2008;453:241–245. doi: 10.1038/nature06857. [DOI] [PubMed] [Google Scholar]
- 26.Lanzetti L, Palamidessi A, Areces L, Scita G, Di Fiore PP. Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature. 2004;429:309–314. doi: 10.1038/nature02542. [DOI] [PubMed] [Google Scholar]
- 27.Tkachenko E, Simons M. Clustering induces redistribution of syndecan-4 core protein into raft membrane domains. J Biol Chem. 2002;277:19946–19951. doi: 10.1074/jbc.M200841200. [DOI] [PubMed] [Google Scholar]
- 28.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tkachenko E, Lutgens E, Stan RV, Simons M. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J Cell Sci. 2004;117:3189–3199. doi: 10.1242/jcs.01190. [DOI] [PubMed] [Google Scholar]
- 30.Zajchowski LD, Robbins SM. Lipid rafts and little caves. Compartmentalized signalling in membrane microdomains. Eur J Biochem. 2002;269:737–752. doi: 10.1046/j.0014-2956.2001.02715.x. [DOI] [PubMed] [Google Scholar]
- 31.Swanson J. Fluorescent labeling of endocytic compartments. Methods Cell Biol. 1989;29:137–151. doi: 10.1016/s0091-679x(08)60192-2. [DOI] [PubMed] [Google Scholar]
- 32.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMahon HT. Endocytosis: An assembly protein for clathrin cages. Curr Biol. 1999;9:R332–R335. doi: 10.1016/s0960-9822(99)80206-1. [DOI] [PubMed] [Google Scholar]
- 35.Stan RV. Structure of caveolae. Biochim Biophys Acta. 2005;1746:334–348. doi: 10.1016/j.bbamcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Vecchione A, Cooper HJ, Trim KJ, Akbarzadeh S, Heath JK, Wheldon LM. Protein partners in the life history of activated fibroblast growth factor receptors. Proteomics. 2007;7:4565–4578. doi: 10.1002/pmic.200700615. [DOI] [PubMed] [Google Scholar]
- 37.Ellerbroek SM, Wennerberg K, Arthur WT, Dunty JM, Bowman DR, DeMali KA, Der C, Burridge K. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell. 2004;15:3309–3319. doi: 10.1091/mbc.E04-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blangy A, Vignal E, Schmidt S, Debant A, Gauthier-Rouvière C, Fort P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of RhoG. J Cell Sci. 2000;113(Pt 4):729–739. doi: 10.1242/jcs.113.4.729. [DOI] [PubMed] [Google Scholar]
- 39.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, Matsuda M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647–4656. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M. The FGF system has a key role in regulating vascular integrity. J Clin Invest. 2008;118:3355–3366. doi: 10.1172/JCI35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antoine M, Wirz W, Tag CG, Mavituna M, Emans N, Korff T, Stoldt V, Gressner AM, Kiefer P. Expression pattern of fibroblast growth factors (FGFs), their receptors and antagonists in primary endothelial cells and vascular smooth muscle cells. Growth Factors. 2005;23:87–95. doi: 10.1080/08977190500096004. [DOI] [PubMed] [Google Scholar]
- 43.Dunham-Ems SM, Pudavar HE, Myers JM, Maher PA, Prasad PN, Stachowiak MK. Factors controlling fibroblast growth factor receptor-1’s cytoplasmic trafficking and its regulation as revealed by FRAP analysis. Mol Biol Cell. 2006;17:2223–2235. doi: 10.1091/mbc.E05-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bryant DM, Wylie FG, Stow JL. Regulation of endocytosis, nuclear translocation, and signaling of fibroblast growth factor receptor 1 by E-cadherin. Mol Biol Cell. 2005;16:14–23. doi: 10.1091/mbc.E04-09-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reilly JF, Mizukoshi E, Maher PA. Ligand dependent and independent internalization and nuclear translocation of fibroblast growth factor (FGF) receptor 1. DNA Cell Biol. 2004;23:538–548. doi: 10.1089/dna.2004.23.538. [DOI] [PubMed] [Google Scholar]
- 46.Meller J, Vidali L, Schwartz MA. Endogenous RhoG is dispensable for integrin-mediated cell spreading but contributes to Rac-independent migration. J Cell Sci. 2008;121:1981–1989. doi: 10.1242/jcs.025130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jean S, Mikryukov A, Tremblay MG, Baril J, Guillou F, Bellenfant S, Moss T. Extended-synaptotagmin-2 mediates FGF receptor endocytosis and ERK activation in vivo. Dev Cell. 2010;19:426–439. doi: 10.1016/j.devcel.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 48.Haugsten EM, Malecki J, Bjørklund SMS, Olsnes S, Wesche J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol Biol Cell. 2008;19:3390–3403. doi: 10.1091/mbc.E07-12-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hausott B, Rietzler A, Vallant N, Auer M, Haller I, Perkhofer S, Klimaschewski L. Inhibition of fibroblast growth factor receptor 1 endocytosis promotes axonal branching of adult sensory neurons. Neuroscience. 2011;188:13–22. doi: 10.1016/j.neuroscience.2011.04.064. [DOI] [PubMed] [Google Scholar]
- 50.Citores L, Wesche J, Kolpakova E, Olsnes S. Uptake and intracellular transport of acidic fibroblast growth factor: Evidence for free and cytoskeleton-anchored fibroblast growth factor receptors. Mol Biol Cell. 1999;10:3835–3848. doi: 10.1091/mbc.10.11.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 52.Madshus IH, Stang E. Internalization and intracellular sorting of the EGF receptor: A model for understanding the mechanisms of receptor trafficking. J Cell Sci. 2009;122:3433–3439. doi: 10.1242/jcs.050260. [DOI] [PubMed] [Google Scholar]
- 53.Orth JD, Krueger EW, Weller SG, McNiven MA. A novel endocytic mechanism of epidermal growth factor receptor sequestration and internalization. Cancer Res. 2006;66:3603–3610. doi: 10.1158/0008-5472.CAN-05-2916. [DOI] [PubMed] [Google Scholar]
- 54.Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, Martin P, Humphries MJ. A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev Cell. 2011;21:681–693. doi: 10.1016/j.devcel.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 56.Jones AT. Macropinocytosis: Searching for an endocytic identity and role in the uptake of cell penetrating peptides. J Cell Mol Med. 2007;11:670–684. doi: 10.1111/j.1582-4934.2007.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ridyard MS, Robbins SM. Fibroblast growth factor-2-induced signaling through lipid raft-associated fibroblast growth factor receptor substrate 2 (FRS2) J Biol Chem. 2003;278:13803–13809. doi: 10.1074/jbc.M210245200. [DOI] [PubMed] [Google Scholar]
- 58.Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: Evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horowitz A, Tkachenko E, Simons M. Fibroblast growth factor-specific modulation of cellular response by syndecan-4. J Cell Biol. 2002;157:715–725. doi: 10.1083/jcb.200112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aoki K, Nakamura T, Fujikawa K, Matsuda M. Local phosphatidylinositol 3,4,5-trisphosphate accumulation recruits Vav2 and Vav3 to activate Rac1/Cdc42 and initiate neurite outgrowth in nerve growth factor-stimulated PC12 cells. Mol Biol Cell. 2005;16:2207–2217. doi: 10.1091/mbc.E04-10-0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. HA-FGFR1 construct validation.
Fig. S2. Endocytosis and ERK1/2 activation in RFPECs expressing S4-FcR.