

Abstract
Alveolar macrophages play an essential role in clearing bacteria from the lower airway, as the resident phagocyte alveolar macrophages must both phagocytose and kill bacteria, and if unable to do this completely must co-ordinate an inflammatory response. The decision to escalate the inflammatory response represents the transition between subclinical infection and the development of pneumonia. Alveolar macrophages are well equipped to phagocytose bacteria and have a large phagolysosomal capacity in which ingested bacteria are killed. The rate-limiting step in control of extracellular bacteria, such as Streptococcus pneumoniae, is the capacity of alveolar macrophages to kill ingested bacteria. Therefore, alveolar macrophages complement canonical microbicidal strategies with an additional level of apoptosis-associated killing to help kill ingested bacteria.
Keywords: bacteria, killing, macrophage, Streptococcus pneumoniae
Macrophage adaptation to function
Alveolar macrophages are the resident phagocytes in the lung [1]. They are mononuclear phagocytes that, potentially, can originate from either the replication of resident post-mitotic macrophages or through the migration of bone marrow progenitors and peripheral blood monocytes that are recruited into tissue, where they develop further in response to environmental imprinting [2]. Macrophage transcriptional profiles are highly responsive to the prevailing stimuli and a range of phenotypes are possible that are often grouped as M1, M2a, M2b and M2c types but which, in reality, are likely to result in multiple variations whose real importance lies in adaption of the macrophage to a range of specialized functions [3]. Inflammatory macrophages are adapted to anti-microbial host defence producing proinflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-12 as well as reactive oxygen species (ROS) and nitric oxide (NO), but have the potential to induce tissue injury. The M2 alternatively activated macrophages are characterized by high levels of IL-10 expression, and although some subsets contribute to defence against parasites, subsets also play roles in tissue homeostasis through tissue remodelling and repair and exert anti-inflammatory and immunoregulatory roles [4].
In the lung the alveolar macrophage is the resident phagocyte in the air space, and these have been viewed traditionally as a cell that results from the recruitment of peripheral blood monocytes into the lung, which undergoes differentiation through intermediate lung phenotypes to become a highly specialized macrophage [5]. However, studies suggesting a predominantly monocyte origin for alveolar macrophages have involved the use of irradiation, which has depleted pulmonary macrophage populations capable of replication. When radiosensitive blood monocyte precursors are depleted using methods whose effect is localized to the bone marrow, but that do not alter proliferation of macrophage populations at remote anatomical sites, the numbers of resident alveolar macrophages and indeed pulmonary proliferation of macrophages are unaltered [6]. Murine models employing conditional cell ablation and adoptive cell transfer have refined our understanding by demonstrating that alveolar macrophage numbers are maintained through recruitment of peripheral blood CX3CR1hi/CCR2– monocytes, which differentiate through a parenchymal intermediate to become alveolar macrophages [7,8]. Both the parenchymal intermediate-stage macrophage and the alveolar macrophage have the capacity for local replication and represent the source of alveolar macrophages in the steady state, while during increased turnover, such as may occur during inflammatory states, the recruitment and differentiation of monocytes via the lung intermediate is probably accelerated to allow maintenance of alveolar macrophage numbers. Although not tested formally in the lung, evidence supports the concept that M1 polarized macrophages may arise preferentially from recruitment of mononcyte populations, while replication of tissue macrophages may be the predominant source of M2 polarized macrophages during T helper type 2 (Th2) biased inflammation [9].
Alveolar macrophages are very long-lived cells that may live for very prolonged periods in the steady state [10]. In murine models there was no detectable turnover and little local replication observed over 8 months, so in a steady state there may be only a limited requirement for in-situ replication because of intrinsic macrophage persistence. This reflects, in part, the resistance of differentiated tissue macrophage to apoptosis and their expression of high levels of anti-apoptotic molecules, including the Bcl-2 family members myeloid cell leukaemia sequence (Mcl)-1 and A1 or the inhibitor of death receptor signalling FLICE (Fas-associated death domain-like IL-1β-converting enzyme)-inhibitory protein (FLIP) [11–13].
Alveolar macrophages are highly adapted to the unique environment of the lung, and at steady state their ability to generate inflammatory responses is regulated tightly to ensure that lung injury is kept to a minimum, thus preserving precarious alveolar physiology and gas exchange [14]. Alveolar macrophages with M2 polarization play a key role in lung development [15]. M2 polarized alveolar macrophages are thought to play important roles in lung homeostasis ensuring tissue remodelling and repair, but emerging data suggest that alveolar macrophages may demonstrate simultaneously both M1 and M2 characteristics during acute inflammation and disease [16]. Differentiated macrophages such as alveolar macrophages have a large surface area with a dynamic cell membrane facilitating active phagocytosis or endocytosis of inhaled particles [17,18]. A large range of surface receptors enable ingestion of a diverse range of particles [19]. Alveolar macrophages have a large compliment of both secondary lysosomes, containing the enzymes with which they degrade ingested particles, and mitochondria, ensuring that the energy requirements of the macrophage are supplied adequately [20,21]. Rodent studies suggest that the secondary lysosomes, which fuse with endocytic vacuoles and are a key characteristic of alveolar macrophages, are a specific feature of adaptation to the air breathing environment [22]. Differentiated macrophages of human or rodent origin lack the capacity to generate certain potent ROS species via myeloperoxidase, used by other phagocytes such as neutrophils. However, they can generate hydrogen peroxide through the nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase system [23,24] and ROS via their mitochondria [25]. They can form more potent microbicidal factors though the reaction of ROS with NO generated through the inducible nitric oxide synthase (NOS2/iNOS) system [26]. They also utilize proteases such as lysozyme activated at low pH in lysosomes to degrade ingested bacteria [27]. Macrophage degradation of bacteria contributes to the activation of pattern recognition receptors and resultant proinflammatory signalling, although the extent depends upon the capacity of bacteria to resist degradation with individual enzymes [28].
Alveolar macrophages role in host defence against pulmonary bacteria
We have used the Gram-positive bacterium Streptococcus pneumoniae (the pneumococcus) to probe the macrophage role in pulmonary host defence. S. pneumoniae is the most common cause of pneumonia and may spread beyond the lung, resulting in invasive disease such as meningitis [29]. In murine models of pulmonary infection alveolar macrophages clear bacteria up to a defined threshold without overt features of pneumonia, but when alveolar macrophages fail to control these subclinical infections recruitment of inflammatory cells, predominantly neutrophils, is required to control infection [30,31]. Using defined inocula of S. pneumoniae, depletion of alveolar macrophages with liposomes containing clodronate or mice with genetic modifications that alter intracellular killing of bacteria (as discussed below), we have demonstrated that mice with decreased numbers of alveolar macrophages, or with alveolar macrophages of reduced microbicidal capacity, are more susceptible to development of pneumonia and that the threshold inoculum required to generate pneumonia is reduced significantly [30,32–34]. The capacity of macrophages to clear bacteria is finite, and even in wild-type mice the inoculum can be increased to a ‘tipping-point’ beyond which alveolar macrophages no longer control the bacteria in the airway [30,31]. At these higher inocula alveolar macrophage depletion does not alter bacterial clearance because the macrophage capacity for bacterial clearance is already saturated. Hence neutrophils become critical for bacterial clearance [31]. The finite capacity for macrophages to clear bacteria is a feature of both human and murine cells in vitro.
Once the capacity of alveolar macrophages to control infection directly has been overwhelmed, the macrophage plays alternative roles in regulating the inflammatory response (Fig. 1). Macrophages are key orchestrators of inflammatory responses in the lung. They produce key regulatory cytokines, such as IL-1β, which helps to prime release of the neutrophil chemokine CXCL8 from epithelial cells [35]. They also help to induce apoptosis in target cells such as monocytes, and efferocytosis of these apoptotic cells helps to down-regulate the proinflammatory cytokine network [36,37]. When the threshold at which macrophages can no longer control bacterial numbers in the lung is exceeded, T cells play an important role in enhancing bacterial clearance [38]. Achieving complete clearance of S. pneumoniae while minimizing lung damage requires tight regulation of neutrophilic inflammation. To achieve this, different T cell populations interact closely with phagocyte populations to enhance clearance capacity for S. pneumoniae but with variable effects on the extent of the inflammatory response [39]. T helper type 1 (Th1) and Th17 CD4+ T cell responses play important roles in clearance of S. pneumoniae, contributing to the anti-microbial host response through activation of phagocyte-driven microbial killing, while CD8+ T cells may have important roles regulating the extent of Th17 responses and neutrophilic inflammation [40–43]. Although adaptive immune responses contribute to these responses, innate immunity can amplify responses in the form of monocyte-induced stimulation of memory T cells, and innate responses generated by cells such as invariant natural killer T cells (iNKT) can prime phagocyte bacterial killing [41,43–45]. Dysregulated T cell activation can influence bacterial clearance adversely [38]. Fas ligand, which plays a role in the pathogenesis of a diverse range of infections [46], has an important role in regulating the extent of T cell activation through the induction of T cell apoptosis in susceptible activated cells [38]. This helps to optimize bacterial clearance. Monocytes induce Fas ligand-mediated T cell apoptosis (including CD4+ T cells, CD8+ T cells and CD4+ Th17 T cells) during pneumococcal infection, limiting excessive T cell activation and preventing bacteria-induced proinflammatory necrotic cell death [47]. As macrophages also induce Fas-mediated apoptosis in susceptible T cells [46,48] and induce apoptosis of Fas-susceptible cells during S. pneumoniae infection [34,47], it is likely that a variety of monocyte/macrophage cell types play an important role in regulating the inflammatory response through the regulation of T cell activation via the induction of Fas ligand-mediated apoptosis. This represents an emerging but important aspect of macrophage control over neutrophilic inflammation in the lung [46,48].
Fig. 1.
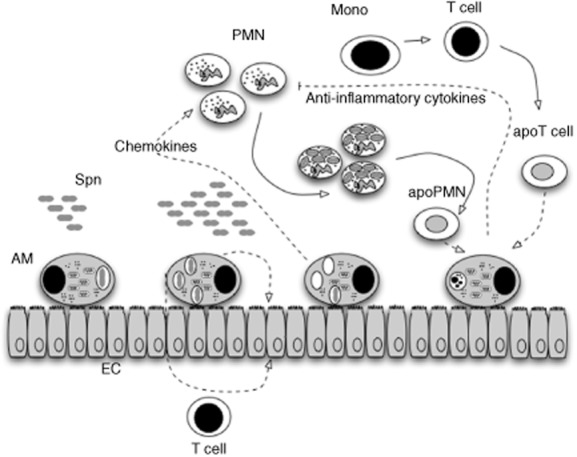
Macrophages play critical roles in host defence against Streptococcus pneumoniae. S. pneumoniae (Spn) are ingested and killed by alveolar macrophages (AM) in the distal airway. When the bacterial inoculum increases above the level at which AM can contain bacteria without recruitment of further phagocytes cross-talk between AM, epithelial cells (EC) and T cells results in the release of chemokines and recruitment of further inflammatory cells, in particular polymorphonuclear leucocytes (PMN). PMN become critical effectors of bacterial clearance. Ultimately, PMN undergo apoptosis. Monocytes may play an important role in regulating inflammatory cells and may induce apoptosis in activated T cells. Ingestion of apoptotic cells (apoPMN and apoT cells) as well as apoptotic monocytes/macrophages by macrophages, a process termed efferocytosis, helps to down-regulate the inflammatory response and results in the generation of anti-inflammatory cytokines and other factors that help to down-regulate the inflammatory response.
Microbicidal mechanisms by which macrophages kill S. pneumoniae
Although ROS generated by the NADPH oxidase system contributes to the killing of many bacteria, it is not required for killing of S. pneumoniae by alveolar macrophages [33,49]. Neutrophils, which have a greater reliance on NADPH oxidase-generated ROS for bacterial killing in general than macrophages, also do not utilize this microbicidal strategy to control S. pneumoniae, instead requiring the neutrophil granule serine proteases, such as cathepsin G and neutrophil elastase, for the effective killing of ingested S. pneumoniae [50,51]. These proteases are not expressed in macrophages. Analysis of S. pneumoniae, which is a catalase-negative bacterium, reveals that it has a variety of adaptations to withstand oxidative stress (Fig. 2), and that it can use the production of hydrogen peroxide to inhibit other bacteria with which it must compete for the environmental niche it exploits in the upper airway. Three enzymes are involved primarily in the production of hydrogen peroxide during pneumococcal metabolism; pyruvate oxidase encoded by spxB, lactate oxidase encoded by lox, which converts lactate to pyruvate and carbamoyl phosphate synthase encoded by carB [52–54]. S. pneumoniae has evolved to cope with the resultant oxidative stress in several ways; sodA encodes the manganese superoxide dismutase (MnSOD) that removes superoxide, the NADH oxidase encoded by nox removes O2 to H2O, preventing its conversion to superoxide, SpxB enables acetyl phosphate to be converted to adenosine triphosphate (ATP), preventing ATP depletion during oxidative stress, psaA encodes a manganese permease and psaD a putative glutathione reductase, which together alter redox status and limit hydrogen peroxide production via SpxB, while AdhC is an alcohol dehydrogenase that generates the reduced glutathione required for PsaD [55–60]. Other factors contributing to resistance to oxidative stress include the heat shock-induced protease HtrA, a putative alkylhydroperoxidase AhpD, the ClpP protease, a putative transcriptional regulator Rgg and a putative thioredoxin-like protein TlpA [61–65]. Although arising primarily as a mechanism by which S. pneumoniae can withstand the ROS generated by their own metabolism, the range of strategies employed to withstand oxidative stress suggest that the bacteria should be relatively more resistant to the ROS, produced by macrophages after ingestion of bacteria, than bacteria that lack these adaptations.
Fig. 2.
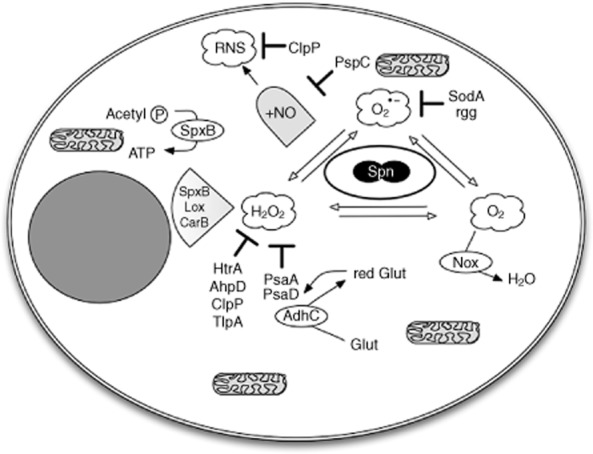
Streptococcus pneumoniae resists anti-microbial killing. Streptococcus pneumoniae (Spn) express several genes, which help them resist reactive oxygen species, generated within phagocytes. Spn express several enzymes that generate hydrogen peroxide (H2O2); SpxB encodes a pyruvate oxidase, Lox a lactate oxidase and CarB a carbamoyl phosphate synthase. Pneumococcal proteins which help resist H2O2 include high temperature requirement A (HtrA), a heat shock-stimulated serine protease, the alkylhydroperoxidase D (AhpD), the caseinolytic peptidase P (ClpP), another heat shock-stimulated serine protease, the thioredoxin-like protein A (TlpA), the pneumococcal surface antigen A (PsaA) a manganese permease, PsaD a putative glutathione peroxidase and an alcohol dehydrogenase (AdhC) that generates the reduced glutathione required by PsaD. Resistance to superoxide (O2•−) involves the superoxide dismutase A (SodA) and the transcriptional regulator Rgg. Nox is a nicotinamide adenine dinucleotide (NADH) oxidase that converts O2 to H2O, limiting the availability of O2 to form O2•−. Spx allows acetyl phosphate (Acetyl P) to provide a source of adenosine triphosphate (ATP) during oxidative stress. In addition, pneumococcal surface protein C (PspC) helps Spn to withstand nitric oxide (NO) and ClpP-reactive nitrogen species (RNS), produced by macrophages.
Macrophages have evolved additional anti-microbial strategies, including generation of NO via NOS2, and NO can react to form S-nitrosothiols, such as S-nitrosoglutathione (GSNO) or can react with with ROS to generate reactive nitrogen species (RNS), such as peroxynitrite formed between NO and superoxide, which are potent anti-microbicidal molecules [26]. Therefore, although ROS may of itself be dispensable as an anti-microbicidal, it can still contribute to pneumococcal killing through formation of RNS. Microbial components, including the toxin pneumolysin and pneumococcal cell wall, stimulate macrophage NO production and NO-mediated bacterial killing [66–68]. In keeping with this, we and others have shown that mice that lack NOS2 are less able to clear S. pneumoniae from the lung [33,69]. Nevertheless, there are also factors which restrict the capacity of NO or RNS to kill ingested bacteria. First, S. pneumoniae also has genes that allow it to adapt to nitrosative stress; pneumococcal surface protein C (PspC) reduces NO production, AdhC is an S-nitrosoglutathione reductase, while ClpP has been implicated in resistance to RNS [70–72]. To compound these challenges imposed by the bacterial genome, human macrophages appear to generate lower levels of NO than murine macrophages and levels of production are lower in differentiated tissue macrophages [47,73]. Much less is known about the capacity of proteases to contribute to pneumococcal killing in macrophages. Although RNS are likely to make a significant contribution to early phagolysosomal killing of S. pneumoniae and proteases have the potential to add additional killing capacity, it is clear that the macrophage is likely to have a finite capacity for bacterial killing and needs to employ an additional microbicidal strategy to control S. pneumoniae. The fact that bacteria such as S. pneumoniae are not believed to demonstrate intracellular persistence suggests this additional mechanism must be effective.
Apoptosis-associated killing
Induction of apoptosis has been demonstrated to aid bacterial killing for intracellular pathogens such as Mycobacterium tuberculosis [74,75]. We have demonstrated that macrophage apoptosis is a feature of in-vitro challenge of macrophages to S. pneumoniae and also occurs in vivo [30,36]. It is observed with both human and murine macrophages. The induction of apoptosis is linked to the burden of intracellular bacteria, suggesting that it is activated when the intrinsic capacity for phagolysosomal killing is exhausted [76]. Apoptosis induction is delayed, occurring typically 16–20 h after bacterial ingestion in vitro, and the apoptosis-associated killing occurs after the initial phase of phagolysosomal killing associated with microbicidal molecules in the phagolysosome. Apoptosis-associated killing of S. pneumoniae requires NO generation [68]. Although induced following phagocytosis, apoptosis is not the result of phagocytosis per se nor linked to uptake by any specific receptor that recognizes S. pneumoniae. It is enhanced in response to opsonized bacteria and in response to less virulent unencapsulated strains, but this seems to be because these circumstances enhance numbers of intracellular bacteria [76,77]. It is also facilitated by responses associated with pathogen recognition; Toll-like receptor (TLR)-4 plays a role in enhancing the response [78]. Apoptosis-associated killing is observed with a range of serotypes of S. pneumoniae, but as the efficiency of ingestion of different serotypes can vary and apoptosis induction is influenced by the number of intracellular bacteria it is seen to greater extents for strains that are phagocytosed more efficiently and is seen to the greatest extent for unencapsulated strains [76].
Inhibition of elements of the apoptotic response reduces bacterial killing in vitro and does not result in cell survival, but rather results in pathogen-driven death by necrosis with impaired bacterial killing [32,34,68]. Analysis of the macrophage proteome demonstrates that apoptosis results from a concerted programme that not only induces apoptosis but also inhibits alternative death paradigms, including necroptosis, a form of programmed necrosis, and the endoplasmic reticulum (ER) stress pathway [79]. We have found no evidence that the death process we observe involves pyroptosis, an alternative form of programmed cell death that is caspase-1-dependent, but which is also associated with anti-microbial responses, although with greater degrees of inflammatory response. Although pyroptosis can be induced in monocytes to a range of bacteria we have found that it is observed predominantly with infections associated with greater levels of internalized bacteria and greater levels of ATP depletion than we observe with S. pneumoniae, and in this model S. pneumoniae did not engage pyroptosis [80]. The delayed apoptotic death process seems to be enhanced in the more differentiated tissue macrophage phenotype [76]. Our group also has unpublished evidence that mice that lack caspase-1 do not show any reduction in cell death in response to S. pneumoniae, when rates of cell death are examined in bronchoalveolar lavage 24 h after intratracheal challenge with S. pneumoniae, a finding which appears to suggest that the death process observed does not represent pyroptosis (Marriott and Dockrell unpublished observations). Recent observations provide further insight into why apoptosis may be induced in macrophages exposed to S. pneumoniae in preference to pyroptosis. Caspase-1-dependent pyroptosis following challenge with S. pneumoniae is inhibited by induction of chitinase 3-like-1, a host response to S. pneumoniae which seems to have a conserved role in evolution, allowing bacterial clearance with a death process that results in lower levels of inflammation than occur with pyroptosis [81].
Murine models illustrate that impairment of apoptosis results in decreased bacterial killing in the lung and enhanced tissue invasion, as reflected by greater levels of bacteraemia [30,32,34]. Ultimately, apoptotic macrophages are ingested by lung macrophages and help to down-regulate proinflammatory cytokine expression, in particular TNF-α-dependent signalling, and this is associated with an overall reduction in neutrophil recruitment to the lung and lung inflammation [33,37].
The molecular regulation of apoptosis-associated killing
Apoptosis following ingestion of S. pneumoniae involves an intrinsic mitochondrial-dependent pathway that is independent of death receptors such as Fas ligand [46,68]. The pathway is caspase-dependent [30,36]. Apoptosis is delayed to maximize the time during which the macrophage can kill ingested bacteria, but occurs when intracellular killing by canonical mechanisms becomes exhausted [32]. The onset of apoptosis is carefully scripted. We have shown that the onset of apoptosis is regulated by expression of the anti-apoptotic protein Mcl-1 [32]. The anti-apoptotic protein Mcl-1 is a Bcl-2 family member and has been demonstrated previously to be expressed highly in differentiated macrophages [11]. It has a relatively short half-life, estimated to be 20–30 min, and is well equipped to respond to dynamic changes in the macrophage ingesting and killing bacteria [82]. Initially it is transcriptionally up-regulated, but following prolonged exposure to S. pneumoniae protein translation becomes gradually reduced and proteasomal degradation via ubiquitination is increased [32,34,79].
Apoptosis arises in response to S. pneumoniae localized in phagolysosomes and we have investigated how this might activate an apoptotic programme that involves Mcl-1 down-regulation. Lysosomal membrane permeabilization can activate a mitochondrial pathway of apoptosis [83]. We have found evidence that lysosomal membrane permeabilization precedes Mcl-1 down-regulation and investigated the role of lysosomal proteases in these events. We have shown, using pharmacological inhibition and genetic manipulation, that the aspartic lysosomal protease cathepsin D plays a key role in the induction of apoptosis [34]; see Fig. 3. Cathepsin D is the most abundant lysosomal protease in differentiated macrophages [84,85]. Activation of cathepsin D allows Mcl-1 to interact preferentially with its E3 ubiquitin ligase, Mcl-1 ubiquitin ligase (Mule, also known as ARF-BP1/HectH9), rather than with heat shock protein 70 with which it can also interact [34]. It also results in down-regulation of eukaryotic elongation factor (eEF) 2, a critical component of the translational machinery required to allow Mcl-1 translation [79]. This results in Mcl-1 down-regulation, induction of apoptosis and enhanced bacterial killing. The role of cathepsin D and Mcl-1 in the regulation of apoptosis-associated bacterial killing are conserved between murine and human macrophages.
Fig. 3.
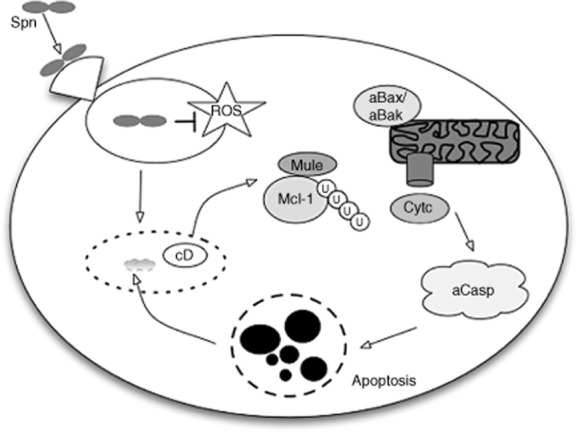
Induction of apoptosis-associated killing complements bacterial killing in macrophages. Streptococcus pneumoniae Spn is internalized following phagocytosis into a phagolysosome, where it can resist killing by reactive oxygen species (ROS). With time, the phagolysosome becomes permeabilized and activation of the lysosomal protease cathepsin D (cD) results in down-regulation of the anti-apoptotic protein myeloid cell leukaemia sequence 1 (Mcl-1). Mcl-1 down-regulation involves ubiquitination mediated by interaction with the Mcl-1 ubiquitin ligase E3 (Mule) and proteasomal degradation. Following Mcl-1 down-regulation, pro-apoptotic Bcl-2 family members result in the ultimate activation of Bcl-2-associated X protein (Bax) and Bcl-2 homologous antagonist/killer (Bak), which interact with the mitochondrion (aBax/aBak) to induce mitochondrial outer membrane permeabilization and cytochrome c (Cytc) release. This results in caspase activation (aCasp) and downstream features of apoptosis, illustrated by nuclear fragmentation. Apoptosis results in macrophage cell death but also killing of the residual viable Spn contained in the phagolysosome.
The molecular basis of lysosomal permeabilization has not yet been established. It seems to arise due to a host response to ingested S. pneumoniae and in particular to the expression of the bacterial toxin pneumolysin [34]. Although the toxin itself causes membrane pores we have no evidence that it causes pores in the lysosomal membrane, as exposure to bacteria expressing toxin lacking pore-forming capability still induce comparable levels of apoptosis to bacteria expressing wild-type toxin (Bewley et al., manuscript submitted). Equally, we have not found any role to date for components of the intracellular recognition systems for pneumolysin, which involve nucleotide-binding oligomerization domain containing protein (Nod)-like receptor (NLR) P3 or the absent in melanoma (AIM) 2 NLR, in the response to pneumolysin that leads to lysosomal membrane permeabilization or to induction of apoptosis [86–88]. The induction of lysosomal membrane permeabilization, cathepsin D activation and induction of apoptosis is not a response limited to S. pneumoniae, as it occurs with a range of other extracellular bacteria [80]. Of some interest, although there are several bacteria in which apoptosis-associated killing appears not to occur, infection with Staphylococcus aureus can result in persistent up-regulation of Mcl-1 and an absence of apoptosis-associated killing [89]. We have found that, in contrast to S. pneumoniae, Staphylococcus aureus fails to induce cathepsin D activation [34]. Furthermore, we have found that Neisseria meningitidis does not induce apoptosis in monocytes or macrophages [80,90]. Thus, some bacteria may have developed strategies to limit induction of apoptosis-associated killing to enable their own intracellular persistence.
Recently it has been suggested that macrophage apoptosis-associated killing during S. pneumoniae infection can be enhanced by TNF-related apoptosis-inducing ligand (TRAIL) and that neutrophils may be a source of this ligand during established pneumonia [91]. Because macrophages undergo apoptosis-associated killing in vitro in the absence of neutrophils, and in murine models in the absence of neutrophil recruitment this mechanism is unlikely to explain the intrinsic mechanism in macrophages, it suggests rather that apoptosis-associated killing can also be engaged by additional pathways involving neutrophils. This may be of relevance during established pneumonia where endogenous activation of apoptosis-associated killing by the macrophage alone is exhausted or inefficient and where additional mechanisms of bacterial killing such as neutrophil-mediated killing are impaired [91]. As cells other than neutrophils also express TRAIL, it remains possible that other cell types such as macrophages, lymphocytes or epithelial cells could contribute to this additional mechanism of apoptosis induction in settings distinct from those described [92].
Concluding remarks
Alveolar macrophages play a key role in host defence, functioning as the resident phagocytes that clear bacteria. They are efficient at phagocytosing bacteria, but the rate-limiting step in bacterial clearance is intracellular killing. Although they utilize a range of anti-microbicidals to kill ingested bacteria, the restrictions on the microbicidal molecules they generate imposed by their localization in the distal airway and the range of survival strategies employed by a pathogen such as S. pneumoniae mean that additional mechanisms are required to kill phagocytosed bacteria. Induction of apoptosis-associated killing is a key component of host defence, which combines bacterial clearance with relatively modest cost to overall pulmonary inflammation. It represents an important and under-recognized component of host defence in the lung. The clearance of intracellular bacteria when initial intraphagolysosomal killing is exhausted is evidence of its efficiency, while the evidence that some bacteria have prevented this host response raises the possibility that selective re-engagement could represent a novel approach to enhance microbial killing, particularly in the face of bacteria that demonstrate high-levels of antimicrobial resistance.
This review results from a talk given at the 3rd Annual Infection and Immunity Meeting, a meeting of the BSI Infection and Immunity Affinity Group held on Thursday 11 April 2013. To view the abstracts from this meeting visit: http://onlinelibrary.wiley.com/doi/10.1111/cei.2013.173.issue-s1/issuetoc.
Disclosure
The authors declare they have no financial or commercial conflict of interest in writing this review. The authors' work is supported by a Wellcome Trust Senior Clinical Fellowship to D.H.D. (076945) and through the MRC-ABPI COPD consortium COPD-MAP.
References
- 1.Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc. 2005;2:403–411. doi: 10.1513/pats.200508-090JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 3.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 5.Thomas ED, Ramberg RE, Sale GE, Sparkes RS, Golde DW. Direct evidence for a bone marrow origin of the alveolar macrophage in man. Science. 1976;192:1016–1018. doi: 10.1126/science.775638. [DOI] [PubMed] [Google Scholar]
- 6.Sawyer RT, Strausbauch PH, Volkman A. Resident macrophage proliferation in mice depleted of blood monocytes by strontium-89. Lab Invest. 1982;46:165–170. [PubMed] [Google Scholar]
- 7.Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol. 2007;179:3488–3494. doi: 10.4049/jimmunol.179.6.3488. [DOI] [PubMed] [Google Scholar]
- 8.Landsman L, Varol C, Jung S. Distinct differentiation potential of blood monocyte subsets in the lung. J Immunol. 2007;178:2000–2007. doi: 10.4049/jimmunol.178.4.2000. [DOI] [PubMed] [Google Scholar]
- 9.Jenkins SJ, Ruckerl D, Cook PC, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol. 2008;38:380–385. doi: 10.1165/rcmb.2007-0224RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages. Role of Mcl-1, independent of nuclear factor (NF)-kappaB, Bad, or caspase activation. J Exp Med. 2001;194:113–126. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pagliari LJ, Perlman H, Liu H, Pope RM. Macrophages require constitutive NF-kappaB activation to maintain A1 expression and mitochondrial homeostasis. Mol Cell Biol. 2000;20:8855–8865. doi: 10.1128/mcb.20.23.8855-8865.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perlman H, Pagliari LJ, Georganas C, Mano T, Walsh K, Pope RM. FLICE-inhibitory protein expression during macrophage differentiation confers resistance to Fas-mediated apoptosis. J Exp Med. 1999;190:1679–1688. doi: 10.1084/jem.190.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piantadosi CA, Schwartz DA. The acute respiratory distress syndrome. Ann Intern Med. 2004;141:460–470. doi: 10.7326/0003-4819-141-6-200409210-00012. [DOI] [PubMed] [Google Scholar]
- 15.Jones C, Williams T, Walker K, et al. M2 macrophage polarisation is associated with alveolar formation during postnatal lung development. Respir Res. 2013;14:41. doi: 10.1186/1465-9921-14-41. doi: 10.1186/1465-9921-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J Immunol. 2012;189:946–955. doi: 10.4049/jimmunol.1200660. [DOI] [PubMed] [Google Scholar]
- 17.Sokol RJ, Hudson G, James NT, Frost IJ, Wales J. Human macrophage development: a morphometric study. J Anat. 1987;151:27–35. [PMC free article] [PubMed] [Google Scholar]
- 18.Steinman RM, Mellman IS, Muller WA, Cohn ZA. Endocytosis and the recycling of plasma membrane. J Cell Biol. 1983;96:1–27. doi: 10.1083/jcb.96.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 20.Cohn ZA, Benson B. The differentiation of mononuclear phagocytes. Morphology, cytochemistry, and biochemistry. J Exp Med. 1965;121:153–170. doi: 10.1084/jem.121.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohn ZA, Fedorko ME, Hirsch JG. The in vitro differentiation of mononuclear phagocytes. V. The formation of macrophage lysosomes. J Exp Med. 1966;123:757–766. doi: 10.1084/jem.123.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kradin RL, McCarthy KM, Preffer FI, Schneeberger EE. Flow-cytometric and ultrastructural analysis of alveolar macrophage maturation. J Leukoc Biol. 1986;40:407–417. doi: 10.1002/jlb.40.4.407. [DOI] [PubMed] [Google Scholar]
- 23.Cohen AB, Cline MJ. The human alveolar macrophage: isolation, cultivation in vitro, and studies of morphologic and functional characteristics. J Clin Invest. 1971;50:1390–1398. doi: 10.1172/JCI106622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–3017. [PubMed] [Google Scholar]
- 25.West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 27.Ip WK, Sokolovska A, Charriere GM, et al. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J Immunol. 2010;184:7071–7081. doi: 10.4049/jimmunol.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf AJ, Arruda A, Reyes CN, et al. Phagosomal degradation increases TLR access to bacterial ligands and enhances macrophage sensitivity to bacteria. J Immunol. 2011;187:6002–6010. doi: 10.4049/jimmunol.1100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.File TM, Jr, Marrie TJ. Burden of community-acquired pneumonia in North American adults. Postgrad Med. 2010;122:130–141. doi: 10.3810/pgm.2010.03.2130. [DOI] [PubMed] [Google Scholar]
- 30.Dockrell DH, Marriott HM, Prince LR, et al. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol. 2003;171:5380–5388. doi: 10.4049/jimmunol.171.10.5380. [DOI] [PubMed] [Google Scholar]
- 31.Knapp S, Leemans JC, Florquin S, et al. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med. 2003;167:171–179. doi: 10.1164/rccm.200207-698OC. [DOI] [PubMed] [Google Scholar]
- 32.Marriott HM, Bingle CD, Read RC, et al. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest. 2005;115:359–368. doi: 10.1172/JCI21766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marriott HM, Hellewell PG, Whyte MK, Dockrell DH. Contrasting roles for reactive oxygen species and nitric oxide in the innate response to pulmonary infection with Streptococcus pneumoniae. Vaccine. 2007;25:2485–2490. doi: 10.1016/j.vaccine.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bewley MA, Marriott HM, Tulone C, et al. A cardinal role for cathepsin D in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog. 2011;7:e1001262. doi: 10.1371/journal.ppat.1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marriott HM, Gascoyne KA, Gowda R, et al. IL-1beta regulates CXCL8 release and influences disease outcome in response to Streptococcus pneumoniae, defining intracellular cooperation between pulmonary epithelial cells and macrophages. Infect Immun. 2012;80:1140–1149. doi: 10.1128/IAI.05697-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dockrell DH, Lee M, Lynch DH, Read RC. Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis. 2001;184:713–722. doi: 10.1086/323084. [DOI] [PubMed] [Google Scholar]
- 37.Marriott HM, Hellewell PG, Cross SS, Ince PG, Whyte MK, Dockrell DH. Decreased alveolar macrophage apoptosis is associated with increased pulmonary inflammation in a murine model of pneumococcal pneumonia. J Immunol. 2006;177:6480–6488. doi: 10.4049/jimmunol.177.9.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marriott HM, Daigneault M, Thompson AA, et al. A decoy receptor 3 analogue reduces localised defects in phagocyte function in pneumococcal pneumonia. Thorax. 2012;67:985–992. doi: 10.1136/thoraxjnl-2012-201591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dockrell DH, Whyte MK, Mitchell TJ. Pneumococcal pneumonia: mechanisms of infection and resolution. Chest. 2012;142:482–491. doi: 10.1378/chest.12-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boucheron N, Sharif O, Schebesta A, et al. The protein tyrosine kinase Tec regulates a CD44highCD62L– Th17 subset. J Immunol. 2010;185:5111–5119. doi: 10.4049/jimmunol.1001734. [DOI] [PubMed] [Google Scholar]
- 41.Olliver M, Hiew J, Mellroth P, Henriques-Normark B, Bergman P. Human monocytes promote Th1 and Th17 responses to Streptococcus pneumoniae. Infect Immun. 2011;79:4210–4217. doi: 10.1128/IAI.05286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber SE, Tian H, Pirofski LA. CD8+ cells enhance resistance to pulmonary serotype 3 Streptococcus pneumoniae infection in mice. J Immunol. 2011;186:432–442. doi: 10.4049/jimmunol.1001963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest. 2009;119:1899–1909. doi: 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brigl M, Tatituri RV, Watts GF, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med. 2011;208:1163–1177. doi: 10.1084/jem.20102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinjo Y, Illarionov P, Vela JL, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol. 2011;12:966–974. doi: 10.1038/ni.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dockrell DH. Apoptotic cell death in the pathogenesis of infectious diseases. J Infect. 2001;42:227–234. doi: 10.1053/jinf.2001.0836. [DOI] [PubMed] [Google Scholar]
- 47.Daigneault M, De Silva TI, Bewley MA, et al. Monocytes regulate the mechanism of T-cell death by inducing Fas-mediated apoptosis during bacterial infection. PLoS Pathog. 2012;8:e1002814. doi: 10.1371/journal.ppat.1002814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Badley AD, Dockrell D, Simpson M, et al. Macrophage-dependent apoptosis of CD4+ T lymphocytes from HIV-infected individuals is mediated by FasL and tumor necrosis factor. J Exp Med. 1997;185:55–64. doi: 10.1084/jem.185.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biggar WD, Buron S, Holmes B. Bactericidal mechanisms in rabbit alveolar macrophages: evidence against peroxidase and hydrogen peroxide bactericidal mechanisms. Infect Immun. 1976;14:6–10. doi: 10.1128/iai.14.1.6-10.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marriott HM, Jackson LE, Wilkinson TS, et al. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am J Respir Crit Care Med. 2008;177:887–895. doi: 10.1164/rccm.200707-990OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Standish AJ, Weiser JN. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol. 2009;183:2602–2609. doi: 10.4049/jimmunol.0900688. [DOI] [PubMed] [Google Scholar]
- 52.Hoffmann O, Zweigner J, Smith SH, et al. Interplay of pneumococcal hydrogen peroxide and host-derived nitric oxide. Infect Immun. 2006;74:5058–5066. doi: 10.1128/IAI.01932-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pericone CD, Overweg K, Hermans PW, Weiser JN. Inhibitory and bactericidal effects of hydrogen peroxide production by Streptococcus pneumoniae on other inhabitants of the upper respiratory tract. Infect Immun. 2000;68:3990–3997. doi: 10.1128/iai.68.7.3990-3997.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taniai H, Iida K, Seki M, et al. Concerted action of lactate oxidase and pyruvate oxidase in aerobic growth of Streptococcus pneumoniae: role of lactate as an energy source. J Bacteriol. 2008;190:3572–3579. doi: 10.1128/JB.01882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yesilkaya H, Kadioglu A, Gingles N, Alexander JE, Mitchell TJ, Andrew PW. Role of manganese-containing superoxide dismutase in oxidative stress and virulence of Streptococcus pneumoniae. Infect Immun. 2000;68:2819–2826. doi: 10.1128/iai.68.5.2819-2826.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J, Bryant AP, Marra A, et al. Characterization of the Streptococcus pneumoniae NADH oxidase that is required for infection. Microbiology. 2001;147:431–438. doi: 10.1099/00221287-147-2-431. [DOI] [PubMed] [Google Scholar]
- 57.Pericone CD, Park S, Imlay JA, Weiser JN. Factors contributing to hydrogen peroxide resistance in Streptococcus pneumoniae include pyruvate oxidase (SpxB) and avoidance of the toxic effects of the Fenton reaction. J Bacteriol. 2003;185:6815–6825. doi: 10.1128/JB.185.23.6815-6825.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McAllister LJ, Tseng HJ, Ogunniyi AD, Jennings MP, McEwan AG, Paton JC. Molecular analysis of the PsA permease complex of Streptococcus pneumoniae. Mol Microbiol. 2004;53:889–901. doi: 10.1111/j.1365-2958.2004.04164.x. [DOI] [PubMed] [Google Scholar]
- 59.Tseng HJ, McEwan AG, Paton JC, Jennings MP. Virulence of Streptococcus pneumoniae: PsaA mutants are hypersensitive to oxidative stress. Infect Immun. 2002;70:1635–1639. doi: 10.1128/IAI.70.3.1635-1639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Potter AJ, Kidd SP, McEwan AG, Paton JC. The MerR/NmlR family transcription factor of Streptococcus pneumoniae responds to carbonyl stress and modulates hydrogen peroxide production. J Bacteriol. 2010;192:4063–4066. doi: 10.1128/JB.00383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ibrahim YM, Kerr AR, McCluskey J, Mitchell TJ. Role of HtrA in the virulence and competence of Streptococcus pneumoniae. Infect Immun. 2004;72:3584–3591. doi: 10.1128/IAI.72.6.3584-3591.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paterson GK, Blue CE, Mitchell TJ. An operon in Streptococcus pneumoniae containing a putative alkylhydroperoxidase D homologue contributes to virulence and the response to oxidative stress. Microb Pathog. 2006;40:152–160. doi: 10.1016/j.micpath.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 63.Robertson GT, Ng WL, Foley J, Gilmour R, Winkler ME. Global transcriptional analysis of clpP mutations of type 2 Streptococcus pneumoniae and their effects on physiology and virulence. J Bacteriol. 2002;184:3508–3520. doi: 10.1128/JB.184.13.3508-3520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bortoni ME, Terra VS, Hinds J, Andrew PW, Yesilkaya H. The pneumococcal response to oxidative stress includes a role for Rgg. Microbiology. 2009;155:4123–4134. doi: 10.1099/mic.0.028282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andisi VF, Hinojosa CA, de Jong A, Kuipers OP, Orihuela CJ, Bijlsma JJ. Pneumococcal gene complex involved in resistance to extracellular oxidative stress. Infect Immun. 2012;80:1037–1049. doi: 10.1128/IAI.05563-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orman KL, Shenep JL, English BK. Pneumococci stimulate the production of the inducible nitric oxide synthase and nitric oxide by murine macrophages. J Infect Dis. 1998;178:1649–1657. doi: 10.1086/314526. [DOI] [PubMed] [Google Scholar]
- 67.Braun JS, Novak R, Gao G, Murray PJ, Shenep JL. Pneumolysin, a protein toxin of Streptococcus pneumoniae, induces nitric oxide production from macrophages. Infect Immun. 1999;67:3750–3756. doi: 10.1128/iai.67.8.3750-3756.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marriott HM, Ali F, Read RC, Mitchell TJ, Whyte MK, Dockrell DH. Nitric oxide levels regulate macrophage commitment to apoptosis or necrosis during pneumococcal infection. FASEB J. 2004;18:1126–1128. doi: 10.1096/fj.03-1450fje. [DOI] [PubMed] [Google Scholar]
- 69.Kerr AR, Wei XQ, Andrew PW, Mitchell TJ. Nitric oxide exerts distinct effects in local and systemic infections with Streptococcus pneumoniae. Microb Pathog. 2004;36:303–310. doi: 10.1016/j.micpath.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 70.Peppoloni S, Colombari B, Neglia R, et al. The lack of Pneumococcal surface protein C (PspC) increases the susceptibility of Streptococcus pneumoniae to the killing by microglia. Med Microbiol Immunol (Berl) 2006;195:21–28. doi: 10.1007/s00430-005-0243-8. [DOI] [PubMed] [Google Scholar]
- 71.Stroeher UH, Kidd SP, Stafford SL, Jennings MP, Paton JC, McEwan AG. A pneumococcal MerR-like regulator and S-nitrosoglutathione reductase are required for systemic virulence. J Infect Dis. 2007;196:1820–1826. doi: 10.1086/523107. [DOI] [PubMed] [Google Scholar]
- 72.Park CY, Kim EH, Choi SY, et al. Virulence attenuation of Streptococcus pneumoniae clpP mutant by sensitivity to oxidative stress in macrophages via an NO-mediated pathway. J Microbiol. 2010;48:229–235. doi: 10.1007/s12275-010-9300-0. [DOI] [PubMed] [Google Scholar]
- 73.Jesch NK, Dorger M, Enders G, et al. Expression of inducible nitric oxide synthase and formation of nitric oxide by alveolar macrophages: an interspecies comparison. Environ Health Perspect. 1997;105(Suppl. 5):1297–1300. doi: 10.1289/ehp.97105s51297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keane J, Balcewicz-Sablinska MK, Remold HG, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 76.Ali F, Lee ME, Iannelli F, et al. Streptococcus pneumoniae-associated human macrophage apoptosis after bacterial internalization via complement and Fcgamma receptors correlates with intracellular bacterial load. J Infect Dis. 2003;188:1119–1131. doi: 10.1086/378675. [DOI] [PubMed] [Google Scholar]
- 77.Weber S, Tian H, van Rooijen N, Pirofski LA. A serotype 3 pneumococcal capsular polysaccharide-specific monoclonal antibody requires Fcgamma receptor III and macrophages to mediate protection against pneumococcal pneumonia in mice. Infect Immun. 2012;80:1314–1322. doi: 10.1128/IAI.06081-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Srivastava A, Henneke P, Visintin A, et al. The apoptotic response to pneumolysin is Toll-like receptor 4 dependent and protects against pneumococcal disease. Infect Immun. 2005;73:6479–6487. doi: 10.1128/IAI.73.10.6479-6487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bewley MA, Pham TK, Marriott HM, et al. Proteomic evaluation and validation of cathepsin D regulated proteins in macrophages exposed to Streptococcus pneumoniae. Mol Cell Proteomics. 2011;10:M111 008193. doi: 10.1074/mcp.M111.008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Webster SJ, Daigneault M, Bewley MA, et al. Distinct cell death programs in monocytes regulate innate responses following challenge with common causes of invasive bacterial disease. J Immunol. 2010;185:2968–2979. doi: 10.4049/jimmunol.1000805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dela Cruz CS, Liu W, He CH, et al. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe. 2012;12:34–46. doi: 10.1016/j.chom.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schubert KM, Duronio V. Distinct roles for extracellular-signal-regulated protein kinase (ERK) mitogen-activated protein kinases and phosphatidylinositol 3-kinase in the regulation of Mcl-1 synthesis. Biochem J. 2001;356:473–480. doi: 10.1042/0264-6021:3560473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boya P, Andreau K, Poncet D, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003;197:1323–1334. doi: 10.1084/jem.20021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jin M, Opalek JM, Marsh CB, Wu HM. Proteome comparison of alveolar macrophages with monocytes reveals distinct protein characteristics. Am J Respir Cell Mol Biol. 2004;31:322–329. doi: 10.1165/rcmb.2004-0080OC. [DOI] [PubMed] [Google Scholar]
- 85.Kato T, Kojima K, Murachi T. Proteases of macrophages in rat peritoneal exudate, with special reference to the effects of actinomycete protease inhibitors. Biochim Biophys Acta. 1972;289:187–193. doi: 10.1016/0005-2744(72)90121-0. [DOI] [PubMed] [Google Scholar]
- 86.Fang R, Tsuchiya K, Kawamura I, et al. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J Immunol. 2011;187:4890–4899. doi: 10.4049/jimmunol.1100381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McNeela EA, Burke A, Neill DR, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Witzenrath M, Pache F, Lorenz D, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 89.Koziel J, Maciag-Gudowska A, Mikolajczyk T, et al. Phagocytosis of Staphylococcus aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS One. 2009;4:e5210. doi: 10.1371/journal.pone.0005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tunbridge AJ, Stevanin TM, Lee M, et al. Inhibition of macrophage apoptosis by Neisseria meningitidis requires nitric oxide detoxification mechanisms. Infect Immun. 2006;74:729–733. doi: 10.1128/IAI.74.1.729-733.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Steinwede K, Henken S, Bohling J, et al. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J Exp Med. 2012;209:1937–1952. doi: 10.1084/jem.20120983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McGrath EE, Lawrie A, Marriott HM, et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligand exacerbates lung injury and fibrosis. Thorax. 2012;67:796–803. doi: 10.1136/thoraxjnl-2011-200863. [DOI] [PMC free article] [PubMed] [Google Scholar]