

Abstract
Amyloid β-peptide (Aβ) deposits and neurofibrillary tangles are key hallmarks in Alzheimer's disease (AD). Aβ stimulates many signal transducers involved in the neuronal death. However, many mechanisms remain to be elucidated because no definitive therapy of AD exists. Some studies have focused on the control of translation which involves eIF2 and eIF4E, main eukaryotic factors of initiation. The availability of these factors depends on the activation of the double-stranded RNA-dependent protein kinase (PKR) and the mammalian target of rapamycin (mTOR), respectively. mTOR positively regulates the translation while PKR results in a protein synthesis shutdown. Many studies demonstrated that the PKR signalling pathway is up-regulated in cellular and animal models of AD and in the brain of AD patients. Interestingly, our results showed that phosphorylated PKR and eIF2α levels were significantly increased in lymphocytes of AD patients. These modifications were significantly correlated with cognitive and memory test scores performed in AD patients. On the contrary, the mTOR signalling pathway is down-regulated in cellular and animal models of AD. Recently, we showed that p53, regulated protein in development and DNA damage response 1 and tuberous sclerosis complex 2 could represent molecular links between PKR and mTOR signalling pathways. PKR could be an early biomarker of the neuronal death and a critical target for a therapeutic programme in AD.
Keywords: Alzheimer's disease, control of translation, PKR, mTOR, apoptosis
Introduction
Control of translation
Dysfunctions of protein synthesis mediated by PKR- and mTOR-dependent signalling pathways
Crosslink between the up-regulation of PKR/eIF2α and the down-regulation of mTOR/RS6K in AD
PKR: a potential biomarker of AD diagnosis
Conclusion and perspectives
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease and the most common form of dementia which causes serious impairments of cognitive functions. The majority of AD cases in people over the age of 65 are of the sporadic (or ‘late onset’) form, suggesting that the disease has no family link. However, about 2–5% per cent of the Alzheimer population have ‘familial’ (FAD) or ‘early onset’ AD via autosomal dominant inheritance. FAD is identical to the sporadic form but it occurs because of the inheritance of certain genes which at some point in the family's history ‘mutated’ from having normal to abnormal characteristics. Most cases of FAD result from mutations in one of the three following genes: the amyloid precursor protein (APP), the presenilin 1 and 2 (PS1, PS2). All mutations result in elevated levels of amyloid-β (Aβ) peptide [1]. The most important genetic risk factor for both familial and sporadic forms of AD is the apoE4 gene [2, 3]. ApoE4 enhances the rate of amyloid fibril formation, interacts with the microtubule-associated proteins (MAPs) [4, 5] and directly affects cholinergic activity in the brains of AD patients [6]. Two studies also confirm the association between genetic variants in sortilin-related receptor (SORL1) and AD [7, 8]. SORL1 binds APP and acts as a sorting receptor for APP. Absence of SORL1 switches APP away from the recycling endosomes pathway and instead leads APP to the β- and γ-secretase cleavage pathways to generate Aβ[7]. However, it is unclear whether these changes are causal or simply reactive to AD.
AD is characterized by three major pathologic hallmarks: extracellular plaques of the 39–43 amino acid Aβ aggregated, intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated τ-protein and neuronal death. Aβ is derived by proteolytic cleavage of APP. The action of β-secretase liberates a C-terminal fragment, which is subsequently processed by γ-secretase to release Aβ. Cleavage by γ-secretase releases a number of Aβ species, which vary in length. Presenilin proteins are an integral part of γ-secretase complex involved in the processing of APP. The major form of Aβ contains 40 amino-acid residues (Aβ 40), although minor C-terminally extended forms (Aβ42 and Aβ43) are also produced. Aβ42 has a higher propensity to aggregate than Aβ40 and evidence from several studies indicates that increased production of Aβ42 is closely associated with the development of AD [9–11]. Besides amyloid plaques, NFTs are composed of arrays of paired helical filaments as described for the first time by Kidd [12]. NFTs are mainly localized in the hippocampus, entorrhinal cortex and amygdala. Paired helical filaments are structures generated by self-aggregation of hyperphosphorylated τ-proteins making a compact filamentous network [13].
Furthermore, increasing evidence suggests that inflammation significantly contributes to the pathogenesis of AD. Research performed in the last 20 years supports the hypothesis of Fisher [14]. Senile plaques might be the result of an inflammation mediated regenerative response of surrounding nerve fibres against extracellular deposits of a foreign substance. In the following years, it was shown that the ‘foreign’ substance, now identified as Aβ fibrils, could indeed induce a local inflammatory response [15, 16]. However, the chronic inflammatory response in AD brains is described in the plaques containing fibrillar Aβ deposits but not in the diffuse plaque with the non-congophilic low-fibrillar Aβ depositions [17, 18]. A spectrum of inflammatory mediators up-regulated in AD has been demonstrated [16, 19–22] and indicates that such inflammation is an important part of pathology and suggests many routes for future therapeutic intervention.
Yet, it is clear that a variety of cellular mechanisms can lead to this neurodegenerative disorder. Elucidation of the factors that trigger the sequence changes in the normal neuronal machinery leading to neurodegeneration and the mechanisms underlying signal transductions that determine neuronal death in AD brains is of utmost importance because no definitive therapy exists for resolution.
Some works reported the mitotic hypothesis in which vulnerable neurons in the AD brain do not only show activation of cell cycle components, but at least in some cases, show evidence of DNA replication (S-phase) prior to neuronal death [23]. Studies of the lysosomal system in AD identified a robust mobilization of this system and its impairment in neurons, which may actively promote disease pathogenesis not only by accelerating amyloidogenesis but also by triggering degeneration [24]. Another mechanism for substrate degradation in eukaryotic cells is the proteasome. In neuritic plaques and tangles in brains of AD patients, the inhibition of the proteasome may result in neuronal death [25]. In addition, several studies have provided evidence implication of the oxidative stress as a major pathogenic mechanism in AD [26].
In the last 8 years, works have focused on pathways of the control of translation, in particular the control of initiation [27–30]. Our research works are related to these molecular signalling mechanisms to explain the neuronal death observed in AD.
In this review, we first describe the control of translation in eukaryotic cells before providing the current knowledge of the control of translation dysfunction in AD. We also examine the crosslink between disturbed PKR/eIF2α and mTOR/RS6K signalling pathways. Then, we discuss the choice of PKR as a biomarker of AD.
Control of translation
In eukaryotes, protein translation includes four consecutive phases: initiation, elongation, termination and ribosome recycling. The initiation phase corresponds to the connection between mRNA and ribosomes. The elongation phase includes the links between amino acids at the ribosomal level, and is followed by the termination phase at the level of stop codon and then by the dissociation of ribosomal subunits. All phases are regulated by proteins called translation factors, which can interact directly with mRNAs but most regulation is exerted at initiation where the start codon is identified and decoded by the methionyl tRNA specialized for initiation (Met-tRNAi). The process of initiation can be divided into three stages: (1) association of Met-tRNAi and several initiation factors with the 40S ribosomal subunit to form the 43S pre-initiation complex (PIC); (2) the binding of this complex to mRNA, followed by its migration to the correct start codon and (3) the addition of the 60S ribosomal subunit to assemble an 80S ribosome at the initiation codon, ready for elongation [31, 32]. In the first stage, the binary eukaryotic initiation factor (eIF)2-GDP complex requires eIF2B, a guanine nucleotide exchange factor to catalyse the regeneration of the eIF2-GTP complex for recruitment of the initiator tRNA and conduct it as a eIF2-GTP-Met-tRNAi ternary complex (TC), [33, 34]. Then, the TC is associated to eIF1, eIF1A, eIF3 and eIF5 to form the PIC. eIF1 collaborates with eIF1A to promote scanning from the 5′ end in order to select the start codon [35, 36]. The multi-subunit factor eIF3 plays a role in the PIC assembly and in the recruitment of PIC to the mRNA [37]. The 43S PIC binds near the cap with the eIF4F complex (eIF4E, eIF4G and eIF4A) which covers the 5′ terminal methyl7GTP cap of the mRNA. In mammals, eIF3 forms a protein bridge to the mRNA by interacting with eIF4G [36]. The poly(A) binding protein interacts with eIF4G and mediates circularization of the mRNA by linking the cap to the poly(A) tail in a ‘closed loop’. The mRNA is activated and the 48S assembly can start the scanning of the start codon in an ATP-dependent reaction and partial hydrolysis of eIF2-bound GTP in the TC to eIF2-GDP-Pi [31]. When start codon is recognized, eIF1 is dissociated from the 40S, allowing the release of Pi and eIF2-GDP. Then, eIF5B-GTP catalyses the joining of the 60S subunit and then this second GTP hydrolysis triggers release of eIF5B-GDP and eIF1A to yield the final 80S initiation complex [36].
Translation of mRNAs into proteins is recognized as an important site of regulation of gene expression, with the initiation stage as the most commonly observed target for physiological control. Two particular steps of the initiation pathway appear to be hot spots for physiological regulation, the binding of Met-tRNAi to the 40S ribosomal subunit, mediated by eIF2, and the initial binding of the 43S PIC to the 5′ end of mRNA, mediated by eIF4E and associated factors (Fig. 1). In addition, various cis-regulatory elements located in the 5′- and 3′ UTRs (untranslated regions) control the synthesis of protein by employing one of the many mechanisms such as alterations in the stability of the mRNA, its accessibility to the ribosomes, its circularization and interaction with the translation machinery [38]. In the 5′ UTR of an mRNA, multiple upstream open reading frames (uORFs) influence translation rate and the impairment of these elements result in several human diseases, for example hereditary thrombocytemia by efficient expression of thrombopoietin [39], AD by enhancing B-site APP-cleaving enzyme 1 (BACE1) expression [40], bipolar affective disorder by mutation in the uORFs of 5-hydroxytryptamine receptor 3 [41]. The exact process by which these inhibitory uORFs are bypassed is currently unknown. The microRNAs (miRNAs) are cis-regulatory elements in the 3′ UTR and also regulate the translation by controlling the localization and stability of the mRNA. Mutations affecting the termination codon, polyadenylation signal and secondary structure of 3′ UTR of mRNA can cause diseases but this association needs to be confirmed [42, 43].
Fig. 1.
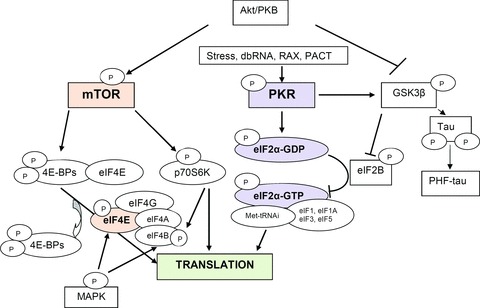
Main molecular signalling pathways involved in the control of the initiation of translation.
The control of eIF2 is linked to the state of phosphorylation at serine 51 site on the α-subunit. When phosphorylated, eIF2α becomes a competitive inhibitor of the 5-subunit guanine nucleotide exchange factor, eIF2B, and decreasing TC assembly [44, 45] (Fig. 1). There are four different eIF2α kinases in mammals activated by different stress, PKR (double-stranded (ds) RNA-dependent protein kinase), PKR-like endoplasmic reticulum kinase, heme regulated kinase and amino-acid regulated kinase, which phosphorylate the same residue in eIF2α, resulting in down-regulation of general translation [46]. However, in mammalian cells there is a reinitiation mechanism as in the yeast which overcomes the inhibitory effects of uORFs and allows the transcriptional activation of stress response genes including the regulatory subunit of an eIF2(αP) phosphatase (GADD34) to provide a negative feedback [46].
Several studies revealed an increased PKR expression with ageing [47] or in neurodegenerative diseases such as AD, Parkinson's disease, Huntington's disease [48–50] or the phosphorylation of eIF2α in cerebral ischemia-reperfusion, hypoxia or zinc toxicity [51–53]. It is demonstrated that these kinases play an important role in the process of cellular apoptosis by interacting with protein synthesis and apoptotic factors. Besides, the β-isoform of the glycogen synthase kinase-3 (GSK3β) can phosphorylate the ε subunit of eIF2B blocking the GDP/GTP exchange eIF2 and thereby leads to the inhibition of translation [54] (Fig. 1). Furthermore, it is well known that GSK3β, the main τ-kinase, can induce memory deficits in vivo and may be involved in neuronal death by inactivation of transcription factors involved in cellular defence systems [55].
PKR is an interferon (IFN) inducible eIF2α kinase that binds to dsRNA and then undergoes homodimer formation and autophosphorylation at several sites, resulting in its activation [56]. Considerable evidence confirmed that PKR, an ubiquitous protein, plays an important role in host defence against virus infection. However, in response to stress signals, an additional dsRNA-independent mechanism of activation of this enzyme is mediated by the human protein PACT (PKR-associated activator) or murine RAX (PKR activator X) in vitro and in vivo[57, 58]. Important roles in the regulation of protein synthesis and the control of cell growth and survival were demonstrated for PKR [44, 59, 60]. Several studies established that activation of PKR can either induce apoptotic cell death or at least enhance this process when apoptosis is initiated by other agents [61, 62]. Conversely, in cells deficient for the kinase or containing a dominant-negative form, there is substantial resistance to apoptosis [30, 61, 63]. It is not clear whether the phosphorylation of eIF2α is sufficient to mediate the pro-apoptotic effects of PKR. The cell expression of an inhibitor of eIF2α phosphorylation or the non-phosphorylable eIF2α mutant does partially protect cells from apoptosis [63]. Many studies performed in virus-infected cells reported that PKR regulates also different signal transducers, in particular, p53 [64], NF-κB (nuclear factor-κB) [65], mitogen-activated protein kinases (MAPK) [66], FADD (Fas-associated protein with a death domain)/caspase-8 [65, 67], signal transducers and activators of transcription (STAT) [68] and activating transcription factor 3 (ATF-3) [69].
The availability of the eIF4E factor is linked to the binding of specific proteins called 4E-BPs. When these proteins are non-phosphorylated, they have a great affinity for eIF4E, which is unable to bind the eIF4F complex, leading to the inhibition of the recognition of the mRNA 5′ cap structure [70], (Fig. 1). These proteins are mainly phosphorylated by a kinase called mTOR (mammalian target of rapamycin) or FRAP (FKBP12-rapamycin complex-associated protein) [71, 72] (Fig. 1). mTOR, is an essential serine/threonine protein kinase that functions in two distinct multiprotein complexes, mTOR complex 1 and 2 [73, 74]. mTOR complex 1 is inhibited by rapamycin and is thought to couple growth cues to cellular metabolism; mTOR complex 2 is not inhibited by rapamycin and appears to regulate spatial aspects of growth such as cell polarity. mTOR can also phosphorylate eIF4G, the ribosomal S6 kinases (RS6K) such as p70S6K which phosphorylate eIF4B, enhancing its interaction with eIF3 and stimulate protein synthesis [75–77] (Fig. 1). The regulation of mTOR activity is important for the availability of eIF4E. mTOR is activated by the phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt/PKB) pathway [70], (Fig. 1). The mTOR signalling is physiologically active and allows protein and ribosome synthesis. Many studies have shown that cellular stress can increase the binding of 4E-BPs to the eIF4E factor and can reduce protein translation [59]. Recently, it has been demonstrated that two tumour suppressor genes, the tuberous sclerosis complex (TSC) gene TSC1 (or Hamartin) and TSC2 (or Tuberin) were negative regulators of mTOR phosphorylation [78] by blocking the small GTPase Rheb activity, which is a positive regulator of mTOR [79, 80].
In addition, the reduction of eIF4E level is not enough to induce cellular apoptosis [81] and PKR can induce dephosphorylation of the proteins 4E-BPs by increasing the activity of the phosphatase 2A after its interaction with the regulatory subunit B56α, leading to cell apoptosis [82–84]. Electrophysiological results have also demonstrated that mTOR could play a role in neuronal plasticity and in the process of learning and memory. In brain slices treated with the mTOR inhibitor rapamycin, the authors have observed a decrease of the late phase of LTP induced by synaptic stimulation or brain-derived neurotrophic factor (BDNF) exposure [85, 86]. Another signalling pathway is the Ras-MAPK pathway, which is responsible for the phosphorylation of eIF4E and eIF4B and strongly impacts translation [76] (Fig. 1).
According to these results, it appears interesting to study these two pathways: PKR (pro-apoptotic) and mTOR (anti-apoptotic), involved in the control of initiation, in different models of neurodegenerative disorders and in AD patients. Both these molecular signalling pathways were largely described in AD. Studies have also reported that brain tissues from patients with Parkinson's and Huntington's diseases displayed strong induction of phosphorylated PKR in hippocampal neurons compared with age-matched disease controls [49].
Dysfunctions of protein synthesis mediated by PKR- and mTOR-dependent signalling pathways
It has been recognized for several years that the mRNA translation was disturbed in the brains of AD patients [87]. The inhibition of the ribosomal translation could be responsible for the modifications of gene expression detected in affected brain regions. An increased phosphorylation of the elongation factor 2 was also noted in the AD brains [88].
For the last 8 years, studies have focused on the control of translation, in particular the control of initiation. Aβ exposure induces a sustained reduction of mTOR/p70S6K signalling in neuroblastoma cell cultures marked by a progressive decrease of phosphorylated mTOR and phosphorylated p70S6K [27, 89]. In addition, the mTOR signalling is reduced in the cortex but not in the cerebellum of APPsl/PS1 mutant transgenic mice. We also studied the control of translation in the newly engineered APPSL/PS1 knock in transgenic mice (APPSL/PS1 KI) which display, in addition to extracellular Aβ deposits, more than 50% CA1 neuron loss at 10 months of age, starting approximately at 6 months combined with intraneuronal Aβ accumulations [90]. In the brains of APPsl/PS1 KI mice at 3 and 6 months of age, we also analysed upstream and downstream factors of mTOR. Although mTOR levels were not modified, we found a great activation of Akt with a robust accumulation of PT308-Akt in non-apoptotic neurons at 6 months of age. By contrast, a significant decrease of the p70/85S6K activation was observed in brains of PS1 KI and APPSL/PS1 KI mice with a very weak or no nucleocytoplasmic PT389-p70/85S6K staining in apoptotic neurons of APPSL/PS1 KI mice. These findings demonstrate a clear dissociation between Akt and ribosomal S6K signalling markers in these mice which could be involved in the AD pathological process [91]. Furthermore, in the lymphocytes of AD patients the PT389-p70S6K/total p70S6K ratio was reduced in AD patients (n= 26) compared to control individuals (n= 28) and levels were positively correlated with Mini Mental Status Examination (MMSE) scores [27]. However, in a larger cohort, our recent results showed no significant difference between AD and control groups for the levels of PT1462-TSC2 (control: 2.39 ± 0.59 [n= 36]; AD: 2.48 ± 0.51 [n= 50]), PS2448-mTOR (control: 0.9 ± 0.1 [n= 38]; AD: 1.34 ± 0.2 [n= 49]) and PT389-p70S6K (Control: 4.83 ± 1.01 [n= 32]; AD: 3.87 ± 0.53 [n= 50]). Other studies showed significant increases of P-p70S6K (T389 and T421/S424), P-mTOR (S2481), P-eIF4E and P-4E-BP1 (T70 and S65) in homogenates of the medial temporal cortex in AD patients as compared to control brains [92, 93]. The levels of the mTOR signalling markers were significantly and positively correlated with total τ and P-t[92, 93]. Taken together, one may propose that there are two different neuronal responses: some neurons are characterized by a highly reactive Akt/mTOR/RS6K signalling pathway involved in τ-hyperphospho-rylation and in future NFTs and other neurons displayed a total inhibition of PT389-p70/85S6K leading to the apoptotic process in AD [91, 94]. This dichotomy of neurons has to be taken into account in future therapeutic strategies.
For the PKR/eIF2α signalling pathway, previous reports revealed that phosphorylation of PKR and eIF2α was associated with degenerating neurons in AD brains [50, 95, 96]. Some data also showed that phosphorylated PKR and eIF2α co-localized with phosphorylated τ-protein in affected neurons in AD brains [50]. Another report demonstrated in vitro that Aβ neurotoxicity was associated with an increased PKR and eIF2α phosphorylation linked to increased intracellular calcium and caspase-8 activation following Aβ exposure [28, 30]. All these results suggest that PKR could be involved in the pathogenesis of AD. Western blot results revealed an increase of Pt451-PKR and PS51-eIF2α levels in the brains of APPsl/PS1 KI transgenic mice. A progressive increase of apoptotic nuclei is also observed at 3, 6 and 12 months of age in these mice and PKR is closely co-localized with DNA strand breaks in apoptotic nuclei of hippocampal neurons [97]. Furthermore, we showed in APPsl, APPsl/PS1 and APPSL/PS1 KI that Pt451-PKR was expressed around amyloid deposits (data not published) as shown by Peel and Bredesen in another APP transgenic mouse model [29]. The PT451-PKR signal was not co-localized with Aβ, but surrounded these amyloid deposits as the terminal membrane attack complex attacking dystrophic neurites in brains of AD patients [17, 21]. In addition, our results showed a great reactive microglia and activated astrocytes by Fluorojade® B staining around amyloid deposits in hippocampus and cortex of APPsl (10 months old), APPsl/PS1 (12 month-old) and APPsl/PS1 KI (starting at 6 months old) [98]. In the last mouse model, Pt451-PKR was localized in nuclei of neurons at 3 months of age before the presence of amyloid deposits and astrogliosis. In brains of AD patients, Pt451-PKR was also localized in nuclei and around amyloid deposits but the Braak score was not indicated in papers [50, 95]. However, the Braak score IV to V is characterized by the increased presence of neuroinflammation and activated microglia cells associated with fibrillar Aβ deposits [99, 100].
Interestingly, our results showed that the activation of PKR and eIF2α was significantly increased in lymphocytes of AD patients. These PKR and eIF2α levels were negatively correlated with cognitive and memory test scores performed in AD patients: the higher expression of PKR with the lower cognitive test score [101, 102].
Crosslink between the up-regulation of PKR/elF2α and the down-regulation of mTOR/RS6K in AD
Besides the eIF2a factor, PKR can phosphorylate other targets, which could explain the role of PKR in the control of translation. It is known that PKR is physically associated with p53 and phosphorylated it in mouse embryo fibroblasts cells treated with IFN [64]. Furthermore, reports showed that p53 activation is able to inhibit the activity of mTOR through a pathway involving the subsequent activation of TSC gene products TSC1 and TSC2, which are tumour suppressors and negative regulators of mTOR [78, 103–105]. In addition, authors have discovered a new transcriptional target of p53, Redd1 (regulated protein in development and DNA damage response) named also DDIT4/RTP 801/dig 2 [106, 107]. Redd1 is a new stress-induced protein and is known as a negative regulator of mTOR through TSC1/TSC2 complex [106–115]. Redd1 appears to function in the regulation of cellular ROS, a pathway linked to both stress responses and modulation of growth factor signalling [110]. Data demonstrated that endogenous Redd1 is induced following energy stress and inhibition of endogenous Redd1 increases cell size in a rapamycin-sensitive manner [111]. The interaction between these factors and PKR was mainly described in IFN-treated or virus-infected models and in hypoxia-induced models. However, the possible link between these two signalling pathways through p53, Redd1 and TSC2 remains yet unknown in AD.
A clinical study was conducted in lymphocytes of AD patients isolated from whole blood. The results showed that the expression of p53 and Redd1 genes, the activation of p53 and the levels of Redd1 protein were significantly higher in lymphocytes of AD patients compared to age-matched controls. Furthermore, statistical correlations between proteins and genes suggest that active PKR could phosphorylate p53, which could induce the transcription of Redd1 gene. However, p53 and Redd1 at transcriptional and translational levels were not correlated with the MMSE scores [102]. Other authors found that a high rate of p53 assumed an unfolded tertiary structure in fibroblasts and blood cells from sporadic AD patients with no point mutations in the p53 gene sequences [116, 117]. Furthermore, they showed that the exposure of non-AD fibroblasts to Aβ40 peptide induced the expression of this unfolded p53 protein isoforms [118]. Same results were observed in human embryonic kidney cells stably transfected with wild-type APP 751 [119]. Other works showed that the expression of the conformationally altered p53 confers a higher resistance threshold to DNA damage by cytotoxic agents than in cells with the wild-type of p53 [119, 120]. Interestingly, PKR activates GSK-3β, which phosphorylates p53 in nucleus and promotes the proteasomal degradation of p53 by the Hdm2-dependent ubiquitination [121]. Furthermore, authors showed that the down-regulation of p53 by double-stranded RNA was not a consequence of apoptosis but rather seemed to sensitize cells to death [122].
p53 can also interrupt the proliferation by detecting abnormal ribosomal biogenesis [123]. In AD, the ribogenesis is altered in apoptotic neurons [27, 91, 124]. Interestingly, p53 is able to inhibit the activity of mTOR through TSC1/TSC2 complex [105]. It is well known that 14–3-3 proteins bind TSC2 protein and this interaction is dependent of the TSC2 phosphorylation and negatively regulates TSC2 [125, 126]. A recent study showed that endogenous Redd1 is required for both dissociation of endogenous TSC2/14–3-3 proteins and inhibition of mTOR in response to hypoxia [127]. In fact, Redd1 can bind 14–3-3 proteins and so releases TSC2 which is dephosphorylated to inhibit the mTOR activity. Authors showed that the antisense Redd1 gene protected the cells from Aβ neurotoxicity [128]. Recently, we showed that the transfection of PKR siRNA to Aβ42-treated SH-SY5Y cells significantly decreased the p66 isoform of p53, the transcriptional expression of Redd1 and increased the levels of phosphorylated TSC2 [129].
Besides, it is known that the neuronal cell cycle regulatory failure, leading to apoptosis, may be a significant component of the pathogenesis of AD [130–132]. Many studies showed the expression of a repertoire of proteins in neurons involved in the control of cell cycle until the phase G2 with the expression of the cyclin B1 for the maturation promoting factor in order to re-enter the cell cycle [133–137]. However, little is known about the possible causes of the neuronal cell cycle re-entry and its subsequent regulatory failure. In neurons that express high levels of Bax (apoptotic mRNA transcriptionally activated by p53), the G2 arrest will lead to apoptosis and in neuronal populations that are not-apoptosis competent, a prolonged G2 arrest could lead to the development of AD-type pathology with hyperphosphorylation of t controlling by various cell cycle kinases [135]. In our experimental conditions, one may propose that the p66 protein can represent an ubiquitinylated form of p53 which will be degraded in the proteasome. Thus, p53 in this state cannot control the cell cycle. Neurons re-enter in the cell cycle and replicate their DNA. p53 protein will carry another post-translational modification which can be involved in the failure of the cell cycle regulation either by loss of its transcriptionally activity of cyclin-dependent kinase (CDK) inhibitor (CDKI) gene or by gain of a new function involving in the abortion of the cell cycle. PKR can also play a role in this failure by stimulation the proteasomal degradation of cyclin D1 [138].
TSC2 negatively regulates CDK2 and positively CDKI. Interestingly, some CDK trigger phosphorylation of TSC2 levels, leading to its proteasomal degradation [125]. In our recent work, the PKR gene silencing induced an increase in the levels of PT1462-TSC2 protein in Aβ42-exposed SH-SY5Y cells. Thus, PKR also controls this state of phosphorylation of TSC2 perhaps through the activation of the proteasomal degradation of cyclin D1 involved in the increased phosphorylation of TSC2 when it is associated to CDK4/6 [139]. Then, TSC2 in PKR gene-silencing conditions can be blocked by sequestration with 14–3-3 proteins to prevent the inhibitory effect of TSC2 in the mTOR signalling pathway.
Finally, these results underline the critical role of PKR in the biochemical cascade leading to death in AD. p53, Redd1 and TSC2 can be molecular links between the up-regulation of PKR and the down-regulation of mTOR largely described in AD (Fig. 2). PKR seems to be the initiator of this deregulation which could be involved in the failure of the re-entry of neurons in cell cycle. The inhibition of PKR activity may represent an effective therapeutic strategy in the treatment of AD.
Fig. 2.
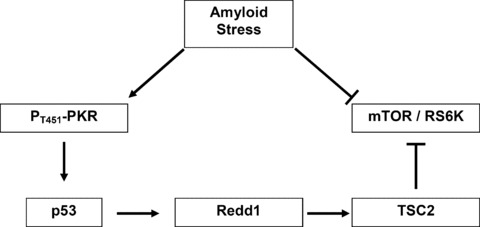
Crosslink between PKR- and mTOR-dependent signalling pathways in apoptotic neurons in AD.
PKR: a potential biomarker of AD diagnosis
At present, AD cannot be diagnosed until dementia appears and the specificity in the discrimination of AD and other dementia with cerebrospinal fluid biomarkers (total-τ, phospho-τ and Aβ 1–42) is not absolute. Thus, the identification of early disease-related targets is crucial to improve diagnostic accuracy and/or to facilitate the development of new drug therapies. Candidate biomarkers should be molecules representing the altered cerebral molecular and cellular processes observed in AD. Furthermore, these biomarkers should be easily accessible. In this regard, many reports underline the great importance of the peripheral cells such as fibroblasts, platelets and mononuclear cells [101, 118, 124, 140–144]. Ray et al.[145] have found 18 signalling proteins in blood plasma that can be used to classify blinded samples from Alzheimer's and control patients with close to 90% accuracy and to identify patients with mild cognitive impairment that progressed to AD 2–6 years later. Biological analysis of the 18 proteins points to systemic deregulation of haematopoiesis, immune responses, apoptosis and neuronal support in pre-symptomatic AD [145]. Thus, peripheral cell's biomarkers, easily accessible, could facilitate an early diagnosis or help following the severity of disease. The control of translation through PKR/eIF2a signalling pathway is altered in AD lymphocytes as it was observed in brains of AD patients [50, 95]. In addition, we reported that PKR represents an initiator of deregulated translation via two consecutive targets p53 and Redd1 in AD lymphocytes [102]. However, our data showed that the number of mononuclear cells was significantly higher (43%) in AD patients than in controls. These results are in accordance with other studies [146, 147]. Paradoxically, the up-regulation of PKR in mononuclear cells do not lead to the cell death observed in cellular models exposed to Ap, in AD transgenic mice and in brains of AD patients [50, 95, 97]. With all these data, we can propose some hypotheses for cellular paradox between neurons and lymphocytes in AD (Fig. 3). First, expression of the conformationaly altered p53 could confer a higher resistance to the activated PKR signalling pathway. Second, Akt signalling pathway is up-regulated in lymphocytes [148]. Third, mTOR suppresses caspase-1 activation and orchestrates the defence programme of innate immune cells [149]. Finally, inflammatory mechanisms represent an important component which, first contribute to defence mechanisms but secondly, stimulated by degeneration, may significantly contribute to disease progression and chronicity [15, 150, 151]. Besides the local innate immunity of the central nervous system (CNS) including the activation of microglia and astrocytes, the involvement of complement factors, pro-inflammatory cytokines and chemokines, increased studies suggest that systemic immune response, including T-cell immune response, may be involved in the inflammation process of AD [152, 153]. Peripheral T cells could exert their effects on the AD process without entering the CNS. Either T cells secrete cytokines such as IFN-γ on activation, and that such pro-inflammatory cytokines enter the CNS and activate microglia and astrocytes, or peripheral activated T cells promote activation of myeloid cells such as monocytes, macrophages and dendritic cells that secrete pro-inflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6. In fact, there is some evidence of elevated IL-6 levels in monocytes derived from AD patients [154–156]. Recently, it was demonstrated that a blockade of TNF-α production at the periphery prevented histological lesions induced by IL-1β in CNS [157].
Fig. 3.
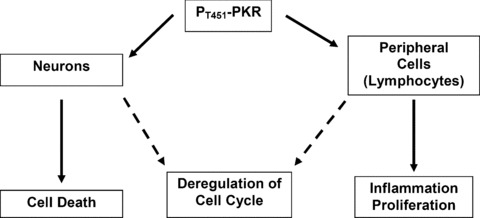
Paradoxal effect of active PKR in neurons and lymphocytes in AD.
Furthermore, activated T cells also exist as infiltrates in the brains of AD patients [158–160]. In vitro data showed a ‘cross-talking’ between microglia and T cells, microglia can serve as antigen-presenting cells for Aβ-reactive T cells and, in turn, T cells themselves can influence microglial differentiation [161–163]. Different types of T cells would be expected to have different effects on microglial cell activation and therefore on amyloid clearance. T-helper 1 cells can promote microglial cell activation through the secretion of IFN-γ, which also up-regulates the expression major histocompatibility complex class II molecules by microglial cells, thereby enhancing the interaction between T cells and microglial cells [164, 165]. A study has shown that the interaction of CD40 (expressed on the surface of microglia) and CD40L (T-cell receptor) promotes pro-inflammatory microglial cell activation in response to Aβ[166]. Furthermore, recent evidence revealed that peripheral T cells of AD patients overexpressed CXCR2 (CXC chemokine receptor 2), which is intracerebral microglial TNF-α-dependent and is associated with T-cell adhesion on brain endothelia and transendothelial migration [167]. It is also known that glial cells can mediate the permeability of blood brain barrier to recruit specialized immune cells from the periphery such as T cells [164]. Besides these roles of peripheral immune cells in AD brains, further studies would be investigated to understand deeply the interactions between glia and peripheral immune cells. These researches may reveal new targets modulating brain inflammation and help in developing novel immunotherapeutic approaches to AD.
Nevertheless, despite these several defence mechanisms, authors showed a cell cycle regulatory failure in lymphocytes as it was observed in neurons [130–132, 168, 169].
PKR is not specific to AD because it is also known to be active in other neurodegenerative diseases like Parkinson's disease [49, 170], Huntington's disease [48] and amyotrophic lateral sclerosis [96]. PKR is not specific to neurodegenerative disease because it is also implied in infectious [61, 171], inflammatory [172, 173] and ischemic [171] pathology without cognitive decline. So, PKR may not be a biomarker of cognitive decline but rather an early biomarker of cell death. Indeed, many studies revealed the role of PKR in apoptosis induction. We can remember that the levels of phosphorylated PKR were significantly increased in brains of APPslPS1 KI mice as early as 3 months of age associated with intraneuronal amyloid peptide before the massive neuronal death observed at 6 months [97]. At present, there are five major effectors, eIF2a, FADD, p53, NF-κB and ATF-3 implicated in mediating PKR-induced apoptosis [171]. In AD, eIF2a and p53 are targets of PKR [28, 30, 97, 102, 129]. NF-kB and ATF-3 activation by PKR is also involved in apoptosis induction as shown in virus-infected cells [69, 174–176]. However, NF-κB could be under the control of PKR in AD because this factor is activated in AD [177, 178]. In addition, FADD and the subsequent caspase-8 are responsible for PKR-induced apoptosis in virus-infected cells [179] and an overexpression of a dominant-negative FADD construct rescued SH-SY5Y cells from either TNF-related apoptosis-inducing ligand or Aβ-induced neurotoxicity [180]. Recently, we demonstrated the physical and functional interactions between PKR and FADD in Aβ neurotoxicity (data submitted). Taken together, PKR could be a critical target in AD because of its early activation and its apoptotic effectors shared with Aβ.
Conclusion and perspectives
In conclusion, data revealed that PKR can represent an initiator of the dysfunction of the control of translation described in AD. In the brain, this disturbance is associated with the neuronal death while in peripheral mononuclear cells of AD patients, PKR can control the cell cycle and the inflammatory reaction. In brains of APPSL/PS1 KI mice, PKR was early up-regulated before the massive neuronal loss [97]. In patients, the PKR levels were correlated to the severity of the disease [101]. Thus, it is now interesting to develop molecules able to cross the blood brain barrier in order to target this early active kinase for the treatment of the disease which represents a great problem of public Health. However, PKR is a constitutive protein and all its physiological roles are yet unknown. Besides its role in the antiviral defence mechanism of the host, PKR controls the cell cycle, the proliferation and differentiation of cells. Thus, further studies will be needed to determine the best concentration of specific inhibitors only inhibiting the excessive active form of PKR or will have to use a therapeutic strategy specifically directed to neurons to prevent the activation of PKR involving in their apoptosis.
Acknowledgments
The authors thank Professor Jacques Hugon for the management of the research group in Poitiers from 2003 to 2006. This study was mainly supported by the grant of AIRMA (Association Internationale pour la Recherche sur la Maladie d’Alzheimer), by the grant of LECMA (Ligue EuropÈenne Contre la Maladie d’Alzheimer), by the French Ministry of Education and Research with a grant to the Research Unit GREVIC, EA 3808 and by the Poitiers University Hospital.
References
- 1.Lage JM. 100 Years of Alzheimer's disease (1906–2006) J Alzheimers Dis. 2006;9:15–26. doi: 10.3233/jad-2006-9s303. [DOI] [PubMed] [Google Scholar]
- 2.Saunders AM, Schmader K, Breitner JC, et al. Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer's disease and in other amyloid-forming diseases. Lancet. 1993;342:710–1. doi: 10.1016/0140-6736(93)91709-u. [DOI] [PubMed] [Google Scholar]
- 3.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's dis-ease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 4.Strittmatter WJ, Saunders AM, Goedert M, et al. Isoform-specific interactions of apolipoprotein E with microtubule-associ-ated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:11183–6. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang DY, Goedert M, Jakes R, et al. Isoform-specific interactions of apolipoprotein E with the microtubule-associated protein MAP2c: implications for Alzheimer's disease. Neurosci Lett. 1994;182:55–8. doi: 10.1016/0304-3940(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 6.Poirier J, Sevigny P. Apolipoprotein E4, cholinergic integrity and the pharmacogenetics of Alzheimer's disease. J Neural Transm Suppl. 1998;53:199–207. doi: 10.1007/978-3-7091-6467-9_18. [DOI] [PubMed] [Google Scholar]
- 7.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–77. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Cheng R, Schupf N, et al. The association between genetic variants in SORL1 and Alzheimer disease in an urban, multiethnic, community-based cohort. Arch Neurol. 2007;64:501–6. doi: 10.1001/archneur.64.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lichtenthaler SF, Wang R, Grimm H, et al. Mechanism of the cleavage specificity of Alzheimer's disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc Natl Acad Sci USA. 1999;96:3053–8. doi: 10.1073/pnas.96.6.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vassar R, Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–22. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 11.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 12.Kidd M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature. 1963;197:192–3. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 13.Goedert M, Klug A, Crowther RA. Tau protein, the paired helical filament and Alzheimer's disease. J Alzheimers Dis. 2006;9:195–207. doi: 10.3233/jad-2006-9s323. [DOI] [PubMed] [Google Scholar]
- 14.Fischer O. Die presbyophrene Demenz, deren anatomische Grundlage und Klinische Abgrenzung. Z Ges Neurol u Psychiat. 1910;3:371–471. [Google Scholar]
- 15.Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 16.Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Itagaki S, McGeer PL, Akiyama H, et al. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–82. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 18.Rozemuller JM, Eikelenboom P, Pals ST, et al. Microglial cells around amyloid plaques in Alzheimer's disease express leucocyte adhesion molecules of the LFA-1 family. Neurosci Lett. 1989;101:288–92. doi: 10.1016/0304-3940(89)90547-8. [DOI] [PubMed] [Google Scholar]
- 19.McGeer PL, Rogers J, McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–6. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- 20.McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–16. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- 21.Webster S, Lue LF, Brachova L, et al. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer's disease. Neurobiol Aging. 1997;18:415–21. doi: 10.1016/s0197-4580(97)00042-0. [DOI] [PubMed] [Google Scholar]
- 22.McGeer PL, Akiyama H, Itagaki S, et al. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett. 1989;107:341–6. doi: 10.1016/0304-3940(89)90843-4. [DOI] [PubMed] [Google Scholar]
- 23.Davies P. A long trek down the pathways of cell death in Alzheimer's disease. J Alzheimers Dis. 2006;9:265–9. doi: 10.3233/jad-2006-9s329. [DOI] [PubMed] [Google Scholar]
- 24.Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis. 2006;9:277–89. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- 25.van Leeuwen FW, Hol EM, Fischer DF. Frameshift proteins in Alzheimer's disease and in other conformational disorders: time for the ubiquitin-proteasome system. J Alzheimers Dis. 2006;9:319–25. doi: 10.3233/jad-2006-9s336. [DOI] [PubMed] [Google Scholar]
- 26.Smith MA. Oxidative stress and iron imbalance in Alzheimer disease: how rust became the fuss! J Alzheimers Dis. 2006;9:305–8. doi: 10.3233/jad-2006-9s334. [DOI] [PubMed] [Google Scholar]
- 27.Lafay-Chebassier C, Paccalin M, Page G, et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem. 2005;94:215–25. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- 28.Suen KC, Yu MS, So KF, et al. Upstream signaling pathways leading to the activation of double-stranded RNA-dependent serine/threonine protein kinase in beta-amyloid peptide neurotoxicity. J Biol Chem. 2003;278:49819–27. doi: 10.1074/jbc.M306503200. [DOI] [PubMed] [Google Scholar]
- 29.Peel AL, Bredesen DE. Activation of the cell stress kinase PKR in Alzheimer's disease and human amyloid precursor protein transgenic mice. Neurobiol Dis. 2003;14:52–62. doi: 10.1016/s0969-9961(03)00086-x. [DOI] [PubMed] [Google Scholar]
- 30.Chang RC, Suen KC, Ma CH, et al. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J Neurochem. 2002;83:1215–25. doi: 10.1046/j.1471-4159.2002.01237.x. [DOI] [PubMed] [Google Scholar]
- 31.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pain VM. Initiation of protein synthesis in eukaryotic cells. Eur J Biochem. 1996;236:747–71. doi: 10.1111/j.1432-1033.1996.00747.x. [DOI] [PubMed] [Google Scholar]
- 33.Pain VM. Initiation of protein synthesis in mammalian cells. Biochem J. 1986;235:625–37. doi: 10.1042/bj2350625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merrick WC. Mechanism and regulation of eukaryotic protein synthesis. Microbiol Rev. 1992;56:291–315. doi: 10.1128/mr.56.2.291-315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fekete CA, Mitchell SF, Cherkasova VA, et al. N- and C-terminal residues of eIF1A have opposing effects on the fidelity of start codon selection. EMBO J. 2007;26:1602–14. doi: 10.1038/sj.emboj.7601613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pestova TV, Lorsch JR, Hellen CUT. The mechanism of translation initiation in eukaryotes. In: Mathews M, Sonenberg N, Hershey JWB, editors. In translational control in biology and medicine. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 87–128. [Google Scholar]
- 37.Hinnebusch AG, Dever TE, Asano K. Mechanism of translation initiation in the yeast Saccharomyces cerevisiae. In: Mathews M, Sonenberg N, Hershey JWB, editors. In translational control in biology and medicine. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 225–68. [Google Scholar]
- 38.Chatterjee S, Pal JK. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol Cell. 2009;101:251–62. doi: 10.1042/BC20080104. [DOI] [PubMed] [Google Scholar]
- 39.Cazzola M, Skoda RC. Translational pathophysiology: a novel molecular mechanism of human disease. Blood. 2000;95:3280–8. [PubMed] [Google Scholar]
- 40.Zhou W, Song W. Leaky scanning and reinitiation regulate BACE1 gene expression. Mol Cell Biol. 2006;26:3353–64. doi: 10.1128/MCB.26.9.3353-3364.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niesler B, Kapeller J, Hammer C, et al. Serotonin type 3 receptor genes: HTR3A, B, C, D, E. Pharmacogenomics. 2008;9:501–4. doi: 10.2217/14622416.9.5.501. [DOI] [PubMed] [Google Scholar]
- 42.Soifer HS, Rossi JJ, Saetrom P. MicroRNAs in disease and potential therapeutic applications. Mol Ther. 2007;15:2070–9. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 43.Conne B, Stutz A, Vassalli JD. The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat Med. 2000;6:637–41. doi: 10.1038/76211. [DOI] [PubMed] [Google Scholar]
- 44.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–20. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 45.Dever TE, Feng L, Wek RC, et al. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68:585–96. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
- 46.Ron D, Harding HP. eIF2α phosphorylation in celluar stress responses and disease. In: Mathews M, Sonenberg N, Hershey JWB, editors. In translational control in biology and medicine. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 345–68. [Google Scholar]
- 47.Ladiges W, Morton J, Blakely C, et al. Tissue specific expression of PKR protein kinase in aging B6D2F1 mice. Mech Ageing Dev. 2000;114:123–32. doi: 10.1016/s0047-6374(00)00097-x. [DOI] [PubMed] [Google Scholar]
- 48.Peel AL, Rao RV, Cottrell BA, et al. Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington's disease (HD) transcripts and is activated in HD tissue. Hum Mol Genet. 2001;10:1531–8. doi: 10.1093/hmg/10.15.1531. [DOI] [PubMed] [Google Scholar]
- 49.Bando Y, Onuki R, Katayama T, et al. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson's disease and Huntington's disease. Neurochem Int. 2005;46:11–8. doi: 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 50.Chang RC, Wong AK, Ng HK, et al. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer's disease. Neuroreport. 2002;13:2429–32. doi: 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- 51.Frerichs KU, Smith CB, Brenner M, et al. Suppression of protein synthesis in brain during hibernation involves inhibition of protein initiation and elongation. Proc Natl Acad Sci USA. 1998;95:14511–6. doi: 10.1073/pnas.95.24.14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alirezaei M, Nairn AC, Glowinski J, et al. Zinc inhibits protein synthesis in neurons. Potential role of phosphorylation of translation initiation factor-2alpha. J Biol Chem. 1999;274:32433–8. doi: 10.1074/jbc.274.45.32433. [DOI] [PubMed] [Google Scholar]
- 53.DeGracia DJ, Adamczyk S, Folbe AJ, et al. Eukaryotic initiation factor 2alpha kinase and phosphatase activity during postischemic brain reperfusion. Exp Neurol. 1999;155:221–7. doi: 10.1006/exnr.1998.6986. [DOI] [PubMed] [Google Scholar]
- 54.Welsh GI, Miller CM, Loughlin AJ, et al. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 1998;421:125–30. doi: 10.1016/s0014-5793(97)01548-2. [DOI] [PubMed] [Google Scholar]
- 55.Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer's disease. J Alzheimers Dis. 2006;9:309–17. doi: 10.3233/jad-2006-9s335. [DOI] [PubMed] [Google Scholar]
- 56.Romano PR, Garcia-Barrio MT, Zhang X, et al. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol Cell Biol. 1998;18:2282–97. doi: 10.1128/mcb.18.4.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bennett RL, Blalock WL, Abtahi DM, et al. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy, and viral infection. Blood. 2006;108:821–9. doi: 10.1182/blood-2005-11-006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–90. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clemens MJ. Translational regulation in cell stress and apoptosis. Roles of the eIF4E binding proteins. J Cell Mol Med. 2001;5:221–39. doi: 10.1111/j.1582-4934.2001.tb00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res. 1997;17:503–24. doi: 10.1089/jir.1997.17.503. [DOI] [PubMed] [Google Scholar]
- 61.Balachandran S, Barber GN. PKR in innate immunity, cancer, and viral oncolysis. Methods Mol Biol. 2007;383:277–301. doi: 10.1007/978-1-59745-335-6_18. [DOI] [PubMed] [Google Scholar]
- 62.Der SD, Yang YL, Weissmann C, et al. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:3279–83. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srivastava SP, Kumar KU, Kaufman RJ. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem. 1998;273:2416–23. doi: 10.1074/jbc.273.4.2416. [DOI] [PubMed] [Google Scholar]
- 64.Cuddihy AR, Wong AH, Tam NW, et al. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene. 1999;18:2690–702. doi: 10.1038/sj.onc.1202620. [DOI] [PubMed] [Google Scholar]
- 65.Gil J, Esteban M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis. 2000;5:107–14. doi: 10.1023/a:1009664109241. [DOI] [PubMed] [Google Scholar]
- 66.Goh KC, deVeer MJ, Williams BR. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000;19:4292–7. doi: 10.1093/emboj/19.16.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balachandran S, Kim CN, Yeh WC, et al. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998;17:6888–902. doi: 10.1093/emboj/17.23.6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramana CV, Grammatikakis N, Chernov M, et al. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J. 2000;19:263–72. doi: 10.1093/emboj/19.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guerra S, Lopez-Fernandez LA, Garcia MA, et al. Human gene profiling in response to the active protein kinase, interferon-induced serine/threonine protein kinase (PKR), in infected cells. Involvement of the transcription factor ATF-3 IN PKR-induced apoptosis. J Biol Chem. 2006;281:18734–45. doi: 10.1074/jbc.M511983200. [DOI] [PubMed] [Google Scholar]
- 70.Raught B, Gingras AC. Signaling to translation initiation. In: Mathews M, Sonenberg N, Hershey JWB, editors. In translational control in biology and medicine. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 369–400. [Google Scholar]
- 71.Raught B, Gingras AC, Sonenberg N. The target of rapamycin (TOR) proteins. Proc Natl Acad Sci USA. 2001;98:7037–44. doi: 10.1073/pnas.121145898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 73.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 74.De Virgilio C, Loewith R. The TOR signalling network from yeast to man. Int J Biochem Cell Biol. 2006;38:1476–81. doi: 10.1016/j.biocel.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 75.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–18. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 76.Holz MK, Ballif BA, Gygi SP, et al. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–80. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 77.Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res. 1999;253:100–9. doi: 10.1006/excr.1999.4683. [DOI] [PubMed] [Google Scholar]
- 78.Krymskaya VP. Tumour suppressors hamartin and tuberin: intracellular signalling. Cell Signal. 2003;15:729–39. doi: 10.1016/s0898-6568(03)00040-8. [DOI] [PubMed] [Google Scholar]
- 79.Long X, Lin Y, Ortiz-Vega S, et al. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–13. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 80.Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28:573–6. doi: 10.1016/j.tibs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 81.Luo H, Duguid W, Chen H, et al. The effect of rapamycin on T cell development in mice. Eur J Immunol. 1994;24:692–701. doi: 10.1002/eji.1830240331. [DOI] [PubMed] [Google Scholar]
- 82.Ruvolo VR, Kurinna SM, Karanjeet KB, et al. PKR regulates B56(alpha)-mediated BCL2 phosphatase activity in acute lymphoblastic leukemia-derived REH cells. J Biol Chem. 2008;283:35474–85. doi: 10.1074/jbc.M800951200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeffrey IW, Bushell M, Tilleray VJ, et al. Inhibition of protein synthesis in apoptosis: differential requirements by the tumor necrosis factor alpha family and a DNA-damaging agent for caspases and the double-stranded RNA-dependent protein kinase. Cancer Res. 2002;62:2272–80. [PubMed] [Google Scholar]
- 84.Xu Z, Williams BR. The B56alpha regulatory subunit of protein phosphatase 2A is a target for regulation by double-stranded RNA-dependent protein kinase PKR. Mol Cell Biol. 2000;20:5285–99. doi: 10.1128/mcb.20.14.5285-5299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang SJ, Reis G, Kang H, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–72. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nguyen PV. Protein synthesis during LTP: linking synaptic activity to translation. Trends Neurosci. 2002;25:180. doi: 10.1016/s0166-2236(02)02166-5. [DOI] [PubMed] [Google Scholar]
- 87.Langstrom NS, Anderson JP, Lindroos HG, et al. Alzheimer's disease-associated reduction of polysomal mRNA translation. Brain Res Mol Brain Res. 1989;5:259–69. doi: 10.1016/0169-328x(89)90060-0. [DOI] [PubMed] [Google Scholar]
- 88.Johnson G, Gotlib J, Haroutunian V, et al. Increased phosphorylation of elongation factor 2 in Alzheimer's disease. Brain Res Mol Brain Res. 1992;15:319–26. doi: 10.1016/0169-328x(92)90124-t. [DOI] [PubMed] [Google Scholar]
- 89.Lafay-Chebassier C, Perault-Pochat MC, Page G, et al. The immunosuppressant rapamycin exacerbates neurotoxicity of Abeta peptide. J Neurosci Res. 2006;84:1323–34. doi: 10.1002/jnr.21039. [DOI] [PubMed] [Google Scholar]
- 90.Casas C, Sergeant N, Itier JM, et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Damjanac M, Rioux Bilan A, Paccalin M, et al. Dissociation of Akt/PKB and ribosomal S6 kinase signaling markers in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2008;29:354–67. doi: 10.1016/j.nbd.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 92.Li X, Alafuzoff I, Soininen H, et al. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 2005;272:4211–20. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 93.Li X, An WL, Alafuzoff I, et al. Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport. 2004;15:2237–40. doi: 10.1097/00001756-200410050-00019. [DOI] [PubMed] [Google Scholar]
- 94.Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer's disease. J Cell Mol Med. 2008;12:2525–32. doi: 10.1111/j.1582-4934.2008.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Onuki R, Bando Y, Suyama E, et al. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer's disease. EMBO J. 2004;23:959–68. doi: 10.1038/sj.emboj.7600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peel AL. PKR activation in neurodegenerative disease. J Neuropathol Exp Neurol. 2004;63:97–105. doi: 10.1093/jnen/63.2.97. [DOI] [PubMed] [Google Scholar]
- 97.Page G, Rioux Bilan A, Ingrand S, et al. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer's disease. Neuroscience. 2006;139:1343–54. doi: 10.1016/j.neuroscience.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 98.Damjanac M, Rioux Bilan A, Barrier L, et al. Fluoro-Jade B staining as useful tool to identify activated microglia and astrocytes in a mouse transgenic model of Alzheimer's disease. Brain Res. 2007;1128:40–9. doi: 10.1016/j.brainres.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 99.Eikelenboom P, Veerhuis R, Scheper W, et al. The significance of neuroinflammation in understanding Alzheimer's disease. J Neural Transm. 2006;113:1685–95. doi: 10.1007/s00702-006-0575-6. [DOI] [PubMed] [Google Scholar]
- 100.Hoozemans JJ, Veerhuis R, Rozemuller AJ, et al. Neuronal COX-2 expression and phosphorylation of pRb precede p38 MAPK activation and neurofibrillary changes in AD temporal cortex. Neurobiol Dis. 2004;15:492–9. doi: 10.1016/j.nbd.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 101.Paccalin M, Pain-Barc S, Pluchon C, et al. Activated mTOR and PKR kinases in lymphocytes correlate with memory and cognitive decline in Alzheimer's disease. Dement Geriatr Cogn Disord. 2006;22:320–6. doi: 10.1159/000095562. [DOI] [PubMed] [Google Scholar]
- 102.Damjanac M, Page G, Ragot S, et al. PKR, a cognitive decline biomarker, can regulate translation via two consecutive molecular targets p53 and Redd1 in lymphocytes of AD patients. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00688.x. [Epub ahead of press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Feng Z, Hu W, de Stanchina E, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 104.Levine AJ, Feng Z, Mak TW, et al. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20:267–75. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 105.Feng Z, Zhang H, Levine AJ, et al. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ellisen LW, Ramsayer KD, Johannessen CM, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10:995–1005. doi: 10.1016/s1097-2765(02)00706-2. [DOI] [PubMed] [Google Scholar]
- 107.Jin HO, An S, Lee HC, et al. Hypoxic condition- and high cell density-induced expression of Redd1 is regulated by activation of hypoxia-inducible factor-1alpha and Sp1 through the phosphatidylinositol 3-kinase/Akt signaling pathway. Cell Signal. 2007;19:1393–403. doi: 10.1016/j.cellsig.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 108.Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Corradetti MN, Inoki K, Guan KL. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem. 2005;280:9769–72. doi: 10.1074/jbc.C400557200. [DOI] [PubMed] [Google Scholar]
- 110.Shoshani T, Faerman A, Mett I, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–93. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sofer A, Lei K, Johannessen CM, et al. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–45. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ellisen LW. Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle. 2005;4:1500–2. doi: 10.4161/cc.4.11.2139. [DOI] [PubMed] [Google Scholar]
- 113.Jozwiak J, Jozwiak S, Grzela T, et al. Positive and negative regulation of TSC2 activity and its effects on downstream effectors of the mTOR pathway. Neuromolecular Med. 2005;7:287–96. doi: 10.1385/NMM:7:4:287. [DOI] [PubMed] [Google Scholar]
- 114.Kimball SR, Do AN, Kutzler L, et al. Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem. 2008;283:3465–75. doi: 10.1074/jbc.M706643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang H, Kubica N, Ellisen LW, et al. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem. 2006;281:39128–34. doi: 10.1074/jbc.M610023200. [DOI] [PubMed] [Google Scholar]
- 116.Lanni C, Racchi M, Mazzini G, et al. Conformationally altered p53: a novel Alzheimer's disease marker? Mol Psychiatry. 2008;13:641–7. doi: 10.1038/sj.mp.4002060. [DOI] [PubMed] [Google Scholar]
- 117.Uberti D, Lanni C, Carsana T, et al. Identification of a mutant-like conformation of p53 in fibroblasts from sporadic Alzheimer's disease patients. Neurobiol Aging. 2006;27:1193–201. doi: 10.1016/j.neurobiolaging.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 118.Lanni C, Uberti D, Racchi M, et al. Unfolded p53: a potential biomarker for Alzheimer's disease. J Alzheimers Dis. 2007;12:93–9. doi: 10.3233/jad-2007-12109. [DOI] [PubMed] [Google Scholar]
- 119.Uberti D, Cenini G, Olivari L, et al. Over-expression of amyloid precursor protein in HEK cells alters p53 conformational state and protects against doxorubicin. J Neurochem. 2007;103:322–33. doi: 10.1111/j.1471-4159.2007.04757.x. [DOI] [PubMed] [Google Scholar]
- 120.Uberti D, Carsana T, Bernardi E, et al. Selective impairment of p53-mediated cell death in fibroblasts from sporadic Alzheimer's disease patients. J Cell Sci. 2002;115:3131–8. doi: 10.1242/jcs.115.15.3131. [DOI] [PubMed] [Google Scholar]
- 121.Baltzis D, Pluquet O, Papadakis AI, et al. The eIF2α kinases PERK and PKR activate glycogen synthase kinase 3 to promote the proteasomal degradation of p53. J Biol Chem. 2007;282:31675–87. doi: 10.1074/jbc.M704491200. [DOI] [PubMed] [Google Scholar]
- 122.Marques JT, Rebouillat D, Ramana CV, et al. Down-regulation of p53 by double-stranded RNA modulates the antiviral response. J Virol. 2005;79:11105–14. doi: 10.1128/JVI.79.17.11105-11114.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Opferman JT, Zambetti GP. Translational research? Ribosome integrity and a new p53 tumor suppressor checkpoint. Cell Death Differ. 2006;13:898–901. doi: 10.1038/sj.cdd.4401923. [DOI] [PubMed] [Google Scholar]
- 124.Paccalin M, Al Khidir F, Barc SP, et al. Peripheral p70S6k levels and emotional memory in patients with Alzheimer's disease. Neurosci Lett. 2006;410:162–4. doi: 10.1016/j.neulet.2006.07.053. [DOI] [PubMed] [Google Scholar]
- 125.Rosner M, Hanneder M, Siegel N, et al. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat Res. 2008;658:234–46. doi: 10.1016/j.mrrev.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 126.Shumway SD, Li Y, Xiong Y. 14–3-3beta binds to and negatively regulates the tuberous sclerosis complex 2 (TSC2) tumor suppressor gene product, tuberin. J Biol Chem. 2003;278:2089–92. doi: 10.1074/jbc.C200499200. [DOI] [PubMed] [Google Scholar]
- 127.DeYoung MP, Horak P, Sofer A, et al. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14–3-3 shuttling. Genes Dev. 2008;22:239–51. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim JR, Lee SR, Chung HJ, et al. Identification of amyloid beta-peptide responsive genes by cDNA microarray technology: involvement of RTP801 in amyloid beta-peptide toxicity. Exp Mol Med. 2003;35:403–11. doi: 10.1038/emm.2003.53. [DOI] [PubMed] [Google Scholar]
- 129.Morel M, Couturier J, Pontcharraud R, et al. Evidence of molecular links between PKR and mTOR signalling pathways in Aβ neurotoxicity: role of p53, Redd1 and TSC2. Neurobiol Dis. 2009 doi: 10.1016/j.nbd.2009.07.004. [Epub ahead of print 22 July 2009] [DOI] [PubMed] [Google Scholar]
- 130.Liu DX, Greene LA. Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell Tissue Res. 2001;305:217–28. doi: 10.1007/s004410100396. [DOI] [PubMed] [Google Scholar]
- 131.Nagy Z. Cell cycle regulatory failure in neurones: causes and consequences. Neurobiol Aging. 2000;21:761–9. doi: 10.1016/s0197-4580(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 132.Neve RL, McPhie DL. The cell cycle as a therapeutic target for Alzheimer's disease. Pharmacol Ther. 2006;111:99–113. doi: 10.1016/j.pharmthera.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 133.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–7. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McShea A, Harris PL, Webster KR, et al. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–9. [PMC free article] [PubMed] [Google Scholar]
- 135.Nagy Z. The last neuronal division: a unifying hypothesis for the pathogenesis of Alzheimer's disease. J Cell Mol Med. 2005;9:531–41. doi: 10.1111/j.1582-4934.2005.tb00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neu-rodegenerative conditions. Acta Neuropathol. 1997;93:294–300. doi: 10.1007/s004010050617. [DOI] [PubMed] [Google Scholar]
- 137.Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease. J Cell Biol. 1996;132:413–25. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Raven JF, Baltzis D, Wang S, et al. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J Biol Chem. 2008;283:3097–108. doi: 10.1074/jbc.M709677200. [DOI] [PubMed] [Google Scholar]
- 139.Zacharek SJ, Xiong Y, Shumway SD. Negative regulation of TSC1-TSC2 by mammalian D-type cyclins. Cancer Res. 2005;65:11354–60. doi: 10.1158/0008-5472.CAN-05-2236. [DOI] [PubMed] [Google Scholar]
- 140.Lanni C, Racchi M, Mazzini G, et al. Conformationally altered p53: a novel Alzheimer's disease marker? Mol Psychiatry. 2008;13:641–7. doi: 10.1038/sj.mp.4002060. [DOI] [PubMed] [Google Scholar]
- 141.Tang K, Hynan LS, Baskin F, et al. Platelet amyloid precursor protein processing: a bio-marker for Alzheimer's disease. J Neurol Sci. 2006;240:53–8. doi: 10.1016/j.jns.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Paccalin M, Pain-Barc S, Pluchon C, et al. The relation between p70S6k expression in lymphocytes and the decline of cognitive test scores in patients with Alzheimer disease. Arch Intern Med. 2005;165:2428–9. doi: 10.1001/archinte.165.20.2428. [DOI] [PubMed] [Google Scholar]
- 143.Eckert A, Schindowski K, Leutner S, et al. Alzheimer's disease-like alterations in peripheral cells from presenilin-1 transgenic mice. Neurobiol Dis. 2001;8:331–42. doi: 10.1006/nbdi.2000.0378. [DOI] [PubMed] [Google Scholar]
- 144.Schindowski K, Kratzsch T, Peters J, et al. Impact of aging: sporadic, and genetic risk factors on vulnerability to apoptosis in Alzheimer's disease. Neuromolecular Med. 2003;4:161–78. doi: 10.1385/NMM:4:3:161. [DOI] [PubMed] [Google Scholar]
- 145.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 146.Lombardi VR, Garcia M, Rey L, et al. Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer's Disease (AD) individuals. J Neuroimmunol. 1999;97:163–71. doi: 10.1016/s0165-5728(99)00046-6. [DOI] [PubMed] [Google Scholar]
- 147.Shalit F, Sredni B, Brodie C, et al. T lymphocyte subpopulations and activation markers correlate with severity of Alzheimer's disease. Clin Immunol Immunopathol. 1995;75:246–50. doi: 10.1006/clin.1995.1078. [DOI] [PubMed] [Google Scholar]
- 148.Juntilla MM, Koretzky GA. Critical roles of the PI3K/Akt signaling pathway in T cell development. Immunol Lett. 2008;116:104–10. doi: 10.1016/j.imlet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schmitz F, Heit A, Dreher S, et al. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol. 2008;38:2981–92. doi: 10.1002/eji.200838761. [DOI] [PubMed] [Google Scholar]
- 150.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–15. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 151.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease – a double-edged sword. Neuron. 2002;35:419–32. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 152.Miscia S, Ciccocioppo F, Lanuti P, et al. Abeta(1–42) stimulated T cells express P-PKC-delta and P-PKC-zeta in Alzheimer disease. Neurobiol Aging. 2009;30:394–406. doi: 10.1016/j.neurobiolaging.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 153.Town T, Tan J, Flavell RA, et al. T-cells in Alzheimer's disease. Neuromolecular Med. 2005;7:255–64. doi: 10.1385/NMM:7:3:255. [DOI] [PubMed] [Google Scholar]
- 154.Huberman M, Sredni B, Stern L, et al. IL-2 and IL-6 secretion in dementia: correlation with type and severity of disease. J Neurol Sci. 1995;130:161–4. doi: 10.1016/0022-510x(95)00016-u. [DOI] [PubMed] [Google Scholar]
- 155.Shalit F, Sredni B, Stern L, et al. Elevated interleukin-6 secretion levels by mononuclear cells of Alzheimer's patients. Neurosci Lett. 1994;174:130–2. doi: 10.1016/0304-3940(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 156.Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol. 1993;47:169–76. doi: 10.1016/0165-5728(93)90027-v. [DOI] [PubMed] [Google Scholar]
- 157.Jiang Y, Deacon R, Anthony DC, et al. Inhibition of peripheral TNF can block the malaise associated with CNS inflammatory diseases. Neurobiol Dis. 2008;32:125–32. doi: 10.1016/j.nbd.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 158.Itagaki S, McGeer PL, Akiyama H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer's disease brain tissue. Neurosci Lett. 1988;91:259–64. doi: 10.1016/0304-3940(88)90690-8. [DOI] [PubMed] [Google Scholar]
- 159.Rogers J, Luber-Narod J, Styren SD, et al. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging. 1988;9:339–49. doi: 10.1016/s0197-4580(88)80079-4. [DOI] [PubMed] [Google Scholar]
- 160.Togo T, Akiyama H, Iseki E, et al. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. 2002;124:83–92. doi: 10.1016/s0165-5728(01)00496-9. [DOI] [PubMed] [Google Scholar]
- 161.Monsonego A, Imitola J, Zota V, et al. Microglia-mediated nitric oxide cytotoxicity of T cells following amyloid betapep-tide presentation to Th1 cells. J Immunol. 2003;171:2216–24. doi: 10.4049/jimmunol.171.5.2216. [DOI] [PubMed] [Google Scholar]
- 162.Monsonego A, Zota V, Karni A, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–22. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Seguin R, Biernacki K, Prat A, et al. Differential effects of Th1 and Th2 lymphocyte supernatants on human microglia. Glia. 2003;42:36–45. doi: 10.1002/glia.10201. [DOI] [PubMed] [Google Scholar]
- 164.Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer's disease. J Cell Mol Med. 2008;12:762–80. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tan J, Town T, Crawford F, et al. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–93. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- 167.Liu YJ, Guo DW, Tian L, et al. Peripheral T cells derived from Alzheimer's disease patients overexpress CXCR2 contributing to its transendothelial migration, which is microglial TNF-alpha-dependent. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.03.024. DOI: 10.1016/j.neurobiolaging.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 168.Nagy Z, Combrinck M, Budge M, et al. Cell cycle kinesis in lymphocytes in the diagnosis of Alzheimer's disease. Neurosci Lett. 2002;317:81–4. doi: 10.1016/s0304-3940(01)02442-9. [DOI] [PubMed] [Google Scholar]
- 169.Stieler JT, Lederer C, Bruckner MK, et al. Impairment of mitogenic activation of peripheral blood lymphocytes in Alzheimer's disease. Neuroreport. 2001;12:3969–72. doi: 10.1097/00001756-200112210-00023. [DOI] [PubMed] [Google Scholar]
- 170.Ryu EJ, Harding HP, Angelastro JM, et al. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–8. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Garcia MA, Gil J, Ventoso I, et al. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70:1032–60. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gern JE, French DA, Grindle KA, et al. Double-stranded RNA induces the synthesis of specific chemokines by bronchial epithelial cells. Am J Respir Cell Mol Biol. 2003;28:731–7. doi: 10.1165/rcmb.2002-0055OC. [DOI] [PubMed] [Google Scholar]
- 173.Matsukura S, Kokubu F, Kurokawa M, et al. Role of RIG-I, MDA-5, and PKR on the expression of inflammatory chemokines induced by synthetic dsRNA in airway epithelial cells. Int Arch Allergy Immunol. 2007;143:80–3. doi: 10.1159/000101411. [DOI] [PubMed] [Google Scholar]
- 174.Gil J, Alcami J, Esteban M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB. Mol Cell Biol. 1999;19:4653–63. doi: 10.1128/mcb.19.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Taddeo B, Luo TR, Zhang W, et al. Activation of NF-kappaB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc Natl Acad Sci USA. 2003;100:12408–13. doi: 10.1073/pnas.2034952100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bonnet MC, Daurat C, Ottone C, et al. The N-terminus of PKR is responsible for the activation of the NF-kappaB signaling pathway by interacting with the IKK complex. Cell Signal. 2006;18:1865–75. doi: 10.1016/j.cellsig.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 177.Fontalba A, Gutierrez O, Llorca J, et al. Deficiency of CARD8 is associated with increased Alzheimer's disease risk in women. Dement Geriatr Cogn Disord. 2008;26:247–50. doi: 10.1159/000160956. [DOI] [PubMed] [Google Scholar]
- 178.Paris D, Patel N, Quadros A, et al. Inhibition of Abeta production by NF-kappaB inhibitors. Neurosci Lett. 2007;415:11–6. doi: 10.1016/j.neulet.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 179.Gil J, Esteban M. The interferon-induced protein kinase (PKR), triggers apoptosis through FADD-mediated activation of caspase 8 in a manner independent of Fas and TNF-alpha receptors. Oncogene. 2000;19:3665–74. doi: 10.1038/sj.onc.1203710. [DOI] [PubMed] [Google Scholar]
- 180.Cantarella G, Uberti D, Carsana T, et al. Neutralization of TRAIL death pathway protects human neuronal cell line from beta-amyloid toxicity. Cell Death Differ. 2003;10:134–41. doi: 10.1038/sj.cdd.4401143. [DOI] [PubMed] [Google Scholar]