

Background: α-Amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase is a key enzyme that controls quinolinic acid levels.
Results: Two arginines, including one from a neighboring subunit, are required for substrate binding.
Conclusion: Dimerization and two arginine residues are required for activity.
Significance: This study provides the first structurally proven example of a functionally active heterodimer hybrid resulting from a simple mixing of two inactive homodimer mutants.
Keywords: Crystal Structure, Decarboxylase, Enzyme Mechanisms, Metabolic Regulation, Protein Assembly, Protein Folding, Dimerization, Hybridization
Abstract
Although the crystal structure of α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase from Pseudomonas fluorescens was solved as a dimer, this enzyme is a mixture of monomer, dimer, and higher order structures in solution. In this work, we found that the dimeric state, not the monomeric state, is the functionally active form. Two conserved arginine residues are present in the active site: Arg-51 and an intruding Arg-239* from the neighboring subunit. In this study, they were each mutated to alanine and lysine, and all four mutants were catalytically inactive. The mutants were also incapable of accommodating pyridine-2,6-dicarboxylic acid, a competitive inhibitor of the native enzyme, suggesting that the two Arg residues are involved in substrate binding. It was also observed that the decarboxylase activity was partially recovered in a heterodimer hybridization experiment when inactive R51(A/K) and R239(A/K) mutants were mixed together. Of the 20 crystal structures obtained from mixing inactive R51A and R239A homodimers that diffracted to a resolution lower than 3.00 Å, two structures are clearly R51A/R239A heterodimers and belong to the C2 space group. They were refined to 1.80 and 2.00 Å resolutions, respectively. Four of the remaining crystals are apparently single mutants and belong to the P42212 space group. In the heterodimer structures, one active site is shown to contain dual mutation of Ala-51 and Ala-239*, whereas the other contains the native Arg-51 and Arg-239* residues, identical to the wild-type structure. Thus, these observations provide the foundation for a molecular mechanism by which the oligomerization state of α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase could regulate the enzyme activity.
Introduction
Arginine residues perform diverse functions in proteins because of their charge distribution, their capability of forming multiple hydrogen bonds, and their long flexible side chains. One popular function is the formation of salt bridges in which the positively charged guanidine head pairs with a negatively charged carboxylate group (1, 2). In enzymes, arginine residues are often proposed to participate in recognizing, binding, and maintaining the orientation of substrates and reaction intermediates (3–5). Arginine is also involved in RNA recognition because its guanidium group is able to form five hydrogen bonds with RNA acceptor groups (6). Although not widespread, arginine residues have also recently been proposed to act as a general base in several enzymes (7–9).
Two arginine residues in α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase (ACMSD)3 caught our attention because they appear to be in an optimal position to bind the substrate α-amino-β-carboxymuconate-ϵ-semialdehyde (ACMS) by interacting with its two carboxyl groups. ACMSD is a metal-dependent enzyme in the amidohydrolase superfamily (10, 11), and its substrate, ACMS, is an unstable tryptophan catabolic intermediate in the kynurenine pathway (11–13). The conversion of ACMS to α-amino muconate-ϵ-semialdehyde is catalyzed by ACMSD; however, ACMS spontaneously forms quinolinic acid (QA) in a nonenzymatic dehydration process in the absence of ACMSD (14, 15) (Scheme 1). Thus, ACMSD is a key enzyme that determines the QA levels. QA is maintained only at the basal level for the de novo synthesis of a small fraction of NAD+ in mammals. Reduced activity of ACMSD leads to accumulation of QA in body fluids, a condition that is known to be related to several neurodegenerative diseases including Alzheimer disease (16), Huntington disease (17), stroke (12), and epilepsy (12, 13, 18). However, the instability of ACMS and the compounds with similar structures makes studying the mechanism of ACMSD substrate binding technically challenging.
SCHEME 1.
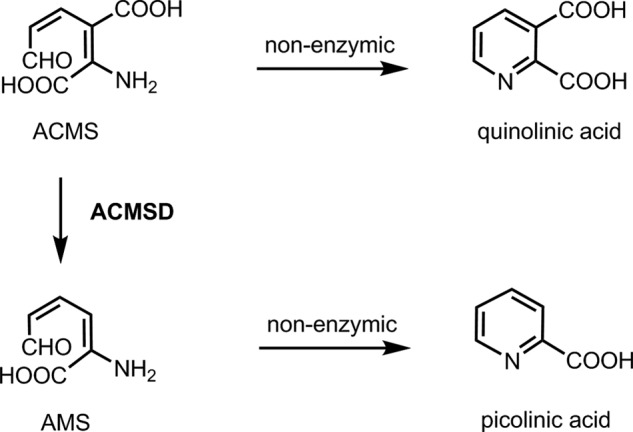
The first crystal structure of ACMSD was solved as a homodimer from Pseudomonas fluorescens (19). Two strictly conserved arginine residues are present at the active site: Arg-51 and Arg-239* (Fig. 1). Arg-51 is located in the mobile insertion domain, which is proposed to undergo conformational changes upon substrate binding because of its unique position and high flexibility (19). Arg-239* is an intruding residue that is inserted from the sixth α-helix of the other subunit, and it is labeled with a * hereafter to distinguish its origin from the chain of a neighboring subunit. Because of its high flexibility, this residue is shown to be present in alternative conformations in chain B of the crystal structure.
FIGURE 1.
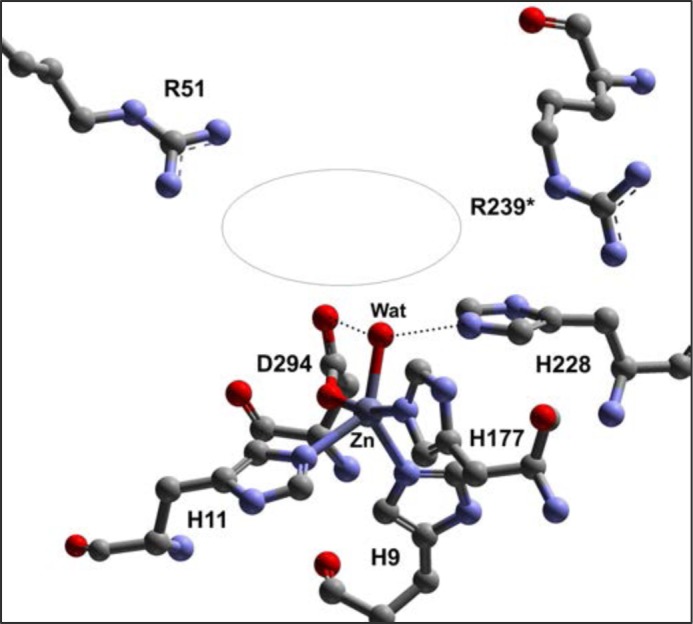
The active site metal center of P. fluorescens ACMSD (Protein Data Bank code 2HBV) and the putative substrate-binding pocket (illustrated with an ellipse). His-228 is a previously identified acid-base catalyst. Arg-51 lies in the putative substrate-binding pocket, and Arg-239* is an intruding residue from the neighboring subunit.
The second ACMSD crystal structure is the human enzyme in complex with 1,3-dihydroxyacetone-phosphate. This inactive complex was assigned as a monomer by Rizzi and co-workers (20). The equivalent residue of Arg-51 is shown in the active site, but the counterpart of Arg-239 is far removed from the catalytic center. The same group previously reported that functional human ACMSD is a monomer because gel filtration chromatography showed the as-isolated enzyme with a molecular mass of ∼50 kDa (21), which is between monomer (38 kDa) and dimer (76 kDa) mass. In fact, it is still unclear whether the catalytically active form of human ACMSD in solution is a monomeric or dimeric enzyme at the present time. All of the P. fluorescens ACMSD structures determined thus far, including wild type and mutants charged with different transition metals, are dimeric (19, 22). If the monomeric form is catalytically active, then only one conserved arginine residue, Arg-51, is present in the active site of the enzyme in both human ACMSD and P. fluorescens ACMSD structures. Arg-239 of the same subunit is not located near the enzyme active site in the monomeric state and would therefore be less likely to be an essential component of catalysis. Thus, point mutation at residue 239 provides a sensible means for probing the effects of protein oligomerization on the catalytic activity.
The present work represents the first attempt to investigate the molecular mechanism by which the oligomerization state of ACMSD could determine the enzyme activity. The quaternary state required for catalytic activity is reported, and the catalytic roles of the two conserved arginine residues, Arg-51 and the intruding Arg-239* from the neighboring subunit, are proposed. Interestingly, we found that the inactive Arg-51 and Arg-239 mutants could form a crystallographically accessible, catalytically active heterodimer through protein hybridization.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
R51A, R51K, R239A, and R239K single mutation mutants were constructed by using the PCR overlap extension mutagenesis technique (23) described previously (10) and by using the plasmid containing ACMSD from P. fluorescens as a template (11). The mutants were verified by DNA sequencing to ensure that base changes were introduced correctly and that no random changes had occurred. After sequencing, the positive clone was used for protein expression in Escherichia coli BL21(DE3).
Protein Preparation, SDS-PAGE, and Size Exclusion Chromatography
Expression and purification of ACMSD proteins followed the protocol established previously (10, 11, 22, 24) with a few minor changes. Rather than using pH 8.0 buffer during affinity chromatography purification, buffer at pH 7.4 was used to increase protein stability of the mutants. Metal-free apo-enzyme was obtained by treating purified protein with 5 mm EDTA for 12 h followed by running a G50 desalting column. Zn(II)- and Co(II)-containing ACMSD were reconstituted as previously described (11). Purified protein samples were mixed with SDS loading buffer and heated for 10 min or with no heating prior to loading to 10% SDS-PAGE. ACMSD was loaded onto a Superdex 75 column pre-equilibrated with buffer containing 25 mm HEPES (pH 7.0) and 5% glycerol (v/v). Protein was eluted with the same buffer.
Enzyme Activity Assay
The substrate ACMS was generated enzymatically by catalyzing the dioxygenation of 3-hydroxyanthranilic acid in oxygen saturated buffer with ferrous 3-hydroxyanthranilate 3,4-dioxygenase containing no free transient metal ion as reported previously (10, 11, 22). Briefly, the ACMSD enzyme assay mixture contained 5–120 μm ACMS and an appropriate amount of ACMSD protein in 25 mm HEPES buffer with 5% glycerol, pH 7.0. Specific activities of ACMSD proteins were measured in triplicate at room temperature on an Agilent 8453 spectrophotometer by monitoring the decrease of ACMS absorbance at 360 nm using a molar extinction coefficient of 47,500 m−1 cm−1 (25) when the substrate concentration was equal to or lower than 20 μm, or at 320 nm using a molar extinction coefficient of 9600 m−1 cm−1 when the substrate concentration was higher than 20 μm (10).
Derivation of Equations for Analysis of the Correlation of Monomer/Dimer Ratio and Catalytic Activity
To show that dimerization is required for catalytic activity, a series of activity assays were performed in which the stock concentration of ACMSD is varied while keeping the final assay concentration constant. If the rate of dissociation of the ACMSD dimers is sufficiently slow on the time scale of the assay, then the measured activity should conform to Equation 1, where kobs is the observed decarboxylation rate, kmax is the maximum rate, and %D is the mole ratio of dimer present in the assay.
To solve for the percent dimer, mass balance was invoked to define M0 as the total protomer concentration, M as the monomer concentration, and D as the dimer concentration.
The dimer percentage can then be written in terms of dimer concentration and total protomer concentration (a quantity that is known in an activity assay) alone.
 |
The association constant for this equilibrium can then be written in terms of dimer concentration and total protomer concentration by substitution of Equation 2, as seen in Equation 4.
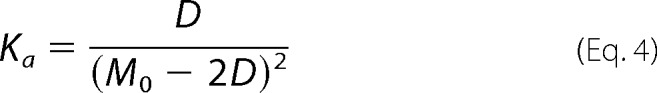 |
Distribution and collection of like terms then gives Equation 5, and Equation 5 can be multiplied by M0−2 to give Equation 6.
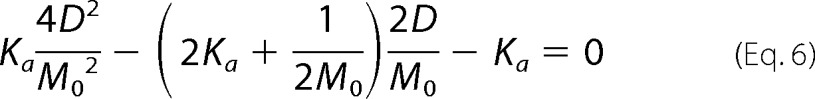 |
The quadratic equation can now be invoked on Equation 6 to solve directly for the dimer percentage and is shown in Equation 7. Multiplying the right side of Equation 7 by a disguised 1, (2 M0/2M0), the expression for the dimer percentage can be simplified to Equation 8.
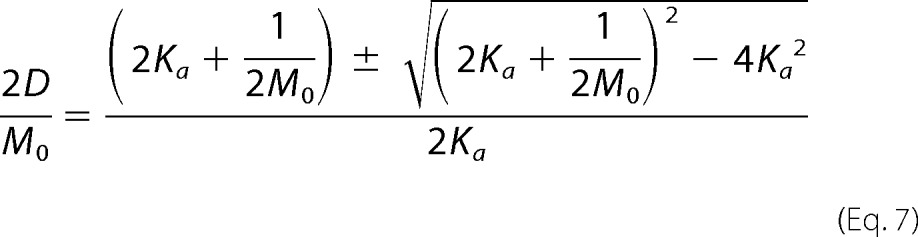 |
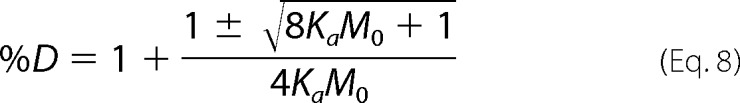 |
Substitution of Equation 8 back into Equation 1 gives kobs as a function of kmax, Ka, and M0 (i.e., Equation 9). Nonlinear least squares fitting can then be used to obtain both Ka and kmax (though both positive and negative square roots should be considered, only the negative roots gave convergent fits of the measured data).
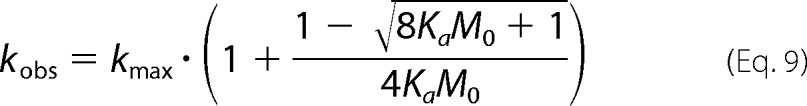 |
Protein Hybridization
Arg-51 and Arg-239 mutants were mixed in a 1:1 ratio in 25 mm HEPES buffer, pH 7.0, containing 5% glycerol. The final protein concentration for each mutant was 12 μm. Specific activity of the mixtures was examined frequently after mixing by a spectrophotometric assay on an Agilent 8453 diode array spectrophotometer as described above.
Identification of a Competitive Inhibitor
Pyridine-2,6-dicarboxylic acid (PDC) inhibition pattern and constant were determined by measuring kcat and Km in the presence of varying inhibitor concentrations (0, 20, 40, and 80 μm). Apparent Km values for each inhibitor concentration were plotted as a function of inhibitor concentration, and inhibition is expressed as Ki values in micromolar (Ki = intercept/slope).
Analysis of PDC Inhibitor Interaction with the Arginine Mutants
PDC in HEPES buffer displays UV absorbance at 270 nm with extinction coefficient of 4,100 m−1 cm−1. ACMSD proteins (1.5 ml, 40 μm in each sample type), including wild type, R51A, R239A, H228G, and premixed R51A and R239A for 48 h, were incubated with one equivalent of PDC for 5 min in separate experiments, followed by filtration in an Amicon ultra centrifugal filter tube with cellulose membrane of 10,000-dalton cutoff. PDC (40 μm) with no protein was used in the control experiment. Samples were centrifuged in 3,000 × g for 8 min. The flow-through solution containing unbound DPA was quantified by an Agilent 8453 spectrophotometer.
X-ray Data Collection and Crystallographic Refinement
Zinc-reconstituted mutant proteins were used for crystallization. All single crystals were obtained following the crystallization conditions described in the previous studies (19, 22). For the heterodimer crystal, R51A and R239A were mixed for more than 48 h at 4 °C before being used for crystallization by hanging drop vapor diffusion in VDX plates (Hampton Research). Mother liquid containing 20% ethylene glycol was used for crystal mounting. X-ray diffraction data were collected at SER-CAT Beamline 22-ID/BM of the Advanced Photon Source (Argonne National Laboratory, Argonne, IL). Data were processed with HKL-2000 (26). Structure solutions were obtained by molecular replacement with MolRep (27) from the CCP4 suite (28) with wild-type ACMSD (Protein Data Bank code 2HBV) as the search model (19). Refinement was carried out using PHENIX software (29), and the model building was carried out in COOT (30). Ala-51 and Ala-239 were modeled based on the 2Fo − Fc and Fo − Fc electron density maps. Refinement was assessed as complete when the Fo − Fc electron density contained only noise.
RESULTS
ACMSD Does Not Exist as a Single Species in Solution
As shown in Fig. 2A, ACMSD presents several bands by SDS-PAGE that correspond to monomer, dimer, and higher order oligomerization states if the sample is not heated before loading onto the gel (Fig. 2A, lane 1). However, if the sample is heated before loading, the bands corresponding to the higher order states and most of the dimer are not observed (Fig. 2A, lane 2). The size exclusion chromatography profile, on the other hand, presents two major peaks. The molecular mass of the protein eluted in the first peak is larger than 100 kDa, and the second peak exhibits a molecular mass of ∼51 kDa. The molecular mass of a P. fluorescens ACMSD monomer, however, is 38 kDa. Also, there is no difference in the specific activity of the two fractions as they elute from the column. The second peak shows two major bands corresponding to ACMSD monomer and dimer molecular masses on SDS-PAGE when not heated before loading (Fig. 2A, lane 3). The band that arises from the dimer is not observed upon heating of the sample before loading (Fig. 2A, lane 4). Hence, we assign the second peak as a combination of ACMSD monomer and dimer and the first peak as a mixture of higher order complexes that are not able to be resolved further.
FIGURE 2.
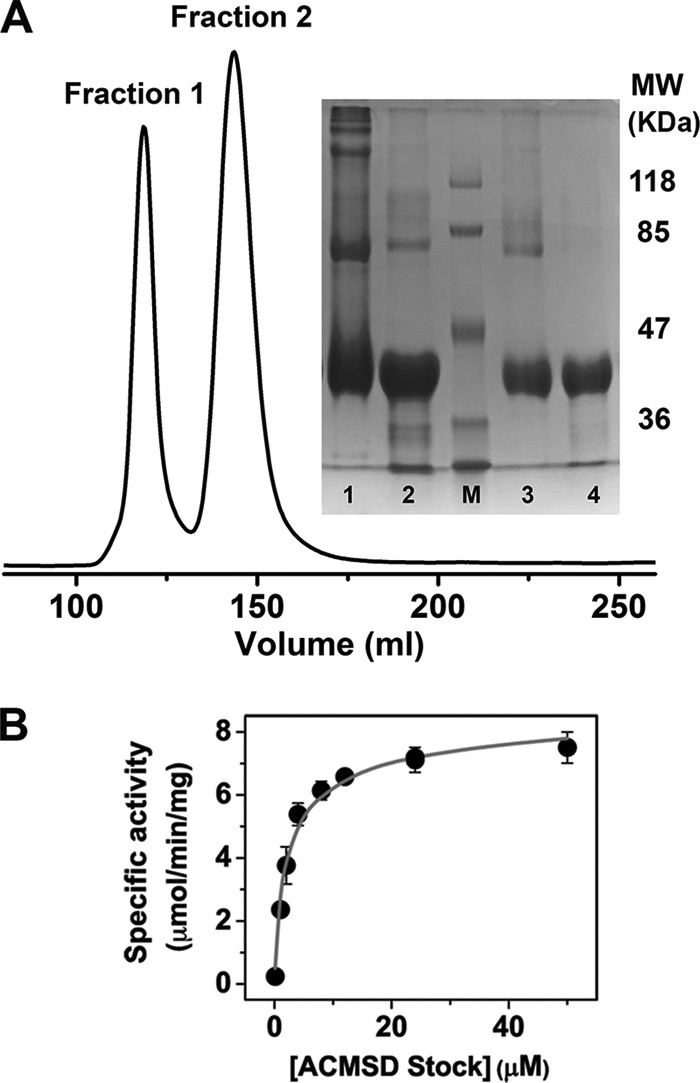
A, the UV absorbance at 280 nm of ACMSD as a function of the elution volume from size exclusion chromatography column: Superdex 75. Fraction 1 corresponds to molecular mass (MW) larger than 100 kDa, and fraction 2 has a molecular mass of ∼51 kDa. The inset is an SDS-PAGE. Lane 1, as-isolated ACMSD with no heating; lane 2, as-isolated ACMSD heated for 10 min; lane M, molecular marker; lane 3, fraction 2 with no heating; lane 4, fraction 2 heated for 10 min. B, the specific activity of ACMSD as a function of different enzyme stock concentration. The final reaction concentration of the enzyme was kept constant at 0.1 μm, and the data were fit to Equation 9.
To estimate the dimer dissociation constant, ACMSD was prepared in stock concentrations ranging from 0.1 to 50 μm (protomer concentration) and allowed to equilibrate at 4 °C for 24 h. Specific activity of the different stock concentrations was determined by keeping the final enzyme and substrate concentrations constant. The specific activity increased with increasing stock concentration. Decarboxylation rates as a function of enzyme stock concentration were plotted in Fig. 2B, and the data were fit to Equation 9, which represents the equilibrium of ACMSD between inactive monomer and active dimer forms as described under “Experimental Procedures.” Fitting of the data to Equation 9 gives a Ka of 3.3 × 106 m−1 and a maximum specific activity of 9.4 μmol·min−1·mg−1.
Mutational Analysis and Catalytic Activity Rescue through Protein Hybridization
Arg-51 and Arg-239 were each mutated to alanine and lysine. All of the four mutants were found to be completely inactive with either zinc or cobalt as the cofactor, the two metals that were previously found to be optimal for catalytic activity in the wild-type enzyme. Chemical rescue performed on R51A and R239A by guanidine derivatives was unsuccessful in restoring enzymatic activity. However, when the two mutants, R51A and R239A, were mixed together, enzyme activity was slowly restored (Fig. 3A). The reaction rates were determined at several time points after mixing. Samples incubated for 1, 7, and 24 h on ice were analyzed by following decay of the substrate ACMS at 360 nm for 1 min. Reaction with no enzyme and the two inactive arginine mutants were used as negative controls. The rate of activity rescue was temperature-dependent, and the rate of recovery was faster when the mixing reaction was carried out at room temperature rather than on ice. The specific activity was calculated and plotted as a function of incubation time as shown in Fig. 3B and fit to a single exponential association, Equation 10, where A is the measured specific activity, Amax is the maximum activity, k is the associative rate constant, and t is time in minutes. According to the fitting, the maximum rate is 152 ± 5 nmol·min−1·mg−1, and the rate constant is 7.2 ± 0.5 × 10−3 min−1. We also determined kinetic parameters for the mixed zinc-reconstituted mutants after 48 h of equilibration on ice. Fig. 3C shows the Michaelis-Menten plot. The Km, 9.3 ± 1 μm, of the mixed mutants is similar to that of the wild-type Zn-ACMSD, 9.6 μm. The kcat, 0.15 ± 0.01 s−1, of the mixed mutants, on the other hand, is dramatically decreased compared with the 6.5 s−1 of the WT Zn-ACMSD (19).
FIGURE 3.
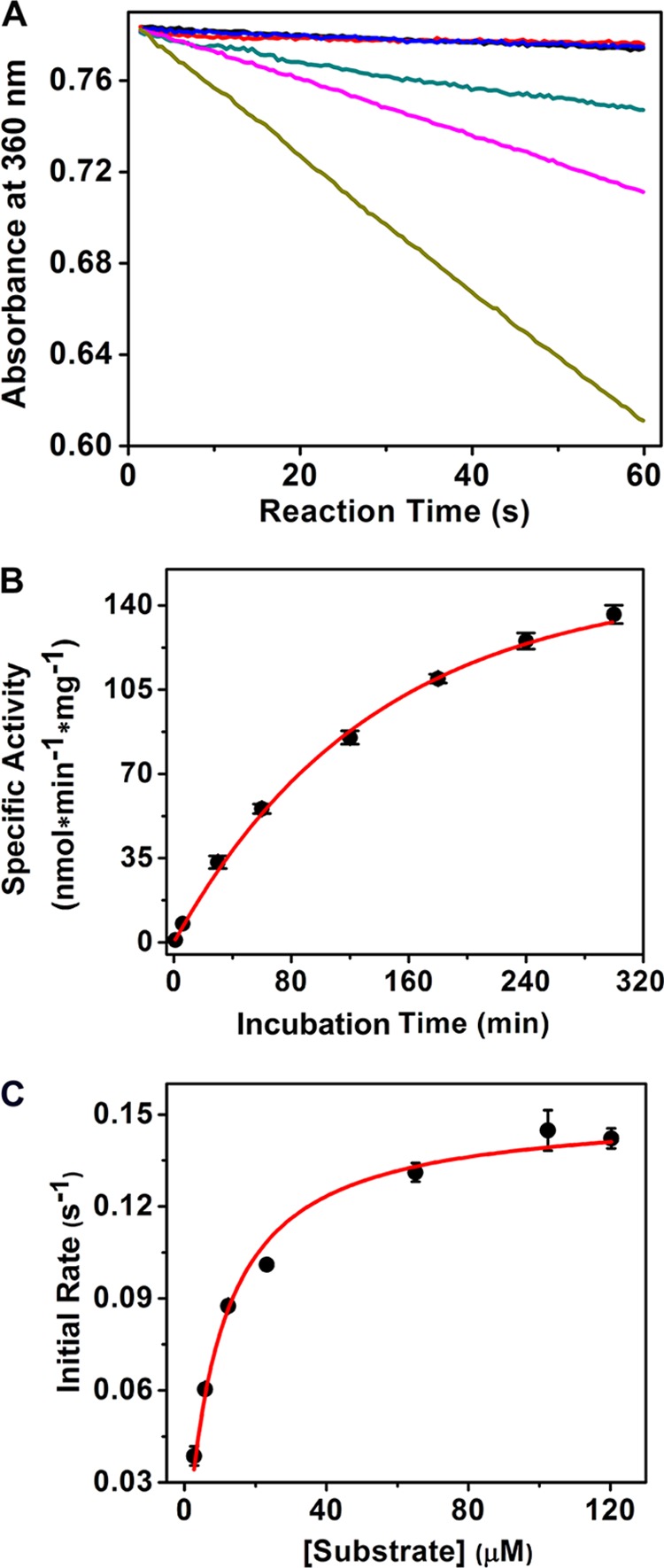
A, catalytic activity rescue of mixed R51A and R239A mutants. Decay of the substrate, ACMS, in the first minute was monitored by UV-visible spectroscopy at 360 nm. Autodecay of substrate (black trace), in the presence of R51A (red trace), and in the presence of R239A (blue trace) are three overlapping lines. R51A and R239A were premixed on ice for 1 h (green trace), 7 h (pink trace), and 24 h (tan trace), respectively. B, the rescued activity increased as a function of incubation time of the two mutants. The data were fit to Equation 10. The incubation and kinetic reaction are done at room temperature. C, Michaelis-Menten kinetics of the 1:1 mixed R51A and R239A after 48 h of equilibration.
Structural Determination of R51A, R239A, and the Hybridized Mixtures of the Two Mutants
All crystals of R51A and R239A ACMSD belong to the P42212 space group. They diffracted poorly and only at relatively low resolutions. The best structures obtained were 2.50 Å resolution for R51A (Protein Data Bank code 4IFO) and 2.40 Å for R239A (Protein Data Bank code 4IFR). Interestingly, crystals from the mixture of these two mutants were crystallized in both the P42212 and C2 space groups.
A total of 20 data sets were collected from individual single crystals obtained from crystallization trays with equal molar ratio mixing of R51A and R239A mutant proteins. Among the 20 crystal structures that we have solved, three of them are R239A homodimer, and one of them is an R51A homodimer. All of these four structures are determined in the P42212 space group. Two single crystals were found to belong to the C2 space group, and they were refined to 1.8 Å (Protein Data Bank entry 4IG2) and 2.0 Å (Protein Data Bank entry 4IFK) resolutions, respectively. The structures determined from these two crystals clearly differ from R51A or R239A, and they fit the proposed heterodimer as illustrated in Fig. 4. Fourteen of the data sets in the P42212 space group cannot be assigned to any homodimer or heterodimer because of incomplete electron density that might be caused by the flexibility of the arginine side chain or low arginine occupancy. The data collection and refinement statistics for both the homodimer single mutants and the heterodimers are listed in Table 1.
FIGURE 4.

Molecular mechanism for catalytic activity rescue through protein hybridization of the two completely inactive mutants, R51A and R239A. Blue represents R239A, whereas pink represents R51A. Circles indicate native residues, and rectangles indicate mutated residues.
TABLE 1.
X-ray crystallography data collection and refinement statistics
| Data collection | R51A-R239A Heterodimer 1 | R51A-R239A Heterodimer 2 | R51A Homodimer | R239A Homodimer |
|---|---|---|---|---|
| Detector type | MAR225 CCD | MAR300 CCD | MAR300 CCD | MAR300 CCD |
| Source | APS, Sector 22-BM | APS, Sector 22-ID | APS, Sector 22-ID | APS, Sector 22-ID |
| Space group | C2 | C2 | P42212 | P42212 |
| Unit cell lengths (Å) | a = 153.10, b = 48.52, c = 109.83 | a = 153.85, b = 49.06, c = 110.55 | a = b = 91.21, c = 170.47 | a = b = 91.16, c = 167.99 |
| Unit cell angles (°) | α = γ = 90, β = 126.80 | α = γ = 90, β = 127.10 | α = β = γ = 90 | α = β = γ = 90 |
| Wavelength (Å) | 1.00 | 0.80 | 0.80 | 0.80 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Resolution (Å)a | 35.00–1.80 (1.86–1.80) | 35.00–2.00 (2.03–2.00) | 35.00–2.50 (2.54–2.50) | 35.00–2.40 (2.44–2.40) |
| Completeness (%)a | 86.1 (42.7) | 97.6 (87.6) | 99.9 (100.0) | 99.6 (95.7) |
| Rmerge (%)a,b | 5.0 (60.0) | 10.7 (47.8) | 7.8 (64.0) | 9.9 (79.3) |
| I/σIa | 37.2 (1.5) | 28.4 (1.9) | 34.9 (2.9) | 50.0 (1.9) |
| Multiplicitya | 6.8 (3.7) | 5.9 (2.9) | 15.8 (11.4) | 22.0 (7.5) |
| Refinement | ||||
| Resolution (Å) | 1.80 | 2.00 | 2.50 | 2.40 |
| No. reflections; working/test | 51829/2590 | 42682/2143 | 25616/1304 | 28612/1452 |
| Rwork (%)c | 23.2 | 20.9 | 19.7 | 21.0 |
| Rfree (%)d | 28.2 | 26.3 | 24.4 | 29.5 |
| No. of protein atoms | 5174 | 5184 | 5162 | 5144 |
| No. of ligand atoms | 2 | 3 | 2 | 2 |
| No. of solvent sites | 141 | 229 | 100 | 26 |
| Average B factor (Å2) | ||||
| Protein | 49.0 | 39.7 | 54.0 | 73.5 |
| Zn (II) | 36.9 | 31.3 | 55.2 | 64.8 |
| Mg (II) | N/A | 60.4 | N/A | N/A |
| Solvent | 40.8 | 38.7 | 42.5 | 59.1 |
| Ramachandran statisticse | ||||
| Preferred (%) | 93.01 | 94.07 | 93.76 | 91.32 |
| Allowed (%) | 4.56 | 4.10 | 4.11 | 5.18 |
| Root mean square deviation | ||||
| Bond lengths (Å) | 0.009 | 0.008 | 0.010 | 0.009 |
| Bond angles (°) | 1.249 | 1.149 | 1.175 | 1.236 |
| PDB entry | 4IG2 | 4IFK | 4IFO | 4IFR |
a The values in parentheses are for the highest resolution shell.
b Rmerge = Σi |Ihkl,i − <Ihkl>|/Σhkl Σi Ihkl,i, where Ihkl,i is the observed intensity, and <Ihkl> is the average intensity of multiple measurements.
c Rwork = Σ||Fo| − |Fc||/Σ|Fo|, where |Fo| is the observed structure factor amplitude, and |Fc| is the calculated structure factor amplitude.
d Rfree is the R factor based on 5% of the data excluded from refinement.
e Based on values attained from refinement validation options in COOT.
Both DNA sequencing and electron density map of the mutant crystal structures demonstrate the successful substitution of arginine residues with alanine in both R51A and R239A variants (Fig. 5A, top and middle panels). The alignment of R51A (Protein Data Bank entry 4IFO) and R239A (Protein Data Bank entry 4IFR) structures with the wild-type structure (Protein Data Bank entry 2HBV) are shown in Fig. 5B (top and middle panels), respectively. Both mutants exhibit homodimeric quaternary structures as previously seen in wild-type ACMSD. Secondary structure changes caused by mutation (colored in blue and cyan) can be observed in both subunits of the two structures, and they are mainly observed in the loop regions. No significant structural deviations, however, were observed for the folding of the overall TIM barrel scaffold, indicating that the impact caused by mutation is limited to specific flexible regions. We further analyzed the dimerization interface using the ACMSD wild-type and mutant structures at an online server: Protein Data Bank Europe Protein Interfaces, Surfaces and Assemblies (PDBePISA) (31). The results indicate that the interface contains a total of 13 H-bonds and 13 salt bridges. Nevertheless, these analyses would not be sufficient to draw a definitive conclusion if there were no intruding residue Arg-239 catalytically important, which allowed us to generate an active heterodimer from mixing the two inactive dimers. In the wild-type structure, Arg-239 contributes two H-bonds by interacting with the main chain molecules of the neighboring monomers to help to stabilize the dimer. In the R239A structure, the dimerization is not interrupted because these two hydrogen bonds are substituted by the following two new H-bonding interactions: Glu-252 of chain A interacts with Lys-189 of chain B and Arg-247 of chain A interacts with Met-191 of chain B.
FIGURE 5.

A, electron density of positions 51 and 239 in the R51A homodimer (top panel), R239A homodimer (middle panel), and R51A-R239A heterodimer (bottom panel). B, superimposed overall structures of WT Zn-ACMSD (Protein Data Bank code 2HBV) with Zn-R51A (Protein Data Bank code 4IFO) (top panel) and Zn-R239A (Protein Data Bank code 4IFR) (middle panel) and heterodimer hybrid (Protein Data Bank code 4IFK) (bottom panel). The well aligned structural components are shown in gray, whereas structural changes for R51A and R239A are highlighted in blue and cyan, respectively. Metal ions are represented by spheres. The graphs were produced using PyMOL. The 2Fo − Fc electron densities are contoured at 1.0 σ.
Fig. 5B (bottom panel) shows the superimposition of the heterodimer structure (Protein Data Bank code 4IFK) and the wild-type Zn-ACMSD structure (Protein Data Bank code 2HBV). The structural deviations of the mutant relative to the wild type are highlighted in red. The comparison clearly shows that major changes can only be observed in chain B. This observation is expected because in subunit A of the heterodimer, Arg-51 and Arg-239* are both in place with electron density similar to that in the wild-type structure (Fig. 5A, bottom panel). The electron density for the side chains of both arginine residues in subunit B is missing, indicating that both residues 51 and 239* are alanines (Fig. 5A, bottom panel). Hence, subunit A represents a complete native-like ACMSD subunit, but subunit B is a “double mutant” containing two alanine residues. It is noted in the structure of the native enzyme that Arg-239* adopts two conformations because of its high flexibility (19). Likewise, this residue in subunit A has alternative conformations that are only partially covered by the electron density map.
Arg-51 and Arg-239 Are Essential for Substrate Binding
Based on the current and previous studies, we propose that the two arginines are critical for proper substrate binding at the enzyme active site and for preventing its autocyclization to QA. Because the substrate, ACMS, is unstable and all arginine mutants are catalytically inactive, studying the postulated role of the two arginines in substrate binding is challenging. Therefore, an efficient competitive inhibitor needs to be found. Unfortunately, the inhibitors reported from previous in vivo studies, phthalate esters for instance (32, 33), were not effective on the pure enzyme in our experiments. Acyclic ACMS analog compounds tend to be unstable in solution; however, PDC, a cyclic compound, is structurally similar to ACMS (Fig. 6A). Both compounds contain conjugate repeating single-bond double-bond structures and two carboxylate groups. The difference is that PDC is a stable compound because of its cyclic, aromatic structure, whereas ACMS tends to dehydratively autocyclize. Fig. 6B shows that PDC behaves kinetically as a competitive inhibitor for the ACMS decarboxylation. The Ki value was determined to be 22.7 ± 1.8 μm. This allows for use of PDC as an analog of ACMS to study substrate binding.
FIGURE 6.
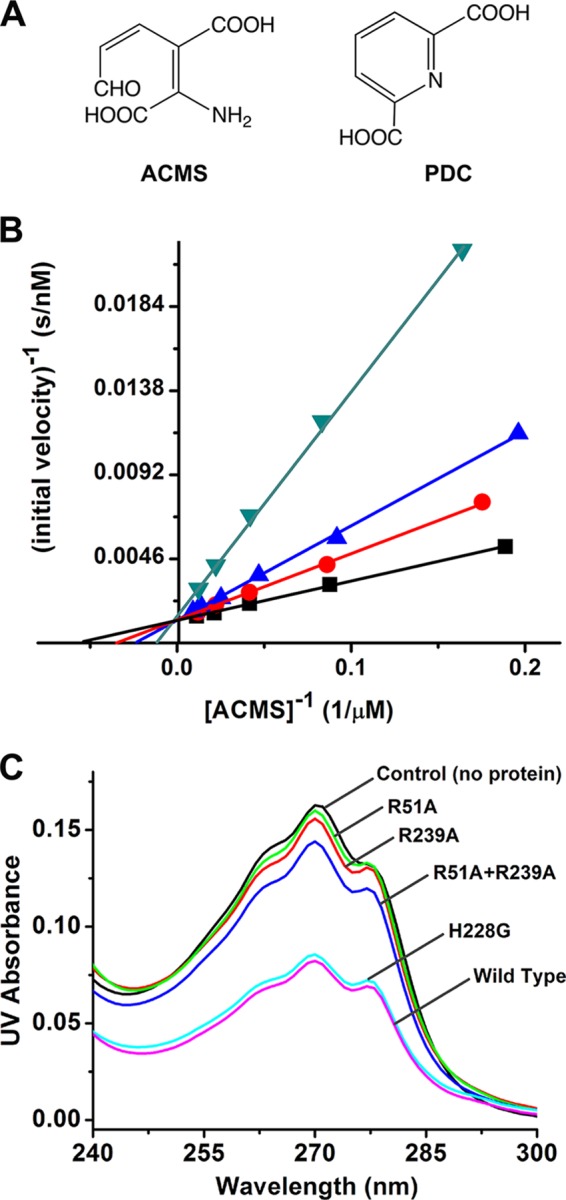
A, chemical structure of ACMS and PDC. B, competitive inhibition of wild-type ACMSD by 0, 20, 40, and 80 μm of PDC as the black, red, blue, and green traces, respectively. C, PDC in flow through after centrifuge-based filtration detected by UV-visible spectroscopy.
The catalytic activity-based kinetic approach, unfortunately, cannot be employed to assess the binding of substrate or substrate analog compounds to the inactive mutant proteins. We took advantage of the fact that PDC has an electronic absorption band at 270 nm (ϵ270 nm = 4100 m−1 cm−1, determined in 25 mm HEPES buffer, pH 7.0) and designed an experiment to test the binding of PDC to the wild-type and mutant proteins of ACMSD utilizing separation by centrifuge filters. The threshold for membranes in centrifuge filters with a 10,000-Da cutoff is smaller than ACMSD but much larger than PDC. If the PDC is bound to ACMSD, it will not be able to pass the filter membrane, whereas unbound PDC molecules can pass freely. The amount of PDC in the flow-through was quantitated by UV absorbance. Wild-type ACMSD was employed as a positive control, whereas the same buffer containing no protein was used as one negative control. A previously well characterized mutant H228G (22) was also employed as a secondary positive control. His-228 is a strictly conserved residue sitting in the interior side of the metal center that has been previously shown to function as a general base to deprotonate the water ligand during catalysis and that is not related with substrate binding. The properties and crystal structure of H228G have been reported previously (22). Fig. 6C shows that WT and H228G ACMSD proteins exhibit similar binding ability toward PDC. In contrast, R51A and R239A are unable to effectively bind PDC as exhibited by the similar absorbance of their flow-throughs to the negative control. This experiment suggests that the two arginine residues play an important role in binding of the substrate analog, which implies that they play a role in substrate binding in the native enzyme.
DISCUSSION
It is often a challenging task to determine whether or not an experimentally determined structure represents the biologically relevant assembly. It is also often difficult to pin down the role of a critical residue in catalysis when its mutants are completely inactive. An additional complexity in the present case is that the two conserved arginine residues that are being studied are proposed to bind the substrate, ACMS, which is unstable, so no binding constant can be easily measured. We have crafted a series of biochemical tests to show that P. fluorescens ACMSD must function as a dimer, and the two conserved arginine residues, including one intruded into the catalytic center from a neighboring subunit, are required for substrate binding in catalysis.
The molecular mechanism by which the catalytic activity is rescued through hybridization is illustrated in Fig. 4. During dimer-monomer equilibrium, three kinds of dimers form in solution when the two inactive homodimers of R51A and R239A are mixed: R51A homodimer, R239A homodimer, and R51A-R239A half-active heterodimer, in which one of the monomers has both Arg-51 and Arg-239 to constitute a complete active site identical to that of the native enzyme. If it is assumed that all homodimers are completely dissociated and rearranged, the percentage of formation of R51A homodimer and R239A homodimer will be both 25%. Consequently, the remaining 50% will be the half-active heterodimer. As a result, the maximum activity that can be rescued theoretically will be 25% of the wild-type ACMSD, assuming there is no cooperative interaction and that each of the dimers has the same association constant. By comparing the kcat values of the equilibrated mixed mutant ACMSD with WT ACMSD, 9.2% of the theoretical maximum activity is recovered. The inability to recover more activity can likely be attributed to unequal equilibrium constants for each of the species (inactive homodimer formation may be thermodynamically preferred to half-active heterodimer formation), or some protein dynamics and cooperativity may have been lost because of the mutations of important active site residues.
To our knowledge, this x-ray crystallographic study provides the first unambiguous experimental evidence for the formation of hybridized active heterodimer by mixing of two inactive homodimers. These structures allow for a molecular level understanding of the correlation of enzymatic activity with the protein quaternary structure and the mechanism of activity rescue from two completely inactive mutants. Together with our previous work, we now can describe the functionally active assembly of the enzyme and the role of the two arginines in a more definitive manner as discussed below.
The homodimer structures contain a ∼2,500 Å2 dimer interface area. Prior to this work, the question of whether dimerization is an artificial aggregation or required for enzyme activity was not known. We showed here two major bands corresponding to monomer and dimer on the polyacrylamide gel, indicating the existence of both dimer and monomer forms in aqueous solution. Enzyme specific activities were measured under identical assay conditions using protein at different stock concentrations. The finding, i.e., increasing specific activity with increasing protein stock concentrations, indicates that ACMSD is active when in the dimeric form and loses activity as it dissociates to the monomeric form.
Dimer formation as a mechanism for enzyme activation is common in nature. Cytochrome c peroxidases (34), cell death protease caspases (35, 36), diaminopimelate epimerase (37), and histidine kinase CheA (38) are just a few examples similar to ACMSD. The reason behind this dimerization-induced activation is usually the generation or extension of binding faces at the dimer interface (39). Hence, protein activation is triggered by an increase in the local protein concentration. As calculated by the dependence of specific activity on stock enzyme concentration, the dissociation constant of ACMSD is 0.3 μm. This is within the Kd range for “weak” transient complexes that show a dynamic mixture of both monomer and dimer states in vivo, which could be tuned by physiological environmental factors such as pH and local protein or ligand concentrations (40).
The mutagenesis and reaction rescue experiments described in this study show that both Arg-51 and Arg-239 are required for enzyme activity. However, their role in catalysis was unknown because of the silent nature of their activity in all the point mutants, including the conservative Lys mutants, until the identification of an effective competitive inhibitor of the enzyme, PDC. By using this competitive inhibitor, the problems that arise from use of an unstable substrate can be mitigated. The inability of PDC to bind to the arginine mutants is demonstrated by the filtration experiment. With proper positive and negative controls, the designed experiment provides a straightforward and reliable conclusion. Hence, we propose that both arginine residues Arg-51 and Arg-239* are involved in substrate binding during catalysis (Fig. 7). It is noteworthy that this scenario resembles a well documented example in a high affinity nitrate transporter where two perfectly conserved arginine residues are required for substrate binding (41). Likewise, arginines play crucial roles in the active sites and subunit interfaces of the ATPase domains of AAA and AAA+ proteins (for a recent review, see Ref. 42).
FIGURE 7.
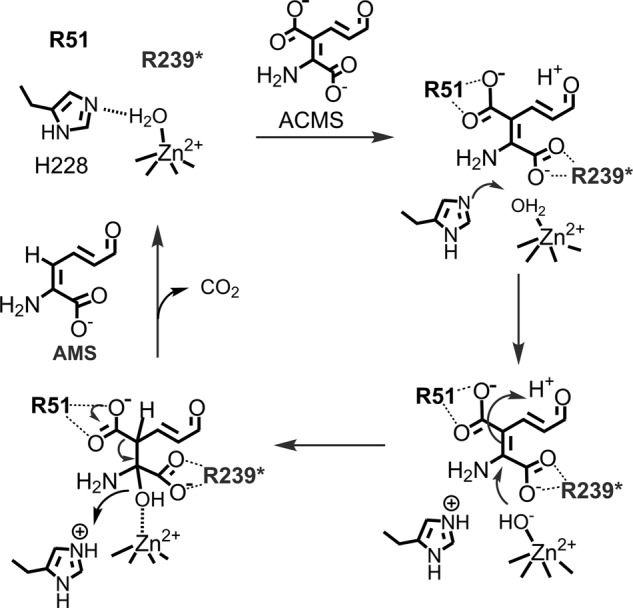
Proposed role of Arg-51 and Arg-239* in ACMS decarboxylation mechanism. The asterisk designates a residue contributed from a neighboring subunit. Currently, it is unknown in which way the substrate is oriented in the active site. Therefore, the presented mechanism represents one possible orientation, not necessarily the correct orientation.
Heterodimer-induced activity restoration has also been previously observed, although not structurally characterized, in histidine protein kinase CheA, a protein essential for bacterial chemotaxis stimulus-response coupling by phosphorylating another two chemotaxis proteins: CheY and CheB (43). CheA must be autophosphorylated at its own His-48 residue before this phosphoryl group can be transferred to CheY and CheB (44, 45). E. coli expresses two kinds of CheA proteins: the full-length CheAL and the N-terminally truncated CheAS, which lacks His-48, the site of autophosphorylation (44). However, in the presence of both CheAS and kinase defective CheAL mutant, CheA470GK, the mutant can still be phosphorylated. Thus, the autophosphorylation accomplished within the CheAS-CheA470GK heterodimer is suggested even in the absence of structural proof as shown in the present study (38).
The results described in this work helped us further refine our working model for the ACMSD catalytic cycle. Previously, the roles of His-228 and the metal-bound water were proposed and demonstrated (22). The two catalytically essential arginine residues Arg-51 and Arg-239* are now added to the model (Fig. 7). They are proposed to work as a team to recognize the substrate and stabilize both the substrate and possibly catalytic intermediates. Because the two residues at each metal center belong to two different subunits, the requirement of protein dimerization for enzyme activity becomes evident.
Among cellular metabolic proteins, monomers are a small fraction compared with multimeric proteins. Homodimers and homotetramers are two major forms of oligomerized proteins (39). Compared with monomeric proteins, oligomerized proteins have some apparent advantages. First, oligomerization enables higher order protein regulation, such as allosteric and concentration sensing regulations. Second, protein oligomerization can help to increase protein stability and the flexibility of activity by subunit cooperation. Lastly, homo-oligomers are more economic because they allow for a smaller genome while still building large proteins. Hence, the oligomerization of ACMSD may function as a mechanism of protein concentration sensing and enzyme activity adjustment in vivo.
Acknowledgments
We thank Dr. B.-C. Wang and the SER-CAT staff scientists of the Section 22 of Argonne National Laboratory for assistance in beamline preparation and x-ray data collection.
This work was supported in part or whole by National Science Foundation Grant MCB-0843537 and funds from the Georgia Research Alliance Distinguished Scholar Program (to A. L.) and by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38.
The atomic coordinates and structure factors (codes 4IG2, 4IFO, 4IFR, and 4IFK) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ACMS
- α-amino-β-carboxymuconate-ϵ-semialdehyde
- ACMSD
- ACMS decarboxylase
- PDC
- pyridine-2,6-dicarboxylic acid
- QA
- quinolinic acid.
REFERENCES
- 1. Donald J. E., Kulp D. W., DeGrado W. F. (2011) Salt bridges. Geometrically specific, designable interactions. Proteins 79, 898–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prell J. S., O'Brien J. T., Steill J. D., Oomens J., Williams E. R. (2009) Structures of protonated dipeptides. The role of arginine in stabilizing salt bridges. J. Am. Chem. Soc. 131, 11442–11449 [DOI] [PubMed] [Google Scholar]
- 3. Ghosh M., Datta A. K. (1994) Probing the function(s) of active-site arginine residue in leishmania-donovani adenosine kinase. Biochem. J. 298, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reczkowski R. S., Taylor J. C., Markham G. D. (1998) The active-site arginine of S-adenosylmethionine synthetase orients the reaction intermediate. Biochemistry 37, 13499–13506 [DOI] [PubMed] [Google Scholar]
- 5. Chaidaroglou A., Brezinski D. J., Middleton S. A., Kantrowitz E. R. (1988) Function of arginine-166 in the active site of Escherichia coli alkaline phosphatase. Biochemistry 27, 8338–8343 [DOI] [PubMed] [Google Scholar]
- 6. Blachnio K., Przykorska A. (2005) Specific arginine mediated RNA recognition. Postepy. Biochem. 51, 339–344 [PubMed] [Google Scholar]
- 7. Charnock S. J., Brown I. E., Turkenburg J. P., Black G. W., Davies G. J. (2002) Convergent evolution sheds light on the anti-β-elimination mechanism common to family 1 and 10 polysaccharide lyases. Proc. Natl. Acad. Sci. U.S.A. 99, 12067–12072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. GuillénSchlippe Y. V., Hedstrom L. (2005) Is Arg418 the catalytic base required for the hydrolysis step of the IMP dehydrogenase reaction? Biochemistry 44, 11700–11707 [DOI] [PubMed] [Google Scholar]
- 9. Doherty M. K., Pealing S. L., Miles C. S., Moysey R., Taylor P., Walkinshaw M. D., Reid G. A., Chapman S. K. (2000) Identification of the active site acid/base catalyst in a bacterial fumarate reductase. A kinetic and crystallographic study. Biochemistry 39, 10695–10701 [DOI] [PubMed] [Google Scholar]
- 10. Li T., Iwaki H., Fu R., Hasegawa Y., Zhang H., Liu A. (2006) α-Amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase (ACMSD) is a new member of the amidohydrolase superfamily. Biochemistry 45, 6628–6634 [DOI] [PubMed] [Google Scholar]
- 11. Li T., Walker A. L., Iwaki H., Hasegawa Y., Liu A. (2005) Kinetic and spectroscopic characterization of ACMSD from Pseudomonas fluorescens reveals a pentacoordinate mononuclear metallocofactor. J. Am. Chem. Soc. 127, 12282–12290 [DOI] [PubMed] [Google Scholar]
- 12. Stone T. W., Darlington L. G. (2002) Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 1, 609–620 [DOI] [PubMed] [Google Scholar]
- 13. Schwarcz R. (2004) The kynurenine pathway of tryptophan degradation as a drug target. Curr. Opin. Pharmacol. 4, 12–17 [DOI] [PubMed] [Google Scholar]
- 14. Mehler A. H., Yano K., May E. L. (1964) Nicotinic acid biosynthesis. Control by an enzyme that competes with a spontaneous reaction. Science 145, 817–819 [DOI] [PubMed] [Google Scholar]
- 15. Colabroy K. L., Begley T. P. (2005) The pyridine ring of NAD is formed by a nonenzymatic pericyclic reaction. J. Am. Chem. Soc. 127, 840–841 [DOI] [PubMed] [Google Scholar]
- 16. Guillemin G. J., Williams K. R., Smith D. G., Smythe G. A., Croitoru-Lamoury J., Brew B. J. (2003) Developments in tryptophan and serotonin metabolism. Quinolinic acid in the pathogenesis of Alzheimer's disease. Adv. Exp. Med. Biol. 527, 167–176 [DOI] [PubMed] [Google Scholar]
- 17. Guidetti P., Schwarcz R. (2003) 3-Hydroxykynurenine and quinolinate. pathogenic synergism in early grade Huntington's disease? Adv. Exp. Med. Biol. 527, 137–145 [DOI] [PubMed] [Google Scholar]
- 18. Reinhard J. F., Jr. (2004) Pharmacological manipulation of brain kynurenine metabolism. Ann. N.Y. Acad. Sci. 1035, 335–349 [DOI] [PubMed] [Google Scholar]
- 19. Martynowski D., Eyobo Y., Li T., Yang K., Liu A., Zhang H. (2006) Crystal structure of α-amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase (ACMSD). Insight into the active site and catalytic mechanism of a novel decarboxylation reaction. Biochemistry 45, 10412–10421 [DOI] [PubMed] [Google Scholar]
- 20. Garavaglia S., Perozzi S., Galeazzi L., Raffaelli N., Rizzi M. (2009) The crystal structure of human α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase in complex with 1,3-dihydroxyacetonephosphate suggests a regulatory link between NAD synthesis and glycolysis. FEBS J. 276, 6615–6623 [DOI] [PubMed] [Google Scholar]
- 21. Pucci L., Perozzi S., Cimadamore F., Orsomando G., Raffaelli N. (2007) Tissue expression and biochemical characterization of human 2-amino 3-carboxymuconate 6-semialdehyde decarboxylase, a key enzyme in tryptophan catabolism. FEBS J. 274, 827–840 [DOI] [PubMed] [Google Scholar]
- 22. Huo L., Fielding A. J., Chen Y., Li T., Iwaki H., Hosler J. P., Chen L., Hasegawa Y., Que L., Jr., Liu A. (2012) Evidence for a dual role of an active site histidine in α-amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase. Biochemistry 51, 5811–5821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horton R. M. (1995) PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol. Biotechnol. 3, 93–99 [DOI] [PubMed] [Google Scholar]
- 24. Li T., Ma J. K., Hosler J. P., Davidson V. L., Liu A. (2007) Detection of transient intermediates in the metal-dependent non-oxidative decarboxylation catalyzed by α-amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase. J. Am. Chem. Soc. 129, 9278–9279 [DOI] [PubMed] [Google Scholar]
- 25. Koontz W. A., Shiman R. (1976) Beef kidney 3-hydroxyanthranilic acid oxygenase. Purification, characterization, and analysis of the assay. J. Biol. Chem. 251, 368–377 [PubMed] [Google Scholar]
- 26. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Method Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 27. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 28. Collaborative Computational Project, Number 4 (1994) The Ccp4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 29. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 32. Fukuwatari T., Ohsaki S., Fukuoka S., Sasaki R., Shibata K. (2004) Phthalate esters enhance quinolinate production by inhibiting α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase (ACMSD), a key enzyme of the tryptophan pathway. Toxicol. Sci. 81, 302–308 [DOI] [PubMed] [Google Scholar]
- 33. Fukuwatari T., Ohsaki S., Suzuki Y., Fukuoka S., Sasaki R., Shibata K. (2003) The effects of phthalate esters on the tryptophan-niacin metabolism. Adv. Exp. Med. Biol. 527, 659–664 [DOI] [PubMed] [Google Scholar]
- 34. Ellis K. E., Frato K. E., Elliott S. J. (2012) Impact of quaternary structure upon bacterial cytochrome c peroxidases. Does homodimerization matter? Biochemistry 51, 10008–10016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S. (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. U.S.A. 98, 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Datta D., McClendon C. L., Jacobson M. P., Wells J. A. (2013) Substrate and inhibitor-induced dimerization and cooperativity in caspase-1 but not caspase-3. J. Biol. Chem. 288, 9971–9981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hor L., Dobson R. C., Downton M. T., Wagner J., Hutton C. A., Perugini M. A. (2013) Dimerization of bacterial diaminopimelate epimerase is essential for catalysis. J. Biol. Chem. 288, 9238–9248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swanson R. V., Bourret R. B., Simon M. I. (1993) Intermolecular complementation of the kinase activity of CheA. Mol. Microbiol. 8, 435–441 [DOI] [PubMed] [Google Scholar]
- 39. Marianayagam N. J., Sunde M., Matthews J. M. (2004) The power of two. Protein dimerization in biology. Trends Biochem. Sci. 29, 618–625 [DOI] [PubMed] [Google Scholar]
- 40. Nooren I. M., Thornton J. M. (2003) Structural characterisation and functional significance of transient protein-protein interactions. J. Mol. Biol. 325, 991–1018 [DOI] [PubMed] [Google Scholar]
- 41. Unkles S. E., Rouch D. A., Wang Y., Siddiqi M. Y., Glass A. D., Kinghorn J. R. (2004) Two perfectly conserved arginine residues are required for substrate binding in a high-affinity nitrate transporter. Proc. Natl. Acad. Sci. U.S.A. 101, 17549–17554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ogura T., Whiteheart S. W., Wilkinson A. J. (2004) Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol. 146, 106–112 [DOI] [PubMed] [Google Scholar]
- 43. Parkinson J. S., Kofoid E. C. (1992) Communication modules in bacterial signaling proteins. Annu. Rev. Genet. 26, 71–112 [DOI] [PubMed] [Google Scholar]
- 44. Hess J. F., Bourret R. B., Simon M. I. (1988) Histidine phosphorylation and phosphoryl group transfer in bacterial chemotaxis. Nature 336, 139–143 [DOI] [PubMed] [Google Scholar]
- 45. Hess J. F., Oosawa K., Matsumura P., Simon M. I. (1987) Protein phosphorylation is involved in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 84, 7609–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]