

Abstract
αβ-tubulin colchicine site inhibitors (CSIs) from four scaffolds that we previously tested for antiproliferative activity were modeled to better understand their effect on microtubules. Docking models, constructed by exploiting the SAR of a pyrrole subset and HINT scoring, guided ensemble docking of all 59 compounds. This conformation set and two variants having progressively less structure knowledge were subjected to CoMFA, CoMFA+HINT, and CoMSIA 3D-QSAR analyses. The CoMFA+HINT model (docked alignment) showed the best statistics: leave-one-out q2 of 0.616, r2 of 0.949 and r2pred (internal test set) of 0.755. An external (tested in other laboratories) collection of 24 CSIs from eight scaffolds were evaluated with the 3D-QSAR models, which correctly ranked their activity trends in 7/8 scaffolds for CoMFA+HINT (8/8 for CoMFA). The combination of SAR, ensemble docking, hydropathic analysis and 3D-QSAR provides an atomic-scale colchicine site model more consistent with a target structure resolution much higher than the ~3.6 Å available for αβ-tubulin.
Keywords: tubulin, 3D-QSAR, microtubule depolymerization, colchicine site inhibitor, HINT, ensemble docking and scoring
Introduction
Microtubules are a validated target for cancer therapies due to their role as one of the major cytoskeletal components in eukaryotic cells and their critical functions in the maintenance of cell shape, protein trafficking, signaling and segregation of chromosomes during mitosis.1 Microtubule-targeting agents function by interfering with microtubule dynamics, a process that controls the balance between microtubule assembly and microtubule disassembly.2 Four major binding sites for these agents have been identified: the taxane site and the laulimalide/peloruside A site, both for microtubule-stablizing agents, and the vinca site and the colchicine site for microtubule-destablizing agents.2,3
Compared to taxanes and vinca alkaloids, which have been used successfully in clinical therapies for cancer, colchicine itself, while a potent compound, has been restrained by its toxicity to normal tissues at effective drug concentrations and has only been approved for the treatment of familial Mediterranean fever and acute gout flares.4 However, owing to the fact that microtubules are important regulators of endothelial cells, the interest in developing colchicine-site agents or colchicine-site inhibitors (CSIs) has recently intensified as angiogensis inhibitors (preventing new blood vessel formation) and vascular disrupting agents (destroy existing vasculature) for cancer treatment.5 In particular, the combretastatin family of colchicine-site agents is progressing through clinical trials for this purpose.1,4 Also of interest is that colchicine site agents might be able to circumvent βIII-tubulin overexpression, which compromises the clinical use of taxanes and vinca alkaloids.6,7
A large number of CSIs, including both natural and synthetic compounds, have been reported and they possess significant structural diversity. So far, the compounds under clinical investigation represent at least 26 different scaffolds, including colchicine, combretastatin, podophyllotoxin and steganacin, and there are even more in preclinical studies.5,8 The ability of the colchicine site to accommodate such structural diversity is due to the inherent flexibility of the site, which has been demonstrated by X-ray crystal structures of the protein complexed with different agents (PDBID: 1SA0, 1SA1, 3HKC, 3HKE, 3HKD, 3N2K and 3N2G)9–11 and molecular dynamics simulations.12
The fact that the available crystallographic structural data for αβ-tubulin are of relatively poor resolution (~3.6 Å) should not be minimized: successes in structure-based drug discovery at this site have been comparatively rare.13 In 2005, Nguyen et al. reported docking and molecular dynamics studies as an approach to compensate for modeling inaccuracies arising from target structure resolution in their study of a structurally diverse set of CSIs.14 A common pharmacophore model was proposed to explain the critical interactions for most of the agents binding to the site. However, the binding conformations of a few were not correctly predicted, likely due to there being only one colchicine site crystal structure (1SA0) available at that time.
We have been developing pyrrole-based compounds as CSIs15–20 and as a result have acquired a rich collection of structure-activity relationship (SAR) data that has allowed us to characterize the colchicine site on αβ-tubulin at a level of detail that is, in effect, consistent with a much higher-resolution target structure. We did this by identifying the pyrrole compounds’ binding modes through ensemble docking, with multiple available crystal structures of the site, guided by detailed SAR comparisons to colchicine.15,16 Here, we have expanded our computational analyses to other scaffolds in an attempt to gain a more comprehensive and universal understanding of CSI ligands and the colchicine binding pocket. The last major computational investigation of multiple CSI scaffolds was nearly a decade ago,14 and the resulting model was more qualitative than quantitative. We collected all antiproliferation (vs. MDA-MB-435 cancer cells) and microtubule depolymerization activity data for colchicine site agents from the several diverse scaffolds that have been tested in our laboratory.15–18,21–26 As will be seen, there are a number of rather subtle but impactful features affecting activity, so we restricted our internal training and test sets set to these assay results to be certain that our analyses were not confounded by inter-laboratory variations.
We performed the same ensemble docking protocol reported earlier to identify internally consistent docked modes of all agents. To obtain a detailed and quantitative view of ligand binding, we used 3D-QSAR (three-dimensional quantitative structure-activity relationships),27,28 which identifies and assesses structural factors affecting activity within a pool of compounds. We also introduce a novel hydropathic map-based (HINT)29 method that mines additional useful information from the 3D hydrophobic and polar feature maps of a pool of characterized compounds. The result, an activity-weighted linear summation of the maps for our collection of CSIs, produced a visual summary for the overall SAR of these compounds. Finally, the validity of our 3D models was tested with an external test set including 24 compounds derived from 8 templates tested outside our laboratory30–37 that had previously been reported to be CSIs.
Results and Discussion
The goal of the present study was to definitively characterize and refine the colchicine binding site of αβ-tubulin. There is a vast array of compounds and templates reported in the literature to be colchicine site inhibitors (CSIs), but there is not a clear consensus on the pharmacophoric requirements of this site and the resulting SAR for bound compounds at the site is not completely consistent. There are two confounding factors that must be taken into account. First, the resolutions of all crystal structures reported to date are such that the protein’s side chain positions and conformations are not resolvable, as is also true with the bound DAMA-colchicine ligand. In fact, there is also measurable uncertainty with the positions of backbone atoms in the 3–4 Å resolution regime.38,39 Second, many, perhaps the majority, of CSIs may have antiproliferative activities caused by inhibiting targets other than microtubules. This problem is somewhat mitigated when both antiproliferative and microtubule depolymerization assays are performed on a putative CSI, as is the common practice in our laboratory. In our recent work, we have performed very detailed analyses of focused subsets15–17 of pyrrole-based ligands that have enabled us to develop precise docking models and rational SARs.
Internal dataset
We developed a set of criteria to select compounds for this study. Most importantly, we wanted to be sure that the dataset was self-consistent in terms of activity measurements; i.e., that they were evaluated in the same assay. Our laboratory has reported antiproliferative IC50 values in MDA-MB-435 cancer cells using the SRB assay15 for many compounds, from multiple classes, over the past several years. We verify that the antiproliferative activity is, at least in part, due to microtubule depolymerization, and that they are binding at the colchicine site if they (or a structural analogue) competitively inhibit [3H]colchicine binding.15 Because this evidence for a CSI is the best available, and we have collected this data on multiple scaffolds (pyrrole analogues, combretastatin analogues, pyrimidine analogues and colchicine itself),15–18,21–26 the training and internal test sets for this report – of 59 total compounds (see Table 1) – was thus restricted to compounds we have studied. The test set was 9 randomly selected compounds covering all three scaffolds other than colchicine, while the remaining 50 compounds comprised the training set. The pIC50 values for both the training set and the test set compounds covered a range of more than 3 log units. The IC50 and pIC50 values for the training and test sets are set out in Table 1. EC50 and pEC50, the microtubule depolymerization activities evaluated in A-10 cells, are also listed in Table 1. The original data, in the format reported in the respective publications, is provided in Supplementary Information (Table S1).
Table 1.
Structures and activities of compounds in the training and internal test sets.
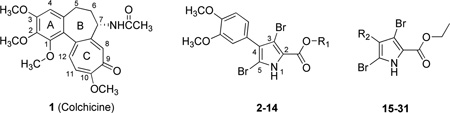 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd. | Ref. | R1 | R2 | IC50 (µM)a | pIC50 | EC50 (µM)b | pEC50 |
| 1† | 16 | N/A | N/A | 0.014 | 7.70 | 0.03 | 7.52 |
| 2† | 16 | Et | — | 0.036 | 7.44 | 0.49 | 6.31 |
| 3 | 16 | Me | — | 0.618 | 6.21 | 5.0 | 5.30 |
| 4* | 16 | n-Pr | — | 0.067 | 7.17 | 3.3c | 5.48 |
| 5 | 16 | i-Pr | — | 0.109 | 6.96 | 3.6c | 5.44 |
| 6 | 16 | t-Bu | — | 1.8 | 5.74 | 25c | 4.60 |
| 7† | 16 | n-Bu | — | 1.3 | 5.89 | 33c | 4.48 |
| 8 | 16 | n-Hex | — | 3.3 | 5.48 | 14c | 4.85 |
| 9 | 16 | Benzyl | — | 5.3 | 5.28 | 100d | 4.00 |
| 10 | 16 | -(CH2)3NMe2 | — | 4.6 | 5.34 | 50c | 4.30 |
| 11 | 16 | -(CH2)2NMe2 | — | 5.2 | 5.28 | 50c | 4.30 |
| 12 | 16 | -(CH2)3NMe2H+Cl− | — | 8.0 | 5.10 | 100d | 4.00 |
| 13 | 16 | -(CH2)2NMe2H+Cl− | — | 11 | 4.97 | 100d | 4.00 |
| 14* | 16 | 4-MeOPh | — | 18 | 4.74 | 100d | 4.00 |
| 15 | 15 | — | Ph | 10 | 4.99 | 167c | 3.78 |
| 16* | 15 | — | 4-MePh | 2.2 | 5.65 | 75c | 4.12 |
| 17 | 15 | — | 4-ClPh | 0.919 | 6.04 | 83c | 4.08 |
| 18 | 15 | — | 4-BrPh | 0.312 | 6.51 | 94c | 4.03 |
| 19 | 15 | — | 4-MeOPh | 0.843 | 6.07 | 7.0 | 5.15 |
| 20 | 15 | — | 3-MeOPh | 0.633 | 6.20 | 2.4 | 5.62 |
| 21 | 15 | — | 3,4,5-(MeO)3Ph | 13 | 4.89 | 71c | 4.15 |
| 22 | 15 | — | 2-Br-4,5-(MeO)2Ph | 2.6 | 5.58 | 14 | 4.85 |
| 23 | 15 | — | 1-Napthyl | 3.2 | 5.49 | 7.0 | 5.15 |
| 24* | 15 | — | 3-Indolyl | 2.0 | 5.70 | 18 | 4.75 |
| 25 | 15 | — | 4-CF3OPh | 1.7 | 5.77 | 27 | 4.57 |
| 26 | 15 | — | 4-MeSPh | 0.626 | 6.20 | 19 | 4.73 |
| 27 | 15 | — | 3,4-Cl2Ph | 0.806 | 6.09 | 9.9 | 5.00 |
| 28 | 15 | — | 3-F-4-MeOPh | 0.539 | 6.27 | 14 | 4.85 |
| 29 | 15 | — | 6-EtO-2-Napthyl | 2.0 | 5.70 | 33c | 4.48 |
| 30 | 15 | — | 1,3-Benzodioxol-6- | 1.8 | 5.74 | 30 | 4.53 |
| 31* | 15 | — | 1,4-Benzodioxan-6- | 4.4 | 5.36 | 21 | 4.68 |
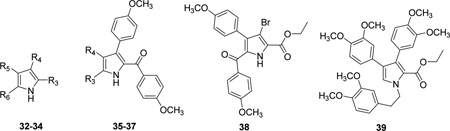 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd. | Ref. | R3 | R4 | R5 | R6 | IC50 (µM)a | pIC50 | EC50 (µM)b | pEC50 |
| 32 | 17 | CO2Et | H | 4-MeOPh | Cl | 5.0 | 5.30 | 75c | 4.12 |
| 33 | 17 | 4-MeOPhCO | 4-MeOPh | CO2Et | H | 100e | 4.00 | 500d | 3.30 |
| 34 | 17 | CO2Et | 4-MeOPh | 4-MeOPh | H | 100e | 4.00 | 500d | 3.30 |
| 35 | 17 | CO2Et | H | — | — | 3.0 | 5.52 | 100c | 4.00 |
| 36 | 17 | CO2Et | 4-MeOPh | — | — | 100e | 4.00 | 500d | 3.30 |
| 37 | 17 | 4-MeOPhCO | 4-MeOPh | — | — | 100 | 4.00 | 500d | 3.30 |
| 38* | 17 | — | — | — | — | 100e | 4.00 | 500d | 3.30 |
| 39 | 17 | — | — | — | — | 100e | 4.00 | 500d | 3.30 |
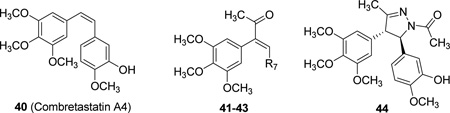 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd. | Ref. | R7 | IC50 (µM)a | pIC50 | EC50 (µM)b | pEC50 | |||
| 40† | 22 | — | 0.003 | 8.52 | 0.007 | 8.15 | |||
| 41 | 23 | 4-MeOPh | 0.35 | 6.46 | 18.6 | 4.73 | |||
| 42 | 23 | 4-MePh | 0.095 | 7.02 | 5.6 | 5.25 | |||
| 43 | 23 | 3-OH-4-MeOPh | 0.182 | 6.74 | 1.8 | 5.74 | |||
| 44* | 21 | — | 0.900 | 6.06 | 4.5 | 5.35 | |||
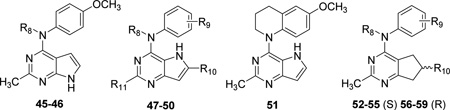 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd. | Ref. | R8 | R9 | R10 | R11 | IC50 (µM)a | pIC50 | EC50 (µM)b | pEC50 |
| 45* | 26 | Me | — | — | — | 0.183 | 6.74 | 5.8 | 5.24 |
| 46 | 26 | H | — | — | — | 20f | 4.70 | 80f | 4.10 |
| 47 | 24 | Me | 4-MeO | H | Me | 0.097 | 7.02 | 1.2 | 5.92 |
| 48 | 24 | Me | 4-MeO | H | H | 0.193 | 6.71 | 1.4 | 5.85 |
| 49 | 24 | Me | 4-MeO | Me | Me | 0.030 | 7.52 | 0.22 | 6.66 |
| 50 | 24 | Me | 4-MeO | Me | NH2 | 0.298 | 6.53 | 8.4 | 5.08 |
| 51* | 24 | — | — | — | — | 0.043 | 7.37 | 0.23 | 6.64 |
| 52 (S)† | 25 | Me | 4-MeO | Me | — | 0.012 | 7.92 | 0.023 | 7.64 |
| 53 (S)g | 26 | H | 4-MeO | Me | — | 12f | 4.92 | 43f | 4.37 |
| 54 (S)g | 26 | Me | 3-MeO | Me | — | 0.095 | 7.02 | 1.6 | 5.80 |
| 55 (S)g | 26 | Me | H | Me | — | 1.67 | 5.78 | 43f | 4.37 |
| 56 (R) | 25 | Me | 4-MeO | Me | — | 0.051 | 7.29 | 0.278 | 6.56 |
| 57 (R)g | 26 | H | 4-MeO | Me | — | 51f | 4.29 | 520f | 3.28 |
| 58 (R)g | 26 | Me | 3-MeO | Me | — | 0.402 | 6.40 | 6.9 | 5.16 |
| 59 | 26 | Me | H | Me | — | 7.1 | 5.15 | 520f | 3.28 |
Member of internal test set.
Compound used as structural prototype for family.
IC50s are antiproliferative activities tested using human MDA-MB-435 cancer cells; pIC50 = −log(IC50).
Loss of interphase microtubules evaluated in A-10 cells.
Depolymerization results reported as x% microtubule loss at y µM; value here is 50 y / x, which assumes a linear relationship between EC50 and cell loss.
Literature reports no microtubule loss up to 10 or 50 µM; we assume 5% loss at 10 or 50 µM and the EC50 is calculated using the relation of note c.
In these compounds, no microtubule effect (EC50) was observed up to 50 µM, and the measured IC50, if any, was assumed to arise from another antiproliferative mechanism; thus IC50s were arbitrarily assigned to be 100 µM.
Literature report was IC50 or EC50 > 10 or 40 µM, assuming 25% loss at 10 or 40 µM using relation of note c.
The activities reported are for racemic mixtures. The IC50 and EC50 ratios of compounds 53, 54 and 55 (the R enantiomers) to compounds 57, 58 and 59 (the S enantiomers) were assumed to be the same as the experimentally determined IC50 and EC50 ratios of compound 52 (R) to compound 56 (S).
Overview of the colchicine site and prediction of binding modes from docking
The colchicine site is located at the interface between α-tubulin and β-tubulin and is mostly buried in the β-tubulin subunit as seen in the crystal structures 1SA0, 1SA1, 3HKB, 3HKC, 3HKD and 3HKE9,10 (Figure 1). The site is surrounded by helices H7 and H8, loop T7, and strands S8 and S9 of β-tubulin, and loop T5 of α-tubulin (our nomenclature of the secondary structure features is as previously reported40). DAMA-colchicine occupies the pocket with its A ring fitting into the subpocket A, close to H7, its C ring in the subpocket C, close to T5 and its B ring in the center of the pocket. As different ligands bind, the T7 and T5 loops can move to adapt to the changes, as indicated by the crystal structures (Figure 1). To take this flexibility into account, we performed ensemble docking using five of the available crystal structures (1SA0, 1SA1, 3HKC, 3HKD and 3HKE). In the unliganded 3HKB structure, the T7 loop closes the entrance and thus occupies most of the site and thus 3HKB was not used in the docking study.
Figure 1.

The colchicine site complexed with DAMA-colchicine (yellow). The loops of different colors from different αβ-tubulin X-ray crystal structures represent the flexibility of the pocket (1sa0: cyan; 3hkc: magenta; 3hkb: green; 3hke: brown; 3hkd: red; 1sa1: blue).
In our earlier docking studies of colchicine (1) and pyrrole compounds 2–39, a second very key influence was the “atomic-scale” SAR available for the compounds,15,16 which enabled mapping the shape and hydropathic properties of the A and C subpockets. For example, we showed that 2, with a ethyl ester at the pyrrole C2 position, was ideal for the C subpocket, while both the methyl and n-propyl ester compounds (3 and 4) made somewhat less favorable interactions. As a result, the pyrrole compounds (2–39) adopted two distinct binding modes.16 Mode I, represented by the most active pyrrole, 2 (JG-03-14), binds most favorably to the 3HKC structure (Figure 2a). The ester chain fits into subpocket C with the carbonyl oxygen forming a hydrogen bond with the backbone nitrogen of Val181α, while the alkyl ends reach the hydrophobic bottom. The pyrrole core is located in subpocket B, with its 1-position NH forming hydrogen bonds with the sidechain carbonyl of Asn258β and the backbone carbonyl of Thr179α. The four-carbon side chains of Leu248β and Leu255β (not shown in figure) in subpocket A clamp the dimethoxyphenyl group of 2, and the two methoxys are locked by other residues deeper within the pocket. In addition, the polar residue Cys241β uses its SH group to form a weak hydrogen bond with either one of the two oxygens from the dimethoxyphenyls. Mode II, represented by compound 7 (Figure 2a), tends to bind the 1SA0 structure more favorably. The dimethoxyphenyl group and the pyrrole core are located in positions similar to their positions in Mode I, but because of its larger size, the ester chain shifts significantly away from subpocket C and exposes itself to solvent while anchored by a new hydrogen bond with Asn101α.
Figure 2.

Binding modes of docked compounds in the colchicine site (colchicine: yellow; 1sa0: cyan; 3hkc: magenta). a) The two binding modes of the pyrrole compounds (2 in thick red; 7 in thick purple; other analogues depicted with thin lines); b) The binding modes of the combretastatin analogues (40, combretastatin A-4, in blue) and the pyrimidine analogues (52 in green).
Mode I ligands (e.g., 2) and the complexed colchicine overlap well. The dimethoxyphenyl group of Mode I is located near the trimethoxylphenyl A ring of colchicine and the ester chain of Mode I mimics the combination of the methoxy and the carboxyl oxygen of colchicine’s C ring. The pyrrole core coincides with half of colchicine’s A ring and half of its B ring. In contrast, the only region where Mode II compounds, e.g., 7, and colchicine overlap is 7’s dimethoxyphenyl and colchicine’s trimethoxylphenyl.
The combretastatin analogues (40–44) and the pyrimidine analogues (45–59) were seen in our models to bind most favorably to 3HKC. They adopt a binding mode similar to Mode I (i.e., like 2 and colchicine), as shown in Figure 2b with the most active compound of each scaffold. Subpocket A is occupied by a hydrophobic moiety with hydrogen bond acceptors interacting with Cys241β. This moiety for 40 (the most active combretastatin analogue, combretastatin A-4) is its trimethoxyphenyl group with the oxygens acting as hydrogen bond acceptors, and for compound 52 (the most active pyrimidine analogue, (6R)-N-(4-methoxyphenyl)-N,2,6-trimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidine) is its cyclopentapyrimidine group with an aromatic nitrogen as the acceptor. Subpocket C is occupied by the other end of these ligands: 40 places the methyl from the methoxy group so as to interact with the hydrophobic bottom of subpocket C with the oxygen from the hydroxyl group forming a hydrogen bond with Val181α; 52’s methoxy is also located in subpocket C, but not as deep as the methoxy of 40. However, this methoxy is appropriately positioned to simultaneously interact with the pocket’s hydrophobic bottom and Val181α. The phenyl rings of both 40 and 52 occupy the B subpocket.
3D-QSAR alignment models and pharmacophore definitions
Three alignment models are shown in Figure 3: i) in the docking-based alignment (Figure 3A), all docked poses were directly used except for compounds 33–34 and 36–37 and 39, as these ligands with poor activities were unable to fit sterically in the binding pocket. To rescue these “negative controls” for use in the docking-based alignment models, we superimposed their structures to the most active pyrrole analogue 2; ii) in the “semi-ligand-based” alignment (Figure 3B), the compounds were superimposed onto docked conformations of their appropriate prototypes. Due to the significant difference between the Mode I and II conformations obtained from docking, the pyrrole analogues were treated in two groups. Mode I compounds 3–5, 15–32, 35 and 38 were superimposed onto compound 2 and Mode II compounds 6–14 were superimposed onto compound 7. Compounds 33–34 and 36–37 and 39 were treated as above. All combretastatins were superposed on 40 and all pyrimidines were superposed on 52; and iii) in the naïve alignment (Figure 3C), we adopted the pharmacophore model of Nguyen et al.14 to align the energy-minimized scaffolds with colchicine as the template. The pharmacophore features we used were a hydrogen bond acceptor interacting with Cys241β, another hydrogen bond acceptor interacting with Val181α, and three hydrophobic centers in subpockets A, B and C, respectively. Compounds 1 (colchicine), 2 (Mode I for the pyrroles), 40 and 52 are good examples that contain all these pharmacophore features. It should be noted that the naïve alignment is ignorant of Mode I and Mode II. Both of the latter two approaches are not traditional ligand-based 3D-QSAR alignments, as some conformation information was gleaned from the receptor. For ii, this was necessary because the common substructure of the scaffolds included only a small portion of chemical space, thus precluding an obvious ligand-based alignment, and for iii, Nguyen’s pharmacophore13 was derived with knowledge of the 1SA0 αβ-tubulin-tubulin X-ray crystal structure.
Figure 3.

Aligned poses for the three CSI ligand alignment models. a) Poses for the training set aligned by docking model; b) poses for the training set aligned by the semi-ligand-based model; c) poses for the training set aligned by the naïve pharmacophore model.
3D-QSAR models
The basic concept of 3D-QSAR is to correlate compound activities with interaction fields surrounding the compounds. The interaction fields include thousands of grid points acting as descriptors. Machine learning is applied to find a set of interaction values that are most related to the changes in activity amongst the compounds. In CoMFA27 and CoMSIA,28 partial least squares (PLS) regression is used to derive and statistically validate models. The logic of building a 3D-QSAR model relies on the assumption that variations of activity can be fully explained by the variations in structure, and that these variations are well represented by interaction fields. The most vexing issue in 3D-QSAR is usually identification of the active conformation for each compound, and proposing how these are superimposed/aligned – i.e., how they are presumed to bind at their receptor, binding or active site. We used the three alignment models described above.
The docked alignment conformations are shown in Figure 3a. The semi-ligand-based conformations are shown, for comparison, in Figure 3b. The major difference between the two sets of conformations originates from the bases of the two approaches. The semi-ligand-based approach aligns all ligands of the same scaffold according to their common structures, while aligning the different scaffold families according to the docked conformations. This tends to emphasize the structural differences between templates, while ameliorating differences within the families. The docking-based alignment, however, allows the ligands to adapt individually by optimizing their individual interactions and complementarities with the receptor. Therefore, the docking-based approach more smoothly distributes the ligands within the binding pocket than the semi-ligand-based approach, where ligand placement and conformations appear to be more step-like. The naïve alignment (Figure 3c) tends to almost completely discount the influence of the receptor, and thus shows the least complexity.
Analysis of 3D-QSAR statistics
All three pose sets were used for 3D-QSAR modeling. 3D-QSAR models were evaluated based on the cross-validated correlation coefficient (q2), the non-cross-validated correlation coefficient (r2), standard error of estimate (SEE), the F test value and by evaluation of the model with an internal test set (r2pred.).
The statistical results listed in Table 2 and the experimental and predicted activity plots in Figure 4 indicate that the CoMFA and CoMSIA models built upon this set of diverse scaffolds with the docking-based alignment are statistically reliable. The q2 values were 0.589 and 0.512 and the r2 values were 0.934 and 0.863. The predictability for the internal test set, indicated by r2pred, was above 0.7 for both CoMFA and CoMSIA. In contrast, the semi-ligand-based and the naïve alignments gave inferior results: all q2 values were below 0.5 and all r2pred values were below 0.7. As expected, r2pred values from the naïve alignment were worse than for the semi-ligand-based alignment (0.590 vs. 0.620 for CoMFA, and 0.395 vs. 0.476 for CoMSIA). We also built CoMFA models by adding the hydrophobic/polar HINT fields as an additional independent column, which we are referring to here as CoMFA+HINT models. These field combinations have been used with previous success for hydrophobic datasets.41–43 The CoMFA+HINT applied to the docking-based alignment gave moderately better statistics compared to the CoMFA model with q2 = 0.616, r2 = 0.949 and r2pred = 0.755 (Table 2). While the CoMFA+HINT models used more components than the standard (steric and electrostatic field) CoMFA models, their higher q2 values indicate that the additional components did not over-fit, but represented “real” variance with new information. The numerical values represented in Figure 4 are in Table S2 (Supporting Information).
Table 2.
Summary of 3D-QSAR model statistics.
| Docking alignment | Semi-ligand alignment | Naïve alignment | |||||
|---|---|---|---|---|---|---|---|
| CoMFA (IC50 / EC50) |
CoMSIA (IC50) |
CoMFA+ HINT (IC50) |
CoMFA (IC50) |
CoMSIA (IC50) |
CoMFA (IC50) |
CoMSIA (IC50) |
|
| No. of Components | 5 / 4 | 4 | 6 | 5 | 5 | 7 | 4 |
| q2 (cross-validated r2) | 0.589 / 0.515 | 0.512 | 0.616 | 0.446 | 0.438 | 0.451 | 0.446 |
| r2 | 0.934 / 0.862 | 0.863 | 0.949 | 0.821 | 0.835 | 0.928 | 0.759 |
| Std. error of estimate | 0.295 / 0.442 | 0.272 | 0.264 | 0.488 | 0.468 | 0.316 | 0.559 |
| F value | 125 / 70 | 71 | 132 | 40 | 44 | 77 | 35 |
| r2pred (for test set) | 0.728 / 0.776 | 0.734 | 0.755 | 0.620 | 0.476 | 0.590 | 0.395 |
Figure 4.

Scatter plots of predicted pIC50 values vs. experimental pIC50 values. The plots for a) CoMFA, b) CoMISA models and c) CoMFA+HINT, based on either the docking alignment are shown. The training set contains 50 compounds (blue diamonds) and the test set contains 9 compounds (red squares).
The relationship between antiproliferative effect (IC50) and microtubule depolymerization (EC50) for a compound informs for off-target effects, i.e., those that do not impact tubulin. Ideally, EC50 and IC50 should be the same.15,18 We showed earlier that the pyrrole compounds with key pharmacophoric features correlate these two activities: pEC50 = 1.10 pIC50 – 1.57 (r2 = 0.79).15 Although microtubule depolymerization data were not available for many compounds, we estimated these values (see Table 1 footnotes) and built a CoMFA model from the docking-based alignment that was reasonable, with q2, r2 and r2pred of 0.515, 0.862 and 0.776, respectively (Table 2).
Analysis of 3D-QSAR contour maps
The docked poses of the ligands revealed several residues that interact with the ligands and should thus be expected to significantly affect activity. We analyzed the resulting coefficient × standard deviation (coeff*stddev) contour maps of the 3D-QSAR models to verify these features and identify potential others. These maps, for the CoMFA, CoMFA+HINT and CoMSIA models from the docking alignment, shown in Figure 5, identify regions (and their causative ligand functional groups) that have significant impact on activity, and thus can be useful for not only rationalizing observed SAR, but also for ligand design.
Figure 5.

3D-QSAR contour maps based on docked poses for IC50 target variable. For reference, colchicine is shown in beige in all maps. (See text for legend to numbered highlights.) a) Contour maps of the CoMFA model. Green and yellow contours indicate favorable and unfavorable steric interactions, respectively. Blue regions favor electropositive groups and red regions favor electronegative groups. b) The contour maps of the CoMFA+HINT model with the contours representing the HINT hydrophobic/polar field. Cyan contours indicate favorable hydrophobic interactions while purple contours indicate favorable polar interactions. c) The contour maps of the hydrogen bond donor and acceptor fields from the CoMSIA model. Black and light green represent regions that favor and disfavor hydrogen bond donors, respectively. Magenta and orange represent regions that favor and disfavor hydrogen bond acceptors, respectively.
The CoMFA experiments represent structure with steric and electrostatic fields. The coeff*stddev maps (Figure 5a) thus indicate regions in space where adding steric bulk or changing the electrostatic charge at specific loci in the core molecule should increase (or decrease) target activity. Briefly, green and yellow contours represent favorable and unfavorable steric substitutions, respectively, while red and blue contours represent a favorable effect from more electronegative and more electropositive substitutions, respectively. (Note that this latter can be interpreted in an alternative way, i.e., that red contours also indicate that lower activity results from a more electropositive substitution.)
Favorable steric interactions (1, green) are suggested near substitutions to the ether oxygen on colchicine’s C ring; electronegative groups (2, red) such as that ether oxygen and the carbonyl oxygen would be favorable as well. The higher activity binding mode for the pyrrole compounds (e.g., 2) typify these observations, as they place their ester chains in these regions, i.e., with the carbonyl and ether oxygen around 2 (interacting with the backbone NH of Val181α) and the alkyl group around 1. The small yellow contour (3) at the top of colchicine’s C ring indicates that a too bulky substitution would be unfavorable for this region. The ethyl of the ester chain of 2 is a perfect fit as reported in our previous study.16 The naïve alignment model also highlights this observation (vide infra). The blue contours (4) between colchicine’s B ring and Thr179α suggest that a positively charged group would be favorable, which could again be related to the NHs for the higher activity pyrrole compounds forming hydrogen bonds with Thr179α. The large green contour (5) around the B ring of colchicine suggests favorable steric interactions with the receptor. For example, the pyrimidine analogues 52 and 56, with a methyl substitution on the amino nitrogen, showed significantly higher activity than the corresponding analogues 53 and 57 without such a substitution. These methyls overlay colchicine’s B ring and are surrounded by the four-carbon side chain of Lys254β and Leu248β. Several notable contours are arrayed around the trimethoxylphenyl group (A ring) of colchicine. The green contours (6) represent the favorable steric interactions between the methyls and the hydrophobic residues at the bottom of the pocket. The complex interleaving of red and blue contours (7) represents the importance (and critical positioning) of hydrogen bond acceptors in this subpocket commonly observed for highly active colchicine site binders. We explored the details of this observation by testing compounds with varying hydrogen bond accepting ability in a previous study15 and designated Cys241β as a key donor. The red regions indicate where the acceptor atom (usually alkoxy oxygens) is properly positioned, while the blue regions indicate where this basicity is unfavorable to activity. Note that βIII-tubulin, a taxane-resistant isoform overexpressed in some cancer cells, possesses the better H-bond donor serine as a mutation to the key cysteine,13 and would thus be expected to more strongly bind such CSI compounds, which may make them more selective for the isoform. The few yellow contours around the ligand indicate unfavorable steric interactions. One notable region (8) is the space between rings A and C. While part of ring C shows favorable steric interactions related to Ala316β, the pocket is constrained by this residue, leaving no room for a substituent that is too bulky. Thus, the methoxyphenyl group at the C-3 (i.e., R4, Table 1) position of 33 is predicted by docking to intrude into the protein, and no antiproliferative activity or microtubule effect was observed for the compound. A variety of other fragmented (mainly blue) contours probably indicate adjustments the different scaffolds make to fit the universal model and will not be discussed further. CoMFA maps from the EC50 models were very similar and are available in Supporting Information (Figure S1).
The CoMFA+HINT model supplemented the steric and electrostatic fields with the HINT hydrophobic/polar field. The resulting steric and electrostatic maps were largely the same as shown in Figure 5a. We focus here on the coeff*stddev map representing the influence of the HINT field (Figure 5b), with hydrophobic/polar information about the model as it indicates regions in space where changing the hydropathy at specific loci of the core molecule should increase (or decrease) target activity. Briefly, cyan and purple contours represent a favorable effect from more hydrophobic and more polar substitutions, respectively. In effect, this field helps characterize the nature of the prescribed steric bulk. The regions related to the partially hydrophobic (Val181α sidechain) subpocket C (1), the hydrophobic alkyl side chain of Lys254β and Leu248β around ring B (5) and the hydrophobic subpocket A (6) surrounded by Leu255β all favor hydrophobic groups. The purple regions indicate favorable polar substitutions. They are related to the hydrogen bond donors from the receptor: the backbone NH group of Val181 (2) and the SH group of Cys241β (7).
Although minor differences exist, the steric, electrostatic and hydrophobic coeff*stddev maps of the CoMSIA model (Supporting Information, Figure S2) show similar features as those of CoMFA and CoMFA+HINT, albeit with more compact and focused contours. Our CoMSIA model also included hydrogen bond donor and acceptor field types not present in CoMFA (Figure 5c). Black and light green represent favorable and unfavorable hydrogen bond donors, and magenta and orange represent favorable and unfavorable hydrogen bond acceptors. The magenta contour (9), representing a favorable hydrogen bond acceptor, near the backbone NH of Leu252β is a new observation that can be related to the hydrogen bond between the NH and one of the aromatic nitrogens of pyrimidines, such as 52. Other contours in this map, e.g., 2 and 7, provide complementary information to the other models. Note that the two lobes of 2 now indicate both favorable donor and unfavorable acceptor properties.
The coeff*stddev maps of the CoMFA and CoMSIA models from the semi-ligand-based alignment (Supporting Information, Figures S3 and S4) are generally similar to maps based on the docking alignment. Concomitant with the poorer statistics of these models, the naïve alignment yielded maps (Supporting Information, Figures S5 and S6) of poorer quality that were difficult to interpret. The major alignment difference, the assumption that all pyrroles would adopt the same binding mode, was shown to be clearly wrong with unfavorable steric contours extending well beyond the boundaries of subpocket C.
The contour maps of the 3D-QSAR models correlate very well with the structure of the colchicine site and identify regions within the site that have significant impact on activity. Importantly, our common pharmacophore for CSIs, similar to that previously reported,14 was validated by the 3D-QSAR models: the hydrogen bond acceptors related to Cys241β and Val181α and the three hydrophobic centers in subpockets A, B and C. Of note is that, while these pharmacophores are present in highly active CSIs, as seen in the prototypes, removing them decreases activity, as seen repeatedly in this data set. Beyond the common pharmacophore, however, other features in the 3D-QSAR maps (Figure 5) were identified that also have significant impact on activity. Cataloguing and understanding these features is important for improving the activities of even already highly active compounds because it is possible that these features have not as of yet been consolidated.
One argument against using docking-based alignments is that, like those crystallographically derived, there can be “noise” present in the docked poses, although that is not always the case.44–49 We did not detect this: 1) most compounds docked quite similarly to their (most active) prototypes (see Figure 2a) and their shapes were complementary to the binding pocket; and 2) the structural changes between scaffolds significantly outweighed the intra-scaffold “noise”. In fact, because many more residues were involved in binding than suggested by the pharmacophore model used in the naïve model, docking was able to place individual ligands more precisely in the colchicine site, with better performance metrics (q2, r2, etc.) in validating the resulting 3D-QSAR models.
LOCKSMITH highlights important features of the data set with a composite HINT map
Statistical 3D-QSAR models (as in CoMFA, etc.) depend on many factors, including accuracy of the input data and variance in the training set compounds. They can, however, obscure the unique features of single compounds that may provide valuable ideas for drug design. We previously suggested50 that calculating composite feature (called LOCKSMITH) maps of sets of active compounds, presumed to bind at the same site, could be a useful visualization tool for evaluating these compounds. Here, we calculated a simple linear combination of HINT maps, which contain hydrophobic/polar fields and acid/base fields, for all members of the compound set. Each map in the sum was weighted by 0.1 µM / IC50 for the compound. The goal is to highlight both uniqueness and commonality. Thus, highly active compounds, even if structurally unique, would contribute more to the overall map and features common to a majority of compounds would also be reinforced, but features that are rare or detrimental to activity would be weakly, if at all, visible. Much as illustrated above for 3D-QSAR, it should be possible to use such composite maps for the design of new compounds that incorporate the best features of multiple templates.
The resulting overall HINT map calculated with all CSIs from the dataset is shown in Figure 6. This map generally agrees with the contour maps from the 3D-QSAR analyses, as it reveals the hydrogen bond acceptors interacting with Cys241β (1) and Val181α (2) and a very large hydrophobic area (3) covering subpockets A, B and C of the site. These features are congruent with the common pharmacophore model, and are thus the commonalities of the active compounds. Other features in the map are interesting: i) an acid (4) representing the hydrogen donating ability of the pyrrole core NH interacting with Thr179α, especially 2; ii) bases (5) representing one of the aromatic nitrogens of 52 and the carbonyl oxygen of 42 (not clearly indicated by 3D-QSAR) forming hydrogen bonds with Asp251β and Leu252β respectively.
Figure 6.

The overall HINT composite map based on the entire set of colchicine site agents. Colchicine is shown in beige. Cyan represents hydrophobic regions of the compound set. Blue and red represent acidic and basic regions, respectively, of the compound set.
The composite HINT map represents an alternative approach to 3D-QSAR. First, it is important to note that CoMFA and related contour features are designed to extend away from occupied space, thus suggesting how extensions to existing compounds should impact activity. In contrast, the HINT contours are centered on the atoms and functional groups of existing compounds. Thusly, the often-fragmented contours in 3D-QSAR models can reveal specific information about a small handful of compounds, and rational insight into possible modifications thereof. LOCKSMITH maps are more limited in scope – only revealing a consensus of what is known about the actives. The HINT map approach may be most useful for hypothesis generation: it is a simple operation to superimpose a putative compound over this map to determine its suitability.
Evaluation of other scaffolds in an external dataset
A vast number of scaffolds for CSIs have been reported in the literature, with some possessing impressive activities. As detailed above, we trained our models using only compounds that had been evaluated in our laboratory to reduce experimental uncertainties. However, the union of the training set provides a comprehensive sampling of the colchicine site, and the models we obtained should, if they are truly valid, be able to evaluate other CSI scaffolds by predicting IC50s consistent with our assays. We therefore collected a second, external, test set (see Table 3) from 8 scaffolds (60–67) by selecting three representatives of each: a) highly active, b) medium active and c) weakly active compounds. Scaffolds 60 and 61 are combretastatin-like, but 62–67 are new. These compounds were evaluated with the CoMFA, CoMFA+HINT and CoMSIA docking-based alignment 3D-QSAR models, after docking each as above (see Supporting Information Figures S7a–h for representative docked conformations). The predicted activities (Table 3) are not necessarily numerically comparable to experiment because the assays were different, but, in fact, we could fairly reasonably correlate our predicted pIC50 to the measured and reported activities with linear regression (see Figure 7). The CoMFA and CoMSIA models produced r2external values of 0.474 and 0.294, respectively, while the CoMFA+HINT model’s r2external was 0.424.
Table 3.
Structures, activities and predictions for compounds in the external test set.
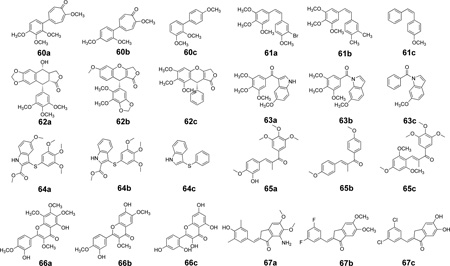 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd. | Ref. | Activity: IC50 or Kd (µM) |
pActivity | 3D-QSAR Predictions from model: |
HINT map score |
||
| CoMFA | CoMSIA | CoMFA+ HINT |
|||||
| 60a | 30 | 2.13 | 5.67 | 6.54 | 7.21 | 6.27 | 242.2 |
| 60b | 30 | 4.76 | 5.32 | 4.79 | 6.18 | 5.24 | 221.8 |
| 60c | 30 | 47 | 4.33 | 4.66 | 5.95 | 5.18 | 167.7 |
| 61a | 31 | 0.001 | 9.00 | 8.10 | 7.54 | 7.93 | 141.5 |
| 61b | 31 | 0.31 | 6.51 | 6.40 | 6.88 | 6.73 | 205.5 |
| 61c | 31 | 5 | 5.30 | 5.06 | 6.14 | 5.40 | 226.8 |
| 62a | 32 | 0.02 | 7.70 | 6.71 | 7.71 | 6.66 | 193.2 |
| 62b | 32 | 0.1 | 7.00 | 6.39 | 6.73 | 6.59 | 164.7 |
| 62c | 32 | 2 | 5.70 | 5.26 | 5.47 | 5.28 | 130.9 |
| 63a | 33 | 0.007 | 8.15 | 6.53 | 6.82 | 6.50 | 319.9 |
| 63b | 33 | 0.59 | 6.23 | 6.39 | 6.64 | 6.49 | 137.5 |
| 63c | 33 | >10a | 4.70 | 5.10 | 5.98 | 5.29 | 132.1 |
| 64a | 34 | 0.013 | 7.89 | 6.76 | 6.66 | 6.67 | 316.5 |
| 64b | 34 | 0.025 | 7.60 | 6.18 | 6.11 | 5.98 | 288.5 |
| 64c | 34 | 2.5 | 5.60 | 4.87 | 6.22 | 4.60 | 397.4 |
| 65a | 35 | 0.00021 | 9.68 | 5.13 | 5.85 | 5.36 | 339.9 |
| 65b | 35 | 1.9 | 5.72 | 4.93 | 6.20 | 5.41 | 173.6 |
| 65c | 35 | >25a | 4.30 | 4.64 | 5.38 | 5.45 | 146.6 |
| 66a | 36 | 0.13 | 6.89 | 5.61 | 6.46 | 5.92 | 289.0 |
| 66b | 36 | 6 | 5.22 | 4.72 | 6.15 | 4.83 | 276.7 |
| 66c | 36 | >100a | 3.70 | 4.18 | 5.21 | 3.80 | 137.7 |
| 67a | 37 | 0.001 | 9.00 | 5.45 | 6.12 | 5.39 | 109.6 |
| 67b | 37 | 19 | 4.72 | 4.77 | 5.90 | 4.76 | 103.1 |
| 67c | 37 | >50a | 4.00 | 4.52 | 5.74 | 4.63 | 108.8 |
| Ranking performance: | 8/8 | 6/8 | 7/8 | 5/8 | |||
For these cases, where activity was reported as > x, we used 2x.
Figure 7.

Scatter plots of predicted pActivity values vs. experimental pActivity values, where Activity is IC50 or Kd, for external test set. The plots for a) CoMFA, b) CoMSIA and c) CoMFA + HINT models based on the docking alignment are shown. The external test set contains 24 compounds from 8 scaffolds. For the CoMFA model, Apred = 0.395 Aexp + 3.013 (r2 = 0.474); for the CoMSIA model, Apred = 0.202 Aexp + 5.039 (r2 = 0.294); for the CoMFA+HINT model, Apred = 0.352 Aexp + 3.486 (r2 = 0.424).
We further analyzed the ranking performance of the models, i.e., whether the models could correctly rank the high, medium and low-activity compounds within the same scaffold. While ranking performance is a more gross evaluation of a model compared to r2external, it is perhaps more appropriate considering the implied uncertainties of mixing results from multiple assays. All three docking models performed very well (Table 3). The CoMFA model was best at this task with all 8 scaffolds correctly ranked; CoMFA+HINT ranked 7/8 of the scaffolds; and CoMSIA ranked 6/8 correctly. As a simple experiment, we also scored each of the external test set members against the HINT composite map by correlating map point characters and intensities; these scores (5/8 scaffolds ranked correctly) are also listed in Table 3. We also tested the semi-ligand-based and naïve alignment models with the external test set (Supporting Information, Table S3), and the results were, not surprisingly, less reliable. Validation against an external test set, with diverse and unique scaffolds, shows that we are able to use 3D-QSAR models built from an alignment based on docking to identify active CSI compounds.
Composite of features identified by 3D-QSAR analyses for CSIs
Two elements of the process are key: 1) the “atomic-scale” SAR we derived from a large collection of pyrrole analogues enabled us to build atomic scale models of the colchicine active site despite the poor resolution of the target X-ray structure; and 2) because the colchicine site is so flexible, only by using ensemble docking with the full set of ligand-bound αβ-tubulin crystal structures, was it possible to obtain self-consistent molecular models for all complexes.
We summarize in Figure 8 the features identified by 3D-QSAR analyses for the studied CSIs. Colchicine is shown for reference as the binding ligand in the site. An optimally favorable ligand would have four hydrogen bond acceptors (red) interacting with Val181α, Leu252β and Cys241β, a hydrogen bond donor (blue) interacting with Thr179α, and a hydrophobic skeleton with four hydrophobic centers (green circles) interacting with all three hydrophobic subpockets. Colchicine lacks the hydrogen bond acceptors for Leu252β and the hydrogen bond donor for Thr179α. Molecular designs for CSIs that pick up these pharmacophoric features might be expected to have higher activity.
Figure 8.

Summary of relevant features identified by SAR, hydropathic and 3D-QSAR analyses for colchicine site agents. Red spheres indicate hydrogen bond acceptors, blue spheres indicate hydrogen bond donors, and green ovals indicate hydrophobic regions. The black curves indicate the shape of the colchicine site with yellow zones indicating particularly relevant steric barriers.
Conclusions
Despite the significance of αβ-tubulin as a target for multiple classes of anticancer therapeutics, it has not yielded to traditional structure-based drug discovery/design because of the quite poor resolution of the available crystal structures. In particular, targeting of the colchicine site has produced many hundreds of compounds from multiple templates with modest to very good activity – however, no approved anticancer drugs – but these leads were found almost exclusively by screening. For the past several years, we have been investigating an extensive set of polysubstituted pyrroles15–18 that have microtubule depolymerization activity. The SAR developed with this set of compounds not only rationalized their activity, but also enabled the creation of atomic resolution docking models.15,16
In this work we expanded the pyrrole set of docking models with additional ligand scaffolds and imported these poses as an alignment rule for a panel of 3D-QSAR analyses. We also used two other alignment rules as controls – one a naïve alignment based only on the previously published pharmacophore of Nguyen et al.14 and the other a hybrid approach where only a prototype for each scaffold was docked while the other members of the family were fit to it. The docking-based, semi-ligand-based and naïve alignment approaches represent typical scenarios in 3D-QSAR modeling. The first is used when the structure of the receptor is known from experiments or homology modeling, so that the putative bioactive conformations of the ligands can be obtained from docking. The other limiting case scenario is when the structure of the receptor is unavailable or unsuitable for docking, so that the bioactive conformations of ligands have to be obtained from conformational search combined with energy minimization; the resulting conformations are overlaid based on substructure similarity and “experience”. These naïve ligand-based alignments are generally regarded as better for detecting the real “signals” from the real variance in the structures of the different ligands, while docking-based alignments may confuse the regression method with “noise”, i.e., the variation in the coordinates of a common substructure. However, for structurally diverse ligands, the docking-based approaches are more practical because the comparisons between different scaffolds are clearly indicated by their docked poses, while simple substructure similarity may not be enough for alignment using ligand-based approaches. In this particular study, we also adopted a semi-ligand-based alignment, where we took information from docking to align the different scaffolds, and thus treated all ligands of the same scaffold with alignment rules based on common substructures to remove “noise”. The resulting statistics from all three alignments are grossly comparable (Table 2), but the docking-based alignment modestly outperformed the others in q2, which was probably due to the fact that docking places individual functional groups more precisely in the pocket. In addition, the purported “noise” from the docking-based approach did not seem to affect the detection of “signal” as seen with satisfactory q2 and r2pred values.
The calculated 3D-QSAR contour maps revealed a pharmacophore model for CSIs with much more detail than previously available. Features in these maps indicating favorable and unfavorable interactions are directly relatable to specific site residues. To summarize, by combining the different approaches highlighted in this work, especially the classic medicinal chemistry tools of chemical synthesis and SAR, we were able to obtain detailed insights into how the CSI ligand structures affect activity and interactions with the colchicine site. We believe that these models will be useful guides for the design and optimization of new and active colchicine-site agents.
Experimental Section
Dataset selection
The compounds used in this study were reported by Mooberry and co-workers to be antitubulin agents.15–18,20–26 We set two criteria to select compounds for the study, including members of both the training and internal test sets: 1) to ensure the consistency of the activity measurements, we only selected compounds with reported antiproliferative IC50 values measured in MDA-MB-435 cancer cells using the SRB assay;14 and 2) we consider a compound as a valid colchicine-site binder for our modeling if: a) the compound showed inhibition of radiolabeled [3H]colchicine binding to tubulin, or b) its structurally-similar parent compound showed the inhibition of [3H]colchicine.
In general, cellular microtubule loss experiments evaluated in A-10 cells were performed to ensure that the antiproliferative activity for each compound corresponds to tubulin binding.15
The external test set was selected from scaffolds for CSIs reported by Nguyen et al.14 We chose scaffolds with at least gross similarity to colchicine in terms of pharmacophore features, and that were diverse. Three compounds, having high, medium and low activity, for each scaffold were extracted from the literature30–37 for the external test set. The reported quantitative measures of activities in these studies are not necessarily predictable with our models since different assays and conditions were used.
Preparation of models
X-ray crystal structure models of αβ-tubulin complexed with different ligands (pdbids: 1SA0, 1SA1, 3HKC, 3HKD and 3HKE; 3N2K and 3N2G are very similar to 3HKD and thus not used in the present study)9–11 were obtained from the RCSB protein data bank. Sybyl 8.151 was used to prepare these models for docking, and for building all-atom models of the small molecule ligands in the study. The latter, and hydrogens added to the protein models, were energy minimized with the Tripos forcefield with Gasteiger-Hückel charges to a gradient of 0.005 kcal mol−1 Å−1. In general, the preparation and docking procedure was the same as reported previously.15,16
Docking of small molecule ligands
The ligands were docked using GOLD 5.152 into the active site, which was defined as a 6.0 Å radius region around the complexed small molecules in the crystal structures. One hundred conformations were generated for each compound in the training and internal test sets, and twenty conformations for each compound in the external test set. The conformations were initially analyzed by GoldScore and further by rescoring with HINT.29,53 To select the final “active” conformation, we considered binding to all five receptor structures and chose the conformation with both a high HINT score and a high degree of similarity to the conformation of the complexed (crystal) small ligand. It should be noted that, because the resolutions of the tubulin crystal structures are so poor, only around 3.6 Å, the crystallographic models for the bound ligands are only guides to their actual conformations. The ligand conformations, as docked, were assumed to be the bioactive conformations for the compounds.
Ligand alignment models
Three alignment models were applied to the training and internal test sets, and the external test set as well. The first alignment model was simply to use the docked conformations for all compounds as generated above. The second was to use the docked conformations of the most active compounds for different scaffolds as prototypes. The remaining compounds from each template set were superimposed to these prototypes according to common structures. Since the bioactive conformations for this alignment were derived using information from docked poses, we are characterizing it as “semi-ligand-based”. The third alignment was naïve in that we energy minimized all compound prototypes and superimposed each on colchicine’s conformation as much as possible with the “Fit Atoms” function of Sybyl, based on the previously proposed pharmacophore model.14 This alignment is more truly ligand-based, although it could be argued that Nguyen’s pharmacophore was constructed with structural knowledge.
3D-QSAR modeling and validation
Sybyl 8.1 was used to perform the 3D-QSAR analyses. Two complementary methods were used: the Comparative Molecular Field Analysis (CoMFA)27 and the Comparative Molecular Similarity Indices Analysis (CoMSIA).28 For CoMFA field generation, we used the standard steric and electrostatic fields and the HINT hydropathic (hydrophobic/polar) field29,41 (vide infra). The steric, electrostatic, hydrophobic, hydrogen bond donor and hydrogen bond acceptor fields were used for CoMSIA. Gasteiger-Hückel charges were assigned to all compounds. Except for HINT fields, the grid resolutions were set to 1.0 Å. Other field settings were default. Partial least squares (PLS) regression methods, as installed in Sybyl, were used to derive models in this study. Leave-one-out (LOO) cross-validation with the sample-distance PLS (SAMPLS) algorithm was used to identify the optimum number of components. LOO approaches evaluate the predictability and over-fitting of a regression model with the crossvalidated correlation coefficient q2. We determined the optimum number of components to be the smallest number below 8 that gave the largest value of q2. The non-cross-validated models were built with the optimum number of components over the entire training set and evaluated with the correlation coefficient (r2), standard error of estimate (SEE) and F-test value. The resulting models were further validated using a test set of 9 compounds from the same scaffolds that had been set aside. The predictive r2 (r2pred) indicates the correlation between the predicted and experimental activities of this internal test set.
HINT fields and composite HINT maps
As described above, HINT was used as a rescoring function for docking and the hydrophobic/polar fields of HINT were combined with the steric and electrostatic fields of CoMFA for 3D-QSAR analyses with options and conditions reported earlier.41,54 The HINT (Hydropathic INTeractions) scoring function29 evaluates atom-atom interactions using a set of parameters derived from the solvation partition coefficients, LogP, of small molecules measured in the 1-octanol/water system. Numerous studies have validated the HINT scoring function in various biological environments.55
In addition to its scoring functions, the HINT program calculates both hydrophobic/polar and (Lewis) acid/base field maps.55 The field value A of each grid point (t) is given by
At = Σ ai Si Rit
where ai and Si are the hydrophobic atom constant and solvent accessible surface area, respectively, for the atom i, and Rit is a function of the distance between the atom i and the test atom (grid map point) t. We used a resolution of 0.5 Å to calculate all HINT field maps in this study.
The two types of HINT maps, hydrophobic/polar and Lewis acid/base, were generated as a pair for all the compounds in the training and test sets. We generated an overall composite map by a linear combination of the individual maps, where each grid point of the map was weighted for the compounds’ activity values (IC50). In this study, we applied 0.1 (µM)/IC50 (µM) as the weight for each compound. This is similar in many respects to the LOCKSMITH concept that we described in 1992.50 To score external test set molecules against the composite map pair, we multiplied the individual by the composite on a point-by-point basis: for the hydrophobic/polar map, each hydrophobic*hydrophobic instance was scored positive, each hydrophobic*polar instance was scored negative and each polar*polar instance was ignored; for the Lewis acid/base map each acid*acid and base*base instance was scored negative and each acid*base instance was scored positive. The total score is the sum of all these point scores.
Supplementary Material
Acknowledgments
This work was partially supported by NIH R01 CA135043 (to D. A. Gewirtz, VCU), NIH R15 CA067236 (to JTG) and the President's Council Research Excellence Award (to SLM).
Footnotes
Associated Content
Supporting Information
Additional tables of experimental and predicted activity for training/test set and external test set compounds. Additional 3D-QSAR stddev*coeff maps. Docked models for external test set molecules. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Jordan MA, Wilson L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.Dumontet C, Jordan MA. Microtubule-Binding Agents: A Dynamic Field of Cancer Therapeutics. Nat. Rev. Drug Discovery. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett MJ, Barakat K, Huzil JT, Tuszynski J, Schriemer DC. Discovery and Characterization of the Laulimalide-Microtubule Binding Mode by Mass Shift Perturbation Mapping. Chem. Biol. 2010;17:725–734. doi: 10.1016/j.chembiol.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 4.Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs That Target Dynamic Microtubules: A New Molecular Perspective. Med. Res. Rev. 2011;31:443–481. doi: 10.1002/med.20242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu Y, Chen J, Xiao M, Li W, Miller DD. An Overview of Tubulin Inhibitors that Interact with the Colchicine Binding Site. Pharm. Res. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sève P, Dumontet C. Is Class III Beta-tubulin a Predictive Factor in Patients Receiving Tubulinbinding Agents? Lancet Oncol. 2008;9:168–175. doi: 10.1016/S1470-2045(08)70029-9. [DOI] [PubMed] [Google Scholar]
- 7.Stengel C, Newman SP, Leese MP, Potter BV, Reed MJ, Purohit A. Class III Betatubulin Expression and in vitro Resistance to Microtubule Targeting Agents. Br. J. Cancer. 2010;102:316–324. doi: 10.1038/sj.bjc.6605489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Liu T, Dong X, Hu Y. Recent Development and SAR Analysis of Colchicine Binding Site Inhibitors. Mini-Rev. Med. Chem. 2009;9:1174–1190. doi: 10.2174/138955709789055234. [DOI] [PubMed] [Google Scholar]
- 9.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-like Domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 10.Dorleans A, Gigant B, Ravelli RB, Mailliet P, Mikol V, Knossow M. Variations in the Colchicine-binding Domain Provide Insight into Structural Switch of Tubulin. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13775–13779. doi: 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbier P, Dorleans A, Devred F, Sanz L, Allegro D, Alfonso C, Knossow M, Peyrot V, Andreu JM. Stathmin and Interfacial Microtubule Inhibitors Recognize a Naturally Curved Conformation of Tubulin Dimers. J. Biol. Chem. 2010;285:31672–31681. doi: 10.1074/jbc.M110.141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborti S, Chakravarty D, Gupta S, Chatterji BP, Dhar G, Poddar A, Panda D, Chakrabarti P, Ghosh DS, Bhattacharyya B. Discrimination of Ligands with Different Flexibilities Resulting from the Plasticity of the Binding Site in Tubulin. Biochemistry. 2012;51:7138–7148. doi: 10.1021/bi300474q. [DOI] [PubMed] [Google Scholar]
- 13.Massarotti A, Coluccia A, Silvestri R, Sorba G, Brancale A. The Tubulin Colchicine Domain: a Molecular Modeling Perspective. ChemMedChem. 2012;7:33–42. doi: 10.1002/cmdc.201100361. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, Wipf P, Hamel E, Gussio R. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J. Med. Chem. 2005;48:6107–6116. doi: 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- 15.Da C, Telang N, Gupton JT, Mooberry SL, Kellogg GE. Developing Novel C-4 Analogues of Pyrrole-Based Antitubulin Agents: Weak but Critical Hydrogen Bonding in the Colchicine Site. Med. Chem. Commun. 2013;4:417–421. doi: 10.1039/C2MD20320K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Da C, Telang N, Barelli P, Jia X, Gupton JT, Mooberry SL, Kellogg GE. Pyrrole-Based Antitubulin Agents: Two Distinct Binding Modalities are Predicted for C-2 Analogues in the Colchicine Site. ACS Med. Chem. Lett. 2012;3:53–57. doi: 10.1021/ml200217u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tripathi A, Fornabaio M, Kellogg GE, Gupton JT, Gewirtz DA, Yeudall WA, Vega NE, Mooberry SL. Docking and Hydrophobic Scoring of Polysubstituted Pyrrole Compounds with Antitubulin Activity. Bioorg. Med. Chem. 2008;16:2235–2242. doi: 10.1016/j.bmc.2007.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mooberry SL, Weiderhold KN, Dakshanamurthy S, Hamel E, Banner EJ, Kharlamova A, Hempel J, Gupton JT, Brown ML. Identification and Characterization of a New Tubulin-Binding Tetrasubstituted Brominated Pyrrole. Mol. Pharmacol. 2007;72:132–140. doi: 10.1124/mol.107.034876. [DOI] [PubMed] [Google Scholar]
- 19.Dalyot-Herman N, Delgado-Lopez F, Gewirtz DA, Gupton JT, Schwartz EL. Interference with endothelial cell function by JG-03-14, an agent that binds to the colchicine site on microtubules. Biochem. Pharmacol. 2009;78:1167. doi: 10.1016/j.bcp.2009.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biggers JW, Nguyen T, Di X, Gupton JT, Henderson SC, Emery SM, Alotaibi M, White KL, Jr, Brown R, Almenara J, Gewirtz DA. Autophagy, cell death and sustained senescence arrest in B16/F10 melanoma cells and HCT-116 colon carcinoma cells in response to the novel microtubule poison, JG-03-14. Cancer Chemother. Pharmacol. 2013;71:441–455. doi: 10.1007/s00280-012-2024-6. [DOI] [PubMed] [Google Scholar]
- 21.Lee M, Brockway O, Dandavati A, Tzou S, Sjoholm R, Satam V, Westbrook C, Mooberry SL, Zeller M, Babu B, Lee M. A Novel Class of trans-Methylpyrazoline Analogues of Combretastatins: Synthesis and in-vitro Biological Testing. Eur. J. Med. Chem. 2011;46:3099–3104. doi: 10.1016/j.ejmech.2011.03.064. [DOI] [PubMed] [Google Scholar]
- 22.Lee L, Robb LM, Lee M, Davis R, Mackay H, Chavda S, Babu B, O'Brien EL, Risinger AL, Mooberry SL, Lee M. Design, Synthesis, and Biological Evaluations of 2,5-Diaryl-2,3-Dihydro-1,3,4-Oxadiazoline Analogues of Combretastatin-A4. J. Med. Chem. 2010;53:325–334. doi: 10.1021/jm901268n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babu B, Lee M, Lee L, Strobel R, Brockway O, Nickols A, Sjoholm R, Tzou S, Chavda S, Desta D, Fraley G, Siegfried A, Pennington W, Hartley RM, Westbrook C, Mooberry SL, Kiakos K, Hartley JA, Lee M. Acetyl analogs of combretastatin A-4: synthesis and biological studies. Bioorg. Med. Chem. 2011;19:2359–2367. doi: 10.1016/j.bmc.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 24.Gangjee A, Pavana RK, Li W, Hamel E, Westbrook C, Mooberry SL. Novel Water-Soluble Substituted Pyrrolo[3,2-d]pyrimidines: Design, Synthesis, and Biological Evaluation as Antitubulin Antitumor Agents. Pharm. Res. 2012;29:3033–3039. doi: 10.1007/s11095-012-0816-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gangjee A, Zhao Y, Hamel E, Westbrook C, Mooberry SL. Synthesis and Biological Activities of (R)- and (S)-N-(4-Methoxyphenyl)-N,2,6-Trimethyl-6,7-Dihydro-5HCyclopenta[ d]pyrimidin-4-Aminium Chloride as Potent Cytotoxic Antitubulin Agents. J. Med. Chem. 2011;54:6151–6155. doi: 10.1021/jm2007722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gangjee A, Zhao Y, Lin L, Raghavan S, Roberts EG, Risinger AL, Hamel E, Mooberry SL. Synthesis and Discovery of Water-Soluble Microtubule Targeting Agents that Bind to the Colchicine Site on Tubulin and Circumvent Pgp Mediated Resistance. J. Med. Chem. 2010;53:8116–8128. doi: 10.1021/jm101010n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cramer RD, III, Patterson DE, Bunce JD. Comparative Molecular Field Analysis (CoMFA). 1Effect of Shape on Binding of Steroids to Carrier Proteins. J. Am. Chem. Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 28.Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 29.Kellogg GE, Abraham DJ. Hydrophobicity: Is LogPo/w More than the Sum of Its Parts? Eur. J. Med. Chem. 2000;35:651–661. doi: 10.1016/s0223-5234(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 30.Andreu JM, Perez-Ramirez B, Gorbunoff MJ, Ayala D, Timasheff SN. Role of the colchicine ring A and its methoxy groups in the binding to tubulin and microtubule inhibition. Biochemistry. 1998;37:8356–8368. doi: 10.1021/bi9728553. [DOI] [PubMed] [Google Scholar]
- 31.Gaukroger K, Hadfield JA, Lawrence NJ, Nolan S, McGown AT. Structural requirements for the interaction of combretastatins with tubulin: how important is the trimethoxy unit? Org. Biomol. Chem. 2003;1:3033–3037. doi: 10.1039/b306878a. [DOI] [PubMed] [Google Scholar]
- 32.Chernysheva NB, Tsyganov DV, Philchenkov AA, Zavelevich MP, Kiselyov AS, Semenov RV, Semenova MN, Semenov VV. Synthesis and comparative evaluation of 4- oxa- and 4-aza-podophyllotoxins as antiproliferative microtubule destabilizing agents. Bioorg. Med. Chem. Lett. 2012;22:2590–2593. doi: 10.1016/j.bmcl.2012.01.128. [DOI] [PubMed] [Google Scholar]
- 33.Liou JP, Chang YL, Kuo FM, Chang CW, Tseng HY, Wang CC, Yang YN, Chang JY, Lee SJ, Hsieh HP. Concise synthesis and structure-activity relationships of combretastatin A-4 analogues, 1-aroylindoles and 3-aroylindoles, as novel classes of potent antitubulin agents. J. Med. Chem. 2004;47:4247–4257. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]
- 34.La Regina G, Edler MC, Brancale A, Kandil S, Coluccia A, Piscitelli F, Hamel E, De Martino G, Matesanz R, Díaz JF, Scovassi AI, Prosperi E, Lavecchia A, Novellino E, Artico M, Silvestri R. Arylthioindole inhibitors of tubulin polymerization. 3. Biological evaluation, structure-activity relationships and molecular modeling studies. J. Med. Chem. 2007;50:2865–2874. doi: 10.1021/jm061479u. [DOI] [PubMed] [Google Scholar]
- 35.Ducki S, Rennison D, Woo M, Kendall A, Chabert JF, McGown AT, Lawrence NJ. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: synthesis and biological evaluation of antivascular activity. Bioorg. Med. Chem. 2009;17:7698–7710. doi: 10.1016/j.bmc.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 36.Beutler JA, Hamel E, Vlietinck AJ, Haemers A, Rajan P, Roitman JN, Cardellina JH, 2nd, Boyd MR. Structure-activity requirements for flavone cytotoxicity and binding to tubulin. J. Med. Chem. 1998;18:41, 2333–2338. doi: 10.1021/jm970842h. [DOI] [PubMed] [Google Scholar]
- 37.Shih H, Deng L, Carrera CJ, Adachi S, Cottam HB, Carson DA. Rational design, synthesis and structure-activity relationships of antitumor (E)-2-benzylidene-1-tetralones and (E)-2-benzylidene-1-indanones. Bioorg. Med. Chem. Lett. 2000;10:487–490. doi: 10.1016/s0960-894x(00)00032-9. [DOI] [PubMed] [Google Scholar]
- 38.DePristo MA, de Bakker PIW, Blundell TL. Heterogeneity and inaccuracy in protein structures solved by X-ray crystallography. Structure. 2004;12:831–838. doi: 10.1016/j.str.2004.02.031. [DOI] [PubMed] [Google Scholar]
- 39.Koparde VN, Scarsdale JN, Kellogg GE. Applying an Empirical Hydropathic Forcefield in Refinement May Improve Low-Resolution Protein X-Ray Crystal Structures. PLoS One. 2011;6:e15920. doi: 10.1371/journal.pone.0015920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Löwe J, Li H, Downing KH, Nogales E. Refined structure of alpha beta-Tubulin at 3.5 A Resolution. J. Mol. Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 41.Kellogg GE, Semus SF, Abraham DJ. HINT - A New Method of Empirical Hydrophobic Field Calculation for CoMFA. J. Computer-Aided Mol. Design. 1991;5:545–552. doi: 10.1007/BF00135313. [DOI] [PubMed] [Google Scholar]
- 42.Kellogg GE. Finding Optimum Field Descriptors for 3D QSAR. Med. Chem. Res. 1997;7:417–427. [Google Scholar]
- 43.Oprea TI, Waller CL, Marshall GR. 3D-QSAR of human immunodeficiency virus (I) protease inhibitors. III. Interpretation of CoMFA results. Drug Des. Discov. 1994;12:29–51. [PubMed] [Google Scholar]
- 44.Waller CL, Oprea TI, Giolitti A, Marshall GR. Three-dimensional QSAR of human immunodeficiency virus (I) protease inhibitors. 1. A CoMFA study employing experimentally-determined alignment rules. J. Med. Chem. 1993;36:4152–4160. doi: 10.1021/jm00078a003. [DOI] [PubMed] [Google Scholar]
- 45.Cho SJ, Garsia ML, Bier J, Tropsha A. Structure-based alignment and comparative molecular field analysis of acetylcholinesterase inhibitors. J. Med. Chem. 1996;39:5064–5071. doi: 10.1021/jm950771r. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Z, Madura JD. CoMFA 3D-QSAR Analysis of HIV-1 RT Nonnucleoside Inhibitors, TIBO Derivatives Based on Docking Conformation and Alignment. J. Chem. Inf. Comput. Sci. 2004;44:2167–2178. doi: 10.1021/ci049893v. [DOI] [PubMed] [Google Scholar]
- 47.Yuan W, Luan L-B, Li Y-N. CoMFA 3D-QSAR Analysis of Epothilones Based on Docking Conformation and Alignment. Ch. J. Chem. 2007;25:453–460. [Google Scholar]
- 48.Clark RD. A ligand’s-eye view of protein binding. J. Comput. Aided. Mol. Des. 2008;22:507–522. doi: 10.1007/s10822-008-9177-8. [DOI] [PubMed] [Google Scholar]
- 49.Scior T, Medina-Franco JL, Do Q-T, Martínez-Mayorga K, Yunes Rojas JA, Bernard P. How to Recognize and Workaround Pitfalls in QSAR Studies: A Critical Review. Curr. Med. Chem. 2009;16:4297–4313. doi: 10.2174/092986709789578213. [DOI] [PubMed] [Google Scholar]
- 50.Kellogg GE, Abraham DJ. KEY, LOCK, and LOCKSMITH: complementary hydropathic map predictions of drug structure from a known receptor-receptor structure from known drugs. J. Mol. Graph. 1992:10, 212–217, 226. doi: 10.1016/0263-7855(92)80070-t. [DOI] [PubMed] [Google Scholar]
- 51.SYBYL 8.1. 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: Tripos International; [Google Scholar]
- 52.Jones G, Willett P, Glen R. Molecular Recognition of Receptor Sites Using a Genetic Algorithm with a Description of Desolvation. J. Mol. Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 53.Spyrakis F, Amadasi A, Fornabaio M, Abraham DJ, Mozzarelli A, Kellogg GE, Cozzini P. The Consequences of Scoring Docked Ligand Conformations using Free Energy Correlations. Eur. J. Med. Chem. 2007;42:921–933. doi: 10.1016/j.ejmech.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 54.Sarkar A, Anderson KC, Kellogg GE. Computational Analysis of Structure-Based Interactions and Ligand Properties Can Predict Efflux Effects on Antibiotics. Eur. J. Med Chem. 2012;52:98–110. doi: 10.1016/j.ejmech.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarkar A, Kellogg GE. Hydrophobicity – Shake Flasks, Protein Folding and Drug Discovery. Curr. Top. Med. Chem. 2010;10:67–83. doi: 10.2174/156802610790232233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.